

Abstract
Early conformational events during folding of acyl-CoA binding protein (ACBP), an 86-residue α-helical protein, were explored by using a continuous-flow mixing apparatus with a dead time of 70 μs to measure changes in intrinsic tryptophan fluorescence and tryptophan-dansyl fluorescence energy transfer. Although the folding of ACBP was initially described as a concerted two-state process, the tryptophan fluorescence measurements revealed a previously unresolved phase with a time constant τ = 80 μs, indicating formation of an intermediate with only slightly enhanced florescence of Trp-55 and Trp-58 relative to the unfolded state. To amplify this phase, a dansyl fluorophore was introduced at the C terminus by labeling an I86C mutant of ACBP with 5-IAEDANS [5-((((2-iodoacetyl)amino)ethyl)amino)naphthalene-1-sulfonic acid]. Continuous-flow refolding of guanidine HCl-denatured ACBP showed a major increase in tryptophan-dansyl fluorescence energy transfer, indicating formation of a partially collapsed ensemble of states on the 100-μs time scale. A subsequent decrease in dansyl fluorescence is attributed to intramolecular quenching of donor fluorescence on formation of the native state. The kinetic data are fully accounted for by three-state mechanisms with either on- or off-pathway intermediates. The intermediate accumulates to a maximum population of 40%, and its stability depends only weakly on denaturant concentration, which is consistent with a marginally stable ensemble of partially collapsed states with ∼1/3 of the solvent-accessible surface buried. The findings indicate that ultrafast mixing methods combined with sensitive conformational probes can reveal transient accumulation of intermediate states in proteins with apparent two-state folding mechanisms.
The existence of submillisecond events during folding of numerous small single-domain proteins has been demonstrated (1–4). In most cases, these fast events can be attributed to the formation of intermediate states during early stages of folding. The importance of these intermediates in guiding the protein along productive channels, however, is a matter of debate. The observation that several small proteins can reach their native state without accumulation of detectable intermediates (5–9) has been used as an argument against the notion that accumulation of intermediates is a prerequisite for fast and efficient folding. It has also been suggested that submillisecond events observed in kinetic experiments may represent a response to the change in solvent conditions rather than the formation of nonrandom structure (10). In contrast to this view, rapid biophysical techniques such as temperature-jump (11) and continuous-flow mixing (1, 12) have shown that early events in protein folding exhibit exponential kinetics and thus represent barrier-limited transitions. Furthermore, the characterization of intermediates by using quenched-flow hydrogen exchange-labeling methods (3, 13–15) has shown that native-like structural elements are often present in the observed intermediate states. These results indicate that rapid formation of partially structured states is important for efficient folding of these proteins. Direct evidence for the existence of a productive folding intermediate was recently obtained by continuous- and stopped-flow kinetic analysis of Im7, a small α-helical protein (4).
The 86-residue acyl-CoA binding protein (ACBP) has been a popular model for folding studies because of its regular four-helix bundle structure (Fig. 1a). Earlier equilibrium and stopped-flow kinetic studies indicated that the folding of ACBP can be described by a two-state mechanism (16). However, T.K. et al. (17) recently reported that protection against hydrogen exchange for certain residues did not follow the global folding of the protein. In particular, residues in the C-terminal helix 4 seem to engage in fluctuating hydrogen bonds, indicating that local structure in this part of the protein forms before the global folding transition (17). Together with the apparent two-state behavior seen in stopped-flow fluorescence experiments, these observations are consistent with the rapid formation of an ensemble of marginally stable intermediate states with optical properties close to the unfolded state.
Figure 1.

(a) Ribbon diagram (MOLSCRIPT; ref. 38) of ACBP, based on the NMR structure coordinates of Andersen and Poulsen (39). The two tryptophan residues and the mutated C-terminal isoleucine are shown in ball and stick. (b) Absorbance spectrum (solid line) and fluorescence emission spectrum with excitation at 280 nm (dashed line) of 15 μM ACBP,I86C labeled with IAEDANS (in 20 mM Na-acetate, pH 5.3).
The rate-determining events in the folding of ACBP are well characterized. Mutation studies have shown that several evolutionarily conserved residues play a key role in the function, stability, and folding of ACBP (18, 19). The observations suggest that the formation of a native-like structure, including eight conserved hydrophobic residues in the first and the fourth helix, is rate-limiting in the folding of ACBP (20).
