

Abstract
Polo-like kinase 4 (PLK4), a member of the serine/threonine protein kinase family, serves as a central regulator of centriole duplication and plays a critical role in eukaryotic mitosis. Overexpression of PLK4 in several cancer types underscores its potential as a therapeutic target. In our previous studies, compound 28t demonstrated acceptable kinase inhibitory activity but exhibited poor cellular activity. Consequently, 28t was selected as a lead compound for further optimization. Through functional group migration and rational drug design strategies, we conducted structural modifications that ultimately yielded 23 novel indazole-based PLK4 inhibitors. Among these, compound C05 exhibited exceptional kinase inhibitory activity (IC50 < 0.1 nM). At the cellular level, C05 demonstrated potent antiproliferative effects against IMR-32 (neuroblastoma), MCF-7 (breast cancer), and H460 (non-small cell lung cancer) cell lines, with IC50 values of 0.948 μM, 0.979 μM, and 1.679 μM, respectively. Notably, compound C05 demonstrated favorable kinase selectivity towards PLK4 among the 10 kinases tested, achieving an inhibition rate of 87.45%. Further pharmacological experimental studies, including apoptosis induction, cell cycle arrest analysis, and clonogenic formation experiments, revealed that C05 outperformed the positive control LCR-263 in both potency and efficacy. Western blot analysis demonstrated that compound C05 effectively suppressed PLK4 autophosphorylation at 4 μM. Unfortunately, compound C05 demonstrated poor metabolic stability in human liver microsomes (HLMs), exhibiting a short half-life (T1/2) of 2.69 minutes under standard incubation conditions. Notwithstanding the suboptimal metabolic stability, the compelling biological activity profile of compound C05 warrants further structural refinement.
Polo-like kinase 4 (PLK4), a member of the serine/threonine protein kinase family, serves as a central regulator of centriole duplication and plays a critical role in eukaryotic mitosis.
1. Introduction
The cell cycle refers to the entire process from the completion of one cell division to the end of the next, consisting of two main phases: interphase and mitotic phase (M phase). During interphase, cells synthesize essential components for division, which is further subdivided into G1 phase, S phase (DNA replication), and G2 phase. The mitotic phase is divided into four distinct stages: prophase, metaphase, anaphase, and telophase.1 Serine/threonine protein kinases play a crucial role in eukaryotic mitosis.2 The Polo-like kinase (PLK) family, a subgroup of serine/threonine kinases, regulates various mitotic processes, including mitotic entry and exit, centrosome duplication, chromosome segregation, and the DNA damage response (DDR).3,4 The PLK family comprises five members: PLK1 localizes to the centrosome during the G2 phase of the cell cycle, where it regulates mitotic spindle assembly.5 PLK2 is localized to the centrosome in early G1 phase and is functionally involved in centriole replication.6 PLK3 primarily resides in the nucleolus and participates in DNA replication processes.7 PLK4 is activated during S phase and acts on the centrosome as a master regulator of centriole duplication.8 PLK5 is also detected in the nucleolus and may play a specialized role in neuronal differentiation.9 All five members have a conserved N-terminal kinase domain and one or more C-terminal Polo-box domains (PBDs).10 Among the PLK family, PLK4 exhibits the most structural divergence.11 In mammalian cells, PLK4 localizes to the centrosome and orchestrates centriole duplication by recruiting centrosomal proteins (e.g., STIL, SAS6) in a stepwise manner, ensuring strictly controlled single-round duplication before mitotic entry.12–14 Beyond its role in centriole biogenesis, PLK4 also maintains centriolar satellite integrity and promotes ciliogenesis, both of which are critical for precise mitotic regulation.15 Studies have demonstrated that PLK4 is overexpressed in several cancer types, including breast cancer,16 lung cancer,17 and neuroblastoma.18 Therefore, the development of PLK4 inhibitors is recognized as a promising therapeutic strategy for cancers driven by centrosome amplification.
Current PLK4 inhibitors are primarily categorized into indazole- and aminopyrazole-based compounds. In 2007, Johnson's team19 first proposed the potential importance of PLK4 as a drug discovery target. They screened a series of known kinase inhibitors targeting PLKs and disclosed an indazole-based compound, axitinib (Fig. 1), which demonstrated potent inhibitory activity against PLK4 (Ki = 4.2 nM). Notably, axitinib was originally developed as a pan-VEGFR inhibitor and received approval from the U.S. Food and Drug Administration (FDA) in 2012 for the treatment of renal cell carcinoma. To develop highly potent and selective PLK4 inhibitors, Pauls et al.20 employed virtual screening of ligand-based libraries and rational drug design strategies to ultimately optimize CFI-400945 (ref. 21) (Fig. 1). It demonstrates exceptional enzymatic potency (PLK4, IC50 = 2.8 nM) and cellular activity, suppressing PLK4 phosphorylation at Ser305 with an EC50 of 12.3 nM in PLK4-overexpressing cells. Notably, CFI-400945 exhibits >20 000-fold selectivity over other PLK family members,22 representing the sole PLK4 inhibitor currently in phase II clinical trials.21
Fig. 1. Major structural classes of PLK4 inhibitors.

In 2007, Johnson et al.19 also reported an aminopyrazole derivative, VX-680 (Fig. 1), which was originally explored as a pan-Aurora kinase (AURK) inhibitor,23 that exhibited nanomolar inhibitory activity against PLK4 (Ki = 7.7 nM). Subsequent systematic structure–activity relationship (SAR) studies by Wong et al.24 on VX-680 led to the design of the PLK4-specific inhibitor centrinone (Fig. 1), which potently inhibited PLK4 with a Ki of 0.16 nM. Notably, centrinone displayed >1000-fold selectivity for PLK4 over Aurora A/B and minimal activity against 442 human kinases in vitro. Recently, Zhao's group25,26 identified aminopyrazole derivative CZS-241 (Fig. 1), with nanomolar PLK4 inhibition (IC50 = 2.6 nM), CZS-241 effectively suppressed leukemia cell proliferation, particularly in chronic myeloid leukemia (CML) cell lines, and exhibited favorable pharmacokinetics with 70.8% relative bioavailability in mice. In vivo, oral administration of CZS-241 (20 mg kg−1 per day) significantly inhibited CML progression.
2. Chemistry
The synthesis of the key intermediate M5 is depicted in Scheme 1. Beginning with commercially available 6-bromoindazole, M5 was constructed through a sequence of functional group transformations comprising NIS-mediated nucleophilic substitution, THP protection, a Sonogashira coupling, and a carbon–sulfur bond-forming reaction.
Scheme 1. Reagents and conditions: (a) NIS, DMF, r.t., 5 h, 72.38%. (b) DHP, p-toluenesulfonic acid, DCM, 0 °C then r.t., 4 h, 56.59%. (c) 3-Ethynylpyridine, CuI, Pd(PPh3)4, TEA, r.t., 5 h, 50.47%. (d) 2-Ethylhexyl 3-mercaptopropionate, DIEA, Pd2(dba)3, Xantphos, toluene, 110 °C, reflux, 5 h, 77.23%.

The preparation of A01–A09 is detailed in Scheme 2. Deprotection of intermediate M5 under alkaline conditions was followed by nucleophilic substitution with intermediates M14a–M14i, and finished with THP deprotection. Notably, the syntheses of A05 and A06 incorporated an iron acid-catalyzed reductive amination step prior to the final deprotection.
Scheme 2. Reagents and conditions: (a) (i) t-BuONa, t-BuOH, r.t., 1 h; (ii) M14a–M14i, THF, r.t., 1 h, 63.16%. (b) Trifluoroacetic acid, r.t., 2 h, 41.17%. (c) Fe, hydrochloric acid, V(EtOH) : V(H2O) = 4 : 1, r.t., 12 h, 51.99%.

The synthetic pathways leading to the preparation of key intermediates M14a through M14h are comprehensively illustrated in Scheme 3. The process was initiated using commercially available aniline derivatives as starting materials. These anilines subsequently underwent reductive amination under appropriate conditions, followed by a nucleophilic acyl substitution reaction employing acyl chlorides to afford the desired amide products. Each step was carefully optimized to ensure satisfactory yields and purity.
Scheme 3. Reagents and conditions: (a) acetaldehyde, NaBH4, MeOH, 0 °C then r.t., 4 h, 54.36%. (b) Chloroacetyl chloride, Et3N, DCM, 0 °C then r.t., 2 h, 80.24%. (c) Acetone, NaBH4, MeOH, 0 °C then r.t., 4 h, 37.26%. (d) Cyclopropylamine, salicylaldoxime, Cu2O, Cs2CO3, 12 h, 14.71%.

In contrast, the synthesis of intermediate M14i was accomplished via an alternative route. Beginning with commercially sourced iodobenzene, a copper-mediated Ullmann coupling reaction was performed with cyclopropylamine to construct the targeted C–N bond. This transformation provided efficient access to the desired intermediate in a convergent manner.
Scheme 4 illustrates the synthesis of intermediates M16a–M16d. The thiol group in intermediate M5 was deprotected to afford intermediate M15, which underwent substitution with ethyl α-bromophenylacetate or its analogues to furnish intermediates M16a–M16d.
Scheme 4. Reagents and conditions: (a) t-BuONa, t-BuOH, r.t., 1 h. (b) Ethyl α-bromophenylacetate and its derivatives, THF, r.t., 1 h. 77.51%.

The synthesis of B01 and B04–B06 is described in Scheme 5. These compounds were liberated from their THP protecting groups via ammonolysis of intermediates M16a–M16d, yielding the final products. Intermediate M16a was subjected to ester hydrolysis, condensation, and THP deprotection to afford BO2 and BO3.
Scheme 5. Reagents and conditions: (a) ethylamine, EtOH, 80 °C, reflux, 12 h, 33.23%. (b) Trifluoroacetic acid, r.t., 2 h, 75.18%. (c) NaOH, V(MeOH) : V(H2O) = 1 : 1, 50 °C, 1 h, 85.87%. (d) Ammonia water or methylamine, HATU, DIEA, DMF, 0 °C then r.t., 12 h, 50.16%–63.23%.

The synthesis of C01–C04 is outlined in Scheme 6. Intermediate M3 underwent Sonogashira coupling and carbon–sulfur coupling to yield M21a–M21d. Subsequent thiol deprotection under alkaline conditions, substitution with M14a, and THP deprotection furnished the final compounds.
Scheme 6. Reagents and conditions: (a) 4-((5-ethynylpyridin-2-yl)methyl)morpholine and its analogues, CuI, Pd(PPh3)4, TEA, r.t., 5 h, 32.28%. (b) 2-Ethylhexyl 3-mercaptopropionate, DIEA, Pd2(dba)3, Xantphos, toluene, 110 °C, reflux, 5 h, 66.48%. (c) (i) t-BuONa, t-BuOH, r.t., 1 h; (ii) 2-chloro-N-(3-fluorophenyl)-N-methylacetamide, THF, r.t., 1 h, 30.23%. (d) p-Toluenesulfonic acid, V(MeOH) : V(H2O) = 3 : 0.5, 80 °C, 5 h, 36.02%.

