

Abstract
The success of nanoparticle-based cancer therapeutics relies on their efficient tumor uptake and retention. Given this, improving nanoparticle localization in tumors is paramount to maximize their therapeutic potential. A common approach to achieve this is to functionalize nanoparticles with active targeting moieties that bind to specific tumor-associated receptors. Among these, arginine-glycine-aspartic acid (RGD) peptides have shown a potential to promote tumor accumulation by targeting the ανβ3 integrin receptor, a receptor commonly overexpressed by tumors owing to its role in promoting angiogenesis, metastasis and proliferation. Yet, its efficacy is commonly assessed using immunocompromised mice models. While useful, these models do not accurately account for immune-related interactions, which could lead to an overestimation of targeting efficacy. In our study, we investigated the efficacy of RGD peptides to improve the tumor accumulation of PEGylated gold nanoparticles (GNPs) using an immunocompetent mouse model. While RGD functionalization increased GNP uptake in cancer cells in vitro, it significantly reduced tumor accumulation in vivo due to enhanced off-target clearance by the mononuclear phagocyte system, with elevated accumulation in the spleen and liver. These findings highlight that RGD functionalization can promote immune-driven clearance in vivo, despite improving GNP uptake in cancer cells in vitro, emphasizing the importance of assessing targeting strategies in immunocompetent models for more physiologically relevant assessments.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-025-22151-7.
Subject terms: Nanomedicine, Nanotechnology in cancer
Introduction
The increasing preclinical and clinical utilization of nanoparticles (NPs) has provided innovative approaches to improve modern oncology, owing to their favorable physicochemical properties that allow them to be tailored for specific applications. This is evidenced by the successful clinical implementation of NPs such as Doxil and Abraxane, which have demonstrated improved therapeutic outcomes compared to their free-form counterparts in chemotherapy treatment1–3. However, the clinical translation of NPs remains limited relative to the number of promising results in preclinical studies. Among other challenges such as complex manufacturing and regulatory hurdles that hinder their translation, the efficacy of NP formulations can also fall short of expectations at the clinical stage4–6. Factors such as rapid clearance by the mononuclear phagocyte system (MPS) and limited tumor penetration hinder tumor delivery, limiting their efficacy7. Therefore, optimizing NP formulations to enhance their tumor-targeting capabilities and improve their accumulation at the tumor site is crucial to realizing their full therapeutic potential8.
The improvement of tumor localization of NPs is commonly achieved through strategic functionalization, modifying their surface chemistry to leverage both passive and active targeting mechanisms. While improving the passive targeting of NPs relies upon increasing their circulation time through stealth molecules such as polyethylene glycol (PEG), active targeting aims to equip NPs with targeting moieties to afford them with a higher degree of specificity towards tumors. Targeting moieties, such as antibodies, peptides or small molecules, are commonly used to target overexpressed surface receptors of cancer cells9. By modifying the NP surface with targeting ligands, NPs can preferentially bind to target receptors, enhancing their cellular internalization and selectivity towards tumors.
Of the many targets, integrin receptors have been investigated as cell surface foci for homing and docking of targeted cancer therapeutics. Integrins, consisting of alpha and beta subunits, form a family of transmembrane receptors that mediate cell adhesion to the extracellular matrix (ECM) and facilitate cell signaling. However, integrins such as ανβ3 are also known to mediate tumor progression, promoting angiogenesis, metastasis and proliferation. ανβ3 facilitates the adhesion of cancer cells to ECM components such as vitronectin, fibronectin, and osteopontin, thereby enhancing tumor invasion and migration10,11. Additionally, its role in angiogenesis, particularly through its synergy with vascular endothelial growth factor (VEGF), supports the formation of new blood vessels to sustain tumor growth12. As a result, ανβ3 is highly expressed in the tumor microenvironment (TME), while its expression is largely absent in normal cells, making it an ideal molecular target for NP formulations for cancer treatment.
To date, numerous studies have targeted the ανβ3 integrin via arginine-glycine-aspartic acid (RGD) peptides to improve the specificity of NP formulations13–17. The RGD motif exhibits a high affinity for the integrin ανβ3, making RGD peptides effective at recognizing ανβ3-expressing tumor cells. Previous in vitro studies demonstrate RGD significantly improves the uptake of NPs by cancer cells18–20. Yet, the translation of these benefits has been shown to vary in vivo14,15,19,21. It has been suggested that RGD can also lead to increased recognition by the MPS. As a result, the circulation time of NPs can be reduced, decreasing the efficacy of RGD as a targeting mechanism against tumors15. Despite this, some studies have reported enhanced tumor localization of RGD-modified NPs14,19. However, studies assessing the efficacy of targeted NPs are often conducted using immunodeficient mouse models. These models lack proper immune-mediated interactions with NPs, which could lead to overstated results22. As such, it is critical to evaluate the efficacy of targeting moieties such as RGD in an immunocompetent environment.
Our goal was to leverage an immunocompetent syngeneic tumor model to provide a more physiologically relevant assessment of RGD-functionalized NPs, offering critical insights into their targeting efficacy and immune interactions. Therefore, we assessed the efficacy of an RGD peptide as a targeting moiety in a syngeneic tumor model (KPCY) (Fig. 1). To investigate this, PEGylated gold nanoparticles (GNPs) were functionalized with either linear (lRGD) or cyclic (cRGD) RGD peptides, two structural formations of the peptide that are commonly used. Prior to conducting in vivo studies, the RGD-mediated uptake was investigated in vitro using both monolayer and spheroid cell culture models to confirm that active targeting of the peptide enhances GNP internalization in KPCY murine pancreatic cancer cells, which are derived from the transgenic KrasG12D/+ Trp53R172H/+ P48-Cre mouse model of pancreatic cancer arising in a C57BL/6 genetic background and express the YFP lineage tag. We then conducted an in vivo biodistribution study to compare the pharmacokinetics and tumor accumulation between RGD-functionalized GNPs and PEGylated GNPs. Additionally, GNP uptake in murine-derived macrophages was assessed in vitro to provide insight into how RGD affects NP recognition and uptake by the MPS. The preceding findings in our study highlight the importance of immunocompetent models in evaluating NP targeting strategies and the potential challenges of employing RGD as a targeting moiety.
