

Abstract
In Drosophila, dosage compensation is controlled by the male-specific lethal (MSL) complex consisting of MSL proteins and roX RNAs. The MSL complex is specifically localized on the male X chromosome to increase its expression ∼2-fold. We recently proposed a model for the targeted assembly of the MSL complex, in which initial binding occurs at ∼35 dispersed chromatin entry sites, followed by spreading in cis into flanking regions. Here, we analyze one of the chromatin entry sites, the roX1 gene, to determine which sequences are sufficient to recruit the MSL complex. We found association and spreading of the MSL complex from roX1 transgenes in the absence of detectable roX1 RNA synthesis from the transgene. We mapped the recruitment activity to a 217 bp roX1 fragment that shows male-specific DNase hypersensitivity and can be preferentially cross-linked in vivo to the MSL complex. When inserted on autosomes, this small roX1 segment is sufficient to produce an ectopic chromatin entry site that can nucleate binding and spreading of the MSL complex hundreds of kilobases into neighboring regions.
Keywords: chromatin remodeling/dosage compensation/non-coding RNAs
Introduction
Transcription of eukaryotic genes is highly sensitive to the way DNA is packaged into chromatin. Recent reports from a number of model systems demonstrate that alteration in chromatin conformation is a major strategy for gene regulation (reviewed in Kingston and Narlikar, 1999). One striking observation is that changes to chromatin architecture often spread from an initiation point, sometimes discontinuously. Examples of repressive chromatin spreading include heterochromatic position effect variegation in flies (Wakimoto, 1998), yeast mating-type cassette silencing (Stone and Pillus, 1998; Donze et al., 1999) and X inactivation in mammals (White et al., 1998; Lyon, 1999). In contrast, male-specific lethal (MSL)-mediated dosage compensation in male Drosophila provides an example of spreading active chromatin from initiation sites to affect flanking genes (Kelley et al., 1999). This spreading requires the two known enzymatic activities of the complex, the Maleless (MLE) ATPase and the Males absent on the first (MOF) acetyltransferase (Lee et al., 1997; Gu et al., 2000). Therefore, the study of cis-acting sites needed to bind the MSL complex and initiate spreading should be of general relevance to the understanding of chromatin-based gene regulation.
The MSL complex is required for X chromosome dosage compensation in Drosophila (Lucchesi, 1998; Kelley and Kuroda, 2000). Dosage compensation occurs primarily by increasing X-linked transcription in males, to make the output of the single male X similar to both female X chromosomes (Mukherjee and Beermann, 1965). A histone H4 isoform specifically acetylated at lysine residue 16 (H4Ac16) is highly concentrated on the male X chromosome and is dependent on MSL complex function, suggesting that hypertranscription of the male X is due, at least in part, to chromatin structure alteration through acetylation of nucleosomes (Turner et al., 1992; Bone et al., 1994; Hilfiker et al., 1997; Akhtar and Becker, 2000; Smith et al., 2000).
The Drosophila MSL complex contains at least five proteins and two RNAs (Lucchesi, 1998; Kelley and Kuroda, 2000). Mutation in any protein member of the MSL complex causes male-specific lethality and lack of enrichment of H4Ac16 or the MSL complex itself on the male X (Bone et al., 1994; Hilfiker et al., 1997). The five identified MSL proteins are MLE, a DExH helicase, MSL1, a novel acidic protein, MSL2, a Ring finger protein, MSL3, a chromodomain protein, and MOF, which has both a chromodomain and the histone H4 acetyltransferase activity. Two non-coding RNAs, RNA on X1 (roX1) and roX2 are also members of the MSL complex (Gu et al., 2000; Meller et al., 2000; Smith et al., 2000). These RNAs are male-specific and paint the male X chromosome (Amrein and Axel, 1997; Meller et al., 1997). roX1 is not essential for male-viability or dosage compensation, but may be functionally redundant with roX2 (Franke and Baker, 1999). A simple mutant allele of roX2 has not been reported. Recently, the JIL-1 protein kinase has been found associated with the MSL complex, although its function in the dosage compensation system is still unknown (Jin et al., 2000).
A complete set of MSL proteins is needed to mediate hypertranscription of the hundreds of active genes along the male X (Kelley and Kuroda, 2000). When any subunit is removed through mutation, the MSL complex is lost from most sites along the X, dosage compensation fails, and males die. However, in some mutant backgrounds, a partial MSL complex remains at ∼35 unusual sites interspersed along the X, which we have called chromatin entry sites (Kelley et al., 1999). These sites have been postulated to be locations of initial MSL recognition of the X or sites of complex assembly. They are most readily observed in msl3 or mof mutants, but a very similar, if weaker, MSL staining pattern can be seen in mle mutants (Palmer et al., 1994; Lyman et al., 1997; Gu et al., 1998). When a chromatin entry site is moved to an autosome, it has the remarkable property of recruiting MSL complexes to the site of insertion, where they can subsequently spread in cis over flanking autosomal genes (Kelley et al., 1999).
Only two of the estimated 35 chromatin entry sites have been cloned, and they harbor the genes coding for roX1 and roX2, the RNA components of the MSL complex (Kelley et al., 1999). This finding presented a number of questions. Is the process of transcription of roX RNA, or the accumulation of this RNA at the site of transcription a hallmark characterizing a functional entry site? And if so, do other chromosome entry sites also encode additional, non-coding RNA species? Do the MSL proteins capture nascent roX transcripts as they are being assembled into MSL complexes or do they recognize a specific DNA sequence within the roX genes? And if such a DNA element could be found, is its presence limited to roX genes or also shared with other entry sites? We have approached these questions by initiating a detailed molecular analysis of the roX1 chromatin entry site. We find that the MSL proteins bind a small DNA region near the middle of the roX1 gene that corresponds to a male-specific DNase I hypersensitive site in chromatin of intact nuclei. No similar DNA sequence could be recognized in the roX2 gene, suggesting the existence of either a rather degenerate DNA target or DNA elements of variable primary sequence but similar function. This short DNA element is sufficient to initiate epigenetic MSL spreading along the flanking chromatin in the absence of active roX1 transcription, suggesting that other chromatin entry sites need not necessarily synthesize roX RNAs. However, we did find that spreading was more consistent in cases where the transgene was transcribed. Furthermore, we found a critical early role for the MLE helicase in recruiting the other MSL proteins to the roX1 gene similar to that previously shown for roX2 (Meller et al., 2000). These observations leave open the possibility that RNA components play an early role in MSL binding, assembly or spreading.
