

Abstract
The place and date of the domestication of the horse has long been a matter for debate among archaeologists. To determine whether horses were domesticated from one or several ancestral horse populations, we sequenced the mitochondrial D-loop for 318 horses from 25 oriental and European breeds, including American mustangs. Adding these sequences to previously published data, the total comes to 652, the largest currently available database. From these sequences, a phylogenetic network was constructed that showed that most of the 93 different mitochondrial (mt)DNA types grouped into 17 distinct phylogenetic clusters. Several of the clusters correspond to breeds and/or geographic areas, notably cluster A2, which is specific to Przewalski's horses, cluster C1, which is distinctive for northern European ponies, and cluster D1, which is well represented in Iberian and northwest African breeds. A consideration of the horse mtDNA mutation rate together with the archaeological timeframe for domestication requires at least 77 successfully breeding mares recruited from the wild. The extensive genetic diversity of these 77 ancestral mares leads us to conclude that several distinct horse populations were involved in the domestication of the horse.
The question of whether horses were domesticated from one wild population or from several is important to prehistoric archaeology, because the domestication of the horse is of central significance for the exploitation of the Eurasian steppe and has been claimed by some as the key to the spread of the Indo-European language family (1). One approach to solving this question relies only on reconstructing the typical mitochondrial (mt)DNA variability expected in former wild horse populations (2, 3). The reasoning is that if modern domestic horses show collectively greater mtDNA diversity than expected for a single wild horse population, then horses must have been drawn from multiple populations for domestication. The difficulty remains in how to estimate the diversity of wild horses, given that not a single wild horse population remains after the last sighting in 1969 of a free-ranging Przewalski's horse in Mongolia (4).
Lister et al. (2) suggested that “the extent of modern haplotype diversity probably reflects an input of wild animals of different areas,” though they concede that “independent domestication of wild animals in very distant parts of the world might have [left] a more coherent [phylogeographic] signature” (2). To provide a more conclusive answer, Vilà et al. (3) sequenced ancient DNA from an indisputably wild horse sample, namely from Alaskan horse remains, preserved in the permafrost, with dates ranging between 12,000 and 28,000 years (y) ago. Compared with modern horses, the Alaskan sample is relatively homogenous: six of the eight ancient mtDNA samples were found to cluster monophyletically. Assuming that this degree of genetic uniformity was typical of wild horse populations, Vilà et al. (3) concluded that “the high diversity of matrilines observed among modern horses suggest the utilization of wild horses from a large number of populations”.
However, two concerns could be leveled at this: although the analysis of ancient Alaskan horse mtDNA is important in its own right, it may legitimately be questioned whether a single horse population, living thousands of kilometers distant from potential domestication centers and up to tens of thousands of years before domestication, is representative of the genetic structure of wild horses at the relevant place(s) and time(s). Conditions during the last glaciation (ending 11,400 y ago) (5) may well have isolated wild horse populations and reduced their mtDNA diversity. The second concern is the caveat noted above by Lister et al. (2): a geographically widespread recruitment of horses should possibly still be visible as a clustering of mtDNA alleles according to breed or geography. Neither Vilà et al. (3) nor Lister et al. (2) had observed such clustering.
In our project, we generated the largest available horse mtDNA sequence database to assess the variation in documented breeds and areas with reputedly indigenous horses. Our new 318 mtDNA sequences were combined with published mtDNA sequences, yielding a total of 652 sequences, which were phylogenetically analyzed. The sample distribution is shown in Fig. 1. The mutation rate was assessed by combining revised palaeontological interpretations (6) with recently published equid mtDNA sequences (7). The number of wild mares contributing their mtDNA to the domestic horse was determined as a minimum estimate, based on the mtDNA mutation rate. The possibility that these wild mares were domesticated from one horse population was assessed by surveying the present geographical distribution of their mtDNA, and comparing their number and diversity with wild Przewalski and Alaskan horse mtDNA types.
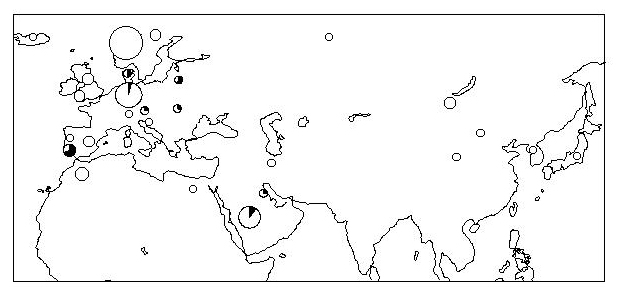
Fig 1.