Fluorescence resonance energy transfer (FRET) involving covalently bound probes is particularly sensitive to tertiary structure changes, even in cases where intrinsic probes are uninformative (21, 22). FRET measurements have been combined with stopped-flow experiments to observe distance changes during protein folding (21, 23–25). In the present study we have combined site-directed fluorescence labeling and ultrarapid mixing to resolve submillisecond events in the folding of ACBP. This approach has allowed us to characterize the kinetics and stability of a transient intermediate populated on the 100-μs time scale, which has eluded detection in previous studies. The findings suggest that the use of techniques with improved temporal resolution and more sensitive conformational probes may reveal early intermediates in other proteins with apparent two-state folding mechanisms.
Materials and Methods
Protein Production.
ACBP-I86C was constructed by site-directed mutagenesis with the strand-overlap extension PCR protocol (26). The mutated ACBP construct was ligated into the pET-3a vector. For expression of wild type and the I86C mutant, Escherichia coli BL21(DE3)/pLysS was transformed with plasmids containing the appropriate construct. E. coli cultures were grown in TB medium (terrific broth). Expression of ACBP was induced by 0.4 mM isopropyl-β-d-thiogalactoside (IPTG) at OD600 = 0.9. After 4 h of induced growth the cultures were harvested and ACBP was purified as described (27).
Fluorescence Labeling.
Lyophilized ACBP-I86C was dissolved in 0.2 M Tris⋅HCl/1 mM EDTA (pH 7.5) to a final protein concentration of 1 mg ml−1. The solution was flushed thoroughly with Ar. A 20-fold molar excess of 5-((((2-iodoacetyl)amino)ethyl)amino) naphthalene-1-sulfonic acid (5-IAEDANS, Molecular Probes) was added from a 20 mM stock. The reaction was left in the dark for 2 h at room temperature and then stopped by addition of a 10-fold excess of DTT. The buffer was changed to 20 mM Tris⋅HCl (pH 8.0) by dialysis. Unmodified protein and side products were removed by ion-exchange chromatography on a MonoQ anion-exchange column. The homogeneity of the purified fluorescence labeled protein was verified by matrix-assisted laser desorption ionization–time-of-flight (MALDI-TOF) MS.
Kinetic Measurements.
For kinetic folding experiments, ACBP and ACBP,I86C-AEDANS were unfolded in 20 mM Na-acetate/3.5 M guanidinium chloride (GuHCl) (pH 5.3) to final concentrations of 220 and 168 μM, respectively. Folding was initiated by dilution with 10 volumes of 20 mM Na-acetate (pH 5.3) with varying concentrations of GuHCl. For kinetic unfolding experiments ACBP,I86C-AEDANS in 20 mM Na-acetate/0.35 M GuHCl (pH 5.3) was diluted 10-fold with 20 mM Na-acetate (pH 5.3) with varying concentrations of GuHCl. All experiments were conducted at 26°C. In fluorescence experiments with both unmodified and fluorescence-labeled protein, the tryptophan residues (Trp-55 and Trp-58) were excited at 280 nm. For the unlabeled protein, tryptophan fluorescence was measured with a 324-nm long-pass filter. For ACBP,I86C-AEDANS, fluorescence from the dansyl group was measured with a 418-nm long-pass filter. Continuous-flow measurements were carried out using the instrument described by Shastry et al. (28) with a total flow-rate of 0.825 ml s−1. The dead time of the capillary mixer, calibrated by measuring the quenching of N-acetyltryptophanamide fluorescence by N-bromosuccinimide at several quencher concentrations, was 70 μs. Stopped-flow measurements were conducted on a Biologic (Grenoble, France) SFM-4 with a measured dead time of 3.5 ms.
Equilibrium Unfolding Measurements.
A 4 μM solution of ACBP,I86C-AEDANS in 20 mM Na-acetate (pH 5.3) was titrated with an identical solution containing 7.56 M GuHCl in steps of ≈0.1 M GuHCl with an OLIS (Jefferson, GA) automatic titration device. After 5 min of equilibration the tryptophans were excited at 280 nm, and fluorescence from the dansyl group was measured above 418 nm with a PTI (South Brunswick, NJ) QM-2000 spectrofluorometer.
Kinetic Modeling.
