

Abstract
The redox reaction between CrO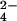 and the fully reduced three-heme cytochrome c7 from Desulfuromonas acetoxidans to give chromium(III) and the fully oxidized protein has been followed by NMR spectroscopy. The hyperfine coupling between the oxidized protein protons and chromium(III), which remains bound to the protein, gives rise to line-broadening effects on the NMR resonances that can be transformed into proton-metal distance restraints. Structure calculations based on these unconventional constraints allowed us to demonstrate that chromium(III) binds at a unique site and to locate it on the protein surface. The metal ion is located 7.9 ± 0.4 Å from the iron of heme IV, 16.3 ± 0.7 Å from the iron of heme III, and 22.5 ± 0.5 Å from the iron of heme I. Shift changes caused by the presence of unreactive MoO
and the fully reduced three-heme cytochrome c7 from Desulfuromonas acetoxidans to give chromium(III) and the fully oxidized protein has been followed by NMR spectroscopy. The hyperfine coupling between the oxidized protein protons and chromium(III), which remains bound to the protein, gives rise to line-broadening effects on the NMR resonances that can be transformed into proton-metal distance restraints. Structure calculations based on these unconventional constraints allowed us to demonstrate that chromium(III) binds at a unique site and to locate it on the protein surface. The metal ion is located 7.9 ± 0.4 Å from the iron of heme IV, 16.3 ± 0.7 Å from the iron of heme III, and 22.5 ± 0.5 Å from the iron of heme I. Shift changes caused by the presence of unreactive MoO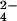 , a CrO
, a CrO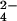 analogue, indicate the involvement of the same protein area in the anion binding. The titration of the oxidation of cytochrome c7 shows a detailed mechanism of action. The presence of a specific binding site supports the hypothesis of the biological role of this cytochrome as a metal reductase.
analogue, indicate the involvement of the same protein area in the anion binding. The titration of the oxidation of cytochrome c7 shows a detailed mechanism of action. The presence of a specific binding site supports the hypothesis of the biological role of this cytochrome as a metal reductase.
Sulfur- and sulfate-reducing bacteria constitute a group of anaerobic organisms, the common feature of which is the use of sulfur and its oxidized forms as electron acceptors. Thiosulfate, sulfur, sulfite, or sulfate “respiration” produces sulfide. The end product of the reaction, hydrogen sulfide, can react with heavy metal ions to form less toxic insoluble metal sulfides (1). These bacteria are also able to enzymatically reduce and precipitate heavy metals (2–8). They therefore are important for the geochemical cycle of metals and become particularly relevant for possible applications in the decontamination of environments polluted by toxic heavy metals through bioreduction coupled with precipitation as insoluble sulfides. Among the metals that can be reduced by these bacteria, chromium(VI) is particularly relevant from the technological and environmental point of view, because it represents one of the most common polluting metals and is highly soluble and toxic.§
The three heme-containing cytochrome c7 from the sulfur-reducing bacterium Desulfuromonas acetoxidans (Cyt c7 hereafter) has been proposed to have a role as electron-transfer protein in the sulfur metabolism of this bacterium, acting as a terminal reductase in the metabolic pathway by directly reducing elemental sulfur to sulfide (9); it has been suggested also that it could be involved in the reduction of iron(III) and manganese(IV) (10). The solution structures of the fully oxidized and fully reduced species are available, followed by the x-ray structure of the oxidized species (11–14). The availability of the assigned NMR spectra and the nuclear Overhauser effects (NOEs; refs. 11 and 15) prompted us to study by NMR the reaction between CrO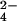 and fully reduced Cyt c7. The products of the reaction are chromium(III) and the oxidized protein with three iron(III) hemes. Because chromium(III) is kinetically inert with respect to ligand exchange, it remains bound to protein residues through which electron transfer occurs. The hyperfine coupling between the unpaired electrons of chromium(III) and the proton nuclei of the protein gives rise to particular features in the NMR spectra (16) that could be used to obtain structural information about the chromium(III) binding site. The redox-inert MoO
and fully reduced Cyt c7. The products of the reaction are chromium(III) and the oxidized protein with three iron(III) hemes. Because chromium(III) is kinetically inert with respect to ligand exchange, it remains bound to protein residues through which electron transfer occurs. The hyperfine coupling between the unpaired electrons of chromium(III) and the proton nuclei of the protein gives rise to particular features in the NMR spectra (16) that could be used to obtain structural information about the chromium(III) binding site. The redox-inert MoO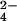 was used to map the binding site of the anion. NMR has allowed us to obtain a clear picture of the mechanism of the reaction and the metal–substrate interaction site around the protein surface. The present results reinforce the hypothesis that Cyt c7 is an enzyme with metal reductase activity.
was used to map the binding site of the anion. NMR has allowed us to obtain a clear picture of the mechanism of the reaction and the metal–substrate interaction site around the protein surface. The present results reinforce the hypothesis that Cyt c7 is an enzyme with metal reductase activity.