Fig. 1.

Schematic figure outlining the assessment of RGD peptides to improve uptake of GNPs. The efficacy of RGD peptides as a targeting moiety was assessed using monolayer cell cultures, spheroid cell cultures, as well as a syngeneic tumor model in vivo. RGD recognizes the ανβ3 integrin that is commonly overexpressed by tumors, facilitating an improvement in GNP uptake in cancer cells.
Results & discussion
Gold nanoparticle functionalization and characterization
Using a citrate reduction method, spherical GNPs with an average core diameter of approximately 12 nm were synthesized, which was confirmed by transmission electron microscopy (TEM) images (Fig. 2A). Following their synthesis, the citrate-capped GNPs were functionalized with 2 kDa thiol terminated PEG at a grafting density of 1 PEG molecule per nm2 of GNP surface area to form the GNP-PEG complex used in our studies. The PEG grafting density was specifically chosen as it significantly reduces protein adsorption onto GNPs, thereby enhancing colloidal stability and minimizing nonspecific interactions23. As a result, PEG minimizes the adsorption of serum proteins onto the NP surface, effectively extending their circulation time24–26. Furthermore, both the GNP-PEG-lRGD and GNP-PEG-cRGD complexes were synthesized by the sequential conjugation of the GNP-PEG complex with a 1.6 kDa linear or cyclic RGD peptide containing a cysteine at the N-terminus, facilitating the conjugation. The RGD peptides were added at a grafting ratio of 1 RGD peptide molecules per 2 PEG molecules, which has been shown to significantly improve the tumor localization of GNPs19.
Fig. 2.

Characterization of GNP complexes. A) TEM image of synthesized 12 nm GNPs. B-C) Measurement of the hydrodynamic diameter and zeta potential of the various GNP complexes. D) Complete summary of characterization measurements performed for the GNP complexes.
To verify the successful conjugation and stability of the various GNP complexes, characterization measurements of each GNP complex were performed. Both Ultraviolet-visible (UV-vis) and dynamic light scattering measurements indicated an increase in the NP size, demonstrating a shift in the peak absorbance wavelength and hydrodynamic diameter of the GNPs with the sequential addition of the PEG and RGD peptides (Fig. 2B, S1A, B). Additional zeta potential measurements further verified the successful conjugation of the PEG and RGD peptides to the GNP surface. With the addition of the PEG molecules, an increase in the GNP surface charge was observed, verifying the replacement of the negative citrate molecules with neutral PEG molecules (Fig. 2C). The GNP-PEG-lRGD and GNP-PEG-cRGD also demonstrated an increase in their surface charge due to the adsorption of positively charged RGD peptide molecules onto the GNP surface. A complete summary of characterization measurements at each stage of the functionalization process is shown in Fig. 2D.
Uptake of GNPs in vitro
Prior to conducting in vivo assays, the targeting efficiency of the linear and cyclic RGD peptides against KPCY cells was assessed using both monolayer and 3D spheroid cell models. Both monolayer and 3D spheroid cell cultures were exposed to the GNP-PEG (PEG), GNP-PEG-lRGD (PEG-lRGD) and GNP-PEG-cRGD (PEG-cRGD) complexes at a concentration of 7.5 µg/mL. Following their exposure, cells were harvested at 1 h, 8 h, and 24 h time points to measure the GNP content via inductively coupled plasma-mass spectrometry (ICP-MS). The GNP content was also further verified qualitatively using confocal imaging to confirm the intracellular accumulation of GNPs within cell cultures.
As shown from the ICP-MS data (Fig. 3A), the addition of an RGD peptide as a targeting moiety significantly increased the intracellular accumulation of GNPs in monolayer cell cultures. Specifically, 1 h after exposure, the targeting potency of the RGD peptide was observed to be the most drastic, with an approximate 100-fold increase in the GNP content for cells treated with the GNP-PEG-lRGD complex and an approximate 150-fold increase for cells treated with the GNP-PEG-cRGD complex (Fig. 3B, C). This increase in uptake was also observed at the 8 h and 24 h timepoints; however, with a decreasing trend in the relative uptake, with an approximate 20-fold increase and 40-fold increase in GNP content being measured in cells 24 h after being treated with the GNP-PEG-lRGD and GNP-PEG-cRGD complexes, respectively.
Fig. 3.

GNP accumulation in monolayer and spheroid cell cultures. A-B) ICP-MS measurements (n = 4) of GNP accumulation in monolayer cell cultures at various time points and the relative increase in GNP accumulation with the addition of RGD peptides compared to GNP-PEG complex. C) Confocal imaging of monolayer cell cultures 1 h after being treated with GNPs complexes (red). Scale bar = 30 μm. D-E) ICP-MS measurements (n = 4) of GNP accumulation in spheroid cell cultures at various time points and the relative increase in GNP accumulation with the addition of RGD peptides compared to GNP-PEG complex. F) Confocal imaging of spheroid cell cultures 1 h and 8 h after being treated with GNP complexes (red). Scale bar = 300 μm. * indicates 0.01 < p < 0.05, ** indicates 0.001 < p < 0.01, *** indicates p < 0.001 and ns indicates nonsignificant.
To further verify the targeting efficacy of the RGD peptides, we also assessed the uptake of the GNP complexes using 3D spheroid models. While monolayer cell cultures demonstrated enhanced GNP accumulation with the addition of RGD peptides, spheroid models provide a more physiologically relevant system for evaluating nanoparticle interactions within the tumor microenvironment (TME), which could lead to altered results. Unlike monolayer cell cultures, 3D spheroid models replicate key in vivo characteristics of the TME, including concentration gradients, cellular heterogeneity, and intricate cell-cell interactions, all of which can influence NP penetration and intracellular accumulation. Spheroids also develop an extracellular matrix (ECM), an important component consisting of a dense network of proteins within the tumor microenvironment that directly influences the distribution of NPs. The composition and density of the ECM can act as a barrier to nanoparticle diffusion, limiting penetration and affecting overall biodistribution27,28, and thus makes spheroid models particularly valuable for studying tumor-targeting efficiency.