Results
roX1 exons are sufficient for recruitment of the MSL complex
In previous studies we found that a 4.9 kb roX1 genomic fragment, inserted into autosomal chromatin, can function as a chromatin entry site for MSL complex nucleation and spreading (Kelley et al., 1999). However, it was not known whether the spreading activity might be a property of the roX1 5′- and 3′-flanking regions, the exons, or the RNA itself. In addition, it was not known whether sequences required for binding the complex were also sufficient for spreading, or whether separate sites were necessary to initiate spreading from a binding site. As a first test, we analyzed a transgene carrying a 3.4 kb roX1 cDNA fragment driven by the constitutive activity of the Hsp83 promoter (Figure 1A). In transgenic animals, strong MSL1 and MLE signals were observed on the autosomal transgenes only in males, indicating that roX1 exons are sufficient for recruitment of the MSL complex (Figure 1B). Since roX1 and roX2 cDNAs can both attract the MSL complex in vivo, we next assessed the importance of a short region of similarity (25/30 bp identity near their 3′ ends, Figure 1A; Franke and Baker, 1999) for attraction of the MSL complex to an autosomal transgene. A Hsp83–roX1Δ30 transgene lacking the 30 bp region of similarity showed strong binding, indistinguishable from a full-length cDNA (Figure 1C), while a multimer of this sequence in the absence of the rest of the roX1 cDNA failed to attract the MSL complex (data not shown). Thus, this short region of sequence similarity between roX1 and roX2 is a poor candidate for the MSL target.

Fig. 1. A cDNA fragment of roX1 recruits the MSL complex. (A) Diagram of the 3.4 kb roX1c20 cDNA driven by the Hsp83 promoter and a deletion derivative lacking a 30 bp segment that shares 25/30 identity with roX2. An intron near the 5′ end is indicated in each construct. The sequence comparison of roX1 and roX2 in the 30 bp region deleted in roX1Δ30 is shown below. (B) Polytene chromosomes from a [Hsp83–roX1]61B male. (C) Polytene chromosomes from a [Hsp83–roX1Δ30]21D male. Chromosome squashes were immuno stained with rabbit anti-MSL1 (red) and counterstained with 4′,6- diamine-2-phenylindole (DAPI) (blue). Both nuclei show a strong site of MSL localization at the insertion site of the respective transgenes (arrowheads). A heavily stained X chromosome is also seen in (B).
De novo synthesis of roX1 RNA is not essential for recruitment and spreading of the MSL complex
One appealing model for MSL binding to roX genes postulates that the roX RNAs are incorporated into MSL complexes cotranscriptionally. MSL proteins might only recognize nascent roX transcripts, not the underlying DNA. This model makes a simple prediction that MSL binding to autosomal roX transgenes should be strictly dependent upon active transcription of roX RNA. As is shown in Figure 2, this is not the case. Two transgenes were assayed for MSL binding when inserted on autosomes. The first contained a constitutive Hsp83 promoter, analogous to Hsp83–roX1 (Figure 1), but transcribing the antisense strand (Hsp83–roX1AS) (Figure 2A). The second carried the same nearly full-length roX1 cDNA without an exogenous promoter (roX1c3.4) (Figure 2B–D). Both of these transgenes provided excellent MSL targets in vivo.

Fig. 2. Localization of MSL complexes to roX1c3.4 transgenes. MSL1 localization in male nuclei, detected by immunostaining with anti-MSL1 antibodies (red). (A) Expressed antisense construct [Hsp83–roX1AS]87A, (B) [roX1c3.4]66C, from which transcription has not been detected, (C) [roX1c3.4]33DE, (D) [roX1c3.4]51A. The [roX1c3.4] transgenes show MSL1 binding at the insertion sites of the transgenes (B–D, arrows), as well as in neighboring regions (C and D, arrowheads). (E) Northern analysis shows fusion RNA expression of roX1 from flanking sequences in the [roX1c3.4]33DE and [roX1c3.4]51A lines, in which relatively frequent spreading was also observed. All transgenic lines carry a mutant allele (roX1ex6) at their endogenous roX1 locus. The [roX1c3.4]66C, 52DE and 47C lines do not express detectable roX1 RNA. The wild-type strain (y w; WT) expresses endogenous roX1 RNA.
We expected that the roX1c3.4 transgene would not be transcribed. However, Drosophila P-elements have a preference for integrating into promoter or regulatory regions and in some cases a neighboring promoter might transcribe into a transgene (O’Kane and Gehring, 1987). Therefore, we performed northern analysis on five lines carrying the transgene at different locations to determine whether any read-through transcription of roX1 sequences occurred. A significant amount of apparent roX1 fusion RNA was detected in two lines, but the other three lines failed to make detectable amounts of roX1 RNA (Figure 2E). The two fusion RNAs were male specific, presumably because roX RNAs require the male-specific MSL complex for stability (Meller et al., 2000). RNA for this northern analysis was isolated from adults, while all MSL binding assays were performed on larval salivary glands. Therefore, we extended our analysis by testing roX1c3.4 expression directly in salivary glands by RNA in situ hybridization. The results of the larval analysis were similar to those in adults; roX1c3.4 lines 33DE and 51A produced roX1 RNA that could be detected at a low level on the X chromosome, while the other three lines did not. Consistent with this result, the 33DE and 51A fusion RNAs were transcribed in the same orientation as authentic roX1 RNA (data not shown).
We examined hundreds of nuclei containing the roX1c3.4 transgene at the five different insertion sites to assay their ability to support spreading of the MSL complex (Figure 2C and D; Table I). MSL spreading was observed in each line, but the fraction of nuclei undergoing spreading varied from 2 up to 57%. The two roX1c3.4 lines exhibiting read-through transcription showed the most consistent spreading of MSL complexes into flanking autosomal chromatin (Table I). We conclude that transcription from the roX1 transgene is not essential for nucleation and spreading of the MSL complex, but it might be a factor that influences the frequency of spreading. Alternatively, the differences in spreading frequency may reflect poorly understood position effects at each insertion site assayed.