Materials and Methods
NMR Sample Preparation.
Cyt c7 from D. acetoxidans was obtained and purified as reported (11). The 1H NMR sample was prepared by dissolving the lyophilized protein in 100 mM phosphate buffer to give a 1 mM solution that was deoxygenated under argon atmosphere and then saturated with hydrogen. Reduction of the protein was achieved by adding a deoxygenated solution of Desulfovibrio vulgaris Hildenborough iron hydrogenase in a catalytic amount. A 10 mM solution of CrO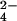 was obtained by dissolving the K2CrO4 in 100 mM phosphate buffer at pH 6.5. At this pH the dominant species is chromate. A 10 mM solution of molybdate was obtained by dissolving the Na2MoO4⋅2H2O in 100 mM phosphate buffer at pH 6.5. The protein solution was titrated by the addition of increasing amounts of the CrO
was obtained by dissolving the K2CrO4 in 100 mM phosphate buffer at pH 6.5. At this pH the dominant species is chromate. A 10 mM solution of molybdate was obtained by dissolving the Na2MoO4⋅2H2O in 100 mM phosphate buffer at pH 6.5. The protein solution was titrated by the addition of increasing amounts of the CrO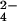 solution directly in the NMR tube. The CrO
solution directly in the NMR tube. The CrO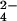 solution was kept under argon atmosphere to avoid the presence of oxygen in the NMR tube. The same procedure was used to prepare the protein–molybdate adduct.
solution was kept under argon atmosphere to avoid the presence of oxygen in the NMR tube. The same procedure was used to prepare the protein–molybdate adduct.
NMR Spectra.
The 1H NMR spectra were recorded on a Bruker AVANCE 700 spectrometer operating at 700.13 MHz and 298 K in H2O solution. One-dimensional 1H NMR spectra were acquired on a spectral width of 50 ppm to detect the hyperfine shifted signals of the heme methyls and iron axial ligands, with presaturation of the solvent resonance during the relaxation delay. Time-proportional phase increment NOE spectroscopy (17, 18) spectra were recorded on a spectral width of 50 ppm with a recycle time of 275 ms and a mixing time of 100 ms. These spectra were obtained by presaturating the residual water resonance during the relaxation delay and mixing time. To optimize the observation of connectivities in the diamagnetic region, time-proportional phase increment NOE spectroscopy (18) spectra were recorded on a spectral width of 22 ppm with a recycle time of 658 ms, a mixing time of 100 ms, and water suppression by gradient-tailored excitation (WATERGATE; ref. 19).
Data processing was performed by using a standard Bruker software package. The two-dimensional maps were analyzed with the aid of the program XEASY (20).
Signals line widths were measured manually at half height with Bruker XWINNMR software.
NMR Constraints and Structure Calculations.
Chromium(III) induces a line-broadening effect on the NMR resonances of nearby nuclei according to the following relationship (16, 21),
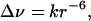 |
1 |
where Δν is the measured increment in line width caused by the presence of chromium(III), and r is the chromium(III)-proton distance. The proportionality constant k can be found experimentally through calibration by treating line broadening as an NOE-derived upper distance limit. To determine upper distance limits between the chromium(III) and protein protons, we used an approach similar to the Caliba procedure designed by Wüthrich and coworkers to calibrate 1H–1H dipolar constraints (22) and extended to the calibration of longitudinal nonselective relaxation times of protons by Bertini et al. (23, 24).
The procedure used in the present case can be summarized as follows:
(i) A first model structure was calculated by imposing distance constraints evaluated assuming that the metal-proton interaction of Eq. 1 is dipolar in origin and that the dominant correlation time for the interaction is the rotational correlation time of the protein estimated from the Stokes–Einstein equation. The 35 NMR solution structures of fully oxidized Cyt c7 (12) were annealed in 10,000 steps with the program DYANA (25) by adding the chromium-proton distance constraints to the other available constraints (12).
(ii) The metal-proton distances thus obtained in the structural model were plotted against (Δν)−1/6. A calibration curve was defined and used for the determination of the new distance constraints. Upper and lower distance limits were chosen in such a way to impose distance constraints with a 20% tolerance. The upper and lower values of the k constant (k = 5.50 × 10−51 Hz⋅m6) differ from the value of the k constant in step i by 25%. Structure calculations were performed starting from random structures.