Scheme 7 presents the preparation of M25a–M25d from commercial precursors M23a–M23dvia Sonogashira coupling and deprotection.
Scheme 7. Reagents and conditions: (a) (triisopropylsilyl)acetylene, PPh3, Pd(OAc)2, TEA, 100 °C, reflux, 10 h, 54.11–68.97%; (b) TBAF, THF, r.t., 2 h, 83.80%.

The synthetic pathways for the preparation of target compounds C05 and C06 are described in detail in Scheme 8. The process commenced with intermediate M3, which first underwent a Heck coupling reaction under palladium catalysis to install the desired olefinic moiety. The resulting intermediate was subsequently subjected to a carbon–sulfur coupling to furnish key intermediates M27a and M27b.
Scheme 8. Reagents and conditions: (a) 3-ethynylpyridine or 2-ethynylpyridine, P(o-tol)3, Pd(OAc)2, DIEA, DMF, 100 °C, 5 h, 44.49%. (b) 2-Ethylhexyl 3-mercaptopropionate, DIEA, Pd2(dba)3, Xantphos, toluene, 110 °C, reflux, 5 h, 66.29%. (c) (i) t-BuONa, t-BuOH, r.t., 1 h; (ii) ethyl α-bromophenylacetate or 2-chloro-N-ethyl-N-phenylacetamide, THF, r.t., 1 h, 26.63%. (d) Ethylamine, EtOH, 80 °C, reflux, 12 h, 33.23%. (e) Trifluoroacetic acid, r.t., 1 h, 21.34%.

The synthetic sequence then proceeded with deprotection of the thiol group under alkaline conditions, followed by nucleophilic displacement with an appropriate electrophile to introduce the requisite functional group. Finally, removal of the tetrahydropyranyl (THP) protecting group under acidic conditions afforded the final compounds C05 and C06.
In contrast to the routes described above, the synthesis of analogues C07 and C08 incorporated an additional ammonolysis step prior to the final deprotection, introducing an amide functionality that diverges from the structural features of C05 and C06 (Fig. 2).
Fig. 2. Design and modification of PLK4 inhibitor types.

3. Compound design and biological activity evaluation
3.1. Compound design
The lead compound 28t,27 identified in our preliminary studies, exhibited acceptable kinase inhibitory activity against PLK4 with an IC50 value of 74 nM. However, this compound demonstrated profoundly weak antiproliferative effects against MCF-7 breast cancer cells and IMR-32 neuroblastoma cells. To address these limitations, we employed compound 28t as a starting point for structural optimization, aiming to enhance antiproliferative potency against both MCF-7 and IMR-32 cell lines while retaining PLK4 inhibitory activity. Preliminary molecular docking studies (Fig. 3) revealed that the indazole core of 28t forms strong hydrogen bond interactions with hinge region residues Glu90 and Cys92. Additionally, the carbonyl group of 28t establishes a robust hydrogen bond with Lys41, while the solvent-exposed 3-ethynylpyridine moiety engages in a strong hydrogen bond interaction with Arg28. This intricate network of interactions effectively anchors 28t within the protein's binding pocket. Based on the consideration that the strong electron-withdrawing nature of the fluorine atom may reduce the hydrophobic interaction of the phenyl ring (by decreasing π–electron density), we removed the fluorine atom to obtain compound A01. The resulting improvement in activity supports our hypothesis. Guided by rational drug design principles, we retained the hydrophilic pharmacophore while modifying the hydrophobic segment extending toward the hinge region, yielding compound A01 (Fig. 3), which demonstrated enhanced kinase inhibitory activity with an IC50 value of 11 nM. This improvement may be attributed to increased electron density at the phenyl ring following deliberate fluorine removal, thereby strengthening hydrophobic interactions within the binding pocket.
Fig. 3. (A and C) Predicted binding patterns of compound 28t at the ATP binding site of PLK4 protein (PDB 4YUR). (B and D) Predicted binding modes of compound A01 at the ATP binding site of PLK4 protein. Blue represents hydrogen bonding interactions, while red represents hydrophobic interactions.

Based on molecular docking studies of A01, we identified that modifying these two moieties could represent an important direction for improving cavity occupation. However, the activity of the resulting compounds did not yield the anticipated improvement. To optimize the occupancy of the protein cavity, alkyl substituents were incorporated to generate compounds A02 and A03. However, neither derivative demonstrated superior activity compared to A01, likely due to steric clashes between the bulky alkyl chains and the binding pocket. Molecular docking analysis revealed residual unoccupied space beneath the benzene ring of A01. Consequently, para- and meta-substitutions (including amino, halogen, and methoxy groups) were explored on this aromatic ring (A04–A09). While the para-amino-substituted analogue A05 exhibited moderate activity (IC50 = 0.043 μM), none of these modifications surpassed the potency of A01. These findings suggest that introducing substituents on the exposed benzene moiety does not enhance activity, potentially owing to the confined nature of the underlying protein cavity. Steric interference arising from substituents on the benzene ring may therefore compromise binding interactions, leading to reduced biological efficacy (Table 1).
Table 1. In vitro PLK4 inhibitor activities of compound A01–A09.

| |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | R1 | R2 | R3 | IC50a (μM) | Entry | R1 | R2 | R3 | IC50a (μM) |
| A01 |

|
H | H | 0.011 | A06 |

|
H |

|
0.062 |
| A02 |

|
H | H | 0.031 | A07 |

|

|
H | 0.090 |
| A03 |

|
H | H | 0.071 | A08 |
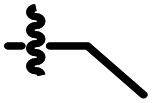
|

|
H | 0.097 |
| A04 |

|
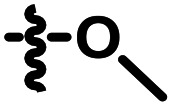
|
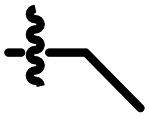
H | 0.266 | A09 |
|
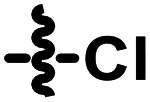
|
H | 0.173 |
| A05 |
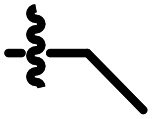
|

|
H | 0.043 | LCR-263b | 0.003 | |||
IC50 values are shown as the mean values from two separate experiments.
Positive control.
In light of the potential metabolic liability associated with the α-position of the carbonyl group, we relocated the phenyl ring. Unfortunately, this modification resulted in a significant decrease in compound activity, indicating that maintaining a flexible linker is crucial for the potency of our compounds. Subsequent to the observed rapid systemic clearance of compound 28tin vivo, we hypothesized that its carbonyl α-carbon represented a metabolically labile hotspot. Motivated by this finding, a functional group relocation strategy was implemented to reposition the phenyl ring from compound A01 to the α-carbon of the carbonyl moiety. This structural modification afforded compound B01, which achieved a PLK4 inhibitory IC50 value of 0.065 μM. Molecular docking analysis of B01 (Fig. 4) revealed that the introduction of the ethyl group induced a conformational change in the compound. Subsequent reduction of alkyl chain substituents on the amide nitrogen afforded compounds B02 and B03 (Table 2), which unexpectedly exhibited decreased kinase inhibitory activity. Furthermore, the docking studies identified unoccupied space adjacent to the DFG motif in the B01–protein complex.
Fig. 4. (A and C) Predicted binding patterns of compound B01 at the ATP binding site of PLK4 protein (PDB 4YUR). (B and D) Predicted binding modes of compound C05 at the ATP binding site of PLK4 protein. Blue represents hydrogen bonding interactions, while red represents hydrophobic interactions.

Table 2. In vitro PLK4 inhibitor activities of compounds.

| |||||
|---|---|---|---|---|---|
| Entry | R1 | IC50a (μM) | Entry | R2 | IC50a (μM) |
| B01 |

|
0.065 | B04 |
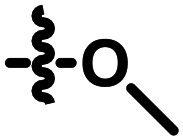
|
0.255 |
| B02 |

|
0.163 | B05 |

|
1.558 |
| B03 | H | 0.68 | B06 |

|
1.526 |
| LCR-263b | 0.003 | ||||
IC50 values are shown as the mean values from two separate experiments.
Positive control.
To exploit this, para-substitutions (F, Cl, OCH3) were introduced. However, these smaller substituents failed to improve activity (Table 2). This may be attributed to the potential conformational shifts in the protein–ligand complex.
Recognizing that modification of the hydrophilic fragment extending into the solvent region is critical for enhancing both the activity and the drug-like properties of the compound, we conducted a systematic investigation of this solvent-exposed moiety. To further enhance kinase inhibitory activity, the hydrophilic fragment of compound A01 was systematically investigated, leading to the design of derivatives C01–C06 (Table 3). Compounds C05 and C06 demonstrated superior kinase inhibitory activity, with IC50 values both below 0.1 nM. Molecular modeling revealed that the vinyl group in C05 adopts a distinct spatial orientation (Fig. 4) compared to the alkyne moiety in A01, enabling optimal filling of the protein cavity while minimizing steric clashes. This conformational advantage likely underpins the dramatic activity improvement. Building on the promising activity observed with 3-vinyl and 2-vinyl fragments, we replaced the 3-ethynylpyridine moiety in compound B01 to generate derivatives C07 and C08. However, these structural modifications did not translate to improved kinase inhibitory activity, yielding disappointing results that fell short of our expectations.
Table 3. In vitro PLK4 inhibitory activities of compounds C01–C08.

