

Abstract
A Saccharomyces cerevisae microarray expression study indicated that an ORF, YER044C, now designated ERG28, was strongly coregulated with ergosterol biosynthesis. Disruption of the ERG28 gene results in slow growth and accumulation of sterol intermediates similar to those observed in erg26 and erg27 null strains, suggesting that the Erg28p may interact with Erg26p and/or Erg27p. In this study, a peptide from human hemagglutinin protein (HA) epitope tag was added to ERG26 and ERG27 genes, and a Myc tag was added to the ERG28 gene to detect interactions between Erg28p and Erg26p/Erg27p. Differential centrifugation showed that Erg26p, Erg27p, and Erg28p are all membrane-associated proteins. Green fluorescent protein–fusion protein localization studies showed that Erg26p, Erg27p, and Erg28p are all located in the endoplasmic reticulum. Solubilized membrane protein coimmunoprecipitation studies using rabbit anti-Erg25p indicated that Erg25p coimmunoprecipitates with both Erg27p and Erg28p. Erg28p was also shown to reciprocally coimmunoprecipitate with Erg27p. However, no coimmunoprecipitation was observed with Erg26p, most likely because of the poor solubilization of this protein. Sucrose gradient ultracentrifugation studies suggested that Erg25p/Erg26p/Erg27p/Erg28p, along with other proteins in sterol biosynthesis, might form a complex between 66 and 200 kDa. Using an anti-HA column with Erg27p-HA and Erg26p-HA as target proteins, a complex containing Erg25p/Erg26p/Erg27p/Erg28p was identified. Thus, we suggest that Erg28p works as a transmembrane scaffold to tether Erg27p and possibly other C-4 demethylation proteins (Erg25p, Erg26p), forming a demethylation complex in the endoplasmic reticulum.
C-4 demethylation of the sterol intermediate, 4,4-dimethylzymosterol, is required in the plant, animal, and fungal sterol biosynthetic pathways. In Saccharomyces cerevisiae, ERG25 encodes the C-4 oxidase, ERG26 encodes the C-4 dehydrogenase/decarboxylase, and ERG27 encodes the C-3 keto-reductase (Fig. 1). All three genes are essential (1–3). Using a microarray expression profile (4), a gene encoding a 148-aa protein with two transmembrane domains (5) was found to be highly coregulated with other ergosterol biosynthetic genes. An ERG28 deletion was found to be slow growing, and gas–chromatographic analyses indicated significant accumulation of sterol precursors that were identified as carboxylic acid sterol and 3-keto sterol intermediates normally found in ERG26 and ERG27 mutant strains. Loss of the Erg28p does not lead to loss of end-product sterol but reduces ergosterol levels to approximately one-third of the wild-type value. It was suggested that Erg28p might anchor the C-3 sterol dehydrogenase/C-4 decarboxylase (Erg26p) and 3-keto-reductase (Erg27p) enzymes to the endoplasmic reticulum (ER) (5), because both of these enzymes lacked transmembrane domains. Other ergosterol biosynthetic enzymes lacking transmembrane domains such as Erg1p, Erg6p, and Erg7p are found in lipid droplet (6).
Figure 1.

Sterol structures and reactions involved in removal of the C-4 α-methyl group by the ERG25, ERG26, and ERG27 enzymes (2). (Copyright 1996, National Academy of Sciences, U.S.A.)
Baudry et al. (7) have studied the interaction of Erg25p with Erg26p and Erg27p by using a yeast two-hybrid system and demonstrated that Erg25p interacted with both Erg26p and Erg27p, but Erg26p and Erg27p did not interact. These authors suggested that Erg25p might not only work as C-4 methyloxidase but also might anchor both Erg26p and Erg27p. In light of the finding that loss of Erg28p leads to the accumulation of C-4 demethylation intermediates, we hypothesized that Erg28p may tether all three C-4 demethylation enzymes (Erg25p, Erg26p, and Erg27p) to the ER and ensure that all three enzymes worked as a complex (5).
By using fluorescent microscopy, Erg4p (8), Erg26p (7), and Erg27p (7) green fluorescent protein (GFP) fusion proteins were located in the ER; Li and Kaplan, using immunofluorescence, demonstrated that Erg25p localized both in the ER and the plasma membrane (9). These results are in accordance with microsomal enzymatic assay location studies (6, 10–13). Erg25p has a predicted transmembrane domain and a KKXX Golgi-to-ER retrieval signal (2); however, no transmembrane domains are predicted in Erg26p or Erg27p (www.cbs.dtu.dk/service/TMHMM), and how they localize in the ER or microsomes is still not known.