For our study, spheroids of ~ 300 μm were formed using Matrigel, an ECM-based hydrogel that provides a scaffold for spheroid formation and introduces in-vivo like characteristics. As expected, a substantial decrease in GNP uptake was observed in spheroid models compared to monolayer cell cultures that were treated with the RGD-functionalized GNPs (Fig. 3D). Interestingly, while the RGD peptides were observed to significantly increase GNP uptake in the spheroid models, the extent of this increase drastically decreased relative to the results observed in monolayer cell cultures (Fig. 3B, E). This suggests that the benefits of the RGD peptides are significantly diminished in more a more physiologically relevant tumor model. Specifically, at the 1 h time point, neither of the RGD peptides were shown to significantly improve the uptake of GNPs, with only an approximate four-fold increase observed 24 h after exposure to GNPs. Furthermore, the maximum relative increase in GNP uptake due to RGD peptides was observed 24 h post-dosing in the spheroid models. In contrast, monolayer cultures displayed the greatest relative enhancement in uptake within the first hour, with the difference between PEGylated and RGD-functionalized GNPs decreasing over time. This trend suggests that, in 2D cultures, cells rapidly reach an equilibrium in GNP accumulation, particularly for RGD-functionalized nanoparticles, as their enhanced binding and internalization can lead to early saturation. On the other hand, GNP diffusion through the ECM is more restricted in the spheroid model29, forming a concentration gradient (Fig. 3F, S2B), leading to a more gradual accumulation over time. This could potentially influence the relative difference between PEGylated and RGD-functionalized GNP uptake. Limited penetration of GNPs reduces the number of RGD-GNPs accessible to cells, preventing the saturation seen in monolayers. Furthermore, spheroid cultures could demonstrate reduced integrin accessibility, which could further narrow the uptake difference between GNP formulations30. It should also be noted that there was an increased accumulation of the GNP-PEG complex in spheroid models compared to their uptake in monolayer cultures, suggesting that a portion of the GNPs may be embedded within the ECM rather than being internalized by cells. The extent of ECM sequestration between the RGD-targeted GNP complexes and GNP-PEG complexes is unlikely to differ significantly, which could contribute to the observed results. Yet, despite this decrease in uptake of RGD functionalized GNPs in spheroid models, our results in both monolayer and spheroid cell culture models still indicate that RGD peptides can significantly improve the uptake of GNPs in KPCY cells compared to GNPs functionalized solely with PEG.
Biodistribution of RGD-functionalized GNPs in immunocompetent model
To assess the targeting efficiency of the RGD-functionalized GNPs using an immunocompetent mouse model, KPCY tumor-bearing C57BL/6J mice were injected with the various GNP complexes at a dosing concentration of 10 µg/g. Twenty-four hours following the injection, the organs, plasma and tumors of the mice were harvested. The gold content contained within the samples was then quantitatively analyzed via ICP-MS, and hyperspectral imaging (HSI) was also performed to qualitatively assess the GNP distribution within the samples. Additionally, blood chemistry analysis was performed that demonstrated no toxicity was induced due to the GNP complexes (Figure S3).
The quantitative results of the GNP biodistribution as measured by ICP-MS are shown in Fig. 4A. Interestingly, the addition of RGD peptides as a targeting moiety significantly reduced the GNP content found in the tumor 24 h post-injection (Fig. 4A, B), with over a 10-fold decrease in the GNP concentration being measured, decreasing the percentage of the injected dose from ~ 0.8% to less than 0.1%. These quantitative results also coincided with hyperspectral images of the tumor tissue, which demonstrated increased localization of the GNP-PEG complex relative to the RGD-functionalized GNPs (Fig. 4C, S4). Furthermore, a significant reduction of GNPs in circulation was observed due to the presence of RGD peptides. Specifically, 24 h post-administration, there was a 99% reduction in GNP concentration found within the plasma for the RGD-functionalized GNPs relative to the GNP-PEG complex (Fig. 4D). This suggests that the RGD peptides enhanced the clearance of the GNPs from the bloodstream, which may explain the drastic decrease of GNP content found in the tumors.
Fig. 4.

In vivo biodistribution of GNP complexes. (A) Percentage of the injected dose of the GNP complexes found in the tumor, kidneys, spleen and liver. (B) Concentrations of GNP complexes measured in KPCY tumors. (C) Hyperspectral images of tumor tissue displaying GNP localization. Images were taken under a 60x magnification. Scale bar = 20 μm. (D) Percentage of injected dose of GNP complexes measured to reside in the plasma per gram of plasma. (E) Comparison of GNP concentration of the various GNP complexes found in the spleen, liver and kidney, respectively. The number of mice used for each experimental condition was n = 4. * indicates 0.01 < p < 0.05, ** indicates 0.001 < p < 0.01 and *** indicates p < 0.001.
As previously discussed, NPs accumulate in the tumor through both passive targeting and active targeting mechanisms. While active targeting promotes the intracellular accumulation of NPs in cells that express specific binding sites, passive targeting relies upon the gradual accumulation of NPs in the TME, promoted by the enhanced permeability and retention (EPR) effect exhibited by tumors. This EPR effect arises from the chaotic nature of the neo-vasculature system found within the TME, often consisting of structural abnormalities that permit the passage of small molecules into tumors31,32. When also paired with the poor lymphatic drainage, it improves the accumulation of circulating NPs into the TME. Given this, it is critical to increase the circulation time of NPs to allow them adequate time to exploit the EPR effect and accumulate in the tumor. Commonly, stealth molecules such as PEG are used as they are known to be effective at prolonging the circulation of NP. However, the addition of RGD peptides was observed to counteract this effect, resulting in their removal from circulation.