| ||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | R1 | IC50a (nM) | Entry | R1 | IC50a (nM) | Entry | R2 | IC50a (nM) |
| C01 |

|
61.3 | C04 |
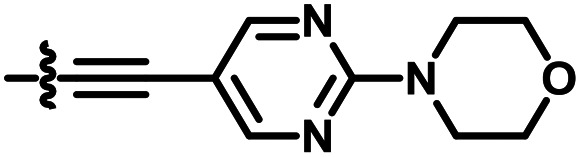
|
247.7 | C07 |

|
30 |
| C02 |
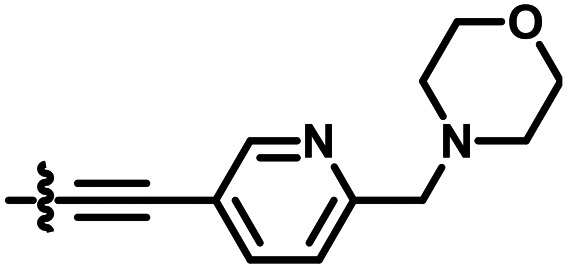
|
90.4 | C05 |

|
<0.1 | C08 |

|
65 |
| C03 |
|
61 | C06 |

|
<0.1 | LCR-263b | 3 | |
IC50 values are shown as the mean values from two separate experiments.
Positive control.
3.2. Biological activity evaluation
3.2.1. In vitro antiproliferative activity evaluation
Substantial evidence has established that PLK4 overexpression is critically involved in the oncogenesis and progression of multiple human cancers, with particularly strong associations observed in neuroblastoma, breast cancer, and lung carcinoma. Based on enzymatic activity screening results, three compounds with IC50 values <20 nM were selected and evaluated for their antiproliferative effects against three cancer cell lines, IMR-32 (neuroblastoma), MCF-7 (breast cancer), and H460 (lung cancer), with LCR-263 as a positive control. As summarized in Table 4, compound C05 demonstrated superior potency to LCR-263 against all three cancer cell lines, with IC50 values of 0.948 μM (IMR-32), 0.979 μM (MCF-7), and 1.679 μM (H460), respectively. These results establish C05 as the superior compound for further pharmacological characterization.
Table 4. In vitro cell anti proliferative activity test of compound C05.
| Entry | IMR-32a (μM) | MCF-7a (μM) | H460a (μM) |
|---|---|---|---|
| A01 | 1.665 | 1.603 | 8.139 |
| C05 | 0.948 | 0.979 | 1.679 |
| C06 | 1.851 | 2.696 | 6.656 |
| LCR-263b | 1.613 | 4.804 | 3.212 |
IC50 values are shown as the mean values from three separate experiments.
Positive control.
3.2.2. Kinase selectivity profile
For kinase inhibitors, selectivity is a critical parameter. We evaluated the selectivity profile against kinases within the same family as well as key cell cycle-related kinases such as CDK4. Notably, our compound demonstrated favorable preliminary kinase selectivity. The kinase selectivity of compound C05 was evaluated by screening against 9 kinases at a concentration of 0.5 μM, with inhibition percentages presented in Fig. 5. Notably, compound C05 exhibited an inhibition rate of 87.45% against PLK4 while demonstrating excellent selectivity over other PLK family members (e.g., PLK1, PLK3). Collectively, these findings indicate that compound C05 possesses a favorable kinase selectivity profile. Concurrently, western blot analysis demonstrated that compound C05 effectively suppressed PLK4 autophosphorylation at 4 μM, consistent with its functional disruption of centriole duplication (Fig. S1).
Fig. 5. Kinase selectivity of compound C05 at a concentration of 0.5 μM.

3.2.3. Flow cytometric analysis
Flow cytometry was employed to evaluate the apoptosis-inducing effects and cell cycle arrest activity of compound C05 on IMR-32 cells. Using LCR-263 as a positive control, the antiproliferative effect of C05 on IMR-32 cells was further assessed by flow cytometric analysis. C05 induced concentration-dependent apoptosis, with total apoptosis rates of 9.7%, 66.7%, and 75.0% at 0.5, 1, and 2 μM, respectively (Fig. 6). This activity significantly surpassed that of LCR-263, which exhibited a 7.9% apoptosis rate at 1 μM. Data are representative of at least 3 independent experiments.
Fig. 6. C05-induced apoptosis in IMR-32 cells.

Concentration-dependent cell cycle modulation by C05 was observed in IMR-32 cells, with increasing S/G2 phase populations (39.6%, 57.9%, and 61.8% at 0.5, 1, and 2 μM, respectively; Fig. 7) accompanied by proportional G1 phase reduction. This S/G2 arrest profile significantly exceeded that of LCR-263 (41.2% at 1 μM). The mitotic arrest was proposed to arise from C05-mediated PLK4 inhibition, thereby blocking centriole duplication and subsequent cell cycle progression. Western blot analysis26 revealed that compound C05 induces cell death by inhibiting the PLK4 signaling pathway. Treatment with 4 μM C05 led to the accumulation of the downstream protein SAS-6, further confirming PLK4 functional disruption. These mechanistic insights align with the compound's superior antiproliferative efficacy compared to the positive control. Data are representative of at least 3 independent experiments.
Fig. 7. C05-mediated cell cycle arrest in IMR-32 cells.

3.2.4. Cell clone formation experiment
Based on its antiproliferative activity, we selected compound C05 for a cell colony formation assay (Fig. 8). The compound demonstrated the ability to inhibit cell proliferation at a concentration of 1 μM. Complete suppression of MCF-7 cell proliferation was observed at a concentration of 4 μM, indicating a concentration-dependent inhibitory effect. Moreover, compound C05 exhibited superior antiproliferative activity against MCF-7 cells compared to the positive control, LCR-263. Data are representative of at least 3 independent experiments.
Fig. 8. C05-mediated cell cloning experiment in MCF-7 cells. Data are representative of at least 3 independent experiments.