Experimental kinetic data were modeled on the basis of a three-state folding scheme with an obligatory on-pathway intermediate. It is assumed that the logarithm of the microscopic rate constants linearly depends on the denaturant concentration:
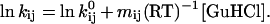 |
1 |
Observed rate constants (λ1 and λ2) and amplitudes were calculated from the corresponding rate matrix. The microscopic rate constants and m-values were varied systematically to give a satisfactory match between calculated and measured data, as described (29).
Results
To introduce a specific fluorescence label, the I86C variant of ACBP, which contains a single Cys residue at the C terminus, was derivatized with IAEDANS, a thiol-specific dansyl derivative. The absorption spectrum of the dansyl group overlaps the tryptophan emission spectrum from 300 to 400 nm (Fig. 1b), and energy transfer from the tryptophans to the dansyl group can occur. Förster distances around 22 Å have been reported for the tryptophan/IAEDANS donor/acceptor pair (30); with a distance between the two Trp residues (residues 55 and 58) and the C terminus of 23–28 Å, the efficiency of the energy transfer is a sensitive probe for conformational changes.
Folding of wild-type ACBP is accompanied by an approximately 3-fold decrease in intrinsic tryptophan fluorescence. The change in fluorescence observed in continuous- and stopped-flow measurements of the folding reaction of unlabeled ACBP at low denaturant concentration (Fig. 2a) can be fitted to a sum of three exponentials: (i) a minor slow phase probably caused by folding of molecules with cis-proline isomers; (ii) a major phase in the 10-ms time range accounting for most of the amplitude, which is the rate-limiting step in the formation of the native structure (20); and (iii) a very fast (τ ∼ 80 μs) and previously unresolved phase observable in continuous-flow mixing experiments. The amplitude of this phase is low and appears almost as a lag in the kinetic fluorescence trace. To characterize this initial phase, a continuous- and stopped-flow kinetic analysis of IAEDANS-labeled ACBP was carried out. There is no indication that the native structure is perturbed by attachment of a dansyl group to the solvent-exposed C-terminal residue. However, the I86C mutation and chemical modification results in a slight decrease in stability; GuHCl-induced unfolding experiments indicate a decrease from 25 kJ mol−1 to 21 kJ mol−1 on modification (data not shown).
Figure 2.

Refolding kinetics of unmodified ACBP (a) and dansyl-labeled ACBP,I86C (b) in 20 mM Na-acetate (pH 5.3) containing 0.34 M GuHCl at 26°C. In a and b, data from continuous-flow (○) and stopped-flow (▿) experiments were matched and combined. (a) The tryptophans were excited at 280 nm and the fluorescence was measured with a 324-nm long-pass filter. The solid line represents a fit to a sum of three exponentials. (b) ACBP,I86C labeled with IAEDANS at the C-terminal Cys residue. The tryptophans were excited at 280 nm, and the fluorescence from the dansyl group was measured with a 418-nm long-pass filter. Refolding at four different GuHCl concentrations are shown. The solid lines represent simulated kinetic traces using kinetic parameters in Table 1, based on a three-state folding mechanism with an on-pathway intermediate (Scheme S1).
By introducing a dansyl group at the C terminus and measuring the fluorescence from this group after excitation of the tryptophans during a kinetic experiment, we were able to observe the initial phase with greatly enhanced amplitude. The continuous-flow measurements show a major increase in dansyl fluorescence on the 100-μs time range, and both the rate and amplitude of this phase could be measured reliably over a range of GuHCl concentrations (Fig. 2b). The time course of folding at GuHCl concentrations from 0.34 M to 1.7 M was measured by following the total fluorescence above 418 nm, with both continuous- and stopped-flow mixing. Time traces from the experiments at the same GuHCl concentration were matched by normalizing the measured fluorescence to the fluorescence of unfolded ACBP at 3.5 M GuHCl. The combined traces over the time window from 70 μs to ∼1 s are very well described by double-exponential fits (data not shown; note that the lines in Fig. 2 represent a global fit of a kinetic model to the data at all denaturant concentrations). In addition, there is a small decrease in fluorescence at longer times (>1 s) indicative of a minor slow phase, which will not be considered further. The observed rate for the initial phase (13,000 s−1 at low GuHCl concentration) is virtually identical to that of the unmodified protein (Fig. 2a), indicating that the introduced fluorescence probe does not perturb the initial conformational event. The large decay in dansyl fluorescence observed by stopped-flow corresponds to the rate-determining step in ACBP folding. The rate of this process (e.g., 111 s−1 at 0.35 M GuHCl) is somewhat slower than that of the corresponding phase in the unmodified protein (238 s−1). Fitting of the combined continuous- and stopped-flow traces resulted in a set of two observable rate constants for the two major folding phases (Fig. 3).