This significant reduction in the GNP content residing in the plasma due to the RGD peptides was also accompanied by a significant increase in the GNP concentration found in the liver and spleen (Fig. 4E), two organs that have been shown to sequester a large portion of the injected dose of NPs in both immunocompromised and immunocompetent animal models33–36. Both organs are integral to the MPS, which functions to clear foreign substances from circulation. In the liver, NP clearance is primarily mediated by Kupffer cells, specialized tissue resident macrophages located within the hepatic sinusoids. These cells have direct contact with NPs from the hepatic artery or hepatic portal vein. Their high phagocytic activity results in significant hepatic accumulation of NPs35. Similarly, the spleen also mediates NP clearance, housing a large population of phagocytic cells concentrated in the red pulp and marginal zone that efficiently sequesters circulating NPs37. The enhancement in GNP concentration found in these organs, coupled with the reduction of GNPs found in the plasma, suggests that the RGD peptide increases NP recognition and clearance by the MPS, despite the GNP complexes also being functionalized with PEG. Without sufficient circulation time to allow for accumulation within the TME, the anticipated benefits of RGD-mediated active targeting to improve intracellular uptake is rendered insignificant, and rather, its presence appears detrimental to improving tumor accumulation.
This finding contrasts with other studies that have reported improved tumor accumulation of RGD-functionalized NPs in immunodeficient mouse models19,38,39. A previous study demonstrated that increasing the grafting density of RGD resulted in increased tumor localization and a decrease in liver accumulation of GNPs in SCID mice, demonstrating RGD’s efficacy to improve GNP targeting. Yet, our results suggest that RGD can be detrimental to tumor localization. One possible explanation for this discrepancy is the functional state of the immune system. Immunocompetent mice, unlike their immunodeficient counterparts, possess a fully active MPS, which may exhibit a heightened ability to recognize and clear RGD-modified NPs. In immunodeficient models, macrophage function and other immune-mediated clearance mechanisms can be impaired40. As such, NPs likely experience prolonged circulation, thereby increasing the opportunity for passive and active tumor accumulation. As demonstrated by our in vitro results, RGD promoted GNP uptake in KPCY cells. RGD has also been demonstrated to improve NP retention within the tumor in vivo15. Thus, when considering the increased accumulation found in the liver and spleen paired with the decrease found residing in the plasma, it is indicative that RGD-functionalized GNPs experienced increased immune cell-related interaction facilitated by the fully functioning immune system present within the mouse model, ultimately causing the decrease in tumor localization.
GNP uptake in macrophages
Since the biodistribution data displayed enhanced accumulation of RGD functionalized GNP complexes in phagocytosing organs, we wanted to further assess their uptake by macrophages. To investigate this, we initially evaluated this using RAW 264.7 macrophages, a murine-derived macrophage cell line that exhibits many functional characteristics of primary macrophages. Like prior in vitro studies, cells were exposed to the GNP complexes at a concentration of 7.5 µg/mL. Following a 1 h and 4 h incubation, the cells were harvested, and the intracellular GNP concentration was measured via ICP-MS (Fig. 5A). As shown, the macrophage cell cultures demonstrated a significant increase in uptake of the RGD-functionalized GNPs compared to the GNP-PEG complex (Fig. 5A). Specifically, there was a ~ 120% and ~ 300% increase for both the GNP-PEG-lRGD and GNP-PEG-cRGD complexes, respectively, relative to the GNP-PEG complex 1 h after dosing, with this differential in uptake increasing 4 h after exposure. Additionally, confocal imaging of the RAW 264.7 macrophages confirmed this intracellular accumulation of GNPs (Fig. 5B).
Fig. 5.

Macrophage uptake of RGD-functionalized GNPs. (A) ICP-MS analysis of GNP uptake in RAW 264.7 macrophages. (B) Confocal imaging of RAW 264.7 cell cultures 1 h after being treated with GNPs complexes (red). Scale bar = 30 μm. (C) Flow cytometry analysis displaying the percentage of GNP-positive BMDMs following a 1 h exposure to the various GNP complexes. (D) Confocal imaging of BMDMs cell cultures 1 h after being incubated with GNP-PEG-cRGD complex at a concentration of 7.5 µg/mL. Scale bar = 30 μm. * indicates 0.01 < p < 0.05, ** indicates 0.001 < p < 0.01 and *** indicates p < 0.001.
While RAW 264.7 macrophages serve as a good model of macrophage interaction with NP formulations, we wanted to further assess the interactions between the GNP complexes and primary murine macrophages in an attempt to provide a better representation of interactions as seen in vivo. Given this, bone marrow-derived macrophages (BMDMs) were isolated from C57BL/6 mice. Following 7 days of culture in M-CSF supplemented growth medium, BMDMs were exposed to the various GNP complexes at a concentration of 0.75 µg/mL. After a 1 h incubation, there was a significant increase in the percentage of GNP-positive cells for BMDMs treated with the RGD-functionalized GNPs (Fig. 5C). Uptake of the GNPs by BMDMs was also confirmed using confocal imaging (Fig. 5D).
The increase in uptake of RGD-functionalized GNPs in BMDMs, paired with the enhanced uptake as shown by the RAW 264.7 macrophages, indicates that RGD modification inadvertently promotes recognition and subsequent phagocytosis by macrophages, which could explain the increased MPS sequestration observed in vivo. The exact mechanism causing this is currently unclear, but our results elicit further investigation to understand the processes causing the enhanced recognition by the MPS. It is possible that the RGD peptide enhances receptor-mediated interactions with macrophages, as macrophages such as Kupffer cells have been shown to express the ανβ3 integrin41. Additionally, the RGD peptide could potentially facilitate an enhancement in the serum protein adsorption, which would consequently lead to enhanced recognition by the MPS42. Nevertheless, it is evident that the RGD peptides as a targeting moiety increases GNP interactions with the MPS, causing a significant decrease in tumor accumulation compared to the GNP-PEG complex. Yet, bypassing this recognition by the MPS could potentially facilitate the expected improvement in tumor localization with RGD peptides as a targeting moiety. A previous study demonstrated that depleting the Kupffer cell population prior to NP injection significantly decreased liver accumulation, subsequently increasing tumor localization owing to their increased bioavailability43. As such, if RGD-functionalized NPs were able to bypass the MPS through strategies such as intratumoral injections or pre-saturation of the MPS44, thereby enabling their accumulation within the TME via the EPR effect, it could provide the means necessary for active targeting to occur and improve tumor uptake relative to PEGylated NPs.
