

Abstract
Overexpression of urokinase plasminogen activator (uPA) in endothelial cells can decrease intravascular thrombosis. However, expression of uPA is increased in atherosclerotic human arteries, which suggests that uPA might accelerate atherogenesis. To investigate whether elevated uPA expression accelerates atherogenesis, we cloned a rabbit uPA cDNA and expressed it in carotid arteries of cholesterol-fed rabbits. uPA gene transfer increased artery-wall uPA activity for at least 1 week, with a return to baseline by 2 weeks. One week after gene transfer, uPA-transduced arteries were constricted, with significantly smaller lumens and thicker walls, but no difference in intimal or medial mass. Two weeks after gene transfer, uPA- and control-transduced arteries were morphologically indistinguishable. By 4 weeks, however, uPA-transduced arteries had 70% larger intimas than control-transduced arteries (P < 0.01) and smaller lumens (P < 0.05). Intimal lesions appeared to be of similar composition in uPA- and control-transduced arteries, except that degradation of elastic laminae was evident in uPA-transduced arteries. These data suggest that elevated uPA expression in atherosclerotic arteries contributes to intimal growth and constrictive remodeling leading to lumen loss. Antagonists of uPA activity might, therefore, be useful in limiting intimal growth and preventing constrictive remodeling. Overexpression of uPA in endothelial cells to prevent intravascular thrombosis must be reconsidered, because this intervention could worsen underlying vascular disease.
Urokinase-type plasminogen activator (uPA; EC 3.4.2.73) is a serine protease that activates the zymogen plasminogen, potentially initiating a cascade of fibrinolysis and extracellular proteolysis (1). uPA is used as a therapeutic agent to lyse occlusive fibrin clots and thrombi in blood vessels and intravascular devices. We previously reported that overexpression of uPA increased fibrinolytic activity in primate endothelial cells both in vitro and in vivo (2, 3), and we proposed that local uPA overexpression might eventually be applied as a gene therapy strategy to prevent intravascular thrombosis (4). However, the consequences of uPA overexpression on the blood vessel wall have never been examined.
Abundant data suggest that uPA may play roles in the vessel wall other than initiating fibrinolysis. uPA is expressed by endothelial cells and smooth muscle cells in normal human arteries (5). uPA expression is elevated in atherosclerotic human aortas, carotid arteries, and coronary arteries (5–8), in which it is expressed by endothelial cells, smooth muscle cells, and macrophages (5). Elevated uPA expression could contribute to vascular lesion formation by facilitating smooth muscle cell migration and proliferation. uPA expression is increased during neointimal formation in injured rat arteries (9), and uPA-deficient mice form smaller neointimas after arterial injury (10). uPA could contribute to proliferative vascular disease by activating growth factors or releasing these factors from binding sites in the extracellular matrix (11, 12). uPA could also promote aneurysm formation by initiating proteolysis of the extracellular matrix (1, 13). However, proteolysis of extracellular matrix could decrease vascular lesion formation by removing intimal mass and by decreasing the number of binding sites for atherogenic lipoproteins (14). uPA could also alter lesion development by actions mediated through its cell surface receptor, uPAR. uPAR is expressed by a variety of vascular and inflammatory cells and its ligation can cause cell activation and stimulate cell migration (1). Through these mechanisms, uPA, as well as plasmin generated by uPA activation of plasminogen, could either accelerate or mitigate atherogenesis.
To determine whether elevated endothelial uPA expression alters atherosclerotic lesion development, we overexpressed uPA in a rabbit model of early atherosclerosis. This animal model, developed in our laboratory, involves intraluminal infusion of adenoviral vectors to carotid arteries of hypercholesterolemic rabbits. Adenoviral vector infusion in this setting initiates subendothelial accumulation of macrophages and smooth muscle cells, leading to formation of intimal lesions that are histologically and ultrastructurally similar to those of early human atherosclerosis (15). Gene transfer in this model is almost exclusively to endothelium (16), which in the present study is genetically modified to secrete increased levels of uPA. Here we report the consequences of increased endothelial uPA expression in atherosclerotic arteries.
Materials and Methods
Cloning of Rabbit uPA.
We cloned a uPA cDNA from a rabbit kidney cDNA library (Stratagene). Purification of a positive plaque yielded a 1.5-kb insert in pBluescript SK (pBSKrbtuPA). Sequencing of the insert confirmed a 1302-bp ORF, 84% identical to human uPA (GenBank accession no. AY122285).
Adenoviral Vectors.
An E1-deleted adenoviral vector that expresses rabbit uPA from the cytomegalovirus (CMV) promoter (AdrbtuPA) was constructed, purified, and propagated using standard methods (17). As a control, we used AdCMVNull, a vector that does not express a transgene (18). The absence of replication-competent virus was confirmed by a PCR-based assay that is capable of detecting one E1A-containing genome per 1 × 106 vector genomes (19).
In Vitro uPA Expression Assays.