Results and Discussion
1H one-dimensional and two-dimensional NOE spectroscopy spectra were recorded for a CrO concentration corresponding to 1/3, 2/3, and 3/3 of the protein concentration. The effect on the one-dimensional spectra is reported in Fig. 1. The reduced protein, containing three iron(II) hemes, undergoes stepwise oxidation after the addition of CrO
concentration corresponding to 1/3, 2/3, and 3/3 of the protein concentration. The effect on the one-dimensional spectra is reported in Fig. 1. The reduced protein, containing three iron(II) hemes, undergoes stepwise oxidation after the addition of CrO . The hemes are labeled I, III, and IV in analogy to the labeling of the four-heme Cyt c3, which is more characterized (26–30). The heme II is missing in the present protein, Cyt c7. Sensible line broadening is observed after oxidation in the hyperfine shifted region of the spectrum, particularly after addition of the first third of the oxidizing agent, with the iron-ligand resonances broadened beyond detection (Fig. 1C). The resonances corresponding to the reduced heme methyls are barely detectable for hemes I and III, whereas those of heme IV show only very minor shifts. Addition of the second third gives rise to very broad resonances between 25 and 10 ppm, with chemical shifts corresponding to those of the methyls of hemes I and III in the fully oxidized protein but again broadened (Fig. 1D). At the same time the resonances of the reduced heme IV are still detectable in the diamagnetic region of the spectrum. Addition of the oxidizing agent to a final concentration equal to that of the protein restores the original spectrum of the fully oxidized protein as far as the number of detectable heme and axial ligand resonances and chemical shifts are concerned (Fig. 1E). However, the line widths of 27 resonances remain larger than those observed when Cyt c7 is oxidized by the air (ref. 31; Fig. 1A).
. The hemes are labeled I, III, and IV in analogy to the labeling of the four-heme Cyt c3, which is more characterized (26–30). The heme II is missing in the present protein, Cyt c7. Sensible line broadening is observed after oxidation in the hyperfine shifted region of the spectrum, particularly after addition of the first third of the oxidizing agent, with the iron-ligand resonances broadened beyond detection (Fig. 1C). The resonances corresponding to the reduced heme methyls are barely detectable for hemes I and III, whereas those of heme IV show only very minor shifts. Addition of the second third gives rise to very broad resonances between 25 and 10 ppm, with chemical shifts corresponding to those of the methyls of hemes I and III in the fully oxidized protein but again broadened (Fig. 1D). At the same time the resonances of the reduced heme IV are still detectable in the diamagnetic region of the spectrum. Addition of the oxidizing agent to a final concentration equal to that of the protein restores the original spectrum of the fully oxidized protein as far as the number of detectable heme and axial ligand resonances and chemical shifts are concerned (Fig. 1E). However, the line widths of 27 resonances remain larger than those observed when Cyt c7 is oxidized by the air (ref. 31; Fig. 1A).
Figure 1.

The 1H one-dimensional spectra of the fully oxidized and fully reduced Cyt c7 recorded at 700 MHz (298 K, pH 6.5) are reported in A and B, respectively. The spectra recorded after the addition of increasing amounts of CrO are reported in C–E. The assignment of some resonances is reported in A (the roman numbers indicate the heme resonances).
are reported in C–E. The assignment of some resonances is reported in A (the roman numbers indicate the heme resonances).
The above observations can be explained as follows. Addition of the first fraction of CrO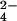 gives rise to one third of cytochrome molecules bearing chromium(III), but all protein molecules are oxidized by one electron. Because the methyl signals of hemes I and III are not detectable, it means that the extra electron essentially is shared by the two hemes, the line broadening being attributed to chemical exchange. From the chemical shifts of hemes I and III of the fully oxidized and fully reduced proteins, an upper limit for the exchange rate of 4.5 × 104 s−1 is estimated. The second fraction of CrO
gives rise to one third of cytochrome molecules bearing chromium(III), but all protein molecules are oxidized by one electron. Because the methyl signals of hemes I and III are not detectable, it means that the extra electron essentially is shared by the two hemes, the line broadening being attributed to chemical exchange. From the chemical shifts of hemes I and III of the fully oxidized and fully reduced proteins, an upper limit for the exchange rate of 4.5 × 104 s−1 is estimated. The second fraction of CrO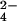 provides a system with two thirds of the protein with a bound chromium(III), and the line width of the methyl signals of hemes I and III decreases. This effect is caused by the disappearance of the intramolecular chemical exchange between hemes I and III. The still large line width could be caused by intermolecular chemical exchange as well as to some chemical exchange with heme IV, the oxidized state of which is partially thermally accessible. Some small line-broadening effect on the resonances of heme III is caused by the presence of chromium(III) (see below). The third fraction of CrO
provides a system with two thirds of the protein with a bound chromium(III), and the line width of the methyl signals of hemes I and III decreases. This effect is caused by the disappearance of the intramolecular chemical exchange between hemes I and III. The still large line width could be caused by intermolecular chemical exchange as well as to some chemical exchange with heme IV, the oxidized state of which is partially thermally accessible. Some small line-broadening effect on the resonances of heme III is caused by the presence of chromium(III) (see below). The third fraction of CrO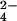 produces the fully chromium(III)-bound oxidized protein.
produces the fully chromium(III)-bound oxidized protein.