In this study, we use epitope tag techniques to demonstrate protein–protein interactions between Erg25p, Erg26p, Erg27p, and Erg28p. Our results indicate that Erg26p, Erg27p, and Erg28p are ER membrane proteins, as determined by differential centrifugation and GFP fusion localization studies. Erg27p and Erg28p coimmunoprecipitate reciprocally. However, we did not find any interactions between Erg26p and Erg28p. Erg25p was also found to interact with Erg27p and Erg28p. Sucrose gradient ultracentrifugation showed that Erg27p/Erg28p and Erg26p/Erg28p are located in a protein complex that sediments between 66 and 200 kDa. Further studies using a mouse anti-HA (HA, peptide from human hemagglutinin protein) column enabled us to isolate a complex containing all four C-4 demethylation proteins (Erg25p/Erg26p/Erg27p/Erg28p). Thus, we conclude that Erg28p works as a scaffold to tether Erg27p with Erg25p and other 4,4-demethylation-related enzymes to form a demethylation enzyme complex in the ER.
Materials and Methods
Yeast Strains and Growth Conditions.
The following yeast strains have been described previously. R712 (MATa erg28∷kanMX4 his3Δ1 leu2Δ0 met15Δ0 ura3Δ0) is an erg28 deletion strain (5); SDG100 (MATα upc2 ade2 his− ura3–52 erg27–1) is an erg27 gene point mutation strain that requires ergosterol for growth (1); SDG200 (MATa ade− his− leu2 erg26Δ∷TRP1 trp1∷hisG Δhem1 ura3–52) is a hem1 erg26-disrupted strain that requires ergosterol and unsaturated fatty acids for viability (3). These strains were transformed with epitope-tagged constructs of ERG28, ERG27, and ERG26. Yeast strains prototrophic for ergosterol were grown at 30°C in yeast extract, peptone, and glucose (YPD) medium (1% yeast extract/2% Bacto-peptone/2% glucose) or YPD medium supplemented with ergosterol [0.002%, dissolved in ethanol/Tween 80 (1:1, vol/vol)] grown anaerobically for ergosterol auxotrophs. Yeast transformants containing epitope-tagged versions of ERG28 and ERG27, and ERG26 plasmids were grown on drop-out synthetic medium containing 0.67% yeast nitrogen base, 2% glucose, amino acids, and nitrogen base (supplements at 0.8%, Bio101, Vista, CA). SMO2826 is an erg28-disrupted strain with plasmids containing Myc-ERG28 and ERG26-HA epitopes, and SMO2827 is an erg28-disrupted strain with plasmids containing Myc-ERG28 and ERG27-HA epitopes. The erg27 erg28 (SD2728) or erg26 erg28 (SD2628) doubly disrupted strains were obtained by mating representative single mutants.
Primer Design and PCR Cloning.
Primers for PCR cloning and epitope tagging of ERG26, ERG27, and ERG28 genes are listed in Table 1. HA- (YPYDVPDYA, peptide from human influenza hemagglutinin protein) and Myc- (SEQKLISEEDL, human c-MYC gene protein epitope) tagged sequences were incorporated into the PCR primer either at the N or C terminus of the target gene. For ERG26 and ERG27 clones, EcoRI and SalI digestion sites were added to the forward and reverse primers, respectively. For the ERG28 clone, BamHI and ClaI digestion sites were added to the forward and reverse primers, respectively.
Table 1.
Primers used for PCR cloning and epitope tagging of ERG26, ERG27, and ERG28 genes
| Genes | Designed primers (5′-3′) |
|---|---|
| ERG26 | ERG26-F: ccGAATTCatgtcaaagatagattcagtttt |
| ERG26-F-HA: ccGAATTCATGTACCCATACGATGTTCCAGATTACGCTatgtcaaagatagattcagtttt | |
| ERG26-R: gcGTCGACttacaaaccttcgtccatcc | |
| ERG26-R-HA: cgcGTCGACTTAAGCGTAATCTGGAACATCGTATGGGTAcaaaccttcgtccatccag | |
| ERG27 | ERG27-F: ccGAATTCatgaacaggaaagtagctatc |
| ERG27-F-HA: ccGAATTCATGTACCCATACGATGTTCCAGATTACGCTatgaacaggaaagtagctatc | |
| ERG27-R: gcGTCGACttaaatgggggttctagtttca | |
| ERG27-R-HA: cgcGTCGACTTAAGCGTAATCTGGAACATCGTATGGGTAaatgggggttctagtttcaac | |
| ERG28 | ERG28-F: ccGGATCCatgttcagcctacaagacgtaata |
| ERG28-F-Myc: ccGGATCCATGTCTGAACAAAAATTGATTTCTGAAGAAGATTTGatgttcagcctacaagacgtaata | |
| ERG28-R: cgcATCGATttaccaagcaacaccagtgtagt | |
| ERG28-R-Myc: cgcATCGATTTACAAATCTTCTTCAGAAATCAATTTTTGTTCAGAccaagcaacaccagtgtagtatt | |
| ERG28-F: ccGAATTCatgttcagcctacaagacgtaata |
F and R indicate forward and reverse primers directly. Underlined sequences represent engineered restriction sites. Nucleotides in capitals represent epitope-coding sequences.