Locations of the 652 horse mtDNAs sampled for this study or previously published. Numbers before brackets represent local sample sizes, and numbers in brackets represent geographically well-documented samples whose breeds agree with their traced maternal origin. The pie charts depict the occurrence of the “pony” mtDNA type C1 in horses with documented ancestry.
Materials and Methods
Samples.
DNA was extracted from the hair-roots of 318 unrelated horses of 25 breeds and varieties from Austria, Britain, Germany, Morocco, Portugal, Spain, and the United States of America. Most of the samples were collected randomly by their breeders, who were able to document the ancestry of each horse, usually for at least five generations. Furthermore we included mtDNA control region sequences, which were available from GenBank or from publications. Altogether, this process gave us mtDNA sequences for 652 horses. The GenBank accession numbers and references for the published sequences are AF132568-AF132594 (8); AF064627-AF064631; D14991, D23665, and D23666 (9); AF168689-AF168698 (11); AF14405-AF14417, and AF056071 (12); AF072975-AF072995 (2); AF169009, AF169010; AF326635-AF326686 (3); and X79547 (13). Additional sequences are described in ref. 10. Whereas the combined sample gives relatively good coverage of Europe, Morocco, and Arabia, only a few central and eastern Asian samples are available (see Fig. 1). Our nucleotide position (np) numbering follows that of Xu et al. (13).
DNA Extraction, Amplification, and Sequencing.
Total DNA was isolated from six hair roots. The control region was amplified by using a two-step seminested asymmetric PCR strategy. For the first PCR, we used the published primers P1 and P2 (9). For the second PCR, we designed a third primer, M13-HMT (5′-TGT AAA ACG ACG GCC AGT ACC ATC AAC ACC CAA AGC-3′). The first 18 nucleotides are a sequencing tag (−21M13), and the remaining primer corresponds to nps 15,425–15,442. The 469-bp PCR product (nps 15,395–15,862) was added without further purification to a second PCR.
For technical details, see the supporting information, which is published on the PNAS web site, www.pnas.org.
DNA sequencing was performed with the Dye Primer Cycle Sequencing Ready Reaction −21-M13 kit (Applied Biosystems) following the supplied protocol. Sequencing products were separated on an ABI 377 DNA Sequencer. The sequences were determined from np 15,428 up to np 15,853.
Phylogenetic Analysis.
Sequences were truncated to nps 15,494–15,740 to accommodate published short sequences. The reduced median network option (14) of NETWORK 3.111 (available at http://www.fluxus-engineering.com) was applied to the (fortunately binary) data to identify obvious parallel mutations. The resulting modified sequence table was then fed into the median-joining algorithm (15). Default settings were chosen (r = 2 and ɛ = 0). Preliminary trials indicated mutational hotspots (nps 15,585, 15,597, 15,650) which we excluded, and we downweighted (weight 0.5) two further hypervariable positions (nps 15,659 and 15,737) in all steps of the analysis. Visual inspection of the resulting network revealed two nonparsimoniously postulated mutations at np15,737 (misplacing samples JIS173 and VEC16), which we reverted manually. Rooting was performed by using equid outgroups (7). The age (ρ statistic) and its standard deviation σ of phylogenetic clusters were calculated with NETWORK 3.111 as published (16, 17).
Mutation Rate Estimation.
The mtDNA tree of modern equids (zebras, asses, horses, hemiones) was compared with the osteological tree of modern and fossil equids, the latter tree being notoriously difficult to unravel (6, 18, 19). A phylogenetic network of the equid mtDNA control region sequences indicates that the mountain zebra split off first, then the asses, the Damara and Grant zebras, the Grevy zebras, the hemiones, and finally the horse (including the Przewalski's horse). The average branch length to the most recent equid mtDNA ancestor is 10.1 mutations in the 244 bp used for the horse phylogeny. The transition-transversion ratio in the equid network is not smaller than in the horse network, indicating that multiple hits at nps have been adequately resolved by the network algorithm. Our maximum fossil benchmark is the generally accepted date for the earliest known Equus fossil, Equus simplicidens, at ≈3.5 million y (20). Our minimum fossil benchmark for Equus is 1 million y, based on Eisenmann and Baylac's (6) latest date for the development of one cranial character common to all extant equid species, that is, their relatively long cranial length. This finding indicates a minimum mutation rate of 1 mutation per 100,000 y and a maximum rate of 1 mutation per 350,000 y, respectively.