Expression of uPA by AdrbtuPA was confirmed by Northern analysis of transduced 293 cells and by zymography and plasminogen activator activity assay of conditioned media, using the plasmin substrate S-2390 (Chromogenix; ref. 2).
Animals.
In vivo experiments were performed with specific-pathogen-free adult male New Zealand White rabbits (2.5–3.5 kg, Charles River Breeding Laboratories). For atherosclerosis studies, hypercholesterolemia (400–700 mg/dl) was induced with a diet containing 0.25% cholesterol and 3% soybean oil (15). All animal protocols were approved by the Committee on Animal Research of the University of California, San Francisco.
In Vivo Gene Transfer.
In vivo gene transfer to carotid artery endothelium was performed as described (15). For the dose–response study, viral stocks were diluted to 1 × 108 to 4 × 109 plaque-forming units (pfu) per ml (2 × 1010 to 8 × 1011 particles per ml). For the atherosclerosis study, we used a final concentration of 8 × 1011 particles per ml for both vectors.
Harvest of Arteries for uPA Expression Assays and Histology.
For expression studies, arteries were harvested at 1–14 days after gene transfer and either used immediately for a plasminogen activation assay or snap frozen in liquid nitrogen for later measurement of mRNA or direct uPA activity. For histologic analyses, arteries were perfusion-fixed in situ at physiologic pressure (15) and harvested 1–4 weeks after gene transfer.
In Vivo Expression of uPA mRNA and Protein.
Northern analysis of carotid segments was performed using cDNA probes for rabbit uPA and glyceraldehyde phosphate dehydrogenase (GAPDH) (19). Bound probe was measured with a phosphoimager.
We used two methods to measure uPA activity in arteries: an in situ plasminogen activation assay on freshly harvested arterial segments and a direct uPA activity assay performed on extracts of snap-frozen arteries. For the in situ plasminogen activation assay, artery segments were incubated with Glu-plasminogen and the plasmin substrate S-2390. For the direct uPA activity assay, frozen artery rings were extracted with Triton X-100, the extract was treated briefly with plasmin to convert single- to two-chain uPA, aprotinin was added to inhibit plasmin, and the uPA-specific substrate S-2444 was added to measure uPA activity.
Morphometric Analysis.
Sections of arteries were stained with Movat's pentachrome, and images were recorded with a digital camera (Diagnostic Instruments, Sterling Heights, MI). An observer blinded to the identity of the specimens performed computer-assisted planimetry to measure the intimal and medial areas, lumen circumference, and the lengths of the internal and external elastic laminae (IEL and EEL). Lumen area (assuming circular lumens) was calculated from lumen circumference (area = circumference2 ÷ 4π). Mean wall (i.e., intimal + medial) thickness was determined by calculating the mean radius of the areas inside both the EEL and the lumen (radius = EEL or lumen circumference ÷ 2π) and subtracting the latter.
Measurements of Intimal Cellularity and Proliferation.
Intimal cell density was measured by counting all intimal nuclei in four high-power fields in each of four sections per artery (16 fields per artery). The mean intimal cell density of the artery was then calculated from these measurements and the number of intimal cells in an arterial section was calculated by multiplying the intimal area by the cell density. Intimal cell proliferation was measured by injecting rabbits with bromodeoxyuridine (BrdUrd; 100 mg s.c.) 1, 9, and 17 h before harvest and staining arterial sections for evidence of BrdUrd incorporation (20). BrdUrd-positive cells were counted in the intimas of four sections per artery and the proliferative index for the artery was calculated as the mean of the four sections.
Assessment of Extracellular Matrix Components.
All Movat-stained sections were examined by an observer blinded to treatment group. Degree and staining pattern of collagen, proteoglycan, and elastin was assessed to determine whether the abundance or distribution of any of these matrix components appeared sufficiently variable to suggest an effect of uPA overexpression. Collagen and proteoglycan staining varied little among the arteries. However, there was a wide range of appearance of elastin. Some arteries had extensive elastic lamina fragmentation and loss, whereas others had largely intact elastic laminae. To test the hypothesis that elastic lamina fragmentation and loss were associated with uPA overexpression, sections from all arteries were examined and assigned to one of two groups: extensive elastic lamina fragmentation and loss present or absent. The vector treatments were then revealed, and the percentage of AdrbtuPA and AdCMVNull arteries in each group was calculated.
Immunohistochemistry.
Macrophages were detected by immunostaining with the RAM-11 antibody (15). Fibrin(ogen) was detected by staining 1-week arteries with a mouse monoclonal antibody to human fibrin(ogen) (#750, American Diagnostica, Greenwich, CT). The specificity of the primary antibodies was confirmed by substituting isotype-matched antibodies. Bound antibodies were detected with biotinylated secondary antibody, streptavidin-peroxidase, and aminoethylcarbazole substrate. Immunohistochemical staining was quantified by computer-assisted image analysis. The percentage of intimal area staining for macrophages was calculated by dividing the stained area by the total intimal area of the same slide. This measurement was performed in four sections per artery, and the mean of the measurements used as a value for the artery.