Table I. Analysis of epigenetic spreading of MSL complexes from roX1c3.4 transgenes.
| Strain | No. of nuclei observed | No. of nuclei with no signal | No. of nuclei with a single band | No. of nuclei with spreading | Spreading frequency (%)a |
|---|---|---|---|---|---|
| 33DE | 288 | 2 | 215 | 71 | 24.7 |
| 47C | 285 | 11 | 266 | 8 | 2.8 |
| 51A | 140 | 0 | 60 | 80 | 57.1 |
| 52DE | 295 | 0 | 271 | 24 | 8.1 |
| 66C | 183 | 0 | 179 | 4 | 2.2 |
aPercentage of observed nuclei with spreading.
The MLE helicase is essential for MSL complex association with the roX1 gene
MSL1 and MSL2 are thought to form a core complex that binds to chromatin entry sites, while MLE and other components including roX RNAs may be added sequentially (Lucchesi, 1998; Gu et al., 2000; Meller et al., 2000). To assess whether all the MSL components are required to bind to the roX1 exons, we analyzed MSL1 localization on the roX1c3.4 transgene in msl mutant animals. Since msl mutant males show poor morphology of polytene chromosomes, we used females carrying a Hsp83–MSL2 transgene. Females normally lack MSL2 and can not form MSL complexes, but when MSL2 is constitutively expressed in females, the complex forms and is localized to both X chromosomes (Kelley et al., 1995). The roX1c3.4 transgene recruits the MSL complex in the absence of functional mof (Figure 3A, arrowhead) as well as in cells lacking msl3 (Figure 3B, arrowhead), as expected for an authentic chromatin entry site. However, in mle mutant animals, surprisingly, the transgene did not bind detectable partial complexes (Figure 3C). By careful re-examination, we found that the endogenous roX1 locus at 3F shows two strong MSL binding sites as a tight doublet in the absence of msl3, but shows a faint, single MSL signal in mle mutants (arrows, Figure 3B and C). This suggests that there are two distinct MSL binding sites located at 3F, one site within the roX1 gene that requires MLE, and an additional site, similar to the majority of entry sites, that can attract partial MSL complexes in the absence of MLE. It should be noted that even at entry sites that attract partial complexes independent of MLE, the strength or stability of binding to those sites is weak (Palmer et al., 1994; Lyman et al., 1997). Since the requirement for MLE is also a characteristic of the endogenous roX2 locus (at 10C) (Meller et al., 2000), it is possible that this is a common feature of entry sites that encode roX RNAs.

Fig. 3. MSL complex binding at roX1 transgenes requires MLE. Females carrying [Hsp83–MSL2] and [roX1c3.4]51A in addition to being homozygous mutant for mof (A), msl-3 (B) or mle (C). Polytene chromosomes were stained with anti-MSL1 antibodies (red) and DAPI (blue). In each of these mutants the MSL complex associates with the X chromosome only at chromatin entry sites. Arrowheads and arrows represent the roX1 transgene and the endogenous roX1 locus at 3F, respectively. Top right inset in each panel shows an enlarged image of anti-MSL1 immunostaining of the endogenous roX1 locus.
Functional msl3 and mof are not required for MSL binding to chromatin entry sites including roX1, but in their absence the complex fails to associate with the additional hundreds of sites seen in its wild-type pattern on the X chromosome (Palmer et al., 1994; Gu et al., 1998). This has been interpreted as an inability of the complex to spread between chromatin entry sites in the absence of either MSL3 or MOF. To test this hypothesis at the level of an individual entry site, we examined a strain in which MSL1 showed frequent spreading from a roX1 transgene (roX1c3.4-51A). We found that lack of functional msl3 or mof prevented spreading, as it does on the endogenous X chromosome (Figure 3A and B). Taken together, the MLE helicase protein is essential for the MSL complex to bind to the roX1 transgene, while the MSL3 chromodomain protein and MOF histone acetyltransferase are required only for spreading.
Male-specific DNase I hypersensitivity within the roX gene
The polytene X chromosome in Drosophila males shows relatively diffuse morphology compared with that of the female, suggesting that the dosage compensation machinery controls chromatin architecture. The interaction of regulatory complexes with chromatin frequently leads to perturbations in chromatin structure that are revealed by the hypersensitivity of associated DNA upon mild digestion of intact nuclei with DNase I. Mapping of DNase I hypersensitive sites allows the identification of regulatory elements, such as transcription enhancers (Nitsch et al., 1990). In order to identify chromatin perturbations caused by the MSL complex we treated nuclei from male and female adult flies with increasing amounts of DNase I and monitored the appearance of hypersensitive sites by indirect end labeling. We found two such sites within roX1 in the chromatin of male, but not in female flies (Figure 4). One of the sites mapped near the beginning of the gene and may therefore correspond to its active promoter. The second site mapped to a region ∼1000–1250 bp into the roX1 gene.

Fig. 4. DNase I hypersensitivity within the roX1 locus. DNase I cleavages were mapped within an EcoRI fragment containing the roX1 gene, schematized to the left (open box; +1 indicates the approximate 5′ end of the gene). Nuclei from female (left) or male (right) adult flies were treated with increasing concentrations of DNase I. DNA was isolated and digested to completion with EcoRI. DNase I cleavages were revealed by Southern blotting using a probe adjacent to an EcoRI site within the opt gene. Arrows highlight male-specific DNase I hypersensitive sites in roX1 chromatin. M, DNA size markers (bp).
A 217 bp roX1 region is sufficient for recruitment and spreading of the MSL complex
To determine whether the male-specific DNase I hypersensitive site in roX1 corresponds to a recognition site for the MSL complex, and to survey the roX1 gene for additional sites, we performed a transgenic deletion analysis. We produced transgenes with the 3.4 kb roX1 cDNA divided into various overlapping fragments, roX1a (1.1 kb), roX1b (1.1 kb), roX1c (1.4 kb), roX1.R5′ (0.9 kb) and roX1.R3′ (2.6 kb) (Figure 5A). Only two of these fragments, roX1a and R3′, showed strong MSL1 binding in transgenic animals (Figure 5B and C). These two constructs share a 300 bp overlap, coincident with the DNase hypersensitive site mapped in the endogenous roX1 gene. When most of this region was deleted from an otherwise full-length transgene (roX1ΔDHS), MSL1 binding to the transgene was significantly reduced (two lines) or undetectable (two lines). To determine which part of the roX1ΔDHS construct was required for the weak MSL binding detected in some lines, we subdivided that construct into its 5′ and 3′ parts. The 5′ portion (R5′+ 100), which is 100 bp longer than construct R5′ at its 3′ end, showed a faint signal, similar to roX1ΔDHS. The 3′ segment (roX1bc), which has the same 5′ end as roX1b but extends to the end of the gene, showed no signal (Figure 5A and data not shown). Thus, the 300 bp overlap between construct roX1a and construct R3′, encompassing the male-specific DNase hypersensitive region, accounts for the major MSL recruiting activity of the roX1 chromatin entry site. A minor activity detected in roX1ΔDHS is derived from the region immediately 5′ to the major activity.