PCR was performed in a Perkin–Elmer 2400 thermocycler by using Pfu DNA polymerase to obtain three constructs for each gene—no epitope tag, tag at the N terminus and tag at the C terminus. Molecular cloning and plasmid manipulations were performed according to standard protocols (14). Digested PCR fragments and plasmid vectors p423GPD (HIS3 marker) and p426GPD (URA3 marker) (15) were purified from agarose gel by using gel purification kit (QIAEX II Gel Extraction System, Qiagen, Chatsworth, CA), then ligated and transformed. Cloned and epitope-tagged genes ERG26 (pERG26, no epitope tag; pHA-ERG26; pERG26-HA), ERG27 (pERG27, no tag; pHA-ERG27; pERG27-HA), and ERG28 (pERG28; no tag; pMyc-ERG28; pERG28-Myc) were obtained. Sequencing of each clone (after subcloning into a pRS306 vector; ref. 16) was performed at the Indiana University School of Medicine Biochemistry Biotechnology Facility (Indianapolis, IN) by using a Perkin–Elmer Biosystems model Applied Biosystems 373 automated DNA sequencer and T3 and T7 promoter primers. ERG26, ERG27, and ERG28 genes were also tagged at the N terminus with the green fluorescent gene harboring a S65A mutation (17). A 735-bp BamHI-EcoRI GFP fragment was subcloned from a pCB74-GFP-ERG27 plasmid and ligated into p416GPD and p413ADH vectors. PCR-amplified ERG26 (EcoRI-SalI), ERG27 (EcoRI-SalI), and ERG28 (EcoRI-ClaI) genes were then cloned into these vectors to obtain GFP-ERG fusion plasmids pGFP-ERG26, pGFP-ERG27, and pGFP-ERG28.
Yeast and Escherichia coli (DH5α) strains were transformed by standard methods (18). The DH5α was grown in Luria–Bertani medium supplemented with ampicillin (50 μg/ml) at 37°C.
Epitope-Tagged Protein Screen.
Yeast cells (1.5 ml) transformed with epitope-tagged plasmids were pelleted after 20–24 h growth and resuspended in 100 μl of 1 × SDS/PAGE loading buffer (0.0625 M Tris⋅HCl, pH 6.8/2% SDS/10% glycerol/1% β-mercaptoethanol/0.001% bromophenol blue, 1× SSB). Cell suspensions were vortexed, boiled for 5 min, and then cooled to room temperature, pelleted, and the supernatant was used for loading onto SDS/PAGE gels and subsequent transfer to nitrocellulose membranes according to standard procedures (14). Nitrocellulose membranes were blocked by using 5% nonfat milk/PBS-T buffer for 1 h and then incubated with anti-HA-peroxidase (clone 12CA5, Roche Diagnostics, Indianapolis, IN) for Erg26p-HA and Erg27p-HA, or anti-Myc-peroxidase (clone 9E10, Roche Diagnostics) for Erg28p-Myc-tagged protein detection.
Differential Centrifugation and Preparation of Microsomes.
The microsome buffer (50 mM Tris⋅HCl, pH 7.5/1 mM EGTA/1 mM β-mercaptoethanol/1 mM phenylmethylsulfonyl fluoride/2 μg/ml of pepstatin A) was prepared according to Gable et al. (19). Cells in late-log or early-stationary phase were pelleted, washed with H2O, and resuspended in microsomal buffer at 2 ml/g of wet weight. Glass beads were added to just below the meniscus, and cells were vortexed six times (1 min each time) with cooling on ice (1 min) between each vortexing. Unbroken cells, beads, and debris were removed by centrifugation (3,000 × g for 10 min). The low-speed supernatant was taken as total protein and was centrifuged at 10,000 × g for 10 min to provide the 10,000 × g (10-K) pellet. The supernatant was again centrifuged at 20,000 × g for 20 min to provide the 20-K microsomal pellet. The supernatant was then centrifuged at 60,000 × g and 130,000 × g for 40 min and 60 min, respectively, to obtain 60- and 130-K pellets. The last supernatant fraction was retained as soluble protein. All of the microsomal pellets were resuspended in microsomal buffer in the same volume as the last soluble protein portion. Protein concentrations were determined by using the Bio-Rad protein assay and BSA as standard. Equal protein concentrations (20 μg/lane) were loaded onto the SDS/PAGE gel.
GFP Fusion Protein Microscopic Observation.
Cells were harvested, pelleted, and washed once in water. One microliter of the residual concentrated cell suspension was placed on a coverslip and covered with a thin sheet of 0.8% agarose (20). Cells immobilized between coverslip and agarose were mounted on a glass slide and observed by using either Nikon E600 fluorescence microscope (Hg lamp; ×100) connected to SPOT charge-coupled device camera or with Carl Zeiss confocal laser scanning microscope (Xe laser; ×100).
Membrane Protein Solubilization and Immunoprecipitation.