Statistical Methods.
Data are presented as mean ± SEM for normally distributed data or median for data not normally distributed. Group means were compared with t tests and medians were compared with the Mann–Whitney rank-sum test. The χ2 test was used to determine whether elastic lamina fragmentation and loss were more common in the AdrbtuPA arteries.
An expanded version of Materials and Methods can be found in Supporting Materials and Methods, which is published as supporting information on the PNAS web site, www.pnas.org.
Results
In Vitro uPA Expression.
Expression of uPA by AdrbtuPA was confirmed by three assays. Northern analysis showed a 2.4-kb uPA transcript in 293 cells transduced with AdrbtuPA but not AdCMVNull (data not shown). Casein-plasminogen zymography revealed lysis zones corresponding to proteins of 45 and 30 kDa (Fig. 1) only in medium conditioned by AdrbtuPA-transduced cells. Similarly, conditioned media from AdrbtuPA-transduced cells contained increased plasminogen activator activity, detected with S-2390 (data not shown).
Fig 1.
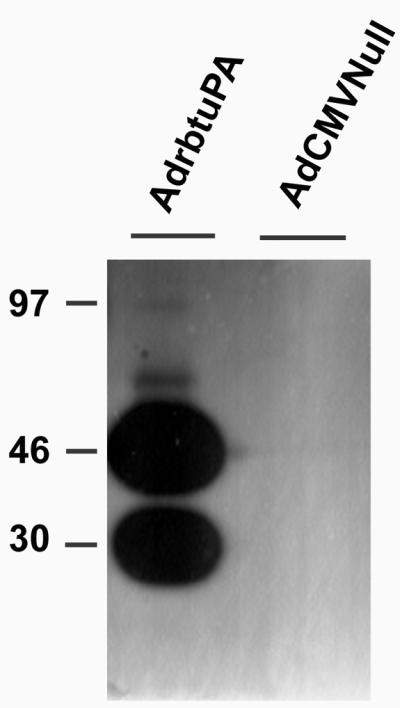
Zymography of medium conditioned by 293 cells transduced with either AdrbtuPA or AdCMVNull. Overnight collections of serum-free medium were analyzed by casein plasminogen zymography. The two large lysis zones likely indicate presence of both high- and low-molecular-mass forms of uPA. The position of migration of molecular mass markers is indicated in kilodaltons.
In Vivo uPA Expression.
We transduced rabbit carotid arteries with AdrbtuPA (1 × 108, 5 × 108, 1.5 × 109, and 4 × 109 pfu/ml), AdCMVNull (1 × 108 and 4 × 109 pfu/ml), or vehicle. Arteries were harvested 1–14 days after gene transfer. Northern analysis revealed dose-dependent expression of the 2.4-kb uPA transgene mRNA, as high as 40-fold above the expression level of the 2.7-kb endogenous uPA transcript (Fig. 2A). Transgene expression peaked at 1–3 days, with lower expression at 7 days and no detectable expression at 14 days (Fig. 2B). An in situ plasminogen activation assay confirmed a dose-responsive increase in plasminogen activator activity in explanted AdrbtuPA arteries (Fig. 3A). Plasminogen activator activity was elevated in AdrbtuPA arteries harvested at 1, 3, and 7 days, but not 14 days (Fig. 3 B–E). To exclude the unlikely possibility that AdCMVNull induced expression of a plasminogen activator inhibitor, arteries (n = 2) were infused with vehicle, harvested 3 days later, and assayed for in situ plasminogen activator activity in parallel with AdCMVNull arteries (n = 4). Vehicle-infused arteries actually had a slightly lower level of plasminogen activator activity than AdCMVNull arteries (data not shown), excluding a significant induction of inhibitor activity by AdCMVNull. To more precisely quantify uPA activity, we used the uPA-specific substrate S-2444 to assay extracts of arteries harvested 3–7 days after gene transfer. The median uPA activity in extracts of AdrbtuPA arteries was 7-fold greater than in AdCMVNull arteries (P = 0.008; Fig. 4).
Fig 2.

In vivo expression of rabbit uPA. (A) Dose-response study. RNA was extracted from arteries harvested 3 days after transduction with either AdCMVNull at 1 × 108 pfu/ml (as a control and to detect endogenous uPA mRNA) or AdrbtuPA at 1 × 108 to 4 × 109 pfu/ml. Phosphorimager analysis of Northern blots was used to calculate the ratio of the signals of uPA transgene mRNA to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA from the same artery. The relative level of endogenous uPA to GAPDH mRNA in the AdCMVNull-transduced arteries was assigned a value of 1.0. (B) Time course of vector-driven uPA expression. Arteries were transduced with AdrbtuPA (4 × 109 pfu/ml) and harvested 1–14 days later. The relative abundance of 2.4-kb uPA transgene mRNA was quantified as in A, except that uPA:GAPDH ratios are presented without normalization to AdCMVNull values. For both panels, data are presented as mean ± SEM of groups of four arteries. *, uPA transgene mRNA was undetectable.