The major difference with respect to the oxidation of Cyt c7 by air consists in an extra line broadening observed for some heme signals and a number of other protein protons clustered around heme IV. This line-broadening effect is attributable to the presence of chromium(III) formed after the three-electron reduction of CrO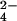 . Chromium(III) is a d3 ion, generally six-coordinate, with three unpaired electrons (S = 3/2). It possesses negligible magnetic anisotropy and therefore does not contribute to the hyperfine shifts of nuclei nearby but broadens the NMR lines because of its long electronic relaxation times (≈4 × 10−8 s at 700 MHz). It is kinetically inert as far as ligand exchange is concerned (16). For all these reasons, it can be used as an efficient relaxation reagent, and indeed it has been used as such to probe specific binding sites in many systems (32–38). However, the present study represents an attempt to exploit these effects to calculate the structure of the metal–protein adduct.
. Chromium(III) is a d3 ion, generally six-coordinate, with three unpaired electrons (S = 3/2). It possesses negligible magnetic anisotropy and therefore does not contribute to the hyperfine shifts of nuclei nearby but broadens the NMR lines because of its long electronic relaxation times (≈4 × 10−8 s at 700 MHz). It is kinetically inert as far as ligand exchange is concerned (16). For all these reasons, it can be used as an efficient relaxation reagent, and indeed it has been used as such to probe specific binding sites in many systems (32–38). However, the present study represents an attempt to exploit these effects to calculate the structure of the metal–protein adduct.
The contribution to the line width of the proton NMR signals caused by the presence of chromium(III) can be analyzed in terms of electron–nucleus dipolar coupling and, to a minor extent, of Curie relaxation (16) as reported in Materials and Methods. Line broadening for the fully oxidized species was observed for heme IV 1-CH3 and 5-CH3, heme III 1-CH3 and 8-CH3, and at least some resonances of the following amino acids: 9–11, 13, 29, 40–42, 46, 47, 50–55, 63, 66. In Fig. 2 the regions of the protein experiencing line broadening are highlighted with a gray-scale code indicating the extent of the effect.
Figure 2.

The solution structure of fully oxidized Cyt c7: the backbone and the three heme groups are shown. The amino acids experiencing signal broadening in the spectra recorded after oxidation with CrO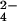 are reported as ball-and-stick models. The atoms are coded according to the observed effects on the line width. Dark gray, nuclei with resonances that experience line broadening >1,000 Hz; gray, those >100 Hz; and light gray, those >10 Hz.
are reported as ball-and-stick models. The atoms are coded according to the observed effects on the line width. Dark gray, nuclei with resonances that experience line broadening >1,000 Hz; gray, those >100 Hz; and light gray, those >10 Hz.
To locate the binding site of chromium(III) on the protein surface, new structural calculations were performed starting from the solution structure of the fully oxidized protein (12) and adding the above-described distance constraints (see Table 1, which is published as supporting information on the PNAS web site, www.pnas.org) to the available NOE constraints. This procedure is justified by the assumption that the overall structure is not modified by the presence of chromium as deduced from the analysis of the two-dimensional NMR maps. The rms deviation of chromium(III) with respect to the average position is 0.97 Å. The target function relative to 27 relaxation constraints is 0.2 ± 0.08 Å2. The low value of this “disagreement function” shows that chromium(III) binds in a single site. A value of 1.00 ± 0.07 Å2 for the disagreement function of the NOE constraints further confirms that the protein structure does not change as an effect of the presence of the bound metal ion, and that NOEs and relaxation data are consistent with one another to provide a unique structural model. Our work shows that chromium(III) is located 7.9 ± 0.4 Å from the iron of heme IV, 16.3 ± 0.7 Å from the iron of heme III, and 22.5 ± 0.5 Å from the iron of heme I. The distances between chromium(III) and the backbone amide protons of Lys-41, Lys-42, Lys-46, Lys-50, and His-66 are 10.8 ± 0.3, 8.7 ± 0.4, 7.1 ± 0.3, 9.3 ± 0.5, 11.8 ± 0.3 Å, respectively. They are reasonable candidates for chromium(III) ligands. The chromium(III) is in a cavity defined on one side by heme IV [the shortest chromium(III)-heme distance being with the CH3 of the thioether-2, 4.9 Å] and on the other side by the backbone segment bearing the four lysines. The same cavity is occupied also by the His-45 axial ligand to iron IV with a short Nδ1-chromium distance.
The above-reported NMR data and the structural information obtained on the site of attachment of chromium(III) can be interpreted as follows. The negatively charged CrO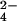 and HCrO
and HCrO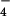 , which constitute the dominant forms of hexavalent chromium at pH 6.5, interact with the positively charged surface area of Cyt c7, which is provided by the above-mentioned lysines located around heme IV (Fig. 3). After reduction it is very likely that the resulting chromium(III) ion remains in the same region, possibly attached to the same lysines. The line-broadening effects induced by the presence of chromium(III) on Lys-41, Lys-42, Lys-46, and Lys-50 are so severe that their resonances are undetectable. This effect does not allow us to determine the conformation of their side chains in the chromium–protein complex but is a definite indication that they are very close to the metal ion. Defining as the “proximal” ligand the histidine residue belonging to the Cys-Xaa-Yaa-Cys-His sequence motif, two different groups of lysines can be identified around heme IV: those on the proximal side of the heme (Lys-9, Lys-10, Lys-61, and Lys-68) and those on the distal side (Lys-41, Lys-42, Lys-46, and Lys-50). The resulting structure indicates that those involved in the metal binding are located on the distal side.
