

Abstract
The mechanism of action of the anti-apoptotic oncogene Bcl-2 is still largely obscure. We have recently shown that the overexpression of Bcl-2 in HeLa cells reduces the Ca2+ concentration in the endoplasmic reticulum ([Ca2+]er) by increasing the passive Ca2+ leak from the organelle. To investigate whether this Ca2+ depletion is part of the mechanism of action of Bcl-2, we mimicked the Bcl-2 effect on [Ca2+]er by different pharmacological and molecular approaches. All conditions that lowered [Ca2+]er protected HeLa cells from ceramide, a Bcl-2-sensitive apoptotic stimulus, while treatments that increased [Ca2+]er had the opposite effect. Surprisingly, ceramide itself caused the release of Ca2+ from the endoplasmic reticulum and thus [Ca2+] increased both in the cytosol and in the mitochondrial matrix, paralleled by marked alterations in mitochondria morphology. The reduction of [Ca2+]er levels, as well as the buffering of cytoplasmic [Ca2+] changes, prevented mitochondrial damage and protected cells from apoptosis. It is therefore concluded that the Bcl-2-dependent reduction of [Ca2+]er is an important component of the anti-apoptotic program controlled by this oncogene.
Keywords: apoptosis/Bcl-2/Ca2+/ceramide/mitochondria
Introduction
Apoptosis, the process that allows multicellular organisms to eliminate unnecessary or damaged cells without evoking inflammation or tissue damage (Hengartner and Horvitz, 1994; Hetts, 1998), takes place under a wide number of physiological and pathological conditions (Ellis et al., 1991; Nagata, 1997). Indeed, it allows elimination of sovrannumerary cells during the development and functional activity of tissues, while its inappropriate activation is supposed to be at the basis of common neurodegenerative disorders (Nicotera et al., 1999; Mattson, 2000). Moreover, its efficacy in removing hazardous cells is circumvented in viral diseases and neoplasia by specific molecular routes. It is thus not surprising that one of the best known oncogenes, Bcl-2, first identified in lymphomas and then found associated to a number of human cancers, has apoptosis as its primary target (Chao and Korsmeyer, 1998). Recent work in experimental oncology has further stressed its importance, by demonstrating that it is the homolog of one of the elementary components of the apoptotic machinery identified in Caenorhabditis elegans and that it belongs to a family of related gene products that includes members with opposite regulatory effects (Boise et al., 1995).
Despite the wide interest and extensive work on this oncogene, its mechanism of action remains debated. The various members of the Bcl-2 family (which include repressors of apoptosis, such as Bcl-x, or activators, such as Bax) dimerize and interact with cofactors of caspases, an obvious route for influencing the molecular machinery of apoptosis (Li and Yuan, 1999). However, converging evidence suggests that an alternative mechanism may be operating, based on the alteration of intracellular ion signaling. Bcl-2 has been demonstrated to act as an ion channel in isolated lipid bilayers (Minn et al., 1997; Schendel et al., 1997) and its complex distribution to intracellular organelles [mitochondria, endoplasmic reticulum (ER)] (Lithgow et al., 1994) could therefore affect the equilibrium of ions across their membranes. Such a possibility is supported by the observation, by us and other groups, that the recombinant expression of Bcl-2 alters the state of filling of intracellular Ca2+ stores and the kinetics and amplitudes of cellular Ca2+ responses (Lam et al., 1994; He et al., 1997; Kuo et al., 1998; Foyouzi-Youssefi et al., 2000; Pinton et al., 2000). In particular, it was recently demonstrated that recombinantly expressed Bcl-2, by increasing the passive leak across the ER membrane, reduces the ER Ca2+ concentration ([Ca2+]er) steady state (Foyouzi-Youssefi et al., 2000; Pinton et al., 2000). Consequently, stimulus-dependent [Ca2+] increases are reduced both in the cytoplasm and mitochondria, an obvious target for an apoptogenic effect of Ca2+. Under those conditions, store-dependent Ca2+ entry is also significantly reduced, thus further dampening the Ca2+ responses of the cells.