Membranes were solubilized at 1 mg/ml with sucrose monolaurate (SML, 2 mM), N-octyl-β-d-glucopyranoside (25 mM) or Triton X-100 (1%) for 20 min at room temperature and then centrifuged at 30,000 × g (30 min). Supernatant and resuspended unsolubilized pellets were applied to a SDS/PAGE gel to determine the most efficient agent to solubilize Erg26p, Erg27p, and Erg28p. The immunoprecipitation procedure was based on Gable et al. (19). Solubilized membrane protein (300 μl) was diluted one time with microsome buffer, incubated with 6 μl of the precipitating antibody, anti-HA (Y-11), and anti-Myc (A-14) (Santa Cruz Biotechnology) for 2 h. Then 40 μl of protein G-agarose (Roche Diagnostics) was added, and samples were further incubated for 2 h. The precipitates were washed three times with 500 μl of 50 mM Hepes (pH 7.5), resuspended in 150 μl of 1 × SDS/PAGE sample loading buffer, and a 15-μl sample was subjected to SDS/PAGE and immunoblot analysis.
Erg26p, Erg27p, and Erg28p Comigrate as a Complex.
Membrane pellets were resuspended at 5 mg/ml and subjected to sucrose gradient (10–60% wt/vol) ultracentrifugation for 6 h at 70,000 rpm at 4°C (NVT90 rotor) to purify Erg27p/Erg28p- and Erg26p/Erg28p-enriched membranes. Fractions enriched in Erg26p, Erg27p, and Erg28p were collected, diluted three times with microsomal buffer, and pelleted again at 70,000 rpm. Pellets were resuspended in microsomal buffer and solubilized by using 2 mM SML at 1 mg/ml. Solubilized membrane proteins were subjected to 30–60% (wt/vol) sucrose gradient ultracentrifugation for 4 h at 70,000 rpm 4°C, and protein fractions (300 μl) were collected. BSA (66 kDa), β-amylase (200 kDa), apoferritin (443 kDa), and thyroglobulin (669 kDa) protein complexes (Sigma) were used as ultracentrifugation markers to determine the size of the Erg25p/Erg26p/Erg27p/Erg28p complex.
Copurification of the Erg25p/Erg26p/Erg27p/Erg28 Complex by Using an Anti-HA Matrix.
Solubilized membrane protein extracted from SMO4701 (SD2728 containing pERG27-HA and pMyc-ERG28) and SMO5001 (SD2628 containing pERG26-HA and pMyc-ERG28) were coimmunoprecipitated overnight by using mouse anti-HA matrix beads. The beads were washed, and copurified proteins were eluted by using 1× SSB.
Results
Complementation of Epitope-Tagged Plasmids.
The ERG28 gene was cloned by PCR either directly into the expression vectors p423GPD (HIS3 marker) and p425GPD (LEU2 marker) (15) or as a N- or C-terminal Myc fusion to generate constructs pERG28, pMyc-ERG28, and pERG28-Myc, respectively. These plasmids were transformed into the erg28 gene-disrupted strain, R712 and complementation were assessed by growth response and sterol profiles. The results indicated that all three constructs complement the slow-growing erg28 deletion strain. The faster growth rates as well as wild-type sterol profiles indicate that the Myc tag did not interfere with the function of ERG28 (data not shown).
Similarly, the ERG27 and ERG26 genes were cloned by PCR and tagged with HA at the N or C terminus of these genes, respectively, and cloned into the p426GPD expression vector (15). ERG27 constructs pERG27 (no tag), pHA-ERG27, and pERG27-HA were transformed into erg27, and ERG26 constructs pERG26 (no tag), pHA-ERG26, and pERG26-HA were transformed into erg26, respectively. All constructs complement, because erg27 and erg26 mutants could grow without ergosterol supplementation, indicating that the HA tag did not interfere with complementation (data not shown).
Protein extracts from complementing yeast colonies were screened for Erg28p-Myc, Erg27p-HA, and Erg26p-HA proteins by using Myc and HA antibody (Fig. 2). The intensity of the Erg28p band using anti-Myc-peroxidase was greater when Myc was located at the N terminus of Erg28p than at the C terminus (Fig. 2A). For Erg27p, when the HA tag was located at the N or C terminus of Erg27p, both showed the same strong bands (36 kDa) using anti-HA-peroxidase (Fig. 2B). However, when HA was located at the N terminus of Erg26p, no HA-Erg26p could be detected, and when HA was located in the C terminus of Erg26p, two bands (42 and 40 kDa) were observed (Fig. 2C).
Figure 2.

(A) Myc-tagged Erg28p (18.4 kDa) was detected by anti-Myc-peroxidase. When Myc is located at the N terminus of Erg28p, a stronger signal was observed than when it was located at the C terminus. Background bands were also observed when using anti-Myc peroxidase. (B) HA-tagged Erg27p (36 kDa) detected by anti-HA-peroxidase. Both N and C terminus-tagged proteins gave the same size bands and signal intensity. (C) HA-tagged Erg26p detected by anti-HA-peroxidase. No band was detected when the HA tag was located at the N terminus. When the HA tag was located at the C terminus, two bands (42 and 40 kDa) were observed.