3.2.5. Human liver microsomal stability assay
The in vitro metabolic stability of a compound constitutes a fundamental prerequisite for achieving and sustaining its therapeutic efficacy in vivo. As depicted in Table 5, the experimental data demonstrate that compound C05 exhibits an exceedingly rapid metabolic turnover in human liver microsomes, with an in vitro half-life (T1/2) of only 2.69 minutes. We hypothesize that the α-position of the carbonyl group and the exposed phenyl ring in C05 represent potential metabolic soft spots. In subsequent efforts, we plan to implement a cyclization strategy around the amide segment and introduce substituents at the para-position of the phenyl ring in an attempt to block these metabolic sites.
Table 5. Liver microsomal stability of compound C05.
| Compound | Parameters | R2 | T 1/2 (min) | CLint,in vitro (mL min−1 kg−1) |
|---|---|---|---|---|
| C05 | Human | 0.9660 | 2.69 | 0.515 |
4. Conclusion
In our previous studies, compound 28t demonstrated acceptable kinase inhibitory activity but exhibited poor cellular activity. Consequently, 28t was selected as the lead compound for subsequent optimization. Through functional group relocation and rational drug design principles, structural modifications were systematically implemented, leading to the discovery of optimized compound C05. This derivative exhibits significantly enhanced kinase inhibitory potency, achieving an IC50 value of less than 0.1 nM.
Cellular evaluation revealed C05's superior antiproliferative activity against IMR-32 (neuroblastoma; IC50 = 0.948 μM), MCF-7 (breast cancer; IC50 = 0.979 μM), and H460 (non-small cell lung cancer; IC50 = 1.679 μM), outperforming the positive control LCR-263. Notably, compound C05 demonstrated favorable kinase selectivity towards PLK4 among the 10 kinases tested, achieving an inhibition rate of 87.45%. Mechanistic studies via flow cytometry demonstrated C05's concentration-dependent apoptosis induction (66.7% at 1 μM) and S/G2-phase cell cycle arrest (57.9% at 1 μM) in IMR-32 cells, consistent with PLK4-mediated mitotic disruption. In the cell colony formation assay, compound C05 at a concentration of 1 μM demonstrated significantly superior inhibitory effects on MCF-7 cells compared to LCR-263. Western blot analysis demonstrated that compound C05 effectively suppressed PLK4 autophosphorylation at 4 μM. Unfortunately, compound C05 demonstrated poor metabolic stability in HLM, exhibiting a short half-life (T1/2) of 2.69 minutes under standard incubation conditions. We hypothesize that the metabolic instability of compound A05 may be attributed to the exposed phenyl ring and the unsubstituted α-position of the carbonyl group. Given its potent PLK4 inhibitory activity and favorable cellular potency, we plan to implement a cyclization strategy on the amide moiety of C05. This structural modification is expected to enhance metabolic stability while maintaining its biological activity.
5. Experimental section
5.1. Synthesis
Starting materials, reagents and solvents were obtained from a commercial supplier and used without further purification unless otherwise indicated. Anhydrous solvents were prepared and stored according to standard procedures. All reactions were monitored by thin-layer chromatography (TLC) on silica gel plates with fluorescence indicator F-254 and visualized with UV light. Column chromatography was performed on silica gel (200–300 mesh). 1H NMR and 13C NMR spectral data were recorded in DMSO-d6 or CDCl3 on a Bruker ARX-600 or a Bruker ARX-400 NMR spectrometer with TMS as an internal standard. Melting points were determined on a BüCHI Melting Point B-540 melting point apparatus. High-resolution accurate mass spectrometry (HRMS) determinations for all final target compounds were obtained on a Bruker micro mass time-of-flight mass spectrometer equipped with an electrospray ionization (ESI) detector.
5.1.1. Synthesis of 6-bromo-3-iodo-1H-indazole (M2)
A mixture of commercially available 6-bromo-1H-indazole (5.0 g, 25.38 mmol) and N-iodosuccinimide (NIS, 8.65 g, 38.06 mmol) in DMF was stirred at room temperature for 4 h. The reaction mixture was poured into water, and saturated aqueous ammonium chloride was added. The resulting precipitate was filtered, washed with water, and dried under vacuum to afford M2 as a white solid (5.93 g, 72.38% yield).
5.1.2. Synthesis of 6-bromo-3-iodo-1-(tetrahydro-2H-pyran-2-yl)-1H-indazole (M3)
A solution of intermediate M2 (3.0 g, 9.29 mmol) and p-toluenesulfonic acid (0.16 g, 0.93 mmol) in anhydrous dichloromethane was cooled to 0 °C. A dichloromethane solution of dihydropyran (DHP) was added dropwise, and the reaction mixture was stirred at room temperature for 4 h. The mixture was then poured into water, adjusted to weakly alkaline pH, and extracted with dichloromethane. The organic phase was concentrated under reduced pressure and purified by silica gel column chromatography. Trituration with methanol afforded M3 as a white solid (2.14 g, 56.59% yield).1H NMR (400 MHz, DMSO-d6) δ 8.11 (d, J = 1.2 Hz, 1H), 7.40 (d, J = 1.0 Hz, 2H), 5.89 (dd, J = 9.8, 2.4 Hz, 1H), 3.92–3.83 (m, 1H), 3.75 (ddd, J = 11.5, 6.2, 3.4 Hz, 1H), 2.34 (tdd, J = 13.2, 10.2, 3.4 Hz, 1H), 2.07–1.90 (m, 2H), 1.78–1.34 (m, 3H).
5.1.3. Synthesis of 6-bromo-3-(pyridin-3-ylethynyl)-1-(tetrahydro-2H-pyran-2-yl)-1H-indazole (M4)
A mixture of intermediate M3 (1.0 g, 2.46 mmol), 3-ethynylpyridine (0.31 g, 2.95 mmol), CuI (0.094 g, 0.49 mmol), and Pd(PPh3)4 (0.26 g, 0.25 mmol) in triethylamine was stirred under an argon atmosphere at room temperature for 5 h. The reaction mixture was extracted with ethyl acetate, and the organic phase was concentrated under reduced pressure. Trituration with a mixture of petroleum ether and ethyl acetate afforded M4 as a pale yellow solid (0.48 g, 50.47% yield). 1H NMR (600 MHz, DMSO-d6) δ 8.90 (s, 1H), 8.65 (d, J = 4.1 Hz, 1H), 8.19 (d, J = 1.1 Hz, 1H), 8.13 (dt, J = 7.9, 1.8 Hz, 1H), 7.89 (d, J = 8.5 Hz, 1H), 7.52 (dd, J = 7.8, 4.9 Hz, 1H), 7.48 (dd, J = 8.5, 1.5 Hz, 1H), 5.98 (dd, J = 9.4, 2.2 Hz, 1H), 3.88 (d, J = 11.5 Hz, 1H), 3.82–3.77 (m, 1H), 2.40–2.32 (m, 1H), 2.05–1.99 (m, 2H), 1.77–1.70 (m, 1H), 1.60 (dd, J = 7.3, 3.7 Hz, 2H). The synthesis of M20a–M20d is modeled after the above method.
5.1.4. Synthesis of 2-ethylhexyl 3-((3-(pyridin-3-ylethynyl)-1-(tetrahydro-2H-pyran-2-yl)-1H-indazol-6-yl)thio)propanoate (M5)
A mixture of intermediate M4 (1.0 g, 2.62 mmol), N,N-diisopropylethylamine (DIEA, 0.91 mL, 5.23 mmol), 2-ethylhexyl 3-mercaptopropanoate (1.07 mL, 4.71 mmol), bis(dibenzylideneacetone)palladium(0) [Pd2(dba)3, 0.12 g, 0.13 mmol], and 4,5-bis(diphenylphosphino)-9,9-dimethylxanthene (Xantphos, 0.15 g, 0.26 mmol) in toluene was heated under reflux at 110 °C for 5 h under an argon atmosphere. The mixture was concentrated under reduced pressure and purified by column chromatography to afford M5 as an orange-yellow oily liquid (1.05 g, 77.23% yield).1H NMR (400 MHz, DMSO-d6) δ 8.91 (d, J = 2.1 Hz, 1H), 8.67–8.65 (m, 1H), 8.09 (dt, J = 8.0, 1.9 Hz, 1H), 7.87–7.79 (m, 2H), 7.50 (dd, J = 7.9, 4.8 Hz, 1H), 7.30 (dd, J = 8.4, 1.4 Hz, 1H), 5.98 (dd, J = 9.3, 2.4 Hz, 1H), 3.98 (dd, J = 5.8, 1.9 Hz, 2H), 3.92 (dt, J = 11.5, 3.6 Hz, 1H), 3.81 (ddt, J = 11.3, 8.4, 4.5 Hz, 1H), 3.37 (t, J = 6.9 Hz, 2H), 2.75 (t, J = 6.9 Hz, 2H), 2.52–2.38 (m, 1H), 2.13–1.99 (m, 2H), 1.83–1.69 (m, 1H), 1.62 (h, J = 5.5, 4.6 Hz, 2H), 1.50 (h, J = 6.1 Hz, 1H), 1.36–1.15 (m, 8H), 0.84 (t, J = 7.5 Hz, 6H). Compounds M21a–M21d, M27a and M27b were synthesized following the aforementioned methodology.
5.1.5. Synthesis of N-ethyl-N-phenyl-2-((3-(pyridin-3-ylethynyl)-1-(tetrahydro-2H-pyran-2-yl)-1H-indazol-6-yl)thio)acetamide (M6a)
Intermediate M5 (0.5 g, 0.96 mmol) and sodium tert-butoxide (0.28 g, 2.89 mmol) were dissolved in tert-butanol and stirred at room temperature for 1 h. The mixture was then concentrated under reduced pressure. Using a one-pot strategy, a solution of 2-chloro-N-ethyl-N-phenylacetamide (0.38 g, 1.92 mmol) in THF was added dropwise to the residue, followed by stirring at room temperature for an additional hour. The crude product was purified by column chromatography to afford the desired compound as a yellow oil (0.40 g, 83.97% yield). The synthesis of intermediates M6b–M6i, M16a–M16d, M22a–M22d, M28a, M28b, M29a and M29b were performed analogously to the procedure described above.
5.1.6. Synthesis of N-ethyl-N-phenyl-2-((3-(pyridin-3-ylethynyl)-1H-indazol-6-yl)thio)acetamide (A01)