, which constitute the dominant forms of hexavalent chromium at pH 6.5, interact with the positively charged surface area of Cyt c7, which is provided by the above-mentioned lysines located around heme IV (Fig. 3). After reduction it is very likely that the resulting chromium(III) ion remains in the same region, possibly attached to the same lysines. The line-broadening effects induced by the presence of chromium(III) on Lys-41, Lys-42, Lys-46, and Lys-50 are so severe that their resonances are undetectable. This effect does not allow us to determine the conformation of their side chains in the chromium–protein complex but is a definite indication that they are very close to the metal ion. Defining as the “proximal” ligand the histidine residue belonging to the Cys-Xaa-Yaa-Cys-His sequence motif, two different groups of lysines can be identified around heme IV: those on the proximal side of the heme (Lys-9, Lys-10, Lys-61, and Lys-68) and those on the distal side (Lys-41, Lys-42, Lys-46, and Lys-50). The resulting structure indicates that those involved in the metal binding are located on the distal side.
Figure 3.

The chromium(III) binding site from solution structure calculations performed with the inclusion of paramagnetic-derived chromium(III)-proton distance constraints. The possible ligands Lys-41, Lys-42, Lys-46, and Lys-50 side chains are reported as black ball-and-stick models. The chromium(III) ion is shown as a black sphere, and the hemes are labeled by roman numbers.
To prove that the CrO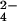 binding site on the protein is the one where chromium(III) is bound, the interaction between the fully reduced Cyt c7 and MoO
binding site on the protein is the one where chromium(III) is bound, the interaction between the fully reduced Cyt c7 and MoO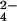 was studied. The latter anion is isostructural and isoelectronic to CrO
was studied. The latter anion is isostructural and isoelectronic to CrO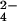 but does not give rise to protein oxidation. The presence of MoO
but does not give rise to protein oxidation. The presence of MoO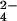 induces small shift changes (up to 0.03 ppm) in some resonances of residues 41–57. These protein amino acids define a single area on the protein surface that corresponds to that found to represent the binding site of chromium(III).
induces small shift changes (up to 0.03 ppm) in some resonances of residues 41–57. These protein amino acids define a single area on the protein surface that corresponds to that found to represent the binding site of chromium(III).
As far as the order of oxidation of the three hemes is concerned, the experimental data presented here show that hemes I and III are the first to undergo oxidation, with the two centers having similar reduction potential (39, 40). One intermediate is identified with hemes I and III largely oxidized and heme IV essentially reduced in accordance with the order of reduction potentials of the unmodified protein. Although no information is available on the intramolecular electron-transfer pathway, it is tempting to propose that heme IV provides the electron to chromium, because they are so close to one another, and hemes I and III reestablish the thermodynamic conditions by electron tunneling to heme IV (41–48). As far as chromium is concerned, it is reasonable that the three electrons provided by the protein reduce chromate(VI) stepwise (49, 50). After reduction the chromium ion expands its coordination number involving one or more protein residues, possibly lysines, to which it remains attached because of its kinetic inertia. The positive lysines therefore play the double role of attracting the chromate(VI) or molybdate(VI) anions and then bind the reduced cation.
Concluding Remarks and Implications for Cyt c3
This research describes the site of interaction for CrO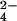 reduction by Cyt c7. The NMR titration of fully reduced Cyt c7 with CrO
reduction by Cyt c7. The NMR titration of fully reduced Cyt c7 with CrO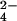 indicates that chromium(III) and fully oxidized Cyt c7 are produced. NMR spectra provide distance constraints between chromium(III) and protein protons to be used in structural calculations together with the already available NOEs. The resulting structure of the protein–chromium adduct shows that chromium(III) is bound to the protein in a specific site close to heme IV, i.e., to the heme with the highest reduction potential. This binding site is formed by a positive patch on the protein surface because of the presence of four lysine residues. In analogy to other chromium(III)–protein adducts (32–34, 37, 38), deprotonated lysine side chains could represent the ligands of the metal ion. The reaction could be interpreted by a mechanism constituted by the following steps: (i) formation of a chromate–fully reduced protein complex (with the chromate anion bound through electrostatic interaction to the “distal” lysines defining the positive surface area close to heme IV), (ii) chromate reduction to chromium(III) with three electrons and binding of chromium(III) to the above lysines, and (iii) intermolecular electron redistribution when Cyt c7 oxidation is only partial. Structural data indicate that heme IV represents the electron sink for the electrons to be transferred to CrO
indicates that chromium(III) and fully oxidized Cyt c7 are produced. NMR spectra provide distance constraints between chromium(III) and protein protons to be used in structural calculations together with the already available NOEs. The resulting structure of the protein–chromium adduct shows that chromium(III) is bound to the protein in a specific site close to heme IV, i.e., to the heme with the highest reduction potential. This binding site is formed by a positive patch on the protein surface because of the presence of four lysine residues. In analogy to other chromium(III)–protein adducts (32–34, 37, 38), deprotonated lysine side chains could represent the ligands of the metal ion. The reaction could be interpreted by a mechanism constituted by the following steps: (i) formation of a chromate–fully reduced protein complex (with the chromate anion bound through electrostatic interaction to the “distal” lysines defining the positive surface area close to heme IV), (ii) chromate reduction to chromium(III) with three electrons and binding of chromium(III) to the above lysines, and (iii) intermolecular electron redistribution when Cyt c7 oxidation is only partial. Structural data indicate that heme IV represents the electron sink for the electrons to be transferred to CrO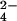 . The NMR titration shows that electron redistribution among the iron centers occurs, and the order of oxidation of the iron ions is that determined by their reduction potentials.