Fig. 5. Deletion analysis used to map MSL complex-recruiting activity within roX1 transgenes. (A) Diagram of the roX1 fragments tested in transgenic assays. (B–G) Male polytene chromosomes from each transgenic strain were immunostained with anti-MSL1 (red) and counterstained by DAPI (blue). The integration site of each transgene is represented by an arrowhead. Two constructs that share a 300 bp overlap and show strong anti-MSL staining were [roX1a]66B (B) and [roX1.R3′]82CD (C). A 217 bp fragment [roX1-S]48AB (D and E) and a 9× tandem repeat of roX1-S [roX1-SM]57C, F and G) also showed MSL1 localization to the transgene insertion site. Spreading from these small roX1 derivatives is seen in (E) and (G) (arrowheads).
To determine whether the 300 bp region required for MSL complex binding and spreading was also sufficient for chromatin entry site activity, we analyzed transgenes containing 302 bp (roX1-L), 217 bp (roX1-S) or nine tandem repeat copies of a 217 bp DHS fragment (roX1-SM). Each of these transgenes showed discrete MSL recruiting activity (Figure 5D–G, data not shown for roX1-L), but failed to produce transcripts on northern blots (data not shown). The construct with nine tandem copies, roX1-SM, showed an especially striking signal of MSL1 staining, even stronger than that of the full-length 3.4 kb construct. In addition, one of three lines of roX1-SM transgenics showed frequent spreading into flanking chromatin (Table II; Figure 5G). These results clearly indicate that a 217 bp roX1 fragment carries the predicted activities for a chromatin entry site, being sufficient for recruitment and spreading of the MSL complex.
Table II. Analysis of epigenetic spreading of MSL complexes from roX1-SM transgenes.
| Strain | No. of nuclei observed | No. of nuclei with no signal | No. of nuclei with a single band | No. of nuclei with spreading | Spreading frequency (%)a |
|---|---|---|---|---|---|
| 35EF | 184 | 25 | 158 | 1 | 0.5 |
| 57C | 115 | 16 | 38 | 61 | 53.0 |
| 86AB | 194 | 2 | 186 | 6 | 3.1 |
aPercentage of observed nuclei with spreading.
Chromatin immunoprecipitation confirms preferential association of the MSL complex within the roX1 gene
We performed chromatin immunoprecipitation experiments in order to physically map the association of the MSL complex with the endogenous roX1 gene (Figure 6). To facilitate in vivo cross-linking, these experiments were conducted in SL2 tissue culture cells, where the MSL complex is known to associate with the X chromosome (Copps et al., 1998; Akhtar et al., 2000; Gu et al., 2000). SL2 cell nuclei were cross-linked with formaldehyde and chromatin was sheared and purified. MSL proteins were immunoprecipitated with specific antibodies and the associated DNA was amplified by ligation-mediated PCR (LM-PCR) (Orlando et al., 1997), labeled by random priming and used as a probe to reveal the presence of roX1 DNA (Figure 6). For this purpose the roX1 cDNA was cleaved into smaller fragments by restriction endonucleases (Figure 6A), which were analyzed by Southern blotting and probing with DNA amplified from the chromatin immunoprecipitate (Figure 6B). Control reactions lacking specific antibodies were performed in parallel (‘mock IP’). Normalization of signal intensities for each fragment revealed a significant enrichment of some roX1 fragments by immunoprecipitation with antibodies directed against MSL proteins (plotted in Figure 6C). Complexes were not enriched on autosomal control genes coding for topoisomerase II, AcfI and hsp26 (data not shown). A HhaI–HindIII digestion was most informative, mapping the dominant site of interaction to a 788 bp fragment containing the male-specific DNase hypersensitive site. Fragmenting the roX1 cDNA with XmnI and RsaI further supported the conclusion that MSL complexes interact with a discrete, DNase I hypersensitive site within roX1 chromatin in male cells.

Fig. 6. Mapping of in vivo interactions of MSL proteins within the roX1 gene by chromatin immunoprecipitation. (A) Restriction map of roX1 cDNA. (B) Raw data of a representative experiment. Left panel: SYBR gold stained agarose gel containing various fragments derived from the roX1 cDNA. The cDNA was excised from pBluescript with XhoI and XbaI and gel purified (lane 8). This fragment was further digested with XmnI (lane 1), RsaI (lane 2), Esp3I–ScaI (lane 3), Esp3I–BstEII (lane 4), BanII–NsiI (lane 5), HhaI–HindIII (lane 6), EcoRI–HindIII (lane 7). Center panel: Southern blot probed with amplified DNA isolated by immunoprecipitation of SL-2 chromatin with anti-MSL2 antibody. Right panel: Southern blot using a DNA probe isolated from a mock immunoprecipitation of SL-2 chromatin (lacking a specific antibody). (C) Summary of one representative experiment. Each column shows the normalized hybridization profile obtained for the chromatin immunoprecipitation of the protein indicated above the column. Each row shows the data derived from one roX1 digest. Row 1, XmnI (see lane 1, B); row 2, RsaI (see lane 2, B); row 3, HhaI–HindIII (see lane 6, B). The y-axes of the diagrams show 1/100th of the normalized PhosphorImager counts. Note the scale changes between columns. DHS, position of the DNase I hypersensitive site as mapped in Figure 4. Asterisks indicate two fragments that could not be separated on the gel. The value indicated assumes that both fragments contribute the same signal, which is not necessarily the case.
Discussion
We recently proposed a model for how the MSL complex might distinguish the male X chromosome from the autosomes. Chromatin entry sites distributed along the X play a critical role in that model as the nucleation points for spreading of the MSL complex into flanking genes. In this report, we define one chromatin entry site within a 217 bp fragment near the middle of the roX1 gene. This region binds MSL complexes tightly in vivo, as shown by cytological analysis of transgenic animals and by immunoprecipitation of roX1 DNA cross-linked to MSL proteins. This MSL binding site overlaps a strong male-specific DNase hypersensitive site within roX1, suggesting that the MSL complex either induces a chromatin conformational change or recognizes a pre-existing open architecture that is male specific. Not only does this fragment recruit MSL complex to ectopic autosomal sites, but it also supports variegated spreading into flanking genes that were never before targets of dosage compensation.