Fig 3.

Plasminogen activator activity in transduced artery segments. (A) Dose–response study. Arteries were transduced with AdrbtuPA or AdCMVNull at the indicated concentrations (pfu/ml). Three days later, segments of transduced arteries were incubated ex vivo with plasminogen and the plasmin substrate S-2390. Plasmin generation is measured as the increase in OD405 over time. Data points represent the mean OD from segments of four separate arteries in each group. For clarity, SEMs are shown at 60 min only. Curves for AdCMVNull arteries are extended toward actual data points measured at 180 min. (B–E) Time course of increased plasminogen activator activity. Arteries were transduced with AdrbtuPA or AdCMVNull (both at 4 × 109 pfu/ml) and segments explanted and assayed for plasminogen activator activity as in A at 1, 3, 7, or 14 days after gene transfer. Data points are means of n = 4 arteries in each group; error bars indicate SEM.
Fig 4.

uPA activity in arterial extracts. Arteries were transduced with AdrbtuPA or AdCMVNull (both at 4 × 109 pfu/ml). Three days later, artery segments were harvested, and proteins were extracted. Extracts were assayed with the uPA-specific chromogenic substrate S-2444. Data points represent individual arteries; bar heights indicate group medians.
Early uPA Overexpression Enhances Later Intimal Growth.
We next sought to determine the consequences of endothelial uPA overexpression on primary atherosclerotic lesion growth. We fed rabbits an atherogenic diet, infused their carotid arteries with AdrbtuPA or AdCMVNull, continued the diet, and harvested arteries 1–4 weeks later. During this period, weekly plasma cholesterol measurements revealed no significant differences between the two groups (Table 1).
Table 1.
Plasma total cholesterol (mg/dl)
|
|
Week after gene transfer | |||
|---|---|---|---|---|
| 1 (n = 19–20) | 2 (n = 9) | 3 (n = 9) | 4 (n = 19) | |
| AdrbtuPA | 523 ± 83 | 484 ± 40 | 472 ± 116 | 474 ± 133 |
| AdCMVNull | 490 ± 47 | 525 ± 160 | 522 ± 87 | 449 ± 89 |
At 1 and 2 weeks after gene transfer, there were no significant differences in intimal area or I:M ratio between the groups (Table 2). At 3 weeks, the intimal area and I/M ratio tended to be higher in AdrbtuPA arteries (24–36%; P = 0.1 and 0.2). At 4 weeks, AdrbtuPA arteries had 70–80% larger intimal areas and I/M ratios than AdCMVNull arteries (P < 0.01; Table 2, Fig. 5 A and B). The larger intimas of the AdrbtuPA arteries were accompanied by increased wall (intima + media) thickness at 3 and 4 weeks (16–24%; P < 0.01) and smaller lumens (13–23%; P = 0.066 and 0.035; Table 2). Medial area did not differ between the groups at any point.
Table 2.
Dimensions of arteries transduced with AdrbtuPA and AdCMVNull
| Week | IEL length, mm | EEL length, mm | Lumen area, μm2 × 105 | Intimal area, μm2 × 104 | Medial area, μm2 × 105 | I/M ratio | Wall thickness, μm | BrdUrd index, % |
|---|---|---|---|---|---|---|---|---|
| 1 | ||||||||
| uPA (n = 18) | 2.7 ± 0.13 | 3.4 ± 0.12 | 5.6 ± .63 | 4.9 ± .51 | 3.3 ± 0.096 | 0.15 ± 0.014 | 130 ± 4.7 | 3.9 ± 1.7 |
| Null (n = 16) | 3.6 ± 0.18 | 4.2 ± 0.16 | 10 ± 1.0 | 5.5 ± .94 | 3.5 ± 0.13 | 0.15 ± 0.024 | 100 ± 5.6 | 7.4 ± 2.6 |
| 2 | ||||||||
| uPA (n = 8) | 3.4 ± 0.20 | 4.0 ± 0.19 | 9.1 ± 1.1 | 7.9 ± 0.77 | 3.5 ± 0.17 | 0.23 ± 0.027 | 110 ± 3.6 | 1.6 ± 0.16 |
| Null (n = 8) | 3.5 ± 0.16 | 4.0 ± 0.14 | 9.1 ± 0.92 | 8.4 ± 1.2 | 3.4 ± 0.066 | 0.25 ± 0.035 | 110 ± 6.3 | 1.8 ± 0.37 |
| 3 | ||||||||
| uPA (n = 8) | 3.7 ± 0.19 | 4.2 ± 0.12 | 10 ± 1.2 | 12 ± 0.97 | 3.6 ± .032 | 0.30 ± 0.033 | 120 ± 4.5 | 0.76 ± 0.10 |
| Null (n = 8) | 4.1 ± 0.10 | 4.7 ± 0.098 | 13 ± 0.74 | 8.8 ± 1.9 | 3.9 ± 0.12 | 0.23 ± 0.048 | 97 ± 5.5 | 0.82 ± 0.26 |
| 4 | ||||||||
| uPA (n = 18) | 4.1 ± 0.11 | 4.7 ± 0.10 | 13 ± 0.70 | 11 ± 1.3 | 4.1 ± 0.16 | 0.28 ± 0.036 | 113 ± 4.8 | 0.50 ± 0.16 |
| Null (n = 18) | 4.4 ± 0.11 | 4.9 ± 0.099 | 15 ± 0.78 | 6.5 ± 0.88 | 4.0 ± 0.095 | 0.15 ± 0.018 | 95 ± 4.8 | 0.52 ± 0.15 |
Values represent mean ± SEM.