. The NMR titration shows that electron redistribution among the iron centers occurs, and the order of oxidation of the iron ions is that determined by their reduction potentials.
Also the isolated tetraheme Cyts c3 from Desulfovibrio and Desulfomicrobium bacteria are able to reduce CrO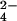 with a catalytic efficiency close to that of Cyt c7 (8). From the structural point of view these cytochromes are very similar to Cyt c7 both in terms of fold and heme-packing geometry, the former protein missing heme II and a protein segment bearing the heme II axial ligands. From the structural comparison of Cyts c3, it results that all of them also contain several lysine residues clustered around heme IV in a region similar to that of Cyt c7. It is interesting to note that heme II has been suggested not to be relevant kinetically in the Cyt c3 intramolecular electron-transfer reactions (8, 51).
with a catalytic efficiency close to that of Cyt c7 (8). From the structural point of view these cytochromes are very similar to Cyt c7 both in terms of fold and heme-packing geometry, the former protein missing heme II and a protein segment bearing the heme II axial ligands. From the structural comparison of Cyts c3, it results that all of them also contain several lysine residues clustered around heme IV in a region similar to that of Cyt c7. It is interesting to note that heme II has been suggested not to be relevant kinetically in the Cyt c3 intramolecular electron-transfer reactions (8, 51).
NMR spectroscopy therefore has shown its potential for proving the occurrence of a single metal binding site of chromium(III) and for determining such interaction sites by using line broadening as structural constraints in established protocols for solution structure determination. Previous studies (32–38) used the same concept to qualitatively establish the binding site of chromium(III) in oxidation reactions involving chromium(II) and assuming that the binding site of chromium(II) is the same as that of chromium(III). However, in such systems the binding of chromium was found often to occur at multiple sites. The use of paramagnetic relaxation for solution structure determination of metalloproteins had been explored by our laboratory (23, 24) and had been used in the presence of mobile paramagnetic tags (52, 53). The present results obtained on Cyt c7 become quite relevant, because they provide the key for the understanding on a structural basis the chromium(VI)-reductase activity of the whole class of multiheme cytochromes.
Supplementary Material
Acknowledgments
This work was supported financially by the European Union RTD Project EVK1-1999-00033, Italian Consiglio Nazionale delle Ricerche Progetto Finalizzato Biotecnologie 99.00509.PF49, and Ministero dell'Universitá e della Ricerca Scientifica e Tecnologica COFIN99 and carried out under the Training and Mobility of Researchers Large Scale Facility Program (European Union contract no. HPRI-CT-1999-00009).
Abbreviations
- Cyt
cytochrome
- NOE
nuclear Overhauser effect
Footnotes
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank, www.rcsb.org (PDB ID code 1LM2).
Chromium(VI) toxicity was made famous in the Oscar-winning film Erin Brockovich.