Erg26p, Erg27p, and Erg28p Are Membrane-Associated Proteins.
Total protein from erg28/pMyc-ERG28, erg27/pERG27-HA, and erg26/pERG26-HA strains were subjected to differential centrifugation, and the fractions were immunoassayed (Fig. 3). These results indicate that Erg28p, Erg27p, and Erg26p are enriched in the 10,000 × g and 20,000 × g pellets, suggesting that these are all membrane-associated proteins. Kar2p (21), an ER protein, was also found to be enriched in the 10- to 60-K membrane pellets (Fig. 3D). The differential centrifugation step removed two strong bands from the total protein of erg28/pMyc-ERG28, facilitating Myc-tagged Erg28p detection (Fig. 3A). This differential centrifugation also facilitated the separation of the two C-terminal HA tagged Erg26p bands. The 40-kDa Erg26p is a membrane-associated protein, and the 42-kDa Erg26p is a soluble protein (Fig. 3C), suggesting that a cleavable signal peptide exists in the N terminus of Erg26p, requisite for Erg26p membrane localization. Interference with this signal peptide would explain why, when HA is located in the N terminus of Erg26p, no HA-Erg26p is observed (Fig. 2C).
Figure 3.

Western blot of total protein (lane 1), 10 K pellet (lane 2), 20 K pellet (lane 3), 60 K pellet (lane 4), 130 K pellet (lane 5), and 130 K supernatant (lane 6) probed with anti-Myc to detect Myc-Erg28p (A) and anti-HA to detect Erg27p-HA and Erg26p-HA (B and C, respectively). (D) Kar2p, an ER protein, is also enriched in the 10–60 K membrane pellets.
Erg26p, Erg27p, and Erg28p Were All Located in the ER.
We tagged the ERG26, ERG27, and ERG28 gene products with GFP to confirm localization of these proteins in vivo. Plasmids expressing GFP-Erg26p, GFP-Erg27p, or GFP-Erg28p fusion proteins were transformed into erg26, erg27, or erg28 mutant strains, respectively. In all three cases, plasmids restored either the growth or the ergosterol requiring phenotype of these strains (results not shown). It was reported previously that GFP-Erg26p and GFP-Erg27p most likely localize to the ER (7), and our results confirmed that the localization of both GFP-Erg26p and GFP-Erg27p fusion proteins are perinuclear in both ERG28 (Fig. 4) and erg28 strains. Nuclear localization was verified by using the DNA-binding dye 4, 6-diamidino-2-phenylindol (data not shown). We also found that as cells approached stationary phase, GFP-Erg27p localizes into cytoplasmic particles, in addition to the ER (Fig. 4C). An ER resident protein, Sur4p (7), fused to GFP localized to the same perinuclear location as the Erg26p, Erg27p, and Erg28p (data not shown).
Figure 4.

The localization of GFP fusion proteins was determined by using fluorescence microscopy. (A) GFP-Erg26p observed in erg26. (B) GFP-Erg28p observed in erg28. (C) GFP-Erg27p observed in erg27. (D) Differential interference contrast of cells observed in C.
Protein–Protein Interactions Among Erg25p, Erg26p, Erg27p, and Erg28p.
To study the interactions of Erg28p-Erg27p and Erg28p-Erg26p, pERG27-HA and pERG26-HA were transformed into erg28/pMyc-ERG28, respectively, and transformed cells were screened on complete synthetic medium-HIS-URA plates. Doubly transformed strains were obtained: SMO2827 (erg28/pMyc-ERG28 and pERG27-HA) and SMO2826 (erg28/pMyc-ERG28 and pERG26-HA). Total protein extracts from SMO2827 and SMO2826 were centrifuged at 20,000 × g to obtain a 20-K membrane pellet. These pellets were resuspended in microsomal buffer at a concentration of 10 mg/ml and solubilized at 1 mg/ml by using SML (2 mM), N-octyl-β-d-glucopyranoside (25 mM), or Triton X-100 (1%) for 20 min at room temperature. Solubilized supernatant and unsolubilized membrane pellets were applied to an SDS/PAGE gel, and immunoassays were performed to test the degree of solubilization of Erg26p, Erg27p, and Erg28p by the three detergents. No detergent was found to completely solubilize all three proteins effectively (Fig. 5A). The most effective detergent for all proteins was 2 mM SML. SMO2827 and SMO2826 membrane proteins, solubilized with 2 mM SML, were immunoprecipitated with anti-HA and anti-Myc. It was found that Erg28p and Erg27p reciprocally coimmunoprecipitated (Fig. 5B); however, no Erg28p-Erg26p coimmunoprecipitation was observed (Fig. 5C). Erg25p also coimmunoprecipitated with Erg27p and Erg28p by using rabbit anti-Erg25p (Fig. 5D). These results indicate that Erg25p, Erg27p, and Erg28p interact with one another. No coimmunoprecipitation was observed with Erg26p, most likely due to the poor solubilization of this protein.