Intermediate M6a (0.035 g, 0.071 mmol) was dissolved in trifluoroacetic acid and stirred at room temperature for 1 h. The reaction mixture was then basified to weakly alkaline pH by addition of 2 N aqueous NaOH solution. The resulting mixture was extracted with ethyl acetate, and the organic layer was concentrated under reduced pressure. Purification by column chromatography afforded the title compound as a white solid (0.012 g, 42.50% yield). m.p. 165.1–166.2 °C. 1H NMR (600 MHz, DMSO-d6) δ 13.59 (s, 1H), 8.87 (s, 1H), 8.64 (d, J = 4.6 Hz, 1H), 8.09 (dt, J = 7.9, 1.9 Hz, 1H), 7.77 (d, J = 8.5 Hz, 1H), 7.52–7.48 (m, 2H), 7.39 (d, J = 5.9 Hz, 2H), 7.31 (s, 1H), 7.23 (s, 1H), 7.16–7.07 (m, 2H), 3.72–3.62 (m, 4H), 1.03 (t, J = 7.1 Hz, 3H). HRMS (ESI, m/z) calcd for C24H20N4NaOS [M + Na]+, 435.1259; found 412.1358. Compounds A02–A09, B01–B06, and C05–C08 were synthesized following an analogous procedure to that described for A01.
5.1.6.1. N-Isopropyl-N-phenyl-2-((3-(pyridin-3-ylethynyl)-1H-indazol-6-yl)thio)acetamide (A02)
White powdery solid, yield: 50.12%. m.p. 158.9–159.9 °C. 1H NMR (600 MHz, DMSO-d6) δ 13.55 (s, 1H), 8.87 (d, J = 2.2 Hz, 1H), 8.63 (dd, J = 4.9, 1.7 Hz, 1H), 8.09 (dt, J = 7.9, 1.9 Hz, 1H), 7.77 (d, J = 8.5 Hz, 1H), 7.52–7.43 (m, 4H), 7.37 (d, J = 1.6 Hz, 1H), 7.32–7.28 (m, 2H), 7.09 (dd, J = 8.5, 1.6 Hz, 1H), 4.81 (p, J = 6.8 Hz, 1H), 3.53 (s, 2H), 1.00 (d, J = 6.8 Hz, 7H). HRMS (ESI, m/z) calcd for C25H22N4NaOS [M + Na]+, 449.1438; found 426.1514.
5.1.6.2. N-Cyclopropyl-N-phenyl-2-((3-(pyridin-3-ylethynyl)-1H-indazol-6-yl)thio)acetamide (A03)
White powdery solid, yield: 73.86%. m.p. 96.9–97.9 °C. 1H NMR (600 MHz, DMSO-d6) δ 13.55 (s, 1H), 8.87 (d, J = 2.2 Hz, 1H), 8.63 (dd, J = 4.9, 1.6 Hz, 1H), 8.10 (dt, J = 7.9, 2.0 Hz, 1H), 7.80 (d, J = 8.5 Hz, 1H), 7.51 (dd, J = 7.9, 4.9 Hz, 2H), 7.41 (d, J = 8.5 Hz, 2H), 7.36–7.28 (m, 1H), 7.28–7.08 (m, 3H), 3.95–3.45 (m, 1H), 3.33 (s, 2H), 0.92–0.46 (m, 4H). HRMS (ESI, m/z) calcd for C25H20N4NaOS [M + Na]+, 447.1258; found 424.1358.
5.1.6.3. N-Ethyl-N-(4-methoxyphenyl)-2-((3-(pyridin-3-ylethynyl)-1H-indazol-6-yl)thio)acetamide (A04)
White powdery solid, yield: 55.79%. m.p. 136.3–137.3 °C. 1H NMR (600 MHz, DMSO-d6) δ 13.54 (s, 1H), 8.87 (d, J = 2.2 Hz, 1H), 8.63 (dd, J = 4.8, 1.7 Hz, 1H), 8.09 (dt, J = 7.9, 2.0 Hz, 1H), 7.77 (d, J = 8.4 Hz, 1H), 7.51 (dd, J = 7.9, 4.8 Hz, 1H), 7.40 (s, 1H), 7.31–7.25 (m, 2H), 7.10 (dd, J = 8.4, 1.6 Hz, 1H), 7.02–6.98 (m, 2H), 3.78 (s, 3H), 3.68–3.59 (m, 4H), 1.01 (t, J = 7.1 Hz, 3H). HRMS (ESI, m/z) calcd for C25H22N4NaO2S [M + Na]+, 465.1369; found 442.1463.
5.1.6.4. N-(4-Aminophenyl)-N-ethyl-2-((3-(pyridin-3-ylethynyl)-1H-indazol-6-yl)thio)acetamide (A05)
Light yellow powdery solid, yield: 62.05%. m.p. 85.2–86.2 °C. 1H NMR (600 MHz, DMSO-d6) δ 13.54 (s, 1H), 8.87 (d, J = 2.3 Hz, 1H), 8.63 (dd, J = 4.8, 1.7 Hz, 1H), 8.09 (dt, J = 7.9, 1.9 Hz, 1H), 7.77 (d, J = 8.5 Hz, 1H), 7.50 (dd, J = 7.9, 4.9 Hz, 1H), 7.40 (d, J = 1.5 Hz, 1H), 7.10 (dd, J = 8.5, 1.6 Hz, 1H), 7.00–6.94 (m, 2H), 6.62–6.54 (m, 2H), 5.32 (s, 2H), 3.65 (s, 2H), 3.57 (q, J = 7.1 Hz, 2H), 1.00 (t, J = 7.2 Hz, 3H). HRMS (ESI, m/z) calcd for C24H21N5NaOS [M + Na]+, 450.1360; found 427.1467.
5.1.6.5. N-(3-Aminophenyl)-N-ethyl-2-((3-(pyridin-3-ylethynyl)-1H-indazol-6-yl)thio)acetamide (A06)
Yellow powdery solid, yield: 43.87%. m.p. 191.4–192.4 °C. 1H NMR (600 MHz, DMSO-d6) δ 13.54 (s, 1H), 8.89–8.83 (m, 1H), 8.63 (dd, J = 4.9, 1.6 Hz, 1H), 8.09 (dt, J = 7.9, 2.0 Hz, 1H), 7.77 (d, J = 8.4 Hz, 1H), 7.50 (ddd, J = 7.9, 4.9, 1.0 Hz, 1H), 7.44 (s, 1H), 7.13–7.06 (m, 2H), 6.58 (dd, J = 7.9, 2.2 Hz, 1H), 6.50 (t, J = 2.1 Hz, 1H), 6.43 (dt, J = 7.4, 1.4 Hz, 1H), 5.34 (s, 2H), 3.69 (s, 2H), 3.61 (q, J = 7.1 Hz, 2H), 1.04 (t, J = 7.1 Hz, 3H). HRMS (ESI, m/z) calcd for C24H21N5NaOS [M + Na]+, 450.1361; found 427.1467.
5.1.6.6. N-Ethyl-N-(4-fluorophenyl)-2-((3-(pyridin-3-ylethynyl)-1H-indazol-6-yl)thio)acetamide (A07)
White powdery solid, yield: 81.55%. m.p. 161.6–162.6 °C. 1H NMR (400 MHz, DMSO-d6) δ 13.53 (s, 1H), 8.88–8.85 (m, 1H), 8.63 (dd, J = 4.9, 1.7 Hz, 1H), 8.09 (dt, J = 7.9, 1.9 Hz, 1H), 7.77 (d, J = 8.5 Hz, 1H), 7.51 (ddd, J = 8.0, 4.9, 1.0 Hz, 1H), 7.47–7.41 (m, 2H), 7.40 (s, 1H), 7.31 (t, J = 8.7 Hz, 2H), 7.10 (dd, J = 8.5, 1.5 Hz, 1H), 3.70–3.60 (m, 4H), 1.02 (t, J = 7.1 Hz, 3H). HRMS (ESI, m/z) calcd for C24H19FN4NaOS [M + Na]+, 453.1167; found 430.1264.
5.1.6.7. N-Ethyl-2-((3-(pyridin-3-ylethynyl)-1H-indazol-6-yl)thio)-N-(p-tolyl)acetamide (A08)
White powdery solid, yield: 45.24%. m.p. 77.3–78.3 °C. 1H NMR (600 MHz, DMSO-d6) δ 13.55 (s, 1H), 8.87 (d, J = 2.2 Hz, 1H), 8.63 (dd, J = 4.9, 1.7 Hz, 1H), 8.09 (dt, J = 7.9, 1.9 Hz, 1H), 7.77 (d, J = 8.5 Hz, 1H), 7.51 (dd, J = 7.9, 4.8 Hz, 1H), 7.40 (d, J = 1.5 Hz, 1H), 7.27–7.21 (m, 4H), 7.11–7.09 (m, 1H), 3.68–3.62 (m, 4H), 2.33 (s, 3H), 1.01 (t, J = 7.1 Hz, 3H). HRMS (ESI, m/z) calcd for C25H22N4NaOS [M + Na]+, 449.1418; found 426.1514.
5.1.6.8. N-(4-Chlorophenyl)-N-ethyl-2-((3-(pyridin-3-ylethynyl)-1H-indazol-6-yl)thio)acetamide (A09)
White powdery solid, yield: 49.80%. m.p. 162.4–163.4 °C. 1H NMR (400 MHz, DMSO-d6) δ 13.54 (s, 1H), 8.87 (d, J = 2.0 Hz, 1H), 8.63 (dd, J = 4.9, 1.7 Hz, 1H), 8.09 (dt, J = 7.9, 1.9 Hz, 1H), 7.77 (d, J = 8.5 Hz, 1H), 7.56–7.49 (m, 3H), 7.42 (d, J = 7.5 Hz, 3H), 7.11 (d, J = 8.5 Hz, 1H), 3.74–3.62 (m, 4H), 1.01 (t, J = 7.1 Hz, 3H). HRMS (ESI, m/z) calcd for C24H19ClN4NaOS [M + Na]+, 469.0865; found 446.0968.
5.1.6.9. N-Ethyl-2-phenyl-2-((3-(pyridin-3-ylethynyl)-1H-indazol-6-yl)thio)acetamide (B01)
White powdery solid, yield: 75.18%. m.p. 184.3–185.3 °C. 1H NMR (400 MHz, DMSO-d6) δ 13.59 (s, 1H), 8.87 (d, J = 2.2 Hz, 1H), 8.63 (dd, J = 4.9, 1.7 Hz, 1H), 8.35 (t, J = 5.5 Hz, 1H), 8.09 (dt, J = 7.9, 2.0 Hz, 1H), 7.81 (d, J = 8.5 Hz, 1H), 7.54–7.46 (m, 4H), 7.37–7.31 (m, 2H), 7.30–7.25 (m, 1H), 7.20 (dd, J = 8.5, 1.5 Hz, 1H), 5.19 (s, 1H), 3.15–2.94 (m, 2H), 0.93 (t, J = 7.2 Hz, 3H). HRMS (ESI, m/z) calcd for C24H20N4NaOS [M + Na]+, 435.1258; found 412.1358.
5.1.6.10. N-Methyl-2-phenyl-2-((3-(pyridin-3-ylethynyl)-1H-indazol-6-yl)thio)acetamide (B02)
White powdery solid, yield: 31.40%. m.p. 199.5–200.5 °C. 1H NMR (400 MHz, DMSO-d6) δ 13.57 (s, 1H), 8.87 (d, J = 2.1 Hz, 1H), 8.63 (dd, J = 4.8, 1.6 Hz, 1H), 8.31 (q, J = 4.5 Hz, 1H), 8.09 (dt, J = 7.9, 1.9 Hz, 1H), 7.86–7.76 (m, 1H), 7.54–7.50 (m, 2H), 7.49 (d, J = 3.5 Hz, 2H), 7.46 (d, J = 9.3 Hz, 1H), 7.33 (s, 1H), 7.31–7.25 (m, 1H), 7.19 (dd, J = 8.5, 1.5 Hz, 1H), 5.20 (s, 1H), 2.58 (dd, J = 4.6, 1.5 Hz, 3H). HRMS (ESI, m/z) calcd for C23H18N4NaOS [M + Na]+, 421.1102; found 398.1201.
5.1.6.11. Synthesis of 2-phenyl-2-((3-(pyridin-3-ylethynyl)-1H-indazol-6-yl)thio)acetamide (B03)
White powdery solid, yield: 51.19%. m.p. 283.6–284.6 °C. 1H NMR (400 MHz, DMSO-d6) δ 13.57 (s, 1H), 8.86 (d, J = 2.2 Hz, 1H), 8.63 (dd, J = 4.9, 1.7 Hz, 1H), 8.09 (dt, J = 7.9, 1.9 Hz, 1H), 7.81 (s, 1H), 7.79 (d, J = 3.7 Hz, 2H), 7.53 (d, J = 1.7 Hz, 1H), 7.52–7.49 (m, 1H), 7.49–7.47 (m, 1H), 7.34 (dd, J = 8.2, 6.3 Hz, 2H), 7.30 (d, J = 7.2 Hz, 1H), 7.27 (d, J = 1.7 Hz, 1H), 7.20 (dd, J = 8.4, 1.5 Hz, 1H), 5.21 (s, 1H). HRMS (ESI, m/z) calcd for C22H16N4NaOS [M + Na]+, 407.0937; found 384.1045.
5.1.6.12. N-Ethyl-2-(4-methoxyphenyl)-2-((3-(pyridin-3-ylethynyl)-1H-indazol-6-yl)thio)acetamide (B04)
White powdery solid, yield: 19.16%. m.p. 213.5–214.5 °C. 1H NMR (400 MHz, DMSO-d6) δ 13.59 (s, 1H), 8.87 (s, 1H), 8.63 (d, J = 4.9 Hz, 1H), 8.30 (t, J = 5.5 Hz, 1H), 8.09 (d, J = 8.0 Hz, 1H), 7.80 (d, J = 8.4 Hz, 1H), 7.56–7.46 (m, 2H), 7.43 (d, J = 8.3 Hz, 2H), 7.20 (d, J = 8.5 Hz, 1H), 6.89 (d, J = 8.3 Hz, 2H), 5.15 (s, 1H), 3.73 (s, 3H), 3.04 (ddt, J = 35.9, 13.0, 6.6 Hz, 2H), 0.92 (t, J = 7.2 Hz, 3H). HRMS (ESI, m/z) calcd for C25H22N4NaO2S [M + Na]+, 465.1381; found 442.1463.
5.1.6.13. N-Ethyl-2-(4-fluorophenyl)-2-((3-(pyridin-3-ylethynyl)-1H-indazol-6-yl)thio)acetamide (B05)
White powdery solid, yield: 55.17%. m.p. 218.6–219.6 °C. 1H NMR (400 MHz, DMSO-d6) δ 13.60 (s, 1H), 8.87 (d, J = 2.1 Hz, 1H), 8.63 (dd, J = 4.9, 1.6 Hz, 1H), 8.34 (t, J = 5.5 Hz, 1H), 8.09 (dt, J = 7.9, 2.0 Hz, 1H), 7.81 (d, J = 8.5 Hz, 1H), 7.56–7.52 (m, 2H), 7.51 (d, J = 3.8 Hz, 1H), 7.49 (d, J = 3.1 Hz, 1H), 7.18 (q, J = 8.8 Hz, 3H), 5.20 (s, 1H), 3.04 (dtd, J = 35.9, 13.3, 6.8 Hz, 2H), 0.93 (t, J = 7.2 Hz, 3H). HRMS (ESI, m/z) calcd for C24H19FN4NaOS [M + Na]+, 453.1173; found 430.1264.
5.1.6.14. 2-(4-Chlorophenyl)-N-ethyl-2-((3-(pyridin-3-ylethynyl)-1H-indazol-6-yl)thio)acetamide (B06)
White powdery solid, yield: 56.73%. m.p. 229.3–230.3 °C. 1H NMR (400 MHz, DMSO-d6) δ 13.60 (s, 1H), 8.87 (dd, J = 2.2, 0.9 Hz, 1H), 8.63 (dd, J = 4.9, 1.7 Hz, 1H), 8.36 (t, J = 5.5 Hz, 1H), 8.09 (dt, J = 7.9, 1.9 Hz, 1H), 7.82 (d, J = 8.4 Hz, 1H), 7.52 (d, J = 2.1 Hz, 1H), 7.51–7.49 (m, 2H), 7.49–7.47 (m, 1H), 7.41 (d, J = 2.1 Hz, 1H), 7.39 (d, J = 2.0 Hz, 1H), 7.20 (dd, J = 8.5, 1.5 Hz, 1H), 5.20 (s, 1H), 3.15–2.95 (m, 2H), 0.94 (t, J = 7.2 Hz, 3H). HRMS (ESI, m/z) calcd for C24H19ClN4NaOS [M + Na]+, 469.0889; found 446.0968.
5.1.6.15. (E)-N-Ethyl-N-phenyl-2-((3-(2-(pyridin-3-yl)vinyl)-1H-indazol-6-yl)thio)acetamide (C05)