Figure 3.

Fit of experimental folding data for ACBP,I86C-AEDANS. Rate constants (a) and kinetic amplitudes (b) for the fast (▴) and rate-determining (▵) folding phases obtained by exponential fitting of matching continuous-flow and stopped-flow measurements of tryptophan-dansyl energy transfer (Fig. 2). Rate constants for the slower phase were also obtained in an independent experiment where total fluorescence above 324 nm was measured (□). Data were fitted to an on-pathway three-state model (Scheme S1). The solid lines indicate the predicted observable rate constants and amplitudes. The corresponding elementary rate constants are shown as dashed lines. (c) Fluorescence above 418 nm (■) measured in an independent equilibrium experiment. The solid line represents the equilibrium fluorescence change predicted from the kinetic parameters in Table 1. The fluorescence of the three states was slightly different in this simulation compared with the simulation of the kinetic data. U (f0 = 1.05; slope = −0.023), I (f0 = 3.04; slope = −0.06), and N states (f0 = 1.028; slope = −0.035).
In Fig. 3a, the observed rate constants of the two main folding phases and the corresponding amplitudes are shown. The rate of the first phase (λ1) depends only weakly on GuHCl concentration, averaging around a value of 13,000 s−1. The rate of the second phase (λ2) decreases sharply with increasing GuHCl concentration, ranging from around 100 s−1 to 2 s−1. Fig. 3b shows the kinetic amplitudes of the two folding phases, scaled relative to the fluorescence of the unfolded state (compare Fig. 2). The emission of the fluorescence acceptor, measured in an independent equilibrium experiment, is shown in Fig. 3c. Because of the small difference in the fluorescence of folded and unfolded ACBP above 418 nm, an independent set of folding and unfolding stopped-flow experiments was performed with detection of total fluorescence above 324 nm where the difference in the signal between the native and unfolded state is larger, making it possible to measure unfolding rates. However, the amplitudes measured with the 324-nm filter cannot be compared with the amplitudes of the folding experiments carried out with the 418-nm filter. The rates obtained in the folding experiments with the 324- and 418-nm filters were identical (Fig. 3a).
The observed rate constants (λ1 and λ2) and amplitudes obtained from fitting of the experimental data were modeled on the basis of a three-state folding scheme with an obligatory intermediate, I, on a direct path between the unfolded (U) to native (N) states (Scheme S1).
Scheme 1.

The elementary rate constants, kij, and their dependence on denaturant concentration are defined by Eq. 1. The four microscopic rate constants and corresponding m-values obtained by the modeling (Table 1; dashed lines in Fig. 3a) account well for the experimentally observed rate constants, λ1 and λ2 (solid lines). In particular, kUI and mUI are constrained by λ1, λ2, and their relative amplitudes at low denaturant concentration where the intermediate is populated (note that mUI determines the slope of λ2 below the midpoint of the unfolding transition), and kIU and mIU are determined by λ1 at higher GuHCl concentration. kIN and mIN are also constrained by the data as λ2 below 1.2 M can be approximated by λ2 = 1/(1 + 1/KUI)kIN, where KUI = kUI/kIU. The rate of unfolding at high GuHCl concentration (λ2) constrains kNI and mNI. Uncertainties in the elementary rate constants and m-values (Table 1) were estimated by systematically exploring the parameter space with a manual procedure (numeric instabilities prevented us from using a least-squares algorithm). The errors are ≈5%, except for mIU, which is less well defined (±16%). The rate parameters obtained by fitting the observed rate constants (Fig. 3a) determine the time-dependent populations of the three states at each denaturant concentration (compare Fig. 4a). The modeled kinetic parameters also describe the effect of GuHCl on the amplitudes of the two phases (Fig. 3b), as well as the equilibrium unfolding transition (Fig. 3c), by using the fluorescence properties for the three states listed in Table 1. The intrinsic fluorescence of the intermediate, f0,I, relative to that of the unfolded state is well constrained by the observed amplitudes (Fig. 3b) because of the dramatic increase in energy transfer efficiency in the folding intermediate. The quality of the fits in Fig. 3 clearly indicates that the observed kinetic behavior is consistent with a three-state folding mechanism involving an on-path intermediate (Scheme S1). However, a nearly identical fit can be obtained by using an alternative kinetic mechanism with an off-pathway intermediate (see Discussion).