Figure 5.

Interactions between Erg25p, Erg26p, Erg27p, and Erg28p. SMO2826 is an erg28-disrupted strain with plasmids containing Myc-ERG28 and ERG26-HA epitopes, and SMO2827 is an erg28-disrupted strain with plasmids containing Myc-ERG28 and ERG27-HA epitopes. (A) Various detergents, N-octyl-β-d-glucopyranoside (25 mM), SML (2 mM), and Triton X-100 (1%), were used to solubilize microsomal proteins. P, unsolubilized protein pellet; S, solubilized supernatant protein. (B) Using anti-Myc or anti-HA antibodies, Erg27p and Erg28p coimmunoprecipitated with each other. (C) No coimmunoprecipitation interaction was observed between Erg26p and other proteins. (D) Using anti-Erg25p, Erg25p coimmunoprecipitate with Erg27p as well as Erg28p.
Erg26p/Erg28p and Erg27p/Erg28p Comigrate in the Same Complex.
The 20-K membrane pellets of SMO2826 and SMO2827 were resuspended in microsomal buffer and subjected to two rounds of sucrose gradient centrifugation; the first gradient (10–60%) was used to obtain purified ER membrane protein rich in Erg26p, Erg27p, and Erg28p. These three proteins were located in the same heavy ER membrane protein fractions (data not shown). These purified fractions enriched in Erg26p, Erg27p, and Erg28p were solubilized and applied to the second sucrose gradient (30–60%) ultracentrifugation to determine the location of Erg26p, Erg27p, and Erg28p complex. Fig. 6 A and B show that Erg26p, Erg27p, and Erg28p are in the same gradient location. When compared with the ultracentrifugation markers (Fig. 6C), these three proteins are in a complex that migrates between the BSA (66-kD) and β-amylase (200-kD) markers.
Figure 6.

Erg26p, Erg27p, and Erg28p are present in a complex between 66 and 200 kD determined by sucrose gradient centrifugation. (A) Solubilized SMO2827 ER proteins after sucrose gradient ultracentrifugation and immunoassayed indicating the location of Erg27p and Erg28p in the same complex. (B) Solubilized SMO2826 ER proteins after sucrose gradient ultracentrifugation and immunoassayed indicating the location of Erg26p and Erg28p. Erg26p is shifted to a slightly larger protein complex. (C) Sucrose gradient ultracentrifugation markers and their relative location in the gradient. Erg26p, Erg27p, and Erg28p are located in the area (dashed line area) between BSA (66 kDa) and β-amylase (200 kDa).
Absence of Erg28p Shifts Erg27p in a Sucrose Gradient to a Lighter ER Fraction.
An erg27 erg28 double-disrupted strain (SD2728) was transformed with pERG27-HA (SMO4700) or transformed with both pERG27-HA and pMyc-ERG28 (SMO4701), to demonstrate that Erg28p influences the location of Erg27p. Sucrose gradient centrifugation of total protein extracts indicates that in SMO4701, the distribution of Erg27p is equally distributed into lighter and heavier ER fractions (Fig. 7D). However, in the pERG27-HA single transformed strain (SMO4700), most of the Erg27p (Fig. 7C) is found in the lighter ER or vacuolar fractions. V-ATPase is a vacuolar marker (Fig. 7A), and Kar2p is an ER marker (Fig. 7B). Erg28p is always found in the heavier ER fraction (Fig. 7E).
Figure 7.

SD2728 (erg27 erg28 double-disrupted strain) transformed with pERG27-HA to form SMO4700, or transformed with pERG27-HA and pMyc-ERG28 to form SMO4701. Sucrose gradient centrifugation of total protein extracts indicates that in SMO4701, the distribution of Erg27p is equally distributed into lighter and heavier ER fraction (D), as indicated by V-ATPase vacuolar marker (A) and ER marker (B). However, in the pERG27-HA single-transformed strain SMO4700, most of the Erg27p (C) is found in the lighter ER or vacuolar fraction using V-ATPase (vacuolar marker, A) and Kar2p (ER marker, B), respectively. Erg28p is always found in the heavier ER fraction (E).
Copurification of the C-4 Demethylation Enzymatic Complex by Using Anti-HA Beads.
Solubilized membrane protein of SMO4701 (erg27 erg28 transformed with pERG27-HA and pMyc-ERG28) and SMO5001 (erg26 erg28 transformed with pERG26-HA and pMyc-ERG28) were coimmunoprecipitated overnight by using mouse anti-HA matrix beads. Immunoprecipitated beads were washed, and the copurified proteins were eluted by using 1× SSB, run on a SDS/PAGE gel, and immunoassayed. The protein profile of the copurified C-4 demethylation enzymatic complex is shown in Fig. 8. Four protein bands were identified as Erg25p, Erg26p, Erg27p, and Erg28p.
Figure 8.