These results resolve a number of questions posed by our earlier model. First, it was unclear whether the MSL proteins were recruited to chromatin entry sites by binding DNA sequences or nascent roX transcripts. Here we show that MSL proteins bind autosomal transgenes carrying even small fragments of roX1 in the absence of detectable transcription in cis (Figure 5 and data not shown). The realization that chromatin entry sites need not produce RNA to function means that the other ∼35 chromatin entry sites may not necessarily encode additional species of roX RNAs. They may function as small DNA targets that recruit mature MSL complexes to initiate local MSL spreading. The presence of only a few distinct roX RNAs is consistent with the report of Franke and Baker (1999) that MSL complexes form in the presence of just roX1 or roX2 RNA, but not if both are absent.
The idea that the roX1 and roX2 genes may be atypical chromatin entry sites, in that they produce RNA components of the MSL complex, is consistent with our results using mle mutants. Partial MSL complexes are readily detectable at all ∼35 chromatin entry sites when either MSL3 or MOF is removed by mutation. In contrast, when the MLE helicase is mutated, partial MSL complexes can be found weakly bound to most chromatin entry sites. However, the 10C site that contains the roX2 gene and is the single brightest staining site in a msl3 mutant, is almost undetectable in a mle mutant (Lyman et al., 1997; Meller et al., 2000). We now find that the same is true for roX1. This was previously overlooked because the 3F band is a composite of two or more very tightly linked entry sites, one due to roX1 and at least one other that has not been characterized. When we separated the roX1 chromatin entry site from its neighbors by moving it to an autosome, we found that MLE was also essential for any partial MSL complex to bind.
These results present a paradox. If production of RNA is not required from the roX1 chromatin entry site, what would be the function of MLE in entry site recognition? MLE possesses not only RNA–RNA but RNA–DNA and DNA–DNA helicase activities in vitro, so it might somehow interact with DNA to stabilize weak interactions of MSL1 and 2 (Lee et al., 1997). Although MLE alone clearly does not have X chromosome specificity (Kuroda et al., 1991; Richter et al., 1996), it might contribute directly to the ability of MSL1 and 2 to associate with DNA. Alternatively, the critical role for MLE may be to incorporate roX2 RNA into MSL complexes, and roX2 may be essential to establish an MSL complex conformation necessary to recognize the roX1 chromatin entry site (Meller et al., 2000).
We propose a model for assembly of MSL complexes at two different classes of chromatin entry sites (Figure 7). MSL1 and 2 promote weak interactions with most chromatin entry sites. However, the two entry sites known to encode RNA components of the MSL complex, roX1 and roX2, additionally require the MLE helicase early, perhaps to package roX RNA into growing complexes. Partial complexes containing at least MSL1, MSL2, MLE and perhaps a roX RNA molecule bind efficiently to all chromatin entry sites. Following addition of the remaining subunits, enzymatically active complexes are able to spread into flanking chromatin to dosage compensate target genes. The discrete assembly steps presented in this model reflect the points where various mutants are arrested. In wild-type males, assembly may proceed in a more concerted fashion, perhaps with roX RNAs being incorporated into partial MSL complexes prior to any interactions with the X chromosome.
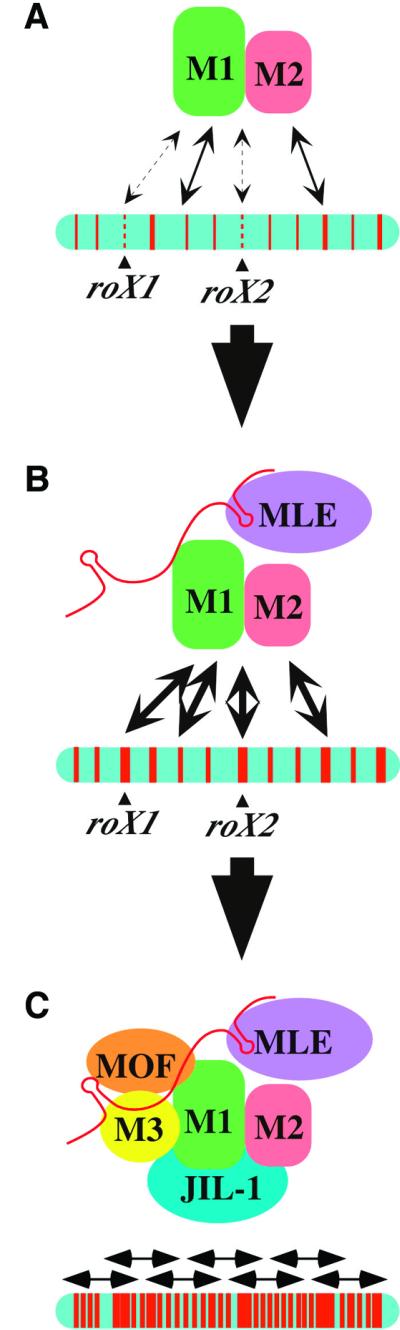
Fig. 7. Model for MSL complex assembly and spreading on the X chromosome. (A) MSL1 and MSL2 show a weak interaction with most chromatin entry sites in the absence of MLE, but roX1 and roX2 are particularly poor targets under these conditions. (B) When MLE is present, roX2 RNA can be incorporated into complexes, potentially altering the conformation to allow strong binding to chromatin entry sites. (C) When complete complexes are present, containing at least five MSL proteins and one or more roX RNAs, binding and spreading occur in a banded pattern along the length of the X chromosome.
Materials and methods
Plasmid DNA construction
To construct Hsp83–roX1 and Hsp83–roX1AS, a 3.4 kb EcoRI fragment containing roX1 cDNA clone c20 (Meller et al., 1997; DDBJ/EMBL/GenBank accession No. AB051842) was subcloned in both orientations into a derivative of pCaSpeR-Hsp83 (Horabin and Schedl, 1993) containing a 470 bp PstI fragment from the tra-2 gene to provide a 3′ poly-A site (W.Mattox, personal communication). The same roX1 fragment was subcloned into pCaSpeR-3, and designated roX1c3.4. For the R5′ and R3′ constructs, 0.9 and 2.6 kb EcoRI fragments of roX1 cDNA clone c3 (Meller et al., 1997) were cloned separately into pCaSpeR (Pirrotta et al., 1988).