Population Size Estimation.
We calculated the smallest possible number of mares ever to have been domesticated from the wild by taking the number of mtDNA types in our modern sample (81 types) and eliminating the proportion of mtDNA types that have arisen (by the fastest conceivable mutation rate) between the earliest conceivable date for horse domestication and the present. The earliest domestication of the horse is uncertain but most probably would have occurred between 9400 BC and 2000 BC [respectively, the end of the Ice Age (5) and the approximate date of the earliest known chariot burials (21)].
Based on the fastest mutation rate (1 mutation per 100,000 y) and earliest domestication date (9400 BC), we eliminated the proportion (11.4%) of newly mutated types that would have arisen within the last 11,400 y. All derived types were considered for elimination (by collapsing into their one-step ancestral types), except for the ancient central cluster types. Although this is a coarse approach in general, in this case the very slow mutation rate renders the absolute error small and irrelevant to our conclusions. The remaining number of mtDNA types is the minimum number of mares for any point in time up to the present. It is a minimum estimate because (i) it ignores the possibility that several mares would have had the same mtDNA type while others would have died out, (ii) the fastest mutation rate estimate was chosen, (iii) frequent (and thus possibly more ancient) mtDNA types were collapsed at the same proportion (determined by ρ) as singleton types, (iv) 9,400 BC is probably an unreasonably early date for the beginnings of horse domestication, and (v) our sample does not include all currently existing mtDNA types.
To obtain an estimate for the effective Holocene population size, we also ran trials with the coalescent-based program batwing (22), one of the few methods to include population models that allow the required inferences to be drawn. However, the current version, amended for mtDNA, was unable to converge reproducibly on replicated runs for this dataset.
Results
We pooled the mtDNA sequences, covering nps 15,494–15,740, of domestic horses (626 sequences), Przewalski's horses (14 sequences), 1,000- to 2,000-y-old samples from archaeological sites in southern Sweden and Estonia (4 sequences) and 12,000- to 28,000-y old Alaskan permafrost remains (8 sequences), a total of 652 sequences (93 mtDNA types). We reconstructed the unrooted evolutionary network (Fig. 2) and then identified the root of the network by using the most closely related equid outgroups, namely Equus grevyi, Equus kiang, and Equus hemionus. The root turned out to be the central horse node A6, which implies that the Alaskan fossil equids are within the Eurasian mtDNA variation. Vilà et al. (3) placed the Alaskan fossil mtDNA as a sister branch to Eurasian mtDNA, but this discrepancy is explained by their choice of an ass (Equus africanus) as an outgroup: the ass's mtDNA (23) is divergent among equids and appears to have undergone a parallel mutation at np 15,703, influencing the root placement. To calculate phylogenetic time estimates, we postulated within the network one most parsimonious tree (shown in bold in Fig. 2) of the 641 modern horses; that is, we disregarded the ancient Alaskan and Scandinavian horses. We dated the mtDNA root type to be between 342,000 ± 74,000 and 1,198,000 ± 260,000 y old (Table 1). The large uncertainty is caused by the poor calibration of the mtDNA mutation rate (see Materials and Methods). This root type is well distinguished from any other modern equid root type (7) and multifurcates into about 13 horse mtDNA branches. This means 300,000 y approximately represents the latest possible date for the first modern caballine horse.
Fig 2.