References
- 1.Kim C, Zhou Q, Deng B, Thornton E, Xu H. Environ Sci Technol. 2001;35:2219–2225. doi: 10.1021/es0017007. [DOI] [PubMed] [Google Scholar]
- 2.Lovley D R, Phillips E J P. Appl Microbiol Biotechnol. 1992;58:850–856. doi: 10.1128/aem.58.3.850-856.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lovley D R, Phillips E J P. Appl Environ Microbiol. 2001;60:726–728. doi: 10.1128/aem.60.2.726-728.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lovley D R, Widman P K, Woodwark J C, Phillips E J P. Appl Environ Microbiol. 1993;59:3572–3576. doi: 10.1128/aem.59.11.3572-3576.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lojou E, Bianco P, Bruschi M. J Electroanal Chem. 1998;452:167–177. [Google Scholar]
- 6.Lojou E, Bianco P, Bruschi M. Electrochim Acta. 1998;43:2005–2013. [Google Scholar]
- 7.Lojou E, Bianco P. J Electroanal Chem. 1999;471:96–104. [Google Scholar]
- 8.Michel C, Brugna M, Aubert C, Bernadac A, Bruschi M. Appl Microbiol Biotechnol. 2001;55:95–100. doi: 10.1007/s002530000467. [DOI] [PubMed] [Google Scholar]
- 9.Pereira I A C, Pacheco I, Liu M-Y, LeGall J, Xavier A V, Tereira M. Eur J Biochem. 1997;248:323–328. doi: 10.1111/j.1432-1033.1997.00323.x. [DOI] [PubMed] [Google Scholar]
- 10.Roden E C, Lovley D R. Appl Environ Microbiol. 1993;59:734–742. doi: 10.1128/aem.59.3.734-742.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Banci L, Bertini I, Bruschi M, Sompornpisut P, Turano P. Proc Natl Acad Sci USA. 1996;93:14396–14400. doi: 10.1073/pnas.93.25.14396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Assfalg M, Banci L, Bertini I, Bruschi M, Turano P. Eur J Biochem. 1998;256:261–270. doi: 10.1046/j.1432-1327.1998.2560261.x. [DOI] [PubMed] [Google Scholar]
- 13.Assfalg M, Banci L, Bertini I, Bruschi M, Giudici-Orticoni M T, Turano P. Eur J Biochem. 1999;266:634–643. doi: 10.1046/j.1432-1327.1999.00904.x. [DOI] [PubMed] [Google Scholar]
- 14.Czjzek M, Arnoux P, Haser R, Shepard W. Acta Crystallogr D. 2001;57:670–678. doi: 10.1107/s0907444901003481. [DOI] [PubMed] [Google Scholar]
- 15.Coutinho I, Turner D L, Liu M-Y, LeGall J, Xavier A V. J Biol Inorg Chem. 1996;1:305–311. [Google Scholar]
- 16.Bertini I, Luchinat C, Parigi G. Solution NMR of Paramagnetic Molecules. Amsterdam: Elsevier; 2001. [Google Scholar]
- 17.Macura S, Wüthrich K, Ernst R R. J Magn Reson. 1982;47:351–357. [Google Scholar]
- 18.Marion D, Wüthrich K. Biochem Biophys Res Commun. 1983;113:967–974. doi: 10.1016/0006-291x(83)91093-8. [DOI] [PubMed] [Google Scholar]
- 19.Piotto M, Saudek V, Sklenar V. J Biomol NMR. 1992;2:661–666. doi: 10.1007/BF02192855. [DOI] [PubMed] [Google Scholar]
- 20.Bartels C, Xia T H, Billeter M, Güntert P, Wüthrich K. J Biomol NMR. 1995;5:1–10. doi: 10.1007/BF00417486. [DOI] [PubMed] [Google Scholar]
- 21.Banci L, Bertini I, Luchinat C. Nuclear and Electron Relaxation: The Magnetic Nucleus-Unpaired Electron Coupling in Solution. Weinheim, Germany: VCH; 1991. [Google Scholar]
- 22.Güntert P, Braun W, Wüthrich K. J Mol Biol. 1991;217:517–530. doi: 10.1016/0022-2836(91)90754-t. [DOI] [PubMed] [Google Scholar]
- 23.Bertini I, Donaire A, Luchinat C, Rosato A. Proteins Struct Funct Genet. 1997;29:348–358. [PubMed] [Google Scholar]
- 24.Bertini I, Couture M M J, Donaire A, Eltis L D, Felli I C, Luchinat C, Piccioli M, Rosato A. Eur J Biochem. 1996;241:440–452. doi: 10.1111/j.1432-1033.1996.00440.x. [DOI] [PubMed] [Google Scholar]
- 25.Güntert P, Mumenthaler C, Wüthrich K. J Mol Biol. 1997;273:283–298. doi: 10.1006/jmbi.1997.1284. [DOI] [PubMed] [Google Scholar]
- 26.Czjzek M, Payan F, Guerlesquin F, Bruschi M, Haser R. J Mol Biol. 1994;243:653–667. doi: 10.1016/0022-2836(94)90039-6. [DOI] [PubMed] [Google Scholar]