To construct Hsp83–roX1Δ30, a pair of PCRs were performed, using roX1 c20 as a template. Primer 13A (5′-GCAAGTAAAAATGCAAACTATAAAAAGAAAATTGAAATC-3′) and M13 reverse primer (5′-CAGGAAACAGCTATGAC-3′) were used in one reaction, while primer 7A′ (5′-ACCAGCAGTTGATTTGCG-3′) and primer 13B (5′-ATAGTTTGCATTTTTACTTGCTTTGGC-3′) in the other. The resulting two fragments were denatured, re-annealed and amplified in a second PCR using primer 7A′ and the M13 reverse primer. The amplified fragment was digested by HindIII and substituted for a 1.2 kb HindIII fragment in pBlueScriptII–roX1c20. A 30 bp deletion of the roX1/roX2 homology region (position 3308–3337) was checked by sequencing, digested by EcoRI (3.4 kb) and subcloned into pCaSpeR-H83 in the correct orientation.
To construct roX1a, roX1b, roX1c, R5′+100 and roX1bc, fragments of roX1 cDNA c20 (positions 1–1121, 1096–2175, 2041–3390, 1–892 and 1096–3390, respectively) were amplified with additional NheI sites introduced at both ends and cloned into pCR-Blunt II TOPO (Invitrogen), followed by sequencing. Clones with the correct sequence were digested with NheI and subcloned into the XbaI site of pCaSpeR.
For the roX1ΔDHS construct, the pBluescript–roX1c20 clone was amplified by using primer 4A (5′-TAGTATGCTAGCACACGACTGCAAAAGCAGC-3′) and 4B (5′-ATATTCGCTAGCCGTCTTCTCGAAACGCAAG-3′), digested by NheI and self-ligated. A resulting plasmid had a 350 bp deletion (positions 868–1202) adjacent to an introduced 6 bp NheI site. The insert was digested by EcoRI and cloned into pCaSpeR.
For roX1-L and S constructs, roX1 fragments from the roX1 c3 cDNA clone (1021–1237 and 980–1281, respectively) were PCR amplified and cloned into a pUC19 variant AvaI site. To make the 9× multimer of roX1-S (roX1-SM), the roX1-S fragment was multimerized by ligation and cloned into a pUC19 variant containing an AvaI site. After digestion by EcoRI and HindIII, those fragments were subcloned into EcoRI–NotI cut pCaSpeR-3 (Pirrotta et al., 1988) using a HindIII–NotI adapter.
Transformation
Transgenic flies are produced by P-element-mediated transformation (Spradling and Rubin, 1982) and, if needed, remobilized from original lines by transposase derived from P[ry+, Δ2-3]99B (Robertson et al., 1988).
Immunostaining of polytene chromosomes
Transgenic flies were grown at 23°C on standard cornmeal–molasses fly media. Preparation of polytene chromosomes and immunostaining using anti-MSL antibodies were as previously described (Kelley et al., 1999).
Genetic crosses
msl mutant females carrying [Hsp83–MSL2] and a [roX1c3.4] transgene were generated by genetic crosses. To generate mof mutant larvae, w cv mof1; [Hsp83–MSL1][Hsp83–MSL2] females were crossed to w cv mof1; [mof t6.8]18H1/[roX1c3.4]51A males. The [mof t6.8]18H1 transgene includes 6.88 kb of the mof genomic region and is able to rescue mof1 mutant males (Hilfiker et al., 1997). Most female larvae carrying the paternal [mof t6.8]18H1 transgene died or showed delayed development at late third-instar larval stage, due to ectopic expression of MSL1 and MSL2. mof female larvae, w cv mof1; [roX1c3.4]51A/+; [Hsp83– MSL1][Hsp83–MSL2]/+, were also distinguished by partial staining of the X chromosome by anti-MSL1 antibodies, due to the absence of the [mof t6.8]18H1 transgene. To generate msl-3 mutant larvae, y w; msl-3 [Hsp83–MSL2] females were crossed to y w; [roX1c3.4]51A; msl-3/TM6C, Sb Tb males. msl-3 female larvae, y w; [roX1c3.4]51A/+; msl-3 [Hsp83–MSL2]/msl-3, were distinguished by absence of the Tb phenotype. To generate mle mutant larvae, y w; pr mle1; [Hsp83– MSL2] females were crossed to y w; pr mle1 [roX1c3.4]51A/CyO, y+ bw males. y w; pr mle1 [roX1c3.4]51A/pr mle1; [Hsp83–MSL2]/+ female larvae were distinguished by the brown color of their mouthhooks.
Northern analysis
RNA extraction was performed using the Trizol™ Reagent (Gibco-BRL) according to the supplier’s instruction. Twenty micrograms of total RNA were applied in each lane for electrophoresis, then transferred to Hybond-N+ (Amersham) and hybridized (Church and Gilbert, 1984). For probe preparation, roX1 cDNA c20 fragments (nt 271–587 or 909–1144) were amplified by PCR and used as templates for random priming.
Mapping of DNase I hypersensitivity
Adult flies were sorted manually according to sex. Two grams of flies were frozen in liquid nitrogen and squashed using mortar and pestle. The powder was suspended in 10 ml of buffer A [60 mM KCl, 15 mM NaCl, 13 mM EDTA, 0.1 mM EGTA, 15 mM Tris pH 7.4, 0.15 mM spermine, 0.5 mM spermidine, 0.5 mM dithiothreitol (DTT)] containing 0.5% NP-40 (buffer ANP). The suspension was homogenized five times in a motor-driven homogenizer (Yamato), dounced 20 times with a tight pestle and filtered through Miracloth (Calbiochem). The filtrate was layered on top of 4 ml of buffer A containing 0.3 M sucrose (buffer AS) and centrifuged for 5 min at 3000 r.p.m. in an HB4 rotor (4°C). The pellet was resuspended in 10 ml of buffer ANP, dounced 10 times with a loose pestle and centrifuged again through 4 ml of buffer AS. The pellet was washed twice in 10 ml of buffer A and once in buffer B (60 mM KCl, 15 mM NaCl, 5 mM MgCl2, 0.1 mM EGTA, 15 mM Tris pH 7.4, 1 mM DTT, 0.3 M sucrose) and finally resuspended in 2 ml of buffer B. The suspension was split into 12 aliquots for DNase I digestion. The nuclei were digested with 0.2–20.5 U of DNase I diluted in buffer B containing 5 mM CaCl2 and 10 mM MgCl2 for 3 min at 25°C, and the reaction stopped by addition of 1/7th volume of 4% SDS, 0.1 M EDTA. DNA was prepared by successive proteinase K and RNase A treatments, phenol– chloroform extraction and ethanol precipitation. Ten micrograms of DNA of each DNase I digest were digested to completion with EcoRI and separated on a 0.8% agarose gel. The DNA was blotted and hybridized as described (Church and Gilbert, 1984). The RNA probe was prepared by in vitro transcription of a 443 bp restriction fragment abutting an EcoRI cleavage site 1053 bp uspstream of the roX1 transcription start site.