It should also be noted that despite the GNP-PEG complex demonstrating greater tumor localization due to their increased circulation, it is questionable whether these GNPs accumulated intracellularly or rather were retained elsewhere within the TME. Our in vitro data demonstrated that PEGylated GNPs had minimal uptake in monolayer cell cultures in the absence of an active targeting ligand, which is typically required to drive receptor-mediated internalization. Without this, tumor-associated GNP accumulation may be largely extracellular, potentially sequestered within collagen-dense regions of the ECM. This distinction is critical when considering nanoparticles for drug delivery or as sensitizers to external therapies (e.g., radiation or photothermal therapy), as intracellular accumulation is often essential for their therapeutic function. As such, active targeting ligands such as RGD could be necessary to improve the therapeutic efficacy of NPs, but alternative strategies to evade initial recognition by the MPS may be required to realize the full potential of RGD peptides as a targeting ligand against tumors.
Conclusion
Equipping nanoparticle NP formulations with active targeting moieties, such as RGD peptides, is a widely explored strategy to enhance tumor specificity and improve the efficacy of NP-based cancer therapeutics. Here, we have specifically tested the efficacy of RGD functionalized GNPs to improve their uptake in a syngeneic tumor model (KPCY). While our in vitro experiments demonstrated a significant improvement in GNP uptake in KPCY cells with the addition of RGD peptides as a targeting moiety, our in vivo studies demonstrated contrasting results. RGD modification was observed to increase GNP clearance and their preferential localization in phagocytosing organs such as the liver and spleen, relative to the PEGylated GNPs. This resulted in a significant decrease in their tumor accumulation, contrasting with previous studies conducted in immunocompromised mice models that have demonstrated improved tumor accumulation with the addition of RGD to NPs. Our results suggest that the presence of a fully functional immune system plays a crucial role in modulating NP biodistribution. Specifically, the increased recognition and clearance of RGD functionalized GNPs by phagocytes may have contributed to their rapid sequestration, as supported by our in vitro data showing enhanced macrophage uptake of RGD-modified GNPs. As such, alternative strategies to mitigate recognition and clearance by the MPS prior to the passive accumulation of NPs in tumors may be required to realize the full potential of RGD peptides as an active targeting moiety.
Materials & methods
Synthesis, functionalization, and analysis of GNP complexes
GNPs with an approximate core diameter of 12 nm were synthesized via a citrate reduction method. Specifically, 1.18 mL of a 1% (w/v) HAuCl₄ solution (Sigma-Aldrich, cat#: 520918) was mixed with 28.82 mL of double-distilled water in an Erlenmeyer flask and heated to boiling under vigorous stirring. Upon reaching a boil, 1.12 mL of 5% (w/v) sodium citrate solution (Sigma-Aldrich, cat#: S4641) was quickly introduced while maintaining continuous stirring. The reaction proceeded until the solution transitioned to a ruby-red color, after which it was allowed to boil for an additional 5 min with continued stirring. The GNP solution was then cooled to room temperature while being continuously stirred.
The GNPs were functionalized with both methoxy- and thiol-terminated 2 kDa polyethylene glycol (mPEG-SH; Nanocs, cat#: PG1-TH-2k) to form the GNP-PEG complex. While being stirred, a solution of 0.2 µg/mL of PEG diluted in double-distilled water was introduced to the GNP colloidal solution at a ratio of one PEG molecule per nm² of GNP surface area. To synthesize the GNP-PEG-lRGD and GNP-PEG-cRGD complexes, either a linear or cyclic RGD peptide (CKKKKKKGGRGDMFG; Anaspec) with a molecular weight of 1.6 kDa was added to the GNP-PEG complex solution. A solution of 0.5 µg/mL RGD peptide diluted in double-distilled water was added to the GNP-PEG complex solution to achieve a ratio of 2:1 PEG to RGD peptide molecules.
The size and shape of the GNPs were characterized using scanning transmission electron microscopy (STEM) (ultrahigh-resolution STEM SU9000, Hitachi). Initial estimations of particle size and concentration were conducted using UV − vis spectrometry (PerkinElmer λ 365 Spectrophotometer). To assess the stability of the colloidal GNP solution, hydrodynamic diameter and ζ-potential measurements were performed using a LiteSizer 500 particle analyzer.
Cell culturing and formation of spheroids
The mouse pancreatic cell line KPCY (cat#: EUP016-FP) was purchased from Kerafast Inc. KPCY cells were cultured in DMEM (Gibco, catalog no. 11966025), supplemented with 4 mM GlutaMax (Gibco, catalog no. 35050061), 10% (v/v) Fetal Bovine Serum (Gibco, catalog no. 12484010), and 1% (v/v) penicillin/streptomycin (Gibco, cat#: 15140148). Cells were incubated at 37 °C with 5% CO2 and were subcultured once 70 − 80% confluency was reached. All washing and dissociation of cell cultures was performed using phosphate-buffered saline (PBS) (Gibco, cat#: 10010023) and a 0.25%Trypsin − EDTA solution (Gibco, cat#: 25200072), respectively.
For the formation of KPCY spheroids, cell medium was supplemented with 3% Matrigel (Corning, cat. no.356234) with the medium being kept on ice. Cells were then seeded into ultra-low attachment 96-well microplates (Corning, Cat#: 3474), with each well being populated with 5000 cells such that the formation of spheroids would result in a size of approximately 300 μm in diameter after 2 days of growth. After cell seeding, microplates were centrifuged at 350 xg for 5 min at 4 °C. Spheroid cell cultures were then incubated at 37 °C with 5% CO2 for 2 days prior to their downstream application.