Although suggestive, these results do not provide a conclusive link between the alteration in Ca2+ signaling and the anti-apoptotic activity of Bcl-2. This is the aim of the current paper, in which we investigated whether the alteration in calcium signaling caused by Bcl-2 is sufficient to prevent cell death triggered by ceramide, an endogenous lipid mediator of apoptosis. We thus mimicked/antagonized the [Ca2+] changes caused by Bcl-2 by different experimental approaches and verified that the level of [Ca2+]er inversely correlates with the efficacy of this apoptotic stimulus.
We then addressed the mechanisms that allow this signaling alteration to be protective. We showed that ceramide induces a rise in cytoplasmic Ca2+ concentration ([Ca2+]c) by releasing Ca2+ from intracellular stores and activating the capacitative Ca2+ entry pathway. These phenomena result in prolonged mitochondrial Ca2+ accumulation and alterations in organelle morphology (swelling and fragmentation) (Duchen, 1999).
A model is discussed where ceramide-induced death is the result of a combined effect of a direct hit of this lipid mediator (or of its metabolites) on mitochondria and of a synergic damaging effect of Ca2+ accumulation by the organelle. Altogether the present data suggest that depletion of Ca2+ from the stores is a key component of the protective action of Bcl-2 against apoptosis.
Results
Ceramide-induced apoptosis is inhibited by lowering the extracellular Ca2+ concentration
HeLa cells were plated in 96-well plates and allowed to grow to ∼80% confluence. At this stage, the medium was changed from Dulbecco’s modified Eagle’s medium (DMEM) + 10% fetal calf serum (FCS) to a modified Krebs-Ringer buffer (KRB) (see Materials and methods), at 37°C in 5% CO2 atmosphere. After ∼4 h in the new medium, cells were treated with 10 µM ceramide. Cell viability was evaluated at different time points through phase contrast microscopy, by counting viable and phase lucent dead cells. Figure 1A shows a representative microscopic field, as derived from the analysis of >10 similar experiments. The percentage of living cells is indicated in the upper right corner; in the whole coverslip (and in the representative field of the figure) ∼90% of the cell population is dead 16 h after the addition of ceramide. The comparison of ceramide-treated (+ Cer) and control cells (control), where the number of dead cells is negligible, indicates that cell death must be entirely ascribed to the effect of ceramide. The direct measurement of caspase activity, showing a marked increase in ceramide-treated cells (Figure 1B), confirms the morphological appearance of apoptotic cell death. In support of this notion, pre-treatment of cells with 50 µM z-VAD, a wide spectrum caspase inhibitor (Nicholson, 1999), increased cell viability upon ceramide treatment to 74% ± 10 (n = 5) (not shown).

Fig. 1. C2 ceramide induces apoptotic cell death. (A) HeLa cells were maintained in KRB supplemented with 1 mM Ca2+ (1 mM Ca2+/KRB), and challenged with 10 µM ceramide (+ Cer). The number of viable cells was determined by phase contrast microscopy after 16 h. The percentage of living cells is reported in the upper right corner. Ceramide induces caspase activation in HeLa cells (B). Cells were incubated with ceramide for 16 h, and caspase-3-like activity of cell lysates was measured as detailed in Materials and methods, and expressed as fluorescence arbitrary units. The caspase inhibitor Ac-DEVD-CHO (inh.) has also been used to show specificity of the caspase-3-like activity. Bcl-2 overexpression protects cells from C2 ceramide-induced cell death (C). HeLa cells were cotransfected with Bcl-2 and with mtGFP expression plasmids. Thirty-six hours after transfection, cells were challenged with increasing ceramide concentrations (from 0 to 20 µM) and viability was assessed by microscope count of living GFP-expressing cells (see text for details). mtGFP alone does not affect cell viability. In order to eliminate possible errors due to the detachment of dead cells during the transfer of the coverslip to the chamber of the fluorescent microscope, the effect of Bcl-2 expression on cell survival was evaluated, and expressed as the percentage of fluorescent cells in the microscope field. Data are averages ± SD of triplicate determinations from experiments repeated at least five times.