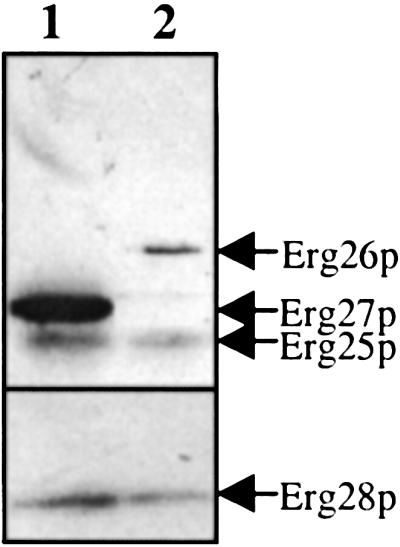
Copurification of the C-4 demethylation complex using anti-HA matrix beads. Solubilized membrane protein of SMO4701 (lane 1) and SMO5001 (lane 2) were coimmunoprecipitated with mouse anti-HA matrix beads. Four protein bands, 18.4, 35, 36, and 40 kD, were identified by immunoassay as Erg28p, Erg25p, Erg27p, and Erg26p, respectively.
Discussion
It appears that all of the yeast ergosterol biosynthetic genes have been cloned and characterized. However, the nature of protein–protein interactions among sterol biosynthetic enzymes is presently unknown. We have chosen the C-4 demethylation enzymes as a starting point to elucidate protein–protein interactions because of the recent finding that a novel transmembrane protein (Erg28p) is involved in C-4 demethylation, and that this protein is found in sterol biosynthetic pathways in fungus, plant, and animal cells (5). Homologues of ERG28 gene have been found in Schizosaccharomyces pombe, Arabidopsis, mouse, and humans. In humans, the ERG28 gene was highly expressed in adult testis and in several cancer lines (22). Because the absence of this gene is not lethal, complete or partial loss of this protein may be one of the causes of inborn errors of sterol biosynthesis (23).
Using epitope tags, we found that Erg26p, Erg27p, and Erg28p are all membrane-associated proteins (Fig. 3), and GFP-fusion protein localization studies confirmed that Erg26p, Erg27p, and Erg28p have the same ER location both in ERG28 and erg28 strains. This is not surprising, because both produce end-product ergosterol, although erg28 strains accumulate only one-third as much as wild type (5). Erg28p interacted with Erg27p, as demonstrated by reciprocal coimmunoprecipitation (Fig. 5B), but no interaction between Erg26p and other proteins was observed (Fig. 5C), most likely due to the poor solubility of this protein. Erg25p was also found to interact with Erg27p and Erg28p (Fig. 5D), consistent with a yeast two-hybrid interaction study (7). Sucrose gradient analysis showed that Erg27p/Erg28p and Erg26p/Erg28p are in the same complex between 66 and 200 kDa (Fig. 6A). Because Erg25p and Erg27p coimmunoprecipitate (Fig. 5C), Erg25p is also present in this complex.
It is now clear why an erg28 strain accumulates carboxylic acid sterol intermediates, 3-keto sterol intermediates, and increased amounts of 4,4-dimethylzymosterol. We suggest that Erg28p works as a scaffold protein for Erg27p and tethers Erg27p to the ER, to ensure that Erg27p works properly as a 3-keto reductase. However, in an erg28 strain, the complex of Erg25p/Erg26p/Erg27p becomes inefficient because neither Erg26p nor Erg27p have transmembrane domains to anchor them to the ER. Our results with ERG26-GFP and ERG27-GFP fusions suggest that both Erg26p and Erg27p are ER bound both in ERG28 and erg28 backgrounds, but the sucrose gradient experiment (Fig. 7) suggests that the distribution of Erg27p is altered in an erg28 background. Thus the 4,4-demethylation reactions may proceed inefficiently in erg28 and C-4 demethylation intermediates accumulate. Although there is an interaction between Erg25p and Erg27p (7), this interaction by itself may not be strong enough to hold the enzymatic complex together. This is consistent with the fact that approximately one-third the wild-type amount of end-product sterols is synthesized in erg28 strains (5). The results in Fig. 8 clearly indicate the existence of a complex containing Erg25p/Erg26p/Erg27p/Erg28p. Further studies of all possible protein–protein interactions are required to elucidate the nature of these interactions as well as the interaction of the C-4 demethylation enzymes with other enzymes in sterol biosynthesis.
Acknowledgments
We thank Drs. Toon de Kroon (CBLE–Utrecht University, The Netherlands) and Jerry Kaplan (University of Utah) for their generous gifts of anti-Erg25p and Dr. Jeff Brodsky for anti-Kar2p. We thank N. Jia for excellent technical assistance. This investigation was supported by National Institutes of Health Grant GM62104 (to M.B.).