Chromatin immunoprecipitation
Chromatin immunoprecipitations were carried out as described previously (Orlando et al., 1997) with modifications. Approximately 2 × 109 Drosophila SL2 cells were fixed in 1.1% formaldehyde (Calbiochem) and sonicated to release soluble chromatin with a modal length of 1 kb. Chromatin was purified by CsCl gradient ultra-centrifugation in a Beckman SW60Ti rotor at 38 kr.p.m. for 40–44 h. Fractionated gradients were dialysed overnight against 1 l of dialysis buffer (5% glycerol, 10 mM Tris pH 8, 1 mM EDTA, 0.5 mM EGTA) using a 28-well microdialysis system (Life Technologies). Chromatin-containing fractions were identified by reversing the cross-links of 50 µl samples, and pooled. The 0.5 ml aliquots were transferred into RIPA buffer (1% Triton X-100, 0.1% sodium deoxycholate, 0.1% SDS, 140 mM NaCl, 10 mM Tris pH 8.0, 1 mM EDTA, 1 mM phenylmethylsulfonyl fluoride) and 2–5 µg of antibodies were added (2 µl anti-MSL1, 2 µl anti-MSL2, 0.5 µl anti-MSL3, 5 µl anti-MOF or 5 µl pre-immune sera for mock immunoprecipitations) and incubated overnight at 4°C. Immunocomplexes were purified using 40 µl of 50% slurry of protein G (MSL3) or protein A (other antibodies) Sepharose. Complexes were washed five times in 1 ml of RIPA buffer, once in 1 ml of LiCl buffer (250 mM LiCl, 10 mM Tris pH 8.0, 1 mM EDTA, 0.5% NP-40, 0.5% sodium deoxycholate) and once in 1 ml of 10 mM Tris pH 8, 1 mM EDTA before reversing cross-links. Purified immunoprecipitated DNA was resuspended in 22 µl of H2O and overhanging ends created by sonication were extended using Pfu DNA polymerase. The DNA was then amplified by ligation-mediated PCR (LM-PCR) to produce sufficient amounts for labeling and use as a probe in Southern blotting. The linker for LM-PCR was created by the annealing of two oligonucleotides (AGAAGC TTGAATTCGAGCAGTCAGp and CTGCTGGAATTCAAGCTTCT), the latter of which was also used for PCR amplification.
Acknowledgments
Acknowledgements
We thank A.Akhtar and X.Bai for critical reading of the manuscript. We thank X.Chu, P.Gordadze and R.Richman for excellent technical assistance. This work was supported by the National Institutes of Health, the Human Frontier Science Program, a TMR fellowship of the European Union, the Welch Foundation, the Howard Hughes Medical Institute and the EU Training and Mobility through Research Programme. M.I.K. is an Investigator of the Howard Hughes Medical Institute.
References
- Akhtar A. and Becker,P.B. (2000) Activation of transcription through histone H4 acetylation by MOF, an acetyltransferase essential for dosage compensation in Drosophila. Mol. Cell, 5, 367–375. [DOI] [PubMed] [Google Scholar]
- Akhtar A., Zink,D. and Becker,P.B. (2000) Chromodomains are protein–RNA interaction modules. Nature, 407, 405–409. [DOI] [PubMed] [Google Scholar]
- Amrein H. and Axel,R. (1997) Genes expressed in neurons of adult male Drosophila. Cell, 88, 459–469. [DOI] [PubMed] [Google Scholar]
- Bone J.R., Lavender,J., Richman,R., Palmer,M.J., Turner,B.M. and Kuroda,M.I. (1994) Acetylated histone H4 on the male X chromosome is associated with dosage compensation in Drosophila. Genes Dev., 8, 96–104. [DOI] [PubMed] [Google Scholar]
- Church G.M. and Gilbert,W. (1984) Genomic sequencing. Proc. Natl Acad. Sci. USA, 81, 1991–1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copps K., Richman,R., Lyman,L.M., Chang,K.A., Rampersad-Ammons,J. and Kuroda,M.I. (1998) Complex formation by the Drosophila MSL proteins: role of the MSL2 RING finger in protein complex assembly. EMBO J., 17, 5409–5417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donze D., Adams,C.R., Rine,J. and Kamakaka,R.T. (1999) The boundaries of the silenced HMR domain in Saccharomyces cerevisiae. Genes Dev., 13, 698–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke A. and Baker,B.S. (1999) The roX1 and roX2 RNAs are essential components of the compensasome, which mediates dosage compensation in Drosophila. Mol. Cell, 4, 117–122. [DOI] [PubMed] [Google Scholar]
- Gu W., Szauter,P. and Lucchesi,J.C. (1998) Targeting of MOF, a putative histone acetyl transferase, to the X chromosome of Drosophila melanogaster. Dev. Genet., 22, 56–64. [DOI] [PubMed] [Google Scholar]
- Gu W., Wei,X., Pannuti,A. and Lucchesi,J.C. (2000) Targeting the chromatin-remodeling MSL complex of Drosophila to its sites of action on the X chromosome requires both acetyl transferase and ATPase activities. EMBO J., 19, 5202–5211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilfiker A., Hilfiker-Kleiner,D., Pannuti,A. and Lucchesi,J.C. (1997) mof, a putative acetyl transferase gene related to the Tip60 and MOZ human genes and to the SAS genes of yeast, is required for dosage compensation in Drosophila. EMBO J., 16, 2054–2060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horabin J.I. and Schedl,P. (1993) Sex-lethal autoregulation requires multiple cis-acting elements upstream and downstream of the male exon and appears to depend largely on controlling the use of the male exon 5′ splice site. Mol. Cell. Biol., 13, 7734–7746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y., Wang,Y., Johansen,J. and Johansen,K.M. (2000) JIL-1, a chromosomal kinase implicated in regulation of chromatin structure, associates with the Male Specific Lethal (MSL) dosage compensation complex. J. Cell Biol., 149, 1005–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley R.L. and Kuroda,M.I. (2000) The role of chromosomal RNAs in marking the X for dosage compensation. Curr. Opin. Genet. Dev., 10, 555–561. [DOI] [PubMed] [Google Scholar]