Animal models and pharmacokinetic tissue sampling
6 to 10 weeks old Female C57BL/6J mice were purchased from Jackson Laboratories and were acclimated for at least 7 days prior to the start of studies. Mice were injected with 1 × 105 KPCY cells subcutaneously into the right flank in a volume of 50 µL using a 28-gauge needle. After tumors reached an average size of 150–200 mm3, mice received intravenous injections of GNP complexes (10 µg/g). Mice were then sacrificed via isoflurane followed by CO2 inhalation 24 h following intravenous injections of GNP complexes. Organs, tumors, and blood were then harvested from mice, with blood being collected via cardiac puncture with a 25-gauge needle and placed into the appropriate microtainer tube (K2 EDTA) for plasma.
ICP-MS analysis of GNP content
For in vitro sample preparation, monolayer cell cultures were seeded with 2 mL of culture medium into 6 well dishes, whereas spheroid cell cultures were seeded into 96-well plates with 100 µL of media. Following a 24 h incubation, monolayer cell cultures were dosed with an additional 1 mL of culture medium containing GNP complexes, whereas spheroid cell cultures were dosed with an additional 100 µL of culture medium containing GNP complexes to achieve a final GNP concentration of 7.5 µg/mL. Cell cultures were then incubated for either 1 h, 8 h, or24 h. Following the incubation periods, cells were washed 3 times with 1 mL of PBS, dissociated, and collected. The number of cells present was then counted using a hemacytometer. For in vivo sample preparation, samples were weighed prior to their disassociation by blending samples in 2 mL of 0.25% Trypsin-EDTA using a handheld homogenizer. Following their disassociation, samples were diluted to 5 mL with double-distilled water.
For both in vitro and in vivo experiments, a portion of each sample (100–500 µL) was transferred to glass tubes and treated with 250 µL of aqua regia a (3:1 molar ratio of HCl and HNO3 (VWR)). Samples were placed into a mineral oil bath at a temperature of 90 °C and left for either 1 h (in vitro samples) or 2 h (in vivo samples). For in vitro samples, an additional 100 µL of hydrogen peroxide was added to the samples after the 1 h time period, with the samples remaining in the mineral oil bath for an additional 1 h. All samples were then diluted to a 2.5% (v/v) acid content with double-distilled water and samples were filtered using a 0.2 μm filter (Sigma-Aldrich, Cat# SLLG025SS). Gold content in the samples was then measured using Inductively coupled plasma-mass spectrometry (ICP-MS; Agilent 8800 Triple Quadrupole, Agilent Technologies).
For in vitro studies, the number of GNPs present in each cell was calculated using the following:
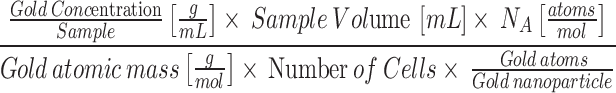 |
Where NA is Avogadro’s number. The number gold atoms per GNP was calculated by the following equation:
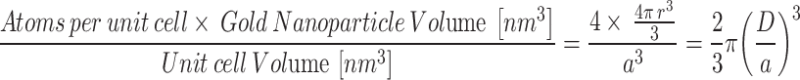 |
Where a = 0.408 nm, the length of a unit cell, and D is the core diameter of the spherical GNPs. The GNPs were synthesized such that it results in a face-centered cubic lattice with 4 atoms per unit cell. For the calculations, it is assumed that the size of the GNPs is homogenous.
Hyperspectral imaging
To prepare in vivo samples for imaging, tissue samples were fixed in 10% neutral-buffered formalin and processed into paraffin using an automated tissue processor. Samples were then embedded and sliced into 4 μm sections onto glass slides. Imaging of the tissue samples was then performed using hyper spectral imaging (HSI; CytoViva) using a dark field microscope under 60x oil immersion objective lens.
Live cell imaging
In preparation for live cell imaging of monolayer cell cultures, cells were seeded with 1.5 mL of cell culture medium in a 35 mm glass coverslip-bottom dishes (MatTek, Cat#: P35GCOL-1.5–14.5-C). For live cell imaging of spheroid cultures, spheroids were cultured as previously described. Both monolayer and spheroid cell cultures were then dosed with GNP complexes to achieve a final concentration of 7.5 µg/mL. Cell cultures were then incubated for either 1 h, 8 h, or 24 h prior to imaging. To visualize the GNPs, thiol terminated 2-kDa Cy5 (excitation 633 nm, emission 650 nm) labelled PEG was used to functionalize the GNPs in place of the PEG previously described. Spheroids were also transferred from the 96-well low attachment microplate to a 35 mm glass coverslip-bottom dish with minimal media to maintain stability but avoid aspiration. Cell cultures were then imaged using a confocal laser scanning microscope (Zeiss LSM 980, Carl ZeissMicroscopy GmbH) under a 10x and 63x objective lens.
Primary cell preparation
Bone marrow derived macrophages (BMDMs) were isolated as previously described before45–47. Briefly, bone marrow was flushed from the femurs and tibia with 1XPBS. Isolated bone marrow cells were then resuspended in red blood cell lysis buffer (1.66% ammonium chloride) for 5 min. Afterwards, cells were cultivated in a 6-well tissue culture plate (Corning, USA) containing 20% supernatant from M-CSF secreting L929 fibroblasts47,48. On days 3 and 5, nonadherent cells were removed and fresh media was added. BMDMs were used for experimental purposes following 7 days in culture and were cultured at 37 °C and 5% CO2. Primary cell cultures were maintained in RPMI (Gibco, Fisher Scientific, Canada) media supplemented with 10% FBS (Gibco, Fisher Scientific, Canada) and 50 µg/mL gentamycin (BioShop, Canada).
GNP uptake and flow cytometry analysis
After 7 days of culture in M-CSF growth media, BMDMs were harvested and seeded (2.5 × 105 cells/well) into a 96-well flat bottom plate (Corning, USA) for evaluation of GNP uptake. The various GNP conditions were used at a concentration of 0.75 µg/mL for 1 h, as this provided the best discrimination between conditions (Figure S5), in RPMI media supplemented with 10% FBS and 50 µg/mL gentamycin and cultured at 37 °C and 5% CO2. Following incubation, cells were washed three times with PBS, then were subsequently acquired on a CytoFLEX flow cytometer (Beckman Coulter, USA) for GNP uptake with the 638 nm excitation laser and the 660-emission detection filter and analyzed using the FlowJo software (BD, USA).