Phylogenetic network of 652 horse mtDNA sequences. The bold lines indicate a postulated most parsimonious tree. Circles are proportional to the number of horses they represent. Links represent mutations at the mtDNA nps indicated alongside. The node A6 is the root of the network according to equid outgroups. Each sample name is composed as follows: the first letter indicates the first author (B = Bowling; D = Dhar, GenBank; I = Ishida; J–Jansen, this study; K = Kavar or Kim; L = Lister; O = Oh, GenBank; V = Vilà), the second and third letter indicates the breed (AB = Arabian–Barb cross, AL = Andalusian, AN = ancient sample according to ref. 2, AR = Arabian, BA = Barb, BE = Belgian, CH = Cheju, DU = Dülmener, EC = according to ref. 2, EX = Exmoor, FE = Fell, FJ = Fjord, FR = Friesian, HO = Holsteiner, IS = Icelandic, JAP = Japanese, KA = Caspian, KO = Konik, KP = Kazakh, LI = Lipizzan, LU = Lusitano, MO = Mongolian domestic, MU = Mustang, NN = no breed given, NO = Noriker, OL = Oldenburger, PL = Pleistocene Alaskan, PR = Przewalski's, QH = Quarter, RD = Rhineland heavy draft, RO = Rottaler, SC = Scottish Highland, SE = Senner, SH = Shire, SO = Sorraia, SP = Shetland, SU = Suffolk punch, TR = Trakehner, TS = Tsushima, VB = Thoroughbred, WB = Warmblood, WP = Welsh, YU = Yunnan), and the final number is taken from the original study. For circles encompassing several horses, only one sample name is given.
Table 1.
Frequencies and ages of the mtDNA clusters
| Cluster | n of central node | n of cluster | Age (1 mutation per 100,000 a) | σ | Age (1 mutation per 350,000 a) | σ |
|---|---|---|---|---|---|---|
| Coalescence | 16 | 639 | 342,000 | 74,000 | 1,198,000 | 260,000 |
| A1 | 33 | 42 | 20,000 | 10,000 | 68,000 | 34,000 |
| A2 | 11 | 14 | 29,000 | 18,000 | 100,000 | 61,000 |
| A3 | 61 | 62 | 5,000 | 3,000 | 17,000 | 10,000 |
| A4 | 16 | 16 | 0 | 0 | 0 | 0 |
| A5 | 15 | 15 | 0 | 0 | 0 | 0 |
| A6 | 13 | 15 | 15,000 | 15,000 | 54,000 | 54,000 |
| B1 | 22 | 22 | 0 | 0 | 0 | 0 |
| B2 | 33 | 37 | 13,000 | 9,000 | 46,000 | 31,000 |
| C1 | 45 | 46 | 2,000 | 2,000 | 8,000 | 8,000 |
| C2 | 37 | 39 | 8,000 | 4,000 | 27,000 | 16,000 |
| D1 | 79 | 83 | 6,000 | 3,000 | 21,000 | 9,000 |
| D2 | 45 | 48 | 6,000 | 5,000 | 22,000 | 16,000 |
| D3 | 37 | 45 | 20,000 | 9,000 | 70,000 | 32,000 |
| E | 16 | 16 | 0 | 0 | 0 | 0 |
| F1 | 21 | 36 | 47,000 | 22,000 | 166,000 | 77,000 |
| F2 | 15 | 17 | 12,000 | 8,000 | 41,000 | 29,000 |
| G | 19 | 22 | 14,000 | 10,000 | 48,000 | 36,000 |
The most striking feature of the mtDNA network (Fig. 2) is the presence of 17 very frequent mtDNA types, most of which are old enough to have developed a star-like branching structure (a phylogenetic cluster). Each cluster comprising over 2% of the sample is listed in Table 1 along with the corresponding age estimates. For sequence motifs defining each cluster, see Table 2. The clusters are historically interesting in that the ancestral mtDNA type of each has had greater breeding success compared with the other ancestral mtDNA types in the network, which are poorly represented or even extinct. To assess whether domestication, climate, or other factors have led to the success of these types, it is necessary to determine where and when they arose. However, the uncertainty in the mtDNA mutation rate calibration and the large standard deviation for each cluster (Table 1) make it impossible to give an accurate date for the expansion of any particular cluster.
Table 2.