, P < 0.05 vs. Null;
, P = < 0.01 vs. Null;
, P = < 0.001 vs. Null.
Fig 5.

Morphometry of transduced arteries. Arteries harvested 4 weeks (A–C) or 1 week (D–F) after gene transfer. Arteries transduced with AdrbtuPA had larger intimas at 4 weeks (B vs. A) and were constricted with smaller lumens at 1 week (E vs. D). Absence of elastic laminae (arrows) and fragmentation of elastic laminae (arrowhead) were more common in AdrbtuPA arteries (C and F and Inset). Movat's pentachrome stain. (Original magnifications: A–C and F, ×40; D–E, ×5.)
uPA Overexpression Causes Arterial Constriction and Lumen Loss.
One week after gene transfer, sections of AdrbtuPA arteries appeared constricted compared with AdCMVNull arteries (Fig. 5 D and E). Planimetry confirmed a 25% decrease in IEL length and a 19% reduction in EEL length in AdrbtuPA arteries (P < 0.001; Table 2). Assuming circular lumens in vivo, we used lumen circumferences measured from the tissue sections to calculate in vivo lumen area. Lumen area was decreased by 44% in AdrbtuPA arteries (P < 0.001; Table 2). Although intimal and medial area did not differ between the two groups, the wall (intima + media) thickness of AdrbtuPA arteries was increased by 30% (P < 0.001; Table 2). There were no significant differences in IEL or EEL length at 2, 3, or 4 weeks (Table 2).
Potential Mechanisms for uPA Enhancement of Lesion Growth.
To investigate the mechanisms by which uPA overexpression enhances lesion growth, we measured intimal fibrin(ogen) deposition, cell density, macrophage content, and cell proliferation. We also used Movat-stained slides to qualitatively assess the abundance and appearance of extracellular matrix components (collagen, proteoglycans, and elastin). There was only faint staining for fibrin(ogen) in the intima and media of the 1-week arteries, with no difference in intensity between the groups. There were also no significant differences between AdrbtuPA and AdCMVNull arteries in intimal cell density, macrophage density, and proliferation at any time point (Table 2 and data not shown). The degree of collagen and proteoglycan accumulation in the intima, media, and adventitia of the AdrbtuPA and AdCMVNull arteries was also similar. However, elastic lamina fragmentation and loss appeared more common in AdrbtuPA arteries (Fig. 5 C and F). Use of elastic lamina fragmentation and loss as a criterion to identify AdrbtuPA arteries permitted correct identification of 72 of 100 arteries (72% overall; range 60–82% at the four time points; P < 0.001 overall).
Discussion
Because endothelial overexpression of plasminogen activators is a potential strategy for antithrombotic therapy (3, 21), we sought to determine whether elevated endothelial uPA activity affects the genesis or progression of atherosclerosis. We cloned a rabbit uPA cDNA and expressed it in rabbit carotid endothelium during atherosclerotic lesion development. Our major findings were: (i) Elevated artery wall uPA expression is detectable 1, 3, and 7 days, but not 14 days after gene transfer. (ii) Elevated uPA expression causes arterial constriction and lumen loss 7 days after gene transfer but not at later time points. (iii) Elevated uPA expression increases intimal growth and arterial wall thickness 3–4 weeks after gene transfer but not at earlier time points. (iv) Elevated uPA expression causes degradation of arterial elastic laminae. These findings diminish enthusiasm for uPA gene therapy as a strategy for preventing intravascular thrombosis.
Our study also suggests that uPA expression, which is elevated in atherosclerotic human arteries (5–8), could contribute to the progression of human atherosclerosis. In the present study, uPA expression is increased in endothelium, whereas uPA is expressed predominantly by macrophages in more advanced human lesions (5). For this reason, we are cautious about extrapolating our results to human atherosclerosis. Nevertheless, because uPA is secreted as an inactive proenzyme (1), and uPA activity is likely determined primarily by the abundance and localization of uPAR and plasminogen (22), elevated uPA expression might alter atherosclerosis progression independently of its cellular source. An aspect of our study that supports its applicability to human atherosclerosis is that the increase in uPA expression in the AdrbtuPA arteries (7-fold; Fig. 4) is similar to the 5–6-fold increase in uPA antigen and activity in atherosclerotic versus normal human arteries (5, 6). Further studies, in which uPA is expressed in macrophages, will be important in confirming an atherogenic role for uPA.