Table 1.
Intrinsic kinetic parameters and fluorescence properties describing the observed kinetics of ACBP,I86C-AEDANS
| U⇌I
|
I⇌N
|
Fluorescence
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| kUI | mUI | kIU | mIU | kIN | mIN | kNI | mNI | f0,U | sU | f0,I | sI | f0,N | sN |
| 11,000 ± 500 | −4.18 ± 0.2 | 7,000 ± 500 | 1.25 ± 0.21 | 650 ± 30 | −5.85 ± 0.21 | 0.07 ± 0.003 | 4.81 ± 0.04 | 1.02 | −0.02 | 3.07 | −0.04 | 1.1 | −0.09 |
Rates are given in s−1, m-values in kJ mol−1 M−1. f, fluorescence in the absence of denaturant; s, change in fluorescence after addition of GuHCl (M−1).
Figure 4.

Populations of unfolded (U), intermediate (I), and native (N) states predicted from the kinetic parameters in Table 1. (a) Time-dependent variation in populations during the first 10 ms of refolding at 0.34 M GuHCl. (b) Populations at equilibrium as function of [GuHCl] predicted by the kinetic parameters.
The fluorescence properties of the three states show that the energy transfer from the tryptophan residues to the dansyl group at the C terminus is approximately three times as efficient in I compared with U and N. This observation indicates that the average distance between tryptophan residues and the C-terminal dansyl group is substantially shorter in I compared with U; the intrinsic tryptophan fluorescence in U and I seems to be very similar, based on the low amplitude of the submillisecond folding phase in wild-type ACBP (Fig. 2a). In N the intensity of the intrinsic tryptophan fluorescence is roughly three-fold smaller than in I and U (Fig. 2a) because of intramolecular quenching of the tryptophans in N. Therefore, the three-fold-higher fluorescence of the dansyl group in I compared with N may reflect the absence of these quenching interactions rather than a significant change in the distance between the tryptophan residues and the dansyl group.
To illustrate the quality of the parameters listed in Table 1, the predicted change in fluorescence during the course of folding was compared with the experimental data (solid lines in Fig. 2b). With minor changes to the fluorescence properties of the three states, the kinetic parameters furthermore describe the fluorescence change in an independent equilibrium unfolding experiment (Fig. 3c).
Discussion
The existence of an early intermediate in the folding reaction of ACBP was suggested by fractional burst-phase protection of the protein in a recent quenched-flow hydrogen-exchange study (17). The continuous-flow fluorescence results reported here indeed revealed a kinetic event on the microsecond time scale (Fig. 2a). Introduction of an acceptor for FRET from the Trp residues resulted in distinct fluorescence properties for the intermediate formed in this fast process and allowed measurement of the kinetics of the fast phase even under destabilizing conditions (1.4 M GuHCl) where the intermediate transiently accumulates to only about 6%.
The kinetic data were modeled by using a kinetic scheme with an obligatory intermediate on a direct path from U to N (Scheme S1). The large difference between λ1 and λ2 renders the U to I transition kinetically uncoupled from the I to N transition, and interconversion between U and I may be regarded as a preequilibrium, as illustrated by the time dependence of the populations of the three states during folding (Fig. 4a). The initial formation of I is directly reflected in the disappearance of U, and during the subsequent formation of N, U and I disappear with the same rate. Under these circumstances, the order of the first two states, U and I, cannot be determined, which makes it impossible to ascertain whether the intermediate is productive (on-path) or not. Because of the low stability of I and the negative slope of log(kIN), there is no significant curvature in λ2 vs. [GuHCl] between 0.35 and 1.1 M GuHCl. Therefore, the folding process of ACBP appears two-state in stopped-flow experiments where only λ2 is observed. The large relative fluorescence of I compared with U and N makes the amplitudes of the two kinetic phases a sensitive measure of the transient population of I, which would not have been possible in unlabeled wild-type ACBP. The fluorescence properties of I in the IAEDANS-labeled ACBP also give an equilibrium stability curve with a distinct contribution from I (Fig. 3c). Calculation of equilibrium populations of the three states indicates that I reaches a maximal population of 3% at the midpoint of the GuHCl-induced unfolding transition (Fig. 4b). Such a low population will not result in significant deviations from a two-state transition if the optical property of I is similar to either U or N. In fact, the change in Trp fluorescence at 356 nm, measured on the same samples used in Fig. 3c, is fully consistent with a two-state model (data not shown).