Sequence motifs of the mtDNA clusters
| Cluster | Sequence motif |
|---|---|
| A1 | 495C 542T 602T 635T 666A 703C 720A |
| A2 | 495C 542T 602T 595G 666A 720A |
| A3 | 495C 666A 720A |
| A4 | 495C |
| A5 | Reference mtDNA sequence |
| A6 | 495C 602T 650G 720A |
| B1 | 495C 538G 596G 602T 709T 720A |
| B2 | 495C 538G 602T 709T 720A |
| C1 | 495C 602T 617C 659C 720A |
| C2 | 495C 601C 602T 720A |
| D1 | 494C 495C 496G 534T 602T 603C 649G |
| D2 | 494C 495C 496G 534T 602C 603C 649G 720A |
| D3 | 494C 495C 496G 534T 602T 603C 604A 649G 720A |
| E | 495C 520G 521A 602T 720A 737C |
| F1 | 495C 602T 604A 667G 703C 720A |
| F2 | 495C 602T 604A 703C 720A 726A 740G |
| G | 594C 598C 602T 615G 616G 659C 703C |
Several clusters are especially interesting from the point of view of geographic distribution and breed. To exclude modern interbreeding as far as possible, we considered in our geographic analysis only horses with a documented geographic origin of their maternal ancestry that corresponds to the origin of the respective breed. For example, if the earliest documented maternal ancestor of a horse sample was Andalusian by breed, but born in Germany, then it was excluded, even though the Spanish origin of Andalusians is not in dispute. Applying this criterion, we included for the geographic analysis only 331 of the 652 horses.
The clearest association between cluster and breed is evidenced by cluster C1 (n = 48): in our sample, it is geographically restricted to central Europe, the British Isles, and Scandinavia, including Iceland (Fig. 1). A total of 17 of 19 documented horses with C1 are northern European ponies (Exmoor, Fjord, Icelandic, and Scottish Highland). Additionally, 14 of 27 undocumented horses (3) with C1 are ponies, including Connemara ponies. The cluster is younger than perhaps 8,000 y (Table 1), but definitely older than 1,500 y, because C1 was also found in two ancient Viking horses. Furthermore, mtDNA cluster E (n = 16) consists entirely of Icelandic, Shetland, and Fjord ponies. Taken together, this suggests a common late glacial or postglacial origin for these pony breeds.
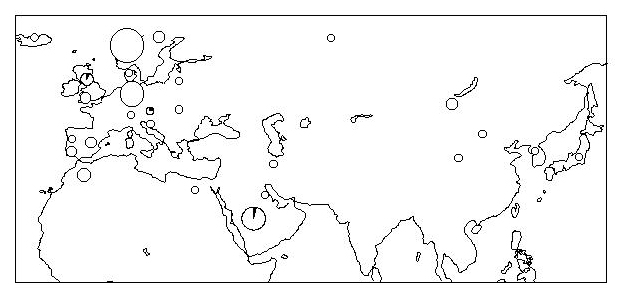
D1 is another geographically striking mtDNA cluster (Fig. 3). It is widespread in our sample, but with a frequency maximum in Iberian (Andalusian and Lusitano) and North African horses (Barbs). As we might expect from the historical presence of the Spanish in North America, a relatively high percentage of American mustangs (31%) also belong to this cluster. It is noteworthy that only a small proportion of the Arabs belong in this cluster (around 5%), regardless of whether we define Arabs by breed or by geography. This genetic result is in accordance with the phenotype; Barb conformation is very similar to that of Iberians, whereas the phenotype of the Arabs is quite distinct from that of Iberians and Barbs (24).
Fig 3.

Geographical distribution of the samples of cluster D1 with documented ancestry. The D1 Breed composition, including samples without documented ancestry, is as follows (% of samples of breed): 5 Arabs (5%), 1 Belgian (100%), 1 Arabian-Barb cross (14%), 7 Andalusians (50%), 7 Barbs (54%), 1 Norwegian Fjord (9%), 1 Friesian (50%), 1 Holsteiner (10%), 1 Icelandic (13%), 2 Caspians (29%), 5 Lusitanos (56%), 24 Mustangs (31%), 2 Rhineland heavy drafts (8%), 3 Rottalers (33%), 2 Shires (20%), 2 Trakehners (40%), 3 Lipizzans (23%), 2 Yunnans (100%), 1 Shetland (20%), 1 historical sample (13%), and 12 EC (according to ref. 3).
Relatively recent bottlenecks are also reflected in the mitochondria, namely in the Senner, Sorraia, and Dülmener. The first two are locally regarded as indigenous.
Senner horses are a small population near Paderborn, Germany. This population is very small and inbred regarding its maternal line, because all horses descend (21 to 24 generations ago) from the mare David, born 1725 (25). mtDNA cluster G, in which we found all sampled Senners, is very rare in other breeds (2 of 94 Arabs, 1 of 24 Rhineland heavy draft, none in others).