Statistical analysis
All results are presented as the mean ± standard deviation (s.d.). Statistical significance of results was assessed using a one-way analysis of variance (ANOVA) with Tukey’s multiple comparison test. P values < 0.05 are considered to be statistically significant.
Ethics approval
The methodology described regarding animal procedures was performed either in accordance with the guidelines and regulations set by the University of British Columbia and approved by the Institutional Animal Care Committee (IACC), or in accordance with the guidelines of the Canadian Council of Animal Use and approved by Queen’s University Animal Care Services. During the study, the care, housing and use of animals was performed in accordance with the Canadian Council on Animal Care Guidelines.
Ethics guidelines
The reporting of animal data in the current study adhered to the recommendations set out by the ARRIVE guidelines.
Supplementary Information
Below is the link to the electronic supplementary material.
Author contributions
Conceptualization: NJ and DBC; methodology: NJ, SB, and DBC; resources: WB, SB, SK, and DBC; data curation: NJ, NB, DC, and SH; writing original draft preparation: NJ; writing review and editing: NJ, NB, DC, SH, KG, WB, SB, SK, and DBC; supervision: KG, WB, SB, SK, and DBC; project administration: KG, WB, SB, SK, and DBC; funding acquisition: WB, SK, and DBC. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded, in part, by Nanomedicines Innovation Network (NMIN) Strategic Initiative (SI) grant 2021-RES SI- 05 from NMIN, Natural Sciences and Engineering Research Council (NSERC) Discovery grant RGPIN-2017 − 04501 by NSERC, Canadian Institutes of Health Research (CIHR) Grant CIHR PJT-399878 by CIHR, and National Institutes of Health (NIH) Grants R01CA257241 from NIH.
Data availability
The datasets used and/or analysed during the current study available from the corresponding author on reasonable request.
Declarations
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.O’Brien, M. E. et al. Reduced cardiotoxicity and comparable efficacy in a phase III trial of pegylated liposomal doxorubicin HCl (CAELYX/Doxil) versus conventional doxorubicin for first-line treatment of metastatic breast cancer. Ann. Oncol.15 (3), 440–449 (2004). [DOI] [PubMed] [Google Scholar]
- 2.Stirland, D. L. et al. Mind the gap: a survey of how cancer drug carriers are susceptible to the gap between research and practice. J. Control Release. 172 (3), 1045–1064 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wu, D. et al. Nanomedicine applications in the treatment of breast cancer: current state of the Art. Int. J. Nanomed.12, 5879–5892 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Đorđević, S. et al. Current hurdles to the translation of nanomedicines from bench to the clinic. Drug Delivery Transl. Res.12(3), 500–525 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hua, S. et al. Current trends and challenges in the clinical translation of nanoparticulate nanomedicines: pathways for translational development and commercialization. Front. Pharmacol.9, 790 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Agrahari, V. & Hiremath, P. Challenges associated and approaches for successful translation of nanomedicines into commercial products. Nanomed. (London England). 12 (8), 819–823 (2017). [DOI] [PubMed] [Google Scholar]
- 7.Ernsting, M. J. et al. Factors controlling the pharmacokinetics, biodistribution and intratumoral penetration of nanoparticles. J. Control Release. 172 (3), 782–794 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cabral, H. et al. Controlling the biodistribution and clearance of nanomedicines. Nat. Reviews Bioeng.2 (3), 214–232 (2024). [Google Scholar]
- 9.Prajapati, A. et al. Receptor-Targeted nanomedicine for cancer therapy. Receptors3 (3), 323–361 (2024). [Google Scholar]
- 10.Chastney, M. R. et al. The role and regulation of integrins in cell migration and invasion. Nat. Rev. Mol. Cell Biol.26 (2), 147–167 (2025). [DOI] [PubMed] [Google Scholar]
- 11.Voura, E. B. et al. Involvement of integrin alpha(v)beta(3) and cell adhesion molecule L1 in transendothelial migration of melanoma cells. Mol. Biol. Cell. 12 (9), 2699 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Valdembri, D. & Serini, G. The roles of integrins in cancer. Fac. Rev.10, 45 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Javid, H. et al. RGD peptide in cancer targeting: Benefits, challenges, solutions, and possible integrin-RGD interactions. Cancer Med.13 (2), e6800 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shan, D. et al. RGD-conjugated solid lipid nanoparticles inhibit adhesion and invasion of αvβ3 integrin-overexpressing breast cancer cells. Drug Delivery Transl. Res.5(1), 15–26 (2015). [DOI] [PubMed] [Google Scholar]
- 15.Shuhendler, A. J. et al. A novel solid lipid nanoparticle formulation for active targeting to tumor α(v) β(3) integrin receptors reveals Cyclic RGD as a double-edged sword. Adv. Healthc. Mater.1(5), 600 (2012). [DOI] [PubMed] [Google Scholar]
- 16.Wu, P. H. et al. Targeting integrins with RGD-conjugated gold nanoparticles in radiotherapy decreases the invasive activity of breast cancer cells. Int. J. Nanomed.12, 5069–5085 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang, T. et al. Multitargeted nanoparticles deliver synergistic drugs across the Blood–Brain barrier to brain metastases of triple negative breast cancer cells and Tumor-Associated macrophages. Adv. Healthc. Mater.8 (18), e1900543 (2019). [DOI] [PubMed] [Google Scholar]
- 18.Cruje, C. & Chithrani, B. D. Integration of peptides for enhanced uptake of pegylayed gold nanoparticles. J. Nanosci. Nanotechnol. 15 (3), 2125–2131 (2015). [DOI] [PubMed] [Google Scholar]