Abbreviations
- ADH
alcohol dehydrogenase
- ER
endoplasmic reticulum
- GPD
glycerol phosphate dehydrogenase
- GFP
green fluorescent protein
- HA
peptide from human hemagglutinin protein
- PCR
polymerase chain reaction
- SML
sucrose monolaurate
- YPD
yeast extract, peptone, glucose medium
References
- 1.Gachotte D, Sen S E, Eckstein J, Barbuch R, Krieger M, Ray B D, Bard M. Proc Natl Acad Sci USA. 1999;96:12655–12660. doi: 10.1073/pnas.96.22.12655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bard M, Bruner D A, Pierson C A, Lees N D, Biermann B, Frye L, Koegel C, Barbuch R. Proc Natl Acad Sci USA. 1996;93:186–190. doi: 10.1073/pnas.93.1.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gachotte D, Barbuch R, Gaylor J, Nickel E, Bard M. Proc Natl Acad Sci USA. 1998;95:13794–13799. doi: 10.1073/pnas.95.23.13794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hughes T R, Marton M J, Jones A R, Roberts C J, Stoughton R, Armour C D, Bennett H A, Coffey E, Dai H, He Y D, et al. Cell. 2000;102:109–126. doi: 10.1016/s0092-8674(00)00015-5. [DOI] [PubMed] [Google Scholar]
- 5.Gachotte D, Eckstein J, Barbuch R, Hughes T, Roberts C, Bard M. J Lipid Res. 2001;42:150–154. [PubMed] [Google Scholar]
- 6.Milla P, Athenstaedt K, Viola F, Oliaro-Bosso S, Kohlwein S D, Daum G, Balliano G. J Biol Chem. 2002;277:2406–2412. doi: 10.1074/jbc.M104195200. [DOI] [PubMed] [Google Scholar]
- 7.Baudry K, Swain E, Rahier A, Germann M, Batta A, Rondet S, Mandala S, Henry K, Tint G S, Edlind T, et al. J Biol Chem. 2001;276:12702–12711. doi: 10.1074/jbc.M100274200. [DOI] [PubMed] [Google Scholar]
- 8.Zweytick D, Hrastnik C, Kohlwein S D, Daum G. FEBS Lett. 2000;470:83–88. doi: 10.1016/s0014-5793(00)01290-4. [DOI] [PubMed] [Google Scholar]
- 9.Li L, Kaplan J. J Biol Chem. 1996;271:16927–16933. doi: 10.1074/jbc.271.28.16927. [DOI] [PubMed] [Google Scholar]
- 10.Paltauf F, Kohlwein S D, Henry S A. Gene Expression. 1992;2:415–500. [Google Scholar]
- 11.Faust J L, Trzaskos J M, Gaylor J M. In: Biology of Cholesterol. Yeagle P L, editor. Boca Raton, FL: CRC Press; 1988. pp. 19–38. [Google Scholar]
- 12.Leber R, Landl K, Zinser E, Ahorn H, Spok A, Kohlwein S D, Turnowsky F, Daum G. Mol Biol Cell. 1998;9:375–386. doi: 10.1091/mbc.9.2.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zinser E, Paltauf F, Daum G. J Bacteriol. 1993;175:2853–2858. doi: 10.1128/jb.175.10.2853-2858.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sambrook J, Fritsch E F, Maniatis T. Molecular Cloning: A Laboratory Manual. 2nd Ed. Plainview, NY: Cold Spring Harbor Lab. Press; 1989. [Google Scholar]
- 15.Mumberg D, Muller R, Funk M. Gene. 1995;156:119–122. doi: 10.1016/0378-1119(95)00037-7. [DOI] [PubMed] [Google Scholar]
- 16.Sikorski R S, Hieter P. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cormack B P, Valdivia R H, Falkow S. Gene. 1996;173:33–38. doi: 10.1016/0378-1119(95)00685-0. [DOI] [PubMed] [Google Scholar]
- 18.Ausubel F, Brent R, Kingston R E, Moore D D, Seidman J G, Smith J A, Struhl K. Short Protocols in Molecular Biology. New York: Wiley; 1995. [Google Scholar]
- 19.Gable K, Slife H, Bacikova D, Monaghan E, Dunn T M. J Biol Chem. 2000;275:7597–7603. doi: 10.1074/jbc.275.11.7597. [DOI] [PubMed] [Google Scholar]
- 20.Kohlwein S D. Microsc Res Tech. 2000;15:511–529. doi: 10.1002/1097-0029(20001215)51:6<511::AID-JEMT3>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 21.Rose M D, Misra L M, Vogel J P. Cell. 1989;57:1211–1221. doi: 10.1016/0092-8674(89)90058-5. [DOI] [PubMed] [Google Scholar]
- 22.Veitia R A, Ottolenghi C, Bissery M C, Fellous A. Cytogenet Cell Genet. 1999;85:217–220. doi: 10.1159/000015296. [DOI] [PubMed] [Google Scholar]
- 23.Kelley R I, Herman G E. Annu Rev Genom Hum Genet. 2001;2:299–341. doi: 10.1146/annurev.genom.2.1.299. [DOI] [PubMed] [Google Scholar]