The Sorraias originate from a small group of 7 mares and 4 stallions obtained by R. d'Andrade near Coruche, Portugal, in around 1930, after he had seen a phenotypically identical wild population there in 1920, which was distinct from the local riding horse. According to d'Andrade (26), 5 of the 7 mares passed down mtDNA lineages. All 18 sampled Sorraias have either of two A1 mtDNA types (61% A1 root type, 39% ancestral JSO41 type), which are quite unrelated to the D1 type predominant in the other Iberians.
German Dülmener ponies come from an area where, in AD 1316, “wild” horses were recorded (25). They are supposed to be a mixed breed, but surprisingly, 9 of 10 Dülmeners in our sample belong in cluster D3. Two of these 9 D3 types have derived mutations, suggesting an ancient origin.
Cluster A2, which is restricted to the Przewalski's horses, is problematical. According to studbook records, four mitochondrial lineages survived, and a sequencing study (4) showed that only two mtDNA types exist in these four lineages. However, in the Przewalski's horses' sequences we found three mtDNA types, the third in the data of Ishida et al. (10). Nevertheless, all three Przewalski's horse's mtDNA types are closely related and, in agreement with Ishida et al. (10), we confirm that these types are not found in any other breed.
For the geographic distribution of all clusters, see the supporting information on the PNAS web site.
We used the mtDNA mutation rate to determine the minimum number of mares domesticated from the wild. Excluding the 3 Przewalski's horse mtDNA types, there are 81 domestic horse mtDNA types within the analyzed mtDNA region. Of these domestic mtDNA types, 43 minor (noncluster) types are only one mutation distant from their immediate ancestors (Fig. 2). Using extremely conservative assumptions (see Materials and Methods), it follows that a maximum of 4.4 of the 43 mtDNA types postdate domestication. In other words, 77 successfully breeding mares is a minimum estimate for the number of wild mares ever domesticated. These 77 ancestral types are nearly as diverse as the entire current horse mtDNA pool, and an order of magnitude more diverse than the monophyletic Alaskan wild horse mtDNA or the Przewalski wild horse mtDNA (Table 1).
Discussion
Does the greater diversity in domesticated horse mtDNA vis-à-vis wild Alaskan and Przewalski‘s horse mtDNA indicate that more than one wild horse population was recruited for domestication, or alternatively, does the difference in diversities imply that the wild Alaskan and Przewalski‘s horses have undergone genetic bottlenecks unrepresentative of ancient wild horses? The former possibility appears more likely; even though Alaskan Pleistocene samples are spread over a time window of 16,000 y, most of them cluster monophyletically (3). As for the Przewalski’s horses, in which only closely related mtDNA types were found, it has been claimed that this low diversity is caused by a recent drastic population decrease (4). This appears unlikely because three of four maternal lines, representing at least two captures separated by 45 y, show an identical mtDNA type. The Przewalski's horse indeed experienced a bottleneck situation, but well-documented bottlenecks in other populations (e.g., Exmoor ponies, which collapsed to 46 mares in the 1940s) have not resulted in such a decrease of mtDNA types. The geographically specific mtDNA clusters which we have found, for example in northern European ponies, agree with a scenario of recruitment of wild mares for domestication from geographically different areas. More evidence might be seen in (controversial) archaeological claims identifying “pony” and “warmblood-like” prehistoric horse phenotypes during and after the Ice Age in Europe (27, 28). Recent research indicated that cattle (29, 30, 31) and goats (32) have had much fewer founder types than horses, but were domesticated more than once. This observation further supports the conclusion that horses were domesticated from several wild populations (2, 3), which individually had a lower genetic diversity.
Assuming our interpretation of multiple genetic horse origins is correct, does it follow that the technique of horse domestication was developed independently by different human communities in different places? From an archaeological and ethological point of view, a single origin of the required human expertise cannot be ruled out. Modern breeding of the wild Przewalski's horse initially encountered problems such as pacing, excessive aggression, impotence, and infanticide (33), leading the Przewalski's horse to the brink of extinction. The Przewalski's horse is not ancestral to domestic horses, but if their wild ancestors were similarly intractable, it is unlikely that the technique was mastered many times independently during prehistory. The ease of domestic horse breeding today may be the genetic consequence of selections of particularly amenable beasts some thousands of years ago (34, 35). Furthermore, if domestication had arisen independently multiple times, one would expect to find archaeological evidence for domestication at very different times and places. This may not be the case. Although there are claims for horse domestication as early as 4500 BC for Iberia and the Eurasian steppe, the earliest undisputed evidence are chariot burials dating to ≈2000 BC from Krivoe Ozero (Sintashta-Petrovka culture) on the Ural steppe (36, 37, 38). Burial, textual, and/or iconographic evidence shows that by 1250 BC, chariots were widespread from Greece to China (37, 39, 40). Such an expansion may suggest a diffusion of the knowledge of horse breeding (34), and possibly a concomitant spread of horses themselves, originally localized both temporally and spacially. In this reading of the archaeological record, the knowledge and the initially domesticated horses themselves would have spread, with local mares incorporated en route, forming our regional mtDNA clusters.