- 27.Haser R, Pierrot M, Frey M, Payan F, Astier J P, Bruschi M, LeGall J. Nature (London) 1979;282:806–810. doi: 10.1038/282806a0. [DOI] [PubMed] [Google Scholar]
- 28.Higuchi Y, Kusunoki M, Matsuura Y, Yasuoka N, Kakudo M. J Mol Biol. 1984;172:109–139. doi: 10.1016/0022-2836(84)90417-0. [DOI] [PubMed] [Google Scholar]
- 29.Matias P M, Frazao C, Morais J, Coll M, Carrondo M A. J Mol Biol. 1993;234:680–699. doi: 10.1006/jmbi.1993.1620. [DOI] [PubMed] [Google Scholar]
- 30.Norager S, Legrand P, Pieulle L, Hatchikian C, Roth M. J Mol Biol. 1999;290:881–902. doi: 10.1006/jmbi.1999.2917. [DOI] [PubMed] [Google Scholar]
- 31.Moura J J G, Moore G R, Williams R J P, Probst I, LeGall J, Xavier A V. Eur J Biochem. 1984;144:433–440. doi: 10.1111/j.1432-1033.1984.tb08484.x. [DOI] [PubMed] [Google Scholar]
- 32.Farver O, Blatt Y, Pecht I. Biochemistry. 1982;21:3556–3561. doi: 10.1021/bi00258a005. [DOI] [PubMed] [Google Scholar]
- 33.Im S-C, Worrall J A R, Liu G, Aliverti A, Zanetti G, Luchinat C, Bertini I, Sykes A G. Inorg Chem. 2000;39:1755–1764. doi: 10.1021/ic991127w. [DOI] [PubMed] [Google Scholar]
- 34.Cho W K, Blair D F, Banerjee U, Hopfield J J, Gray H B, Pecht I, Chan S I. Biochemistry. 1984;23:1858–1862. doi: 10.1021/bi00303a042. [DOI] [PubMed] [Google Scholar]
- 35.Gupta R K, Fung C H, Mildvan A S. J Biol Chem. 1976;251:2421–2430. [PubMed] [Google Scholar]
- 36.Mildvan A S, Sloan D L, Fung C H, Gupta R K, Melamud E. J Biol Chem. 1976;251:2431–2434. [PubMed] [Google Scholar]
- 37.Armstrong F A, Driscoll P C, Hill H A O, Redfield C. J Inorg Biochem. 1986;28:171–180. [Google Scholar]
- 38.Handford P M, Hill H A O, Lee R W K, Henderson R A, Sykes A G. J Inorg Biochem. 1980;13:83–88. [Google Scholar]
- 39.Fiechtner M D, Kassner R J. Biochim Biophys Acta. 1979;579:269–278. doi: 10.1016/0005-2795(79)90054-0. [DOI] [PubMed] [Google Scholar]
- 40.Bruschi M, Loutfi M, Bianco P, Haladjian J. Biochem Biophys Res Commun. 1984;120:384–389. doi: 10.1016/0006-291x(84)91265-8. [DOI] [PubMed] [Google Scholar]
- 41.Beratan D A, Onuchic J N, Betts J N, Bowler B E, Gray H B. J Am Chem Soc. 1990;112:7915–7921. [Google Scholar]
- 42.Gray H B, Ellis W R., Jr . In: Bioinorganic Chemistry. Bertini I, Gray H B, Lippard S J, Valentine J S, editors. Vol. 6. Sausalito, CA: Univ. Sci. Books; 1994. pp. 315–363. [Google Scholar]
- 43.Winkler J R, Gray H B. Chem Rev (Washington, DC) 1992;92:369–379. [Google Scholar]
- 44.Casimiro D R, Richards J H, Winkler J R, Gray H B. J Phys Chem. 1993;97:13073–13077. [Google Scholar]
- 45.Babini E, Bertini I, Borsari M, Capozzi F, Luchinat C, Zhang X, Moura G L C, Kurnikov I V, Beratan D N, Ponce A, et al. J Am Chem Soc. 2000;122:4532–4533. [Google Scholar]
- 46.Canters G W, Van de Kamp M. Curr Biol. 1992;2:859–869. [Google Scholar]
- 47.Langen R, Spielmann H P, Germanas J P, Richards J H, Winkler J R, Gray H B. Science. 1995;268:1733–1735. doi: 10.1126/science.7792598. [DOI] [PubMed] [Google Scholar]
- 48.Langen R, Colon J L, Casimiro D R, Karpishin J R, Winkler J R, Gray H B. J Biol Inorg Chem. 1996;1:221–225. [Google Scholar]
- 49.Nemes A, Bakac A. Inorg Chem. 2001;40:2720–2724. doi: 10.1021/ic0013577. [DOI] [PubMed] [Google Scholar]
- 50. Signorella, S., Daier, V., Santoro, M., Garcia, S., Palopoli, C., Gonzalez, J. C., Korecz, L., Rockenbauer, A. & Sala, F. (2001) Eur. J. Inorg. Chem. 1829–1833.
- 51.Dolla A, Florens L, Bianco P, Haladjian J, Voordouw G, Forest E, Wall J, Guerlesquin F, Bruschi M. J Biol Chem. 2001;269:6340–6346. [PubMed] [Google Scholar]
- 52.Dunham S U, Turner C J, Lippard S J. J Am Chem Soc. 1998;120:5395–5406. [Google Scholar]
- 53.Donaldson L W, Skrynnikov N R, Wing-Yiu C, Muhandiram D R, Sarkar B, Forman-Kay J D, Kay L E. J Am Chem Soc. 2002;123:9843–9847. doi: 10.1021/ja011241p. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.