- Kelley R.L., Solovyeva,I., Lyman,L.M., Richman,R., Solovyev,V. and Kuroda,M.I. (1995) Expression of Msl2 causes assembly of dosage compensation regulators on the X chromosomes and female lethality in Drosophila. Cell, 81, 867–877. [DOI] [PubMed] [Google Scholar]
- Kelley R.L., Meller,V.H., Gordadze,P.R., Roman,G., Davis,R.L. and Kuroda,M.I. (1999) Epigenetic spreading of the Drosophila dosage compensation complex from roX RNA genes into flanking chromatin. Cell, 98, 513–522. [DOI] [PubMed] [Google Scholar]
- Kingston R.E. and Narlikar,G.J. (1999) ATP-dependent remodeling and acetylation as regulators of chromatin fluidity. Genes Dev., 13, 2339–2352. [DOI] [PubMed] [Google Scholar]
- Kuroda M.I., Kernan,M.J., Kreber,R., Ganetzky,B. and Baker,B.S. (1991) The maleless protein associates with the X chromosome to regulate dosage compensation in Drosophila. Cell, 66, 935–947. [DOI] [PubMed] [Google Scholar]
- Lee C.-G., Chang,K.A., Kuroda,M.I. and Hurwitz,J. (1997) The NTPase/helicase activities of Drosophila Maleless, an essential factor in dosage compensation. EMBO J., 16, 2671–2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucchesi J.C. (1998) Dosage compensation in flies and worms: the ups and downs of X-chromosome regulation. Curr. Opin. Genet. Dev., 8, 179–184. [DOI] [PubMed] [Google Scholar]
- Lyman L.M., Copps,K., Rastelli,L., Kelley,R.L. and Kuroda,M.I. (1997) Drosophila male-specific lethal-2 protein: structure/function analysis and dependence on MSL1 for chromosome association. Genetics, 147, 1743–1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyon M.F. (1999) X-chromosome inactivation. Curr. Biol., 9, R235–R237. [DOI] [PubMed] [Google Scholar]
- Meller V.H., Wu,K.H., Roman,G., Kuroda,M.I. and Davis,R.L. (1997) roX1 RNA paints the X chromosome of male Drosophila and is regulated by the dosage compensation system. Cell, 88, 445–457. [DOI] [PubMed] [Google Scholar]
- Meller V.H., Gordadze,P.R., Park,Y., Chu,X., Stuckenholz,C., Kelley,R.L. and Kuroda,M.I. (2000) Ordered assembly of roX RNAs into MSL complexes on the dosage compensated X chromosome in Drosophila. Curr. Biol., 10, 136–143. [DOI] [PubMed] [Google Scholar]
- Mukherjee A.S. and Beermann,W. (1965) Synthesis of ribonucleic acid by the X-chromosomes of Drosophila melanogaster and the problem of dosage compensation. Nature, 207, 785–786. [DOI] [PubMed] [Google Scholar]
- Nitsch D., Stewart,A.F., Boshart,M., Mestril,R., Weih,F. and Schütz,G. (1990) Chromatin structures of the rat tyrosine aminotransferase gene relate to the function of its cis-acting elements. Mol. Cell. Biol., 10, 3334–3342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Kane C.J. and Gehring,W.J. (1987) Detection in situ of genomic regulatory elements in Drosophila. Proc. Natl Acad. Sci. USA, 84, 9123–9127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlando V., Strutt,H. and Paro,R. (1997) Analysis of chromatin structure by in vivo formaldehyde cross-linking. Methods, 11, 205–221. [DOI] [PubMed] [Google Scholar]
- Palmer M.J., Richman,R., Richter,L. and Kuroda,M.I. (1994) Sex-specific regulation of the male-specific lethal-1 dosage compensation gene in Drosophila. Genes Dev., 8, 698–706. [DOI] [PubMed] [Google Scholar]
- Pirrotta V., Bickel,S. and Mariani,C. (1988) Developmental expression of the Drosophila zeste gene and localization of zeste protein on polytene chromosomes. Genes Dev., 2, 1839–1850. [DOI] [PubMed] [Google Scholar]
- Richter L., Bone,J.R. and Kuroda,M.I. (1996) RNA-dependent association of the Drosophila maleless protein with the male X chromosome. Genes Cells, 1, 325–336. [DOI] [PubMed] [Google Scholar]
- Robertson A.M., Preston,C.R., Phillis,R.W., Johnson-Schlitz,D., Benz,W.K. and Engels,W.R. (1988) A stable genomic source of P element transposase in Drosophila melanogaster. Genetics, 118, 461–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith E.R., Pannuti,A., Gu,W., Steurnagel,A., Cook,R.G., Allis,C.D. and Lucchesi,J.C. (2000) The Drosophila MSL complex acetylates histone H4 at lysine 16, a chromatin modification linked to dosage compensation. Mol. Cell. Biol., 20, 312–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spradling A.C. and Rubin,G.M. (1982) Transposition of cloned P-elements into Drosophila germ line chromosomes. Science, 218, 341–347. [DOI] [PubMed] [Google Scholar]
- Stone E.M. and Pillus,L. (1998) Silent chromatin in yeast: an orchestrated medley featuring Sir3p. BioEssays, 20, 30–40. [DOI] [PubMed] [Google Scholar]
- Turner B.M., Birley,A.J. and Lavender,J. (1992) Histone H4 isoforms acetylated at specific lysine residues define individual chromosomes and chromatin domains in Drosophila polytene nuclei. Cell, 69, 375–384. [DOI] [PubMed] [Google Scholar]
- Wakimoto B.T. (1998) Beyond the nucleosome: epigenetic aspects of position-effect variegation in Drosophila. Cell, 93, 321–324. [DOI] [PubMed] [Google Scholar]
- White W.M., Willard,H.F., Van Dyke,D.L. and Wolff,D.J. (1998) The spreading of X inactivation into autosomal material of an x;autosome translocation: evidence for a difference between autosomal and X-chromosomal DNA. Am. J. Hum. Genet., 63, 20–28. [DOI] [PMC free article] [PubMed] [Google Scholar]