Further resolution of ancient horse domestication may best be achieved by better calibration of the equid mtDNA mutation rate using whole mtDNA and more fossil data. This will allow molecular genetic data to discriminate between the genetic events occurring in the Holocene (e.g., 2000 BC) and those in the Pleistocene. We would claim that a finer resolution of domestic horse origins will be achieved within the analytical context outlined here.
Supplementary Material
Acknowledgments
We thank S. Baker, M. Bowling, V. Eisenmann, L. Kaagan, K.-I. Kim, H. Macgregor, E. A. Oakenfull, and C. Vilà for additional information on their publications, I. Wilson for amending batwing for mtDNA, and the breeders for their assistance with collecting the samples. We are indebted to D. Bradley (Trinity College, Dublin) and J. Mallory (Queen's University, Belfast) for critically evaluating a previous version of the manuscript.
Abbreviations
mt, mitochondrial
y, years
np, nucleotide position
References
- 1.Mallory J. P., (1989) In Search of the Indo-Europeans (Thames and Hudson, London).
- 2.Lister A. M., Kadwell, M., Kaagan, L. M., Jordan, W. C., Richards, M. B. & Stanley, H. F. (1998) Ancient Biomol. 2, 267-280. [Google Scholar]
- 3.Vilà C., Leonard, J. A., Götherstöm, A., Marklund, S., Sandberg, K., Lindén, K., Wayne, R. K. & Ellegren, H. (2001) Science 291, 474-477. [DOI] [PubMed] [Google Scholar]
- 4.Oakenfull E. A. & Ryder, O. A. (1998) Anim. Genet. 29, 456-459. [DOI] [PubMed] [Google Scholar]
- 5.Björck S., Kromer, B., Johnsen, S., Bennike, O., Hammarlund, D., Lemdahl, G., Possnert, G., Rasmussen, T. L., Wohlfarth, B., Hammer, C. U. & Spurk, M. (1996) Science 274, 1155-1160. [DOI] [PubMed] [Google Scholar]
- 6.Eisenmann V. & Baylac, M. (2000) Zool. Scripta 29, 89-100. [Google Scholar]
- 7.Oakenfull E. A., Lim, H. N. & Ryder, O. A. (2000) Conserv. Genet. 1, 341-355. [Google Scholar]
- 8.Bowling A. T., Del Valle, A. & Bowling, M. (2000) Anim. Genet. 25, 1-7. [DOI] [PubMed] [Google Scholar]
- 9.Ishida N., Hasegawa, T., Takeda, K., Sakagami, M., Onishi, A., Inumaru, S., Komatsu, M. & Mukoyama, H. (1994) Anim. Genet. 25, 215-221. [DOI] [PubMed] [Google Scholar]
- 10.Ishida N., Oyunsuren, T., Mashima, S., Mukoyama, H. & Saitou, N. (1995) J. Mol. Evol. 41, 180-188. [DOI] [PubMed] [Google Scholar]
- 11.Kavar T., Habe, F., Brem, G. & Dovc, P. (1999) Anim. Gent. 30, 423-430. [DOI] [PubMed] [Google Scholar]
- 12.Kim K.-I., Yang, Y.-H., Lee, S.-S., Park, C., Bouzat, J. L. & Lewin, H. A. (1999) Anim. Genet. 30, 102-108. [DOI] [PubMed] [Google Scholar]
- 13.Xu X. & Arnason, U. (1994) Gene 148, 357-362. [DOI] [PubMed] [Google Scholar]
- 14.Bandelt H.-J., Forster, P., Sykes, B. C. & Richards, M. B. (1995) Genetics 141, 743-753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bandelt H.-J., Forster, P. & Röhl, A. (1999) Mol. Biol. Evol. 16, 37-48. [DOI] [PubMed] [Google Scholar]
- 16.Forster P., Harding, R., Torroni, A. & Bandelt, H.-J. (1996) Am. J. Hum. Genet. 59, 935-945. [PMC free article] [PubMed] [Google Scholar]
- 17.Saillard J., Forster, P., Lynnerup, N., Bandelt, H.-J. & Nørby, S. (2000) Am. J. Hum. Genet. 67, 718-726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Forsten A. (1998) Q. Sci. Rev. 17, 1097-1100. [Google Scholar]
- 19.Schreiber A., Eisenmann, V. & Zimmermann, W. (2000) in EEP, Asiatic Equids, Husbandry Guidelines, ed. Zimmerman, W. (Zoologischer Garten Köln, Köln, Germany), pp. 1–34.