- 19.Yang, C., Bromma, K. & Chithrani, D. Peptide mediated in vivo tumor targeting of nanoparticles through optimization in single and multilayer in vitro cell models. Cancers10 (3), 84 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang, C. et al. Peptide modified gold nanoparticles for improved cellular uptake, nuclear transport, and intracellular retention. Nanoscale6 (20), 12026–12033 (2014). [DOI] [PubMed] [Google Scholar]
- 21.Cui, Y. et al. Renal clearable H-Dots leveraging ligand complexation for enhanced active tumor targeting. Small Sci.4 (11), 2400246 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jackson, N. et al. Dual enhancement in the radiosensitivity of prostate cancer through nanoparticles and chemotherapeutics. Cancer Nanotechnol.14 (1), 75–22 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Walkey, C. D. et al. Nanoparticle size and surface chemistry determine serum protein adsorption and macrophage uptake. J. Am. Chem. Soc.134 (4), 2139–2147 (2012). [DOI] [PubMed] [Google Scholar]
- 24.Jokerst, J. V. et al. Nanoparticle pegylation for imaging and therapy. Nanomed. (Lond). 6 (4), 715–728 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shi, L. et al. Effects of polyethylene glycol on the surface of nanoparticles for targeted drug delivery. Nanoscale13 (24), 1748–1764 (2021). [DOI] [PubMed] [Google Scholar]
- 26.Suk, J. S. et al. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv. Drug Deliv. Rev.99(Pt A), 28–51 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Engin, A. B. et al. Mechanistic Understanding of nanoparticles’ interactions with extracellular matrix: the cell and immune system. Part. Fibre Toxicol.14 (1), 22–16 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stylianopoulos, T. et al. Diffusion of particles in the extracellular matrix: the effect of repulsive electrostatic interactions. Biophys. J.99 (5), 1342–1349 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yohan, D. et al. Size-Dependent gold nanoparticle interaction at Nano–Micro interface using both monolayer and Multilayer(Tissue-Like) cell models. Nano-micro Lett.8 (1), 44–53 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Judmann, B. et al. Are 3D tumor cell spheroids a utile system for the in vitro evaluation of diagnostic radiotracers? ACS Omega. 9 (52), 51349–51362 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hashizume, H. et al. Openings between defective endothelial cells explain tumor vessel leakiness. Am. J. Pathol.156 (4), 1363–1380 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Iyer, A. K. et al. Exploiting the enhanced permeability and retention effect for tumor targeting. Drug Discov Today. 11 (17–18), 812–818 (2006). [DOI] [PubMed] [Google Scholar]
- 33.Fleischmann, D. & Goepferich, A. General sites of nanoparticle biodistribution as a novel opportunity for nanomedicine. Eur. J. Pharm. Biopharm.166, 44–60 (2021). [DOI] [PubMed] [Google Scholar]
- 34.Kumar, M. et al. Nanoparticle biodistribution coefficients: A quantitative approach for Understanding the tissue distribution of nanoparticles. Adv. Drug Deliv. Rev.194, 114708 (2023). [DOI] [PubMed] [Google Scholar]
- 35.Liu, L. Y. et al. Nanoparticle uptake in a spontaneous and immunocompetent woodchuck liver cancer model. ACS Nano. 14 (4), 4698–4715 (2020). [DOI] [PubMed] [Google Scholar]
- 36.Si-Mohamed, S. et al. Evaluation of spectral photon counting computed tomography K-edge imaging for determination of gold nanoparticle biodistribution in vivo. Nanoscale9 (46), 18246–18257 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang, Y. W. & Luo, W. H. Cellular biodistribution of polymeric nanoparticles in the immune system. J. Control Release. 227, 82–93 (2016). [DOI] [PubMed] [Google Scholar]
- 38.Benezra, M. et al. Multimodal silica nanoparticles are effective cancer-targeted probes in a model of human melanoma. J. Clin. Invest.121 (7), 2768–2780 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang, X. et al. cRGD-functionalized, DOX-conjugated, and ⁶⁴Cu-labeled superparamagnetic iron oxide nanoparticles for targeted anticancer drug delivery and PET/MR imaging. Biomaterials32 (17), 4151–4160 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shultz, L. D. et al. Multiple defects in innate and adaptive Immunologic function in NOD/LtSz-scid mice. J. Immunol. (1950). 154 (1), 180–191 (1995). [PubMed] [Google Scholar]
- 41.Turaga, R. C. et al. Targeting integrin alphavbeta3 by a rationally designed protein for chronic liver disease treatment. Commun. Biol.4 (1), 1087 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bellis, S. L. Advantages of RGD peptides for directing cell association with biomaterials. Biomaterials32 (18), 4205–4210 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tavares, A. J. et al. Effect of removing Kupffer cells on nanoparticle tumor delivery. Proc. Natl. Acad. Sci. - PNAS. 114 (51), pE10871–E10880 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zelepukin, I. V., Shevchenko, K. G. & Deyev, S. M. Rediscovery of mononuclear phagocyte system Blockade for nanoparticle drug delivery. Nat. Commun.15 (1), 4366–4314 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Barilo, J. et al. Polarized macrophage functions are affected differentially after CSF-1R Inhibition with PLX5622. Eur. J. Pharmacol.984, 177059 (2024). [DOI] [PubMed] [Google Scholar]
- 46.Alothaimeen, T. et al. Differential TLR7-mediated cytokine expression by R848 in M-CSF- versus GM-CSF-derived macrophages after LCMV infection. J. Gen. Virol. 102(3) (2021). [DOI] [PMC free article] [PubMed]
- 47.Alatery, A. & Basta, S. An efficient culture method for generating large quantities of mature mouse Splenic macrophages. J. Immunol. Methods. 338 (1–2), 47–57 (2008). [DOI] [PubMed] [Google Scholar]
- 48.Alothaimeen, T. et al. Granulocyte/Macrophage Colony-Stimulating Factor-Derived macrophages exhibit distinctive early immune response to lymphocytic choriomeningitis virus infection. Viral Immunol.33 (6), 477–488 (2020). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets used and/or analysed during the current study available from the corresponding author on reasonable request.