Although the on-pathway model (Scheme S1) can fully account for all of our observations, alternative schemes with nonproductive or nonobligatory intermediates cannot be ruled out, as discussed elsewhere (4, 31, 32). The observation that the intermediate contains some native-like structural elements in previous H/D exchange labeling experiments (17) argues in favor of the on-pathway model. The protection from hydrogen exchange during the dead time of the quenched-flow experiments was particularly pronounced in the segments corresponding to the N- and C-terminal helices of the native structure (17). These helices have further been shown to be involved in the rate-limiting step during formation of the native structure (20). In addition, a recent NMR study of the acid unfolding transition of ACBP revealed an intermediate that is structurally close to the unfolded state, but contains residual helical structure in the C-terminal part (33). As noted above, donor quenching in the native state largely accounts for the apparent decrease in dansyl emission during the rate-limiting folding phase. Thus, the efficiency of FRET seems to be similar in the native and intermediate states, implying comparable distances between the two tryptophan residues on helix 3 and the C-terminal dansyl group. This surprising finding suggests that, despite the lack of persistent structure, the overall dimensions of the intermediate are similar to that of the native state.
From the parameters in Table 1, we calculated the free energies of the intermediate and unfolded states relative to the native state, the intervening energy barriers, and their location along the reaction coordinate (Fig. 5). As a measure of the reaction coordinate we used the α-value (α‡2 = mUI/mtot; αI = (mUI + mIU)/mtot; α‡1 = (mUI + mIU + mIN)/mtot; αN = 1, where mtot = mUI + mIU + mIN + mNI), which provides a measure of the burial of solvent-accessible surface relative to the unfolded state. The transition from U to I is accompanied by burial of about 1/3 of the total solvent-accessible surface area, indicating that the intermediate represents a relatively loosely packed ensemble of states. In contrast, with an α-value of 0.7, the rate-limiting transition state (TS1) represents a substantially more compact ensemble of states. The low α-value of I is consistent with the kinetic parameters in Table 1, which indicate that in the absence of denaturant, I is stabilized relative to U by only 1.1 kJ mol−1 (KUI = 1.57). Thus, the intermediate is marginally more stable than the unfolded state only under the most favorable conditions and appears rather unstructured in terms of solvent-accessible surface area (Fig. 5). Nevertheless, it contains some native-like structural elements, as mentioned above. With an α-value of 0.34, the ACBP folding intermediate observed in the present study is among the least-structured intermediates reported [e.g., α = 0.85 for protein G (1), 0.70 for phosphoglycerate kinase (34), 0.73 for Im7 (4), and 0.59 for RNase H (3)].
Figure 5.

Free energy levels of intermediate and transition states encountered during ACBP folding relative to the native state. The energy levels were calculated from the fitted kinetic parameters (Table 1) with a preexponential factor of 4.8 × 108 s−1. The reaction coordinate represents the fractional burial of solvent-accessible surface area calculated from the fitted m-values.
Our characterization of the intermediate in the folding reaction of ACBP permits a refined description of the structural events associated with the folding of ACBP. The first observable event is the formation of a highly dynamic ensemble of intermediate states within 100 μs of refolding from the GuHCl-denatured state. This structural ensemble shows slight protection from hydrogen exchange consistent with the existence of fluctuating helices in the N-terminal and especially in the C-terminal part of the molecule (17). The rate-limiting process in the formation of the native state involves the subsequent formation of tight packing interactions between the side chains in the N- and C-terminal helices (20).
By introducing a sensitive conformational probe and using an observation technique with a very short dead time, we were able to characterize a new state in the folding of ACBP. This finding raises the possibility that, given the right probe and detection technique, marginally stable intermediates may be detectable in other proteins previously thought to follow a two-state mechanism. Studies on peptides have shown that α-helices, β-hairpin, and loop structures may form on the 0.1–10 μs time scale (35, 36). Temperature-jump studies of apomyoglobin suggest that the initial formation of a collapsed state occurs within 15 μs (37). If these conformational changes do not give rise to substantial changes in the spectroscopic probe monitored, they cannot be resolved from the structurally heterogeneous and highly dynamic ensemble of denatured states. To understand the significance of such rapid conformational events for protein folding, we need not only a suitable conformational probe and sufficient temporal resolution, but also a means for initiating the folding reaction under conditions where the native state is stable. We have solved this problem by combining ultrafast mixing with tryptophan-dansyl FRET measurements. This approach promises to be generally applicable for exploring the role of early conformational events in protein folding.