This apoptosis protocol was chosen because Bcl-2 is supposed to be highly efficient in protecting cells from death induced by ceramide and its metabolites (Zhang et al., 1996; Rippo et al., 2000). In order to obtain direct experimental evidence for this notion, in our experimental conditions HeLa cells were transiently transfected with Bcl-2 and treated with increasing ceramide concentrations. By using this protocol, however, it was not possible to verify directly that the surviving cells were those expressing the oncogene. In order to address this issue directly, cells were co-transfected with Bcl-2 and mtGFP/pcDNAI (where GFP is green fluorescent protein), an expression plasmid encoding the fluorescent marker mtGFP (Rizzuto et al., 1995b). Transfected cells were identified 36 h after transfection, by visualizing mtGFP with a fluorescence microscope. As shown in Figure 1C, Bcl-2 overexpressing cells displayed an enhanced survival. Indeed, due to the higher mortality of the cells that do not overexpress Bcl-2, the percentage of living GFP (thus Bcl-2) expressing cells gradually increased with ceramide concentration. No change in the percentage of living fluorescent cells was observed when HeLa cells were transfected with mtGFP alone, as all cells (mtGFP expressing and untransfected) are equally sensitive to the apoptotic agent.
In a previous report we showed that Bcl-2 overexpression decreased [Ca2+]er and we hypothesized that this effect could account, at least partially, for the anti-apoptotic role of this oncoprotein (Pinton et al., 2000). The experiments presented in Figures 2–5 were aimed at testing this hypothesis directly. In the experiment of Figure 2A and B, HeLa cells were maintained in KRB solution supplemented with [Ca2+] ranging from 1 mM (1 mM Ca2+/KRB) to 20 µM (20 µM Ca2+/KRB). This procedure caused the decrease of steady state [Ca2+]er levels from 310 µM (±87 µM, n = 5) for 1 mM Ca2+/KRB to 54 µM (±15 µM, n = 5) for 20 µM Ca2+/KRB, as measured with a targeted aequorin chimera (see Materials and methods for details). Under these conditions, the efficacy of 10 µM ceramide was evaluated as described in Figure 1A. Figure 2A shows representative microscopic fields of the experiments carried out at different extracellular [Ca2+] ([Ca2+]e). In each image, the [Ca2+]e employed and the [Ca2+]er attained are indicated in the lower right and upper left corner, respectively. Figure 2B shows the averages obtained from the analysis of >50 fields in five independent experiments. The percentage of cells surviving the ceramide treatment showed a biphasic correlation with the [Ca2+] of the incubation medium. Ceramide was highly cytotoxic at a [Ca2+]e of ∼20 µM, then survival was enhanced at 40–50 µM [Ca2+]e, and dropped again when [Ca2+]e approached physiological values.

Fig. 2. C2 ceramide-induced cell death is dependent on [Ca2+]e (A and B). Cells were incubated in KRB supplemented with different [Ca2+]e and treated with 10 µM C2 ceramide. (A) Representative microscopic fields. The inset in the upper left and lower right corners report the [Ca2+]er and [Ca2+]e of each condition, respectively. (B) Average values of cell viability obtained from analyzing >50 fields (including >500 cells) in five independent experiments. (C) tBuBHQ mimics the effect of [Ca2+]e reduction on cell viability. Cells were incubated in 1 mM Ca2+/KRB and treated with different [tBuBHQ]. Cell viability was evaluated as in (B).