White powdery solid, yield: 54.25%. m.p. 160.7–161.7 °C. 1H NMR (400 MHz, DMSO-d6) δ 13.19 (s, 1H), 8.88 (d, J = 2.2 Hz, 1H), 8.47 (dd, J = 4.8, 1.6 Hz, 1H), 8.16 (dt, J = 8.0, 2.0 Hz, 1H), 8.10 (d, J = 8.5 Hz, 1H), 7.65 (d, J = 16.8 Hz, 1H), 7.52 (s, 1H), 7.47 (d, J = 7.1 Hz, 2H), 7.42 (dd, J = 8.0, 4.5 Hz, 2H), 7.38–7.33 (m, 3H), 7.06 (d, J = 8.5 Hz, 1H), 3.74–3.61 (m, 4H), 1.03 (t, J = 7.1 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 167.09, 148.85 (2C), 142.49, 142.16, 141.85, 136.32, 135.00, 133.31, 133.10, 130.23, 128.82, 128.64, 126.47, 124.22, 122.83, 122.41, 121.53, 119.52, 109.49, 44.31, 37.16, 13.19. HRMS (ESI, m/z) calcd for C24H22N4NaOS [M + Na]+, 437.1416; found 414.1514.
5.1.6.16. (E)-N-Ethyl-N-phenyl-2-((3-(2-(pyridin-2-yl)vinyl)-1H-indazol-6-yl)thio)acetamide (C06)
White powdery solid, yield: 70.33%. m.p. 137.5–138.5 °C. 1H NMR (600 MHz, DMSO-d6) δ 13.24 (s, 1H), 8.60 (dd, J = 4.9, 1.8 Hz, 1H), 8.08 (d, J = 8.5 Hz, 1H), 7.91 (d, J = 16.3 Hz, 1H), 7.81 (td, J = 7.6, 1.9 Hz, 1H), 7.66 (d, J = 7.8 Hz, 1H), 7.54 (d, J = 16.3 Hz, 1H), 7.48 (t, J = 7.6 Hz, 2H), 7.41 (t, J = 7.4 Hz, 1H), 7.37 (d, J = 6.8 Hz, 3H), 7.27 (ddd, J = 7.6, 4.7, 1.1 Hz, 1H), 7.07 (d, J = 8.5 Hz, 1H), 3.72–3.64 (m, 4H), 1.03 (t, J = 7.1 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 167.09, 155.38, 149.99, 142.27 (2C), 141.85, 137.30, 135.03, 130.23, 129.49, 128.82, 128.64, 124.27, 123.05, 122.89, 122.58, 121.43, 119.62, 109.57, 44.31, 37.17, 13.20. HRMS (ESI, m/z) calcd for C24H22N4NaOS [M + Na]+, 437.1427; found 414.1514.
5.1.6.17. (E)-N-Ethyl-2-phenyl-2-((3-(2-(pyridin-3-yl)vinyl)-1H-indazol-6-yl)thio)acetamide (C07)
White powdery solid, yield: 45.37%. m.p. 221.3–222.3 °C. 1H NMR (400 MHz, DMSO-d6) δ 13.23 (s, 1H), 8.88 (d, J = 2.3 Hz, 1H), 8.47 (dd, J = 4.7, 1.6 Hz, 1H), 8.34 (t, J = 5.5 Hz, 1H), 8.18–8.10 (m, 2H), 7.65 (d, J = 16.8 Hz, 1H), 7.55–7.47 (m, 3H), 7.44–7.39 (m, 2H), 7.34 (dd, J = 8.2, 6.4 Hz, 2H), 7.30–7.23 (m, 1H), 7.16 (dd, J = 8.5, 1.5 Hz, 1H), 5.17 (s, 1H), 3.14–2.95 (m, 2H), 0.94 (t, J = 7.2 Hz, 3H). HRMS (ESI, m/z) calcd for C24H22N4NaOS [M + Na]+, 437.1452; found 414.1514.
5.1.6.18. (E)-N-Ethyl-2-phenyl-2-((3-(2-(pyridin-2-yl)vinyl)-1H-indazol-6-yl)thio)acetamide (C08)
White powdery solid, yield: 43.17%. m.p. 231.1–232.1 °C. 1H NMR (400 MHz, DMSO-d6) δ 13.27 (s, 1H), 8.60 (dd, J = 5.0, 1.7 Hz, 1H), 8.35 (t, J = 5.5 Hz, 1H), 8.11 (d, J = 8.5 Hz, 1H), 7.91 (d, J = 16.4 Hz, 1H), 7.81 (td, J = 7.7, 1.9 Hz, 1H), 7.66 (d, J = 7.8 Hz, 1H), 7.58–7.50 (m, 3H), 7.45 (d, J = 1.6 Hz, 1H), 7.38–7.31 (m, 2H), 7.30–7.24 (m, 2H), 7.17 (dd, J = 8.5, 1.6 Hz, 1H), 5.17 (s, 1H), 3.15–2.96 (m, 2H), 0.94 (t, J = 7.2 Hz, 3H). HRMS (ESI, m/z) calcd for C24H22N4NaOS [M + Na]+, 437.1445; found 414.1514.
5.1.7. Synthesis of N-(4-aminophenyl)-N-ethyl-2-((3-(pyridin-3-ylethynyl)-1-(tetrahydro-2H-pyran-2-yl)-1H-indazol-6-yl)thio)acetamide (M7a)
A mixture of M6h (0.055 g, 0.10 mmol) in a solvent system of methanol/water (2 : 0.5, v/v) was treated with iron powder (0.057 g, 1.01 mmol) and concentrated hydrochloric acid (0.016 mL, 0.20 mmol) under an argon atmosphere. The reaction mixture was stirred at room temperature for 12 h, then concentrated under reduced pressure. Purification by column chromatography afforded the title compound as a yellow oil (0.027 g, 51.99% yield). The synthesis of M7b was conducted analogously to the protocol described for M7a.
5.1.8. Synthesis of N-ethylaniline (M9a)
Commercially available M8a (1.96 mL, 21.46 mmol) was dissolved in methanol and cooled in an ice bath. Acetaldehyde (6.02 mL, 107.36 mmol) was added dropwise, and the mixture was stirred at 0 °C for 2 h. Subsequently, sodium borohydride (NaBH4, 2.44 g, 64.38 mmol) was added portionwise under ice-bath conditions, and stirring was continued for an additional 2 h. The reaction was quenched by slowly adding a mixture of methanol and water (1 : 1 v/v) to the mixture. The aqueous layer was extracted with ethyl acetate, and the combined organic layers were dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure. The crude product was purified by column chromatography to afford 1.08 g of a white solid (41.56% yield). The synthesis of M9b–M9g and M11 were conducted analogously to the protocol described for M9a.
5.1.9. Synthesis of N-cyclopropylaniline (M13)
A mixture of commercially available M12 (0.6 g, 2.94 mmol), cyclopropylamine (0.24 mL, 3.52 mmol), salicylaldoxime (0.081 g, 0.59 mmol), and cesium carbonate (2.40 g, 7.35 mmol) in acetonitrile was degassed and placed under an argon atmosphere. Copper(i) oxide (Cu2O, 0.042 g, 0.29 mmol) was added, and the reaction mixture was heated to 110 °C and refluxed for 12 h. After cooling, the mixture was concentrated under reduced pressure, diluted with ethyl acetate, and filtered through Celite® to remove insoluble residues. The filtrate was concentrated and purified by flash column chromatography to afford M13 as a white solid (0.057 g, 14.71% yield).
5.1.10. Synthesis of 2-chloro-N-ethyl-N-phenylacetamide (M14a)
A solution of M9a (1.5 g, 12.37 mmol) and triethylamine (3.44 mL, 24.76 mmol) in anhydrous dichloromethane (DCM) was cooled in an ice bath. A solution of chloroacetyl chloride in DCM was added dropwise under ice cooling, and the mixture was stirred at room temperature for 2 h after complete addition. The reaction mixture was concentrated under reduced pressure, and the crude product was purified by flash column chromatography to afford M14a as a yellowish-green oily liquid (1.96 g, 80.24% yield). Compounds M14b–M14i were synthesized analogously using the corresponding acyl chlorides.
5.1.11. Synthesis of N-ethyl-2-phenyl-2-((3-(pyridin-3-ylethynyl)-1-(tetrahydro-2H-pyran-2-yl)-1H-indazol-6-yl)thio)acetamide (M17a)
Intermediate M16a (0.26 g, 0.52 mmol) was dissolved in ethanol, and ethylamine (0.29 mL, 5.2 mmol) was added. The mixture was heated to 80 °C under reflux for 12 h, followed by concentration under reduced pressure. Purification by column chromatography (silica gel, gradient elution with hexane/ethyl acetate) afforded M18b as an off-white solid (0.085 g, 33.23% yield). Compounds M17b–M17d, M30a and M30b were synthesized following the aforementioned methodology.
5.1.12. Synthesis of 2-phenyl-2-((3-(pyridin-3-ylethynyl)-1-(tetrahydro-2H-pyran-2-yl)-1H-indazol-6-yl)thio)acetic acid (M18)
A solution of intermediate M16a (0.62 g, 1.24 mmol) in a 1 : 1 (v/v) methanol/water mixture was treated with NaOH (0.50 g, 12.4 mmol) and heated to 50 °C with stirring for 1 h. The reaction mixture was acidified to pH 4 with acetic acid, followed by concentration of the organic phase under reduced pressure to afford the title compound as a yellow viscous oil (0.50 g, 85.87% yield).
5.1.13. Synthesis of N-methyl-2-phenyl-2-((3-(pyridin-3-ylethynyl)-1-(tetrahydro-2H-pyran-2-yl)-1H-indazol-6-yl)thio)acetamide (M19a)
A mixture of intermediate M18 (0.25 g, 0.53 mmol) and HATU (0.41 g, 1.07 mmol) in dry DMF was cooled to 0 °C under an inert atmosphere. DIEA (0.28 mL, 1.59 mmol) was added dropwise, and the reaction was stirred at 0 °C for 30 min. Methylamine (0.21 mL, 5.32 mmol) was then introduced, and the mixture was allowed to warm to room temperature and stirred for 12 h. The crude product was extracted with ethyl acetate and water, and the organic layer was concentrated under reduced pressure. Purification by column chromatography (silica gel, gradient elution) afforded M19a as a yellow viscous oil (0.13 g, 50.16% yield). Compound M19b was prepared following an analogous procedure.
5.1.14. Synthesis of N-ethyl-2-((3-((4-(morpholinomethyl)phenyl)ethynyl)-1H-indazol-6-yl)thio)-N-phenylacetamide (C01)
A solution of M22a (0.11 g, 0.19 mmol) in a mixed solvent system of MeOH/H2O (3 : 0.5, v/v) was treated with p-toluenesulfonic acid (0.48 g, 2.78 mmol). The reaction mixture was heated at 80 °C under reflux conditions for 4 h. After cooling, the pH was adjusted to weakly basic conditions using saturated aqueous NaHCO3 solution. The resulting mixture was extracted with ethyl acetate (3 × 15 mL), and the combined organic layers were concentrated under reduced pressure. Purification by column chromatography (silica gel, ethyl acetate/hexane gradient elution) afforded the title compound as a white solid (0.034 g, 36.02% yield). m.p. 171.6–172.6 °C. 1H NMR (400 MHz, DMSO-d6) δ 13.44 (s, 1H), 7.71 (d, J = 8.5 Hz, 1H), 7.61 (d, J = 7.8 Hz, 2H), 7.48 (t, J = 7.5 Hz, 2H), 7.42 (d, J = 7.5 Hz, 2H), 7.38 (d, J = 4.2 Hz, 3H), 7.36 (s, 1H), 7.07 (d, J = 8.5 Hz, 1H), 3.66 (d, J = 5.5 Hz, 3H), 3.59 (t, J = 4.6 Hz, 5H), 3.51 (s, 2H), 2.37 (t, J = 4.7 Hz, 4H), 1.03 (t, J = 7.1 Hz, 3H). HRMS (ESI, m/z) calcd for C30H30N4NaO2S [M + Na]+, 533.1979; found 510.2089. Compounds C02–C04 were synthesized analogously following the procedure described for C01.