There are several potential mechanisms through which uPA could promote intimal growth, including direct actions on cells and substrates in the artery wall and indirect actions mediated by activation of plasminogen or other proenzymes. Potential direct actions of uPA include uPAR-mediated stimulation of migration and proliferation of smooth muscle cells and macrophages (1). Indirect actions of uPA would most likely be mediated through activation of plasminogen and could include plasmin-mediated enhancement of cell migration (23), activation of latent growth factors (11), release of growth factors from extracellular matrix (12), or degradation and alteration of extracellular matrix proteins (1). Alternatively, uPA could activate members of the matrix metalloproteinase (MMP) family of enzymes. MMPs can contribute to cell migration (24) and matrix degradation (25). Finally, uPA could accelerate intimal growth by up-regulating the expression or bioavailabilty of a chemoattractant or binding protein that increases adhesion or chemotaxis of macrophages or smooth muscle cells.
Although our experiments do not identify mechanisms for uPA's enhancement of intimal growth, they cast doubt on certain mechanisms and provide clues to others. For example, we found no difference in cell proliferation or in the density of intimal macrophages between the two groups. Because the intimal lesions are made up nearly exclusively of macrophages and smooth muscle cells (15), our data also exclude a large effect of uPA on smooth muscle cell density. Thus, the enhanced intimal growth does not appear to be caused by an isolated cell-specific effect, such as enhanced smooth muscle cell or macrophage proliferation. It is possible that migration of either cell type is increased in the setting of uPA overexpression; however, direct measurement of cell migration in vivo is difficult and was not performed in the present study. A role for fibrinolysis is also not apparent, because of the low-level, equivalent fibrin(ogen) staining in the two groups. A large net effect on matrix abundance is excluded by the equivalent intimal cell densities. The temporal gap between elevated uPA expression (peaking at 3–7 days, gone by 2 weeks) and the acceleration of intimal growth (absent at 1–2 weeks, present at 3–4 weeks) is surprising and may be an important clue to the mechanisms of uPA-induced intimal growth. Because uPA activity is not elevated during the period of accelerated intimal growth, neither uPA nor plasmin (which would not be elevated in the absence of persistent uPA activity) can be the proximal mediator of intimal growth. More likely, an early action of uPA or plasmin initiates a relatively slow series of events that eventually leads to intimal growth. As discussed below, the destruction of elastic laminae (visible at 1 week and persistent through 4 weeks) may be one of these events.
The vascular constriction in the uPA arteries at 1 week was unanticipated. This constriction could represent constrictive remodeling or it might reflect an acute vasomotor effect. There is substantial experimental support for a role of the uPA/plasminogen system in vascular remodeling (25) and a small amount of data that support a role for uPA in acute vasoconstriction. For example, Haj-Yehia et al. recently reported that two-chain uPA (tc-uPA), but not single-chain uPA (sc-uPA) had vasoconstrictor activity in vitro (26). However, sc-uPA (which is the primary protein product of the AdrbtuPA vector) acted as a vasodilator in this in vitro study, and tc-uPA had vasoconstrictor activity only in conjunction with phenylephrine. For these reasons, vasoconstrictor activity of tc-uPA is an unlikely explanation for our findings. Nevertheless, it is possible that uPA, plasmin, or an activated MMP might cause vasoconstriction by cleaving a vasodilator peptide. For example, MMP-2 and -9 can cleave and inactivate the vasodilator peptides CGRP and substance P, respectively (27, 28). It will be interesting to determine whether the constriction observed in the uPA arteries at 1 week can be reversed by direct application of a vasodilator. However, even if vascular constriction in the 1-week arteries can be acutely reversed, this finding would not rule out a role for uPA in constrictive remodeling. Acute, vasodilator-responsive constriction can be the first step in long-term constrictive arterial remodeling (29). When vector systems are available that yield prolonged, high-level transgene expression in arteries, it will be important to determine whether uPA overexpression causes persistent arterial constriction and irreversible lumen loss. Until then, it is tempting to speculate that overexpression of uPA in this model will be a useful experimental setting for elucidating molecular mechanisms of atherosclerotic constrictive remodeling, a poorly understood but clinically significant phenomenon (30).