- 20.Prado J. L. & Alberdi, M. T. (1996) Palaeontology 39, 663-680. [Google Scholar]
- 21.Raulwing P., (2000) Horses, Chariots and Indo-Europeans. (Archaeolingua, Budapest).
- 22.Wilson I. J. & Balding, D. J. (1998) Genetics 150, 499-510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xu X., Gullberg, A. & Arnason, U. (1996) J. Mol. Evol. 43, 438-446. [DOI] [PubMed] [Google Scholar]
- 24.Epstein H., (1971) The Origin of the Domestic Animals of Africa (African Publishing, New York), Vol. 2.
- 25.Nissen J., (1997) Enzyklopädie der Pferderassen: Europa, Band 1 (Kosmos, Lisbon).
- 26.d'Andrade R. (1945) Boletim Pecuário XIII, 1-13. [Google Scholar]
- 27.Ebhardt H. (1962) Säugetierkundl. Mitteil. X, 145-168. [Google Scholar]
- 28.Uerpmann H.-P. (1990) Madrider Mitteil. 31, 109-153. [Google Scholar]
- 29.Loftus R. T., Mac Hugh, D. E., Bradley, D. G., Sharp, P. M. & Cunningham, P. (1994) Proc. Natl. Acad. Sci. USA 91, 2757-2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bradley D. G., MacHugh, D. E., Cunningham, P. & Loftus, R. T. (1996) Proc. Natl. Acad. Sci. USA 93, 5131-5135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Troy C. S., MacHugh, D. E., Bailey, J. F., Magee, D. A., Loftus, R. T., Cunningham, P., Chamberlain, A. T., Sykes, B. C. & Bradley, D. G. (2001) Nature (London) 410, 1088-1091. [DOI] [PubMed] [Google Scholar]
- 32.Luikart G., Gielly, L., Excoffier, L., Vigne, J.-D., Bouvet, J. & Taberlet, P. (2001) Proc. Natl. Acad. Sci. USA 98, 5927-5932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Boyd L. & Houpt, K. A. (1994) in Przewalski's Horse, the History and Biology of an Endangered Species, eds. Boyd, L. & Houpt, K. A. (State Univ. of New York Press, Albany), pp. 195–228.
- 34.Levine M. A. (1999) J. Anthropol. Archaeol. 18, 29-78. [Google Scholar]
- 35.Hemmer H., (1990) Domestication: The Decline of Environmental Appreciation (Cambridge Univ. Press, Cambridge, UK).
- 36.d'Andrade F., (1973) A Short History of the Spanish Horse and of the Iberian “Gineta” Horsemanship for which this Horse Is Adapted (Lisbon).
- 37.Levine M. A. (1999) in Late Prehistoric Exploitation of the Eurasian Steppe, eds. Levine, M. A., Rassamakin, Y., Kislenko, A. & Tatarintseva, N. (McDonald Institute for Archaeological Research, Cambridge, UK), pp. 5–58.
- 38.Anthony D. W. (1995) Antiquity 69, 554-565. [Google Scholar]
- 39.Linduff, K. M., in Steppe Adaptation and the Horse, eds. Levine, M., Renfrew, C., Boyle, K. (McDonald Institute for Archaeological Research, Cambridge, UK), in press.
- 40.Renfrew C. (1998) in Creativity in human Evolution and Prehistory, ed. Mithen, S. (Routledge, London), pp. 260–284.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}