5.1.14.1. N-Ethyl-2-((3-((6-morpholinopyridin-3-yl)ethynyl)-1H-indazol-6-yl)thio)-N-phenylacetamide (C02)
Light yellow powdery solid, yield: 42.18%. m.p. 203.4–204.4 °C. 1H NMR (600 MHz, DMSO-d6) δ 13.37 (s, 1H), 8.41 (d, J = 2.4 Hz, 1H), 7.79 (dd, J = 8.9, 2.4 Hz, 1H), 7.70 (d, J = 8.5 Hz, 1H), 7.48 (t, J = 7.6 Hz, 2H), 7.42 (s, 1H), 7.37 (d, J = 9.3 Hz, 3H), 7.05 (d, J = 8.5 Hz, 1H), 6.90 (d, J = 8.9 Hz, 1H), 3.72–3.69 (m, 5H), 3.67 (s, 1H), 3.65 (s, 2H), 3.55 (d, J = 5.0 Hz, 4H), 1.02 (t, J = 7.1 Hz, 3H). HRMS (ESI, m/z) calcd for C28H27N5NaO2S [M + Na]+, 520.1781; found 497.1885.
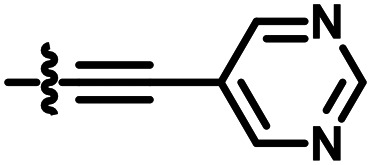
5.1.14.2. N-Ethyl-N-phenyl-2-((3-(pyrimidin-5-ylethynyl)-1H-indazol-6-yl)thio)acetamide (C03)
White powdery solid, yield: 39.97%. m.p. 166.4–167.4 °C. 1H NMR (400 MHz, DMSO-d6) δ 13.62 (s, 1H), 9.23 (s, 1H), 9.13 (s, 2H), 7.80 (d, J = 8.5 Hz, 1H), 7.49 (t, J = 7.4 Hz, 2H), 7.42 (d, J = 9.2 Hz, 2H), 7.38 (s, 1H), 7.36 (s, 1H), 7.11 (d, J = 8.6 Hz, 1H), 3.68 (d, J = 8.4 Hz, 4H), 1.03 (t, J = 7.1 Hz, 3H). HRMS (ESI, m/z) calcd for C23H19N5NaOS [M + Na]+, 436.1213; found 413.1310.
5.1.14.3. N-Ethyl-2-((3-((2-morpholinopyrimidin-5-yl)ethynyl)-1H-indazol-6-yl)thio)-N-phenylacetamide (C04)
White powdery solid, yield: 29.90%. m.p. 185.7–186.7 °C. 1H NMR (600 MHz, DMSO-d6) δ 13.43 (s, 1H), 8.68 (s, 2H), 7.71 (d, J = 8.5 Hz, 1H), 7.48 (t, J = 7.6 Hz, 2H), 7.41 (t, J = 7.4 Hz, 1H), 7.37 (d, J = 7.5 Hz, 3H), 7.06 (d, J = 8.5 Hz, 1H), 3.82–3.75 (m, 4H), 3.67 (dd, J = 11.1, 6.5 Hz, 8H), 1.02 (t, J = 7.1 Hz, 3H). HRMS (ESI, m/z) calcd for C27H26N6NaO2S [M + Na]+, 521.1724; found 498.1838.
5.1.15. Synthesis of 4-(4-((triisopropylsilyl)ethynyl)benzyl)morpholine (M24a)
Commercially available M23a (0.42 g, 1.64 mmol) was dissolved in triethylamine under an argon atmosphere. To this solution were added triphenylphosphine (PPh3, 0.042 g, 0.16 mmol), palladium(ii) acetate (Pd(OAc)2, 0.019 g, 0.082 mmol), and triisopropylsilylacetylene (0.42 g, 1.64 mmol). The reaction mixture was heated at 100 °C under reflux for 10 h. After cooling, the mixture was extracted with ethyl acetate, and the combined organic layers were concentrated under reduced pressure. Purification by column chromatography (silica gel, ethyl acetate/hexane gradient elution) afforded the title compound as a yellow oily liquid (0.32 g, 54.27% yield). Compounds M24b–M24d were prepared following the analogous procedure described for M24a.
5.1.16. Synthesis of 4-(4-ethynylbenzyl)morpholine (M25a)
A solution of M24a (0.32 g, 0.89 mmol) in THF was treated with TBAF (0.70 g, 2.67 mmol) and stirred at room temperature for 2 h. The mixture was extracted with ethyl acetate, and the organic phase was concentrated under reduced pressure to afford the title compound as an orange oil (0.15 g, 83.8% yield). Compounds M25b–M25d were synthesized following an analogous protocol.
5.1.17. Synthesis of (E)-6-bromo-3-(2-(pyridin-3-yl)vinyl)-1-(tetrahydro-2H-pyran-2-yl)-1H-indazole (M26a)
A mixture of intermediate M3 (3.0 g, 7.37 mmol) and DIEA (3.84 mL, 22.11 mmol) in dry DMF was degassed under argon. To this solution were added 3-vinylpyridine (1.02 mL, 9.57 mmol), P(o-tol)3 (0.26 g, 0.737 mmol), and Pd(OAc)2 (0.083 g, 0.37 mmol). The reaction was heated to 100 °C and stirred for 5 h under argon. The mixture was extracted with ethyl acetate, and the organic layer was concentrated under reduced pressure. Purification by column chromatography afforded the title compound as a pale yellow solid (1.26 g, 44.5% yield). Compound M26b was synthesized following an analogous procedure.
6. Pharmacological assay
6.1. PLK4 LanthaScreeen Eu kinase binding assay
PLK4 recombinant human protein, kinase tracer 236 and LanthaScreen Eu-antiGST Antibody were purchased from Thermo Fisher Scientific (USA), and LCR-263 (TargetMol, Catalog No. T14927) was used as the positive control. LanthaScreeen Eu kinase binding assays were performed according to the manufacturer's instructions. The tested compounds were initially dissolved in 100% DMSO to prepare 4 mM stock solutions and then diluted to the desired concentrations with 1× kinase buffer to generate concentration gradients. Secondly, PLK4 protein, Eu-anti-GST antibody and Kinase Tracer 236 were diluted with 1× kinase buffer to the concentration of 2 nM, 4 nM, 4 nM, respectively. For each assay, 4 μL of diluted compound, 8 μL of kinase/antibody mixture and 4 μL of tracer solution were added to the assay wells in sequence. The assay plates were incubated at 25 °C for 60 min and then the signals were obtained by reading plates using an Infinite® F500 microplate reader (Tecan, Switzerland). The acceptor/tracer emission (665 nM) was divided by the antibody/donor emission (615 nM) to calculate the “emission ratio”. The data were analyzed using GraphPad Prism 8. Each experiment was repeated at least two times.
6.2. Cell antiproliferation assay
MCF-7, IMR-32 and H460 cells were seeded into 96-well plates at respective densities of 1000, 10 000, and 1000 cells per well in 100 μL of corresponding complete culture medium and maintained at 37 °C in a humidified atmosphere with 5% CO2 for 24 h. Serially diluted test compounds were added into each well and incubated for an additional 5 days. Then, cell viability was determined using metabolic assays: MTT for MCF-7 and H460 cells, and CCK-8 for IMR-32 cells, following with absorbance measurements using an Infinite® F500 microplate reader (Tecan, Switzerland) (MTT, λ = 492 nm; CCK8, λ = 450 nm). The IC50 values were calculated by concentration–response curve fitting using GraphPad Prism 8.
6.3. Cell apoptosis analysis
MCF-7 cells (2.0 × 106 cells per well) were initially seeded in 6-well plates and cultured for 24 h, then treated with DMSO or test compounds at the corresponding concentrations for 48 h. Following treatment, cells were harvested and washed with cold PBS before undergoing dual staining with annexin V–FITC and propidium iodide (PI) using a commercial apoptosis detection kit (Beyotime Biotechnology) according to the manufacturer's instructions. After staining, flow cytometry (Guava easyCyte, Merck) was performed to quantify the number of apoptotic cells (FITC–annexin V: Abs/Em = 494/518 nm, PI: Abs/Em = 535/617 nm).
6.4. Cell cycle analysis
MCF-7 cells (2.0 × 106 cells per well) were initially seeded in 6-well plates and incubated under standard culture conditions for 24 h, then treated with DMSO or test compounds at the corresponding concentrations for 48 h. After treatment, cells were harvested, washed twice with cold PBS and fixed with ethanol (70%) at 4 °C overnight. The cells were centrifuged to remove the fixative solution and washed twice with cold PBS, then subsequently stained with a cell-cycle and apoptosis analysis kit (Beyotime Biotechnology) according to the manufacturer's instructions at 37 °C in the dark for 30 min. Finally, the cells were analyzed using a flow cytometer (Guava easyCyte, Merck).
6.5. Clonogenic assay
MCF-7 cells were seeded in 6-well plates at a density of 2000 cells per well and incubated at 37 °C in a humidified atmosphere with 5% CO2 for 24 h, then treated with 0.5, 1, 2 and 4 μM compound C05 or 1 μM compound LCR-263, and 0.1% DMSO was the vehicle control. The treatment medium containing compound or DMSO was refreshed every 3 days. After 14 days of incubation, the cells were washed twice with 1× PBS, fixed with formalin for 30 min, stained with cresyl violet (Beyotime Biotechnoligy) for 20 min, and rinsed thoroughly with distilled water before being photographed. The colony number was counted using ImageJ.
6.6. Western blot analysis
IMR-32 cells were treated with 4 μM compound C05 for 48 h, followed by preparation of whole-cell lysates. The lysates were resolved by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and electrotransferred onto nitrocellulose membranes. Membranes were probed with primary antibodies against PLK4 (1 : 1500), SAS-6 (1 : 1500), and GAPDH (1 : 10 000), then incubated with horseradish peroxidase (HRP)-conjugated secondary antibody (anti-rabbit, 1 : 10 000). Immunoreactive proteins were visualized using enhanced chemiluminescence (ECL) detection reagents.
6.7. Human liver microsomal stability assay
Human liver microsomal working solution (445 μL) was added to a prewarmed T60/NCF60 assay plate and incubated at 37 °C with orbital shaking for 10 min. Subsequently, 5 μL of the test compound was introduced, followed by thorough mixing and immediate transfer of 54 μL aliquots to a black polypropylene plate. The reaction was initiated by sequential addition of 6 μL NADPH regenerating system and 180 μL quenching solution (acetonitrile : methanol, 50 : 50 (v/v) containing internal standard). For parallel control incubations, NCF60 plates received 50 μL buffer replacement, while T60 plates were supplemented with 44 μL NADPH prior to 37 °C incubation for 1 h.