A unifying hypothesis that could explain constriction as well as intimal growth is that both are secondary effects resulting from proteolytic destruction of elastic laminae due to uPA activation of plasmin and elastases. Indeed, the 1-week uPA arteries resemble arteries of mice that lack elastin: constriction with lumen loss (31). Moreover, the 4-week uPA arteries resemble arteries of mice and humans hemizygous for elastin: they have thicker walls (32). Arteries of mice and humans hemizygous for elastin are thicker because they have additional medial elastic laminae. These laminae are thought to develop prenatally as a reaction to the increased wall stress that is caused by elastin deficiency. In the uPA-transduced rabbit arteries, remodeling that increases intimal thickness, and therefore total wall thickness, may be a postdevelopmental response to increased wall stress due to damaged elastin. The hypothesis that intimal growth leading to arterial wall thickening is an adaptive response to elastin destruction is supported by data from a study in which the elastase MMP-9 was overexpressed in rat arteries (33). These arteries also had evidence of matrix destruction and underwent both intimal growth and wall thickening over a 28-day course. Elastolysis mediated by plasmin-activated MMP-12 also appears to contribute to alterations in arterial structure in atherosclerotic mice (34). Additional studies are required to confirm a role for MMP-mediated elastolysis in this rabbit model.
In conclusion, increased expression of uPA alters arterial elastic laminae, causes vascular constriction with lumen narrowing, and enhances the growth of early atherosclerotic lesions. Although our results discourage use of uPA for vascular gene therapy, they implicate uPA as a causal agent of vascular constriction and intimal growth. Further studies in animal models and humans may reveal whether antagonism of uPA or its downstream mediators can reduce constrictive remodeling and intimal growth.
Supplementary Material
Acknowledgments
We thank Darek Jones for assistance with planimetry and Gary Howard and Stephen Ordway for editorial assistance. This work was supported in part by National Institutes of Health Grant HL60504 and University of California Tobacco-Related Disease Research Program Grant 7RT-0016 (to D.A.D.), and a Fellowship from the American Heart Association Western States Affiliate (to M.F.).
Abbreviations
AdrbtuPA, recombinant adenovirus expressing rabbit urokinase
CMV, cytomegalovirus
AdCMVNull, recombinant adenovirus containing the CMV promoter but with no transgene
EEL, external elastic lamina
MMP, matrix metalloprotease
pfu, plaque-forming unit
uPA, urokinase plasminogen activator
uPAR, urokinase plasminogen activator receptor
This paper was submitted directly (Track II) to the PNAS office.
Data deposition: The rabbit urokinase cDNA sequence has been deposited in the GenBank database (accession no. AY122285).
References
- 1.Bachmann F. (2001) in Hemostasis and Thrombosis: Basic Principles and Clinical Practice, eds. Colman, R. W., Hirsh, J., Marder, V. J., Clowes, A. W. & George, J. N. (Lippincott, Philadelphia), pp. 275–320.
- 2.Dichek D. A., Lee, S. W. & Nguyen, N. H. (1994) Blood 84, 504-516. [PubMed] [Google Scholar]
- 3.Dichek D. A., Anderson, J., Kelly, A. B., Hanson, S. R. & Harker, L. A. (1996) Circulation 93, 301-309. [DOI] [PubMed] [Google Scholar]
- 4.Vassalli G. & Dichek, D. A. (1997) Cardiovasc. Res. 35, 459-469. [DOI] [PubMed] [Google Scholar]
- 5.Kienast J., Padro, T., Steins, M., Li, C. X., Schmid, K. W., Hammel, D., Scheld, H. H. & van de Loo, J. C. (1998) Thromb. Haemostasis 79, 579-586. [PubMed] [Google Scholar]
- 6.Padró T., Emeis, J. J., Steins, M., Schmid, K. W. & Kienast, J. (1995) Arterioscler. Thromb. Vasc. Biol. 15, 893-902. [DOI] [PubMed] [Google Scholar]
- 7.Lupu F., Heim, D. A., Bachmann, F., Hurni, M., Kakkar, V. V. & Kruithof, E. K. O. (1995) Arterioscler. Thromb. Vasc. Biol. 15, 1444-1455. [DOI] [PubMed] [Google Scholar]
- 8.Raghunath P. N., Tomaszewski, J. E., Brady, S. T., Caron, R. J., Okada, S. S. & Barnathan, E. S. (1995) Arterioscler. Thromb. Vasc. Biol. 15, 1432-1443. [DOI] [PubMed] [Google Scholar]