Acknowledgments
This work was supported by National Science Foundation Grant MCB-079148, National Institutes of Health Grants GM56250 (to H.R.) and core CA06927, an appropriation by the Commonwealth of Pennsylvania to the Institute for Cancer Research, and by the Spectroscopy Support Facility.
Abbreviations
- ACBP
acyl-CoA binding protein
- IAEDANS
5-((((2-iodoacetyl)amino)ethyl)amino)naphthalene-1-sulfonic acid
- GuHCl
guanidinium chloride
- FRET
fluorescence resonance energy transfer
References
- 1.Park S-H, Shastry M C R, Roder H. Nat Struct Biol. 1999;6:943–947. doi: 10.1038/13311. [DOI] [PubMed] [Google Scholar]
- 2.Dalby P A, Oliveberg M, Fersht A R. J Mol Biol. 1998;276:625–646. doi: 10.1006/jmbi.1997.1546. [DOI] [PubMed] [Google Scholar]
- 3.Raschke T M, Marqusee S. Nat Struct Biol. 1997;4:298–304. doi: 10.1038/nsb0497-298. [DOI] [PubMed] [Google Scholar]
- 4.Capaldi A P, Shastry M C, Kleanthous C, Roder H, Radford S E. Nat Struct Biol. 2001;8:68–72. doi: 10.1038/83074. [DOI] [PubMed] [Google Scholar]
- 5.Jackson S E, Fersht A R. Biochemistry. 1991;30:10428–10435. doi: 10.1021/bi00107a010. [DOI] [PubMed] [Google Scholar]
- 6.Clarke J, Hamill S J, Johnson C M. J Mol Biol. 1997;270:771–778. doi: 10.1006/jmbi.1997.1147. [DOI] [PubMed] [Google Scholar]
- 7.Plaxco K W, Spitzfaden C, Campbell I D, Dobson C M. J Mol Biol. 1997;270:763–770. doi: 10.1006/jmbi.1997.1148. [DOI] [PubMed] [Google Scholar]
- 8.Grantcharova V P, Baker D. Biochemistry. 1997;36:15685–15692. doi: 10.1021/bi971786p. [DOI] [PubMed] [Google Scholar]
- 9.Viguera A R, Martinez J C, Filimonov V V, Mateo P L, Serrano L. Biochemistry. 1994;33:2142–2150. doi: 10.1021/bi00174a022. [DOI] [PubMed] [Google Scholar]
- 10.Sosnick T R, Shtilerman M D, Mayne L, Englander S W. Proc Natl Acad Sci USA. 1997;94:8545–8550. doi: 10.1073/pnas.94.16.8545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hagen S J, Eaton W A. J Mol Biol. 2000;301:1019–1027. doi: 10.1006/jmbi.2000.3969. [DOI] [PubMed] [Google Scholar]
- 12.Shastry M C, Roder H. Nat Struct Biol. 1998;5:385–392. doi: 10.1038/nsb0598-385. [DOI] [PubMed] [Google Scholar]
- 13.Gladwin S T, Evans P A. Folding Des. 1996;1:407–417. doi: 10.1016/S1359-0278(96)00057-0. [DOI] [PubMed] [Google Scholar]
- 14.Jennings P A, Wright P E. Science. 1993;262:892–896. doi: 10.1126/science.8235610. [DOI] [PubMed] [Google Scholar]
- 15.Sauder J M, Roder H. Folding Des. 1998;3:293–301. doi: 10.1016/S1359-0278(98)00040-6. [DOI] [PubMed] [Google Scholar]
- 16.Kragelund B B, Robinson C V, Knudsen J, Dobson C M, Poulsen F M. Biochemistry. 1995;34:7217–7224. doi: 10.1021/bi00021a037. [DOI] [PubMed] [Google Scholar]
- 17.Teilum K, Kragelund B B, Knudsen J, Poulsen F M. J Mol Biol. 2000;301:1307–1314. doi: 10.1006/jmbi.2000.4003. [DOI] [PubMed] [Google Scholar]
- 18.Kragelund B B, Poulsen K, Andersen K V, Baldursson T, Kroll J B, Neergård T B, Jepsen J, Roepstorff P, Kristiansen K, Poulsen F M, et al. Biochemistry. 1999;38:2386–2394. doi: 10.1021/bi982427c. [DOI] [PubMed] [Google Scholar]