Time-dependent sampling was performed at 5, 15, 30 and 60 min by adding 180 μL ice-cold quenching solution to each well, followed by extraction of 60 μL supernatant after centrifugation (4000 rpm, 4 °C, 20 min). The collected supernatant (20 μL) was diluted with 60 μL deionized water, vortex-mixed for 10 min, and subjected to automated injection for ultra-performance liquid chromatography–tandem mass spectrometry (UPLC-MS/MS) analysis.
6.8. Molecular docking studies
The molecular docking experiment was performed using Schrodinger's Maestro13.5 for pre-docking, and the crystal structure of PLK4 protein (PDB 4YUR) was downloaded from the PDB database. During the docking process, water molecules are excluded and other parameters are set by software default. Firstly, the protein structure (PDB 4YUR) was imported and the Protein Preparation Wizard module was used for protein preparation. Then, the Ligprep module was used to perform conformational optimization on the imported subsets, and docking boxes were generated using the ReceptorGridGeneration module. Finally, LigandDocking molecular docking was used. The results after docking are visualized using PyMOL3.0.
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Supplementary Material
Acknowledgments
We gratefully acknowledge the Program for Innovative Research Team of the Ministry of Education and the Program for Liaoning Innovative Research Team in University.
Data availability
Supplementary information is available. See DOI: https://doi.org/10.1039/D5MD00654F.
Part of the data supporting this article have been included in the SI. All the data in this article are true.
References
- Sheaff R. J., Kennedy S. and Berrett H., Disruption of Cell Cycle Machinery in Pancreatic Cancer, In Pancreatic Cancer – Molecular Mechanism and Targets, ed. S. K. Srivas-tava, IntechOpen, Rijeka, 2012 [Google Scholar]
- Manning G. Whyte D. B. Martinez R. Hunter T. Sudarsanam S. The Protein Kinase Complement of the Human Genome. Science. 2002;298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- de Cárcer G. Gerard M. Malumbres M. From Plk1 to Plk5. Cell Cycle. 2011;10:2255–2262. doi: 10.4161/cc.10.14.16494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elia A. E. H. Cantley L. C. Yaffe M. B. Proteomic Screen Finds pSer/pThr-Binding Domain Localizing Plk1 to Mitotic Substrates. Science. 2003;299:1228–1231. doi: 10.1126/science.1079079. [DOI] [PubMed] [Google Scholar]
- Clay F. J. McEwen S. J. Bertoncello I. Wilks A. F. Dunn A. R. Identification and cloning of a protein kinase-encoding mouse gene, Plk, related to the polo gene of Drosophila. Proc. Natl. Acad. Sci. U. S. A. 1993;90:4882–4886. doi: 10.1073/pnas.90.11.4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma S. Jean C. Erikson R. L. Role of Plk2 (Snk) in Mouse Development and Cell Proliferation. Mol. Cell. Biol. 2003;23:6936–6943. doi: 10.1128/MCB.23.19.6936-6943.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtrich U. Wolf G. Bräuninger A. Karn T. Böhme B. Rübsamen-Waigmann H. Induction and down-regulation of PLK, a human serine/threonine kinase expressed in proliferating cells and tumors. Proc. Natl. Acad. Sci. U. S. A. 1994;91:1736–1740. doi: 10.1073/pnas.91.5.1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donohue P. J. Alberts G. F. Guo Y. Winkles J. A. Identification by Targeted Differential Display of an Immediate Early Gene Encoding a Putative Serine/Threonine Kinase. J. Biol. Chem. 1995;270:10351–10357. doi: 10.1074/jbc.270.17.10351. [DOI] [PubMed] [Google Scholar]
- Hudson J. W. Chen L. Fode C. Binkert C. Dennis J. W. Sak kinase gene structure and transcriptional regulation. Gene. 2000;241:65–73. doi: 10.1016/s0378-1119(99)00467-9. [DOI] [PubMed] [Google Scholar]
- Lowery D. Lim D. Yaffe M. Structure and function of Polo-like kinases. Oncogene. 2005;24:248–259. doi: 10.1038/sj.onc.1208280. [DOI] [PubMed] [Google Scholar]
- Fode C. Motro B. Yousefi S. Heffernan M. Dennis J. W. Sak, a murine protein-serine/threonine kinase that is related to the Drosophila polo kinase and involved in cell proliferation. Proc. Natl. Acad. Sci. U. S. A. 1994;91:6388–6392. doi: 10.1073/pnas.91.14.6388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettencourt-Dias M. Rodrigues-Martins A. Carpenter L. Riparbelli M. Lehmann L. Gatt M. K. SAK/PL-K4 is required for centriole duplication and flagella development. Curr. Biol. 2005;15:2199–2207. doi: 10.1016/j.cub.2005.11.042. [DOI] [PubMed] [Google Scholar]
- Habedanck R. Stierhof Y.-D. Wilkinson C. J. Nigg E. A. The Polo kinase Plk4 functions in centriole duplication. Nat. Cell Biol. 2005;7:1140–1146. doi: 10.1038/ncb1320. [DOI] [PubMed] [Google Scholar]
- Arquint C. Nigg E. A. The PLK4–STIL–SAS-6 module at the core of centriole duplication. Biochem. Soc. Trans. 2016;44:1253–1263. doi: 10.1042/BST20160116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hori A. Barnouin K. Snijders A. P. Toda T. A non-canonical function of Plk4 in centriolar satellite int-egrity and ciliogenesis through PCM1 phosphorylation. EMBO Rep. 2016;17:326–337. doi: 10.15252/embr.201541432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtani N. Yamakoshi K. Takahashi A. Hara E. Real-time in vivo imaging of p16Ink4agene expressio-n: a new approach to study senescence stress signaling in living animals. Cell Div. 2010;5:1. doi: 10.1186/1747-1028-5-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakami M. Mustachio L. M. Zheng L. Chen Y. Rodriguez-Canales J. Mino B. Polo-like kinase 4 inhibition produces polyploidy and apoptotic death of lung cancers. Proc. Natl. Acad. Sci. U. S. A. 2018;115:1913–1918. doi: 10.1073/pnas.1719760115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lara G. G. Cipreste M. F. Andrade G. F. Silva W. M. Sousa E. M. Response of Fibroblasts MRC-5 to Flufenamic Acid-Grafted MCM-41 Nanoparticles. Bioengineering. 2018;5:1–38. doi: 10.3390/bioengineering5010004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson E. F. Stewart K. D. Woods K. W. Giranda V. L. Luo Y. Pharmacological and functional comparison of the polo-like kinase family: insight into inhibitor and substrate specificity. Biochemistry. 2007;46:9551–9563. doi: 10.1021/bi7008745. [DOI] [PubMed] [Google Scholar]
- Laufer R. Forrest B. Li S.-W. Liu Y. Sampson P. Edwards L. et al., The Discovery of PLK4 Inhibitors: (E)-3-((1H-Indazol-6-yl)methylene)indolin-2-ones as Novel Antiproliferative Agents. J. Med. Chem. 2013;56:6069–6087. doi: 10.1021/jm400380m. [DOI] [PubMed] [Google Scholar]
- Sampson P. B. Liu Y. Patel N. K. Feher M. Forrest B. Li S.-W. The Discovery of Polo-Like Kinase 4 Inhibitors: Design and Optimization of Spiro[cyclopropane-1,3′[3H]indol]-2′(1′H)-ones as Orally Bioavailable Antitumor Agents. J. Med. Chem. 2015;58:130–146. doi: 10.1021/jm5005336. [DOI] [PubMed] [Google Scholar]
- Mason J. M. Lin D. C. Wei X. Che Y. Yao Y. Kiarash R. Functional characterization of CFI-400945, a Polo-like kinase 4 inhibitor, as a potential anticancer agent. Cancer Cell. 2014;26:163–176. doi: 10.1016/j.ccr.2014.05.006. [DOI] [PubMed] [Google Scholar]
- Harrington E. A. Bebbington D. Moore J. Rasmussen R. K. Ajose-Adeogun A. O. Nakayama T. VX-680, a potent and selective small-molecule inhibitor of the Aurora kinases, suppresses tumor growth in vivo. Nat. Med. 2004;10:262–267. doi: 10.1038/nm1003. [DOI] [PubMed] [Google Scholar]
- Wong Y. L. Anzola J. V. Davis R. L. Yoon M. Motamedi A. Kroll A. Cell biology. Reversible centriole depletion with an inhibitor of Polo-like kinase 4. Science. 2015;348:1155–1160. doi: 10.1126/science.aaa5111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y. Sun Y. Wang L. Wu T. Yin W. Wang J. et al., Design, synthesis, and biological evaluation of novel pyrazolo [3,4-d]pyrimidine derivatives as potent PLK4 inhibitors for the treatment of TRIM37-amplified breast cancer. Eur. J. Med. Chem. 2022;238:114424. doi: 10.1016/j.ejmech.2022.114424. [DOI] [PubMed] [Google Scholar]
- Sun Y. Xue Y. Liu H. Mu S. Sun P. Sun Y. et al., Discovery of CZS-241: A Potent, Selective, and Orally Available Polo-Like Kinase 4 Inhibitor for the Treatment of Chronic Myeloid Leukemia. J. Med. Chem. 2023;66:2396–2421. doi: 10.1021/acs.jmedchem.2c02124. [DOI] [PubMed] [Google Scholar]
- Liu N. Hu N. Fan C. Tong M. Shi X. Wang H. et al., Rational Design of CZL-S092: A Novel Indazole-Based PLK4 Inhibitor Targeting Neuroblastoma through Virtual Screening and Fragment-Based Drug Design Strategies. Eur. J. Med. Chem. 2025:117867. doi: 10.1016/j.ejmech.2025.117867. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Supplementary information is available. See DOI: https://doi.org/10.1039/D5MD00654F.
Part of the data supporting this article have been included in the SI. All the data in this article are true.