- 9.Clowes A. W., Clowes, M. M., Au, Y. P. T., Reidy, M. A. & Belin, D. (1990) Circ. Res. 67, 61-67. [DOI] [PubMed] [Google Scholar]
- 10.Carmeliet P., Moons, L., Herbert, J.-M., Crawley, J., Lupu, F., Lijnen, R. & Collen, D. (1997) Circ. Res. 81, 829-839. [DOI] [PubMed] [Google Scholar]
- 11.Lyons R. M., Gentry, L. E., Purchio, A. F. & Moses, H. L. (1990) J. Cell Biol. 110, 1361-1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saksela O. & Rifkin, D. B. (1990) J. Cell Biol. 110, 767-775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vassalli J.-D., Sappino, A.-P. & Belin, D. (1991) J. Clin. Invest. 88, 1067-1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Williams K. J. & Tabas, I. (1995) Arterioscler. Thromb. Vasc. Biol. 15, 551-561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schneider D. B., Vassalli, G., Wen, S., Driscoll, R. M., Sassani, A. B., DeYoung, M. B., Linnemann, R., Virmani, R. & Dichek, D. A. (2000) Arterioscler. Thromb. Vasc. Biol. 20, 298-308. [DOI] [PubMed] [Google Scholar]
- 16.Schneider D. B., Sassani, A. B., Vassalli, G., Driscoll, R. M. & Dichek, D. A. (1999) J. Vasc. Surg. 29, 543-550. [DOI] [PubMed] [Google Scholar]
- 17.Lee S. W., Trapnell, B. C., Rade, J. J., Virmani, R. & Dichek, D. A. (1993) Circ. Res. 73, 797-807. [DOI] [PubMed] [Google Scholar]
- 18.Wen S., Schneider, D. B., Driscoll, R. M., Vassalli, G., Sassani, A. B. & Dichek, D. A. (2000) Arterioscler. Thromb. Vasc. Biol. 20, 1452-1458. [DOI] [PubMed] [Google Scholar]
- 19.DeYoung M. B., Zamarron, C., Lin, A. P., Qiu, C., Driscoll, R. M. & Dichek, D. A. (1999) Hum. Gene Ther. 10, 1469-1478. [DOI] [PubMed] [Google Scholar]
- 20.Schulick A. H., Taylor, A. J., Zuo, W., Qiu, C.-B., Dong, G., Woodward, R. N., Agah, R., Roberts, A. B., Virmani, R. & Dichek, D. A. (1998) Proc. Natl. Acad. Sci. USA 95, 6983-6988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Waugh J. M., Kattash, M., Li, J., Yuksel, E., Kuo, M. D., Lussier, M., Weinfeld, A. B., Saxena, R., Rabinovsky, E. D., Thung, S., et al. (1999) Proc. Natl. Acad. Sci. USA 96, 1065-1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ellis V., Behrendt, N. & Danø, K. (1991) J. Biol. Chem. 266, 12752-12758. [PubMed] [Google Scholar]
- 23.Kenagy R. D., Vergel, S., Mattsson, E., Bendeck, M., Reidy, M. A. & Clowes, A. W. (1996) Arterioscler. Thromb. Vasc. Biol. 16, 1373-1382. [DOI] [PubMed] [Google Scholar]
- 24.Kenagy R. D., Hart, C. E., Stetler-Stevenson, W. G. & Clowes, A. W. (1997) Circulation 96, 3555-3560. [DOI] [PubMed] [Google Scholar]
- 25.Lijnen H. R. (2001) Thromb. Haemostasis 86, 324-333. [PubMed] [Google Scholar]
- 26.Haj-Yehia A., Nassar, T., Sachais, B. S., Kuo, A., Bdeir, K., Al-Mehdi, A. B., Mazar, A., Cines, D. B. & Higazi, A. A.-R. (2000) FASEB J. 14, 1411-1422. [DOI] [PubMed] [Google Scholar]
- 27.Fernandez-Patron C., Stewart, K. G., Zhang, Y., Koivunen, E., Radomski, M. W. & Davidge, S. T. (2000) Circ. Res. 87, 670-676. [DOI] [PubMed] [Google Scholar]
- 28.Backstrom J. R. & Tokes, Z. A. (1995) J. Neurochem. 64, 1312-1318. [DOI] [PubMed] [Google Scholar]
- 29.Langille B. L., Bendeck, M. P. & Keeley, F. W. (1989) Am. J. Physiol. 256, H931-H939. [DOI] [PubMed] [Google Scholar]
- 30.Pasterkamp G., de Kleijn, D. P. & Borst, C. (2000) Cardiovasc. Res. 45, 843-852. [DOI] [PubMed] [Google Scholar]
- 31.Li D. Y., Brooke, B., Davis, E. C., Mecham, R. P., Sorensen, L. K., Boak, B. B., Eichwald, E. & Keating, M. T. (1998) Nature (London) 393, 276-280. [DOI] [PubMed] [Google Scholar]
- 32.Li D. Y., Faury, G., Taylor, D. G., Davis, E. C., Boyle, W. A., Mecham, R. P., Stenzel, P., Boak, B. & Keating, M. T. (1998) J. Clin. Invest. 102, 1783-1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mason D. P., Kenagy, R. D., Hasenstab, D., Bowen-Pope, D. F., Seifert, R. A., Coats, S., Hawkins, S. M. & Clowes, A. W. (1999) Circ. Res. 85, 1179-1185. [DOI] [PubMed] [Google Scholar]
- 34.Carmeliet P., Moons, L., Lijnen, R., Baes, M., Lemaitre, V., Tipping, P., Drew, A., Eeckhout, Y., Shapiro, S., Lupu, F. & Collen, D. (1997) Nat. Genet. 17, 439-444. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.