- 19.Kragelund B B, Højrup P, Jensen M S, Schjerling C K, Juul E, Knudsen J, Poulsen F M. J Mol Biol. 1996;256:187–200. doi: 10.1006/jmbi.1996.0076. [DOI] [PubMed] [Google Scholar]
- 20.Kragelund B B, Osmark P, Neergård T B, Schiødt J, Kristiansen K, Knudsen J, Poulsen F M. Nat Struct Biol. 1999;6:594–601. doi: 10.1038/9384. [DOI] [PubMed] [Google Scholar]
- 21.Scalley M L, Nauli S, Gladwin S T, Baker D. Biochemistry. 1999;38:15927–15935. doi: 10.1021/bi991136g. [DOI] [PubMed] [Google Scholar]
- 22.Hammarstrom P, Owenius R, Martensson L G, Carlsson U, Lindgren M. Biophys J. 2001;80:2867–2885. doi: 10.1016/S0006-3495(01)76253-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lillo M P, Szpikowska B K, Mas M T, Sutin J D, Beechem J M. Biochemistry. 1997;36:11273–11281. doi: 10.1021/bi970789z. [DOI] [PubMed] [Google Scholar]
- 24.Nishimura C, Riley R, Eastman P, Fink A L. J Mol Biol. 2000;299:1133–1146. doi: 10.1006/jmbi.2000.3804. [DOI] [PubMed] [Google Scholar]
- 25.Nishimura C, Uversky V N, Fink A L. Biochemistry. 2001;40:2113–2128. doi: 10.1021/bi000861k. [DOI] [PubMed] [Google Scholar]
- 26.Ho S N, Hunt H D, Horton R M, Pullen J K, Pease L R. Gene. 1989;77:51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- 27.Mandrup S, Højrup P, Kristiansen K, Knudsen J. Biochem J. 1991;276:817–823. doi: 10.1042/bj2760817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shastry M C R, Luck S D, Roder H. Biophys J. 1998;74:2714–2721. doi: 10.1016/S0006-3495(98)77977-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Khorasanizadeh S, Peters I D, Roder H. Nat Struct Biol. 1996;3:193–205. doi: 10.1038/nsb0296-193. [DOI] [PubMed] [Google Scholar]
- 30.Wu P, Brand L. Anal Biochem. 1994;218:1–13. doi: 10.1006/abio.1994.1134. [DOI] [PubMed] [Google Scholar]
- 31.Wildegger G, Kiefhaber T. J Mol Biol. 1997;270:294–304. doi: 10.1006/jmbi.1997.1030. [DOI] [PubMed] [Google Scholar]
- 32.Roder H, Colon W. Curr Opin Struct Biol. 1997;7:15–28. doi: 10.1016/s0959-440x(97)80004-8. [DOI] [PubMed] [Google Scholar]
- 33.Thomsen J K, Kragelund B B, Teilum K, Knudsen J, Poulsen F M. J Mol Biol. 2002;318:805–814. doi: 10.1016/S0022-2836(02)00159-6. [DOI] [PubMed] [Google Scholar]
- 34.Parker M J, Spencer J, Clarke A R. J Mol Biol. 1995;253:771–786. doi: 10.1006/jmbi.1995.0590. [DOI] [PubMed] [Google Scholar]
- 35.Callender R H, Dyer R B, Gilmanshin R, Woodruff W H. Annu Rev Phys Chem. 1998;49:173–202. doi: 10.1146/annurev.physchem.49.1.173. [DOI] [PubMed] [Google Scholar]
- 36.Munoz V, Thompson P A, Hofrichter J, Eaton W A. Nature (London) 1997;390:196–199. doi: 10.1038/36626. [DOI] [PubMed] [Google Scholar]
- 37.Ballew R M, Sabelko J, Gruebele M. Proc Natl Acad Sci USA. 1996;93:5759–5764. doi: 10.1073/pnas.93.12.5759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kraulis P J. J Appl Crystallogr. 1991;24:946–950. [Google Scholar]
- 39.Andersen K V, Poulsen F M. J Biomol NMR. 1993;3:271–284. doi: 10.1007/BF00212514. [DOI] [PubMed] [Google Scholar]