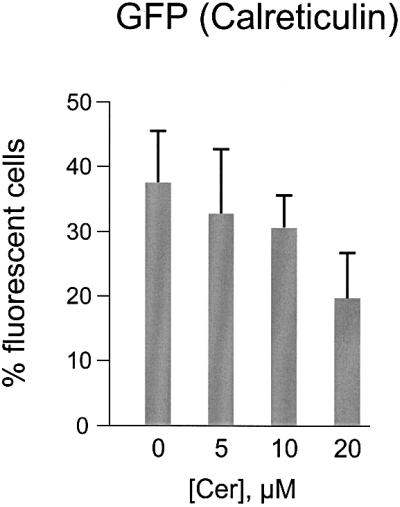
Fig. 5. HeLa cells overexpressing calreticulin are more sensitive to ceramide-induced cell death. Cells were transfected with a calreticulin-expressing plasmid as detailed in Materials and methods. Transfected cells were identified by visualizing co-expressed mtGFP as specified in Figure 1C. Ceramide concentrations were as in Figure 1. Data are expressed as in Figure 1C. Experiments were repeated at least five times.
The increase of cell survival was paralleled by the disappearance of the typical morphological hallmarks of apoptosis, such as chromatin condensation. Figure 3 shows the staining of nuclei with propidium iodide. Nuclei of control cells show normal, dispersed chromatin (Figure 3A). Figure 3B shows cells treated with ceramide in 1 mM Ca2+/KRB. Nuclear shrinkage is evident in most cells. Conversely, when cells were maintained in 50 µM Ca2+/KRB, treatment with ceramide caused no major alteration of nuclear morphology (Figure 3C).
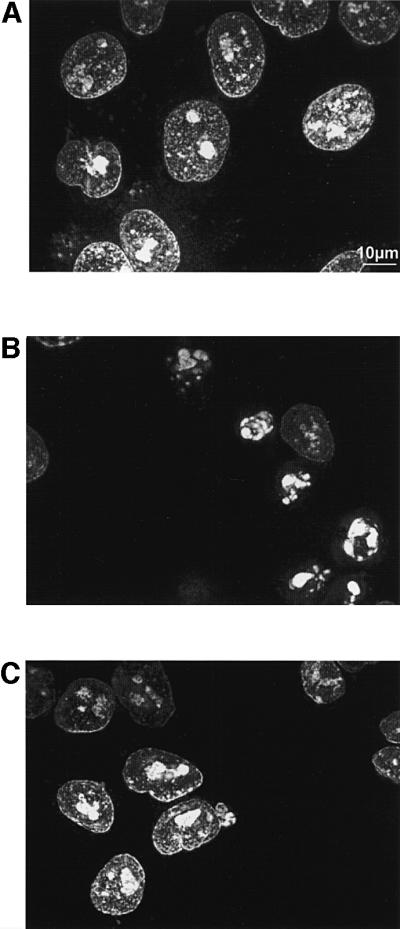
Fig. 3. C2 ceramide induces chromatin condensation, and nuclear shrinkage in cells maintained in 1 mM Ca2+/KRB but not in 50 µM Ca2+/KRB. (A) Control; (B) 1 mM Ca2+/KRB + Cer; (C) 50 µM Ca2+/KRB + Cer. The figure shows a representative microscopic field taken 16 h after the addition of 10 µM ceramide. Cells were permeabilized with 100 µM digitonin and nuclei were stained with 1 µM propidium iodide, as specified in Materials and methods.
The anti-apoptotic effect of Bcl-2 is mimicked by a variety of experimental conditions that reduce [Ca2+]er
Alterations in [Ca2+]e are a rather crude, but efficient, way to reduce the [Ca2+]er. A more direct approach to achieve the same result, while maintaining [Ca2+]e at physiological levels, is to interfere with the Ca2+ pump of the ER (SERCA). HeLa cells were thus treated with different concentrations of a specific SERCA blocker, tert-butyl-benzohydroquinone (tBuBHQ) (Kass et al., 1989). By this approach, [Ca2+]er can be reduced proportionally to the inhibitor concentration. In the experiment presented in Figure 2C, cells were treated with concentrations of tBuBHQ ranging from 0.1 to 30 µM, which caused a reduction of steady state [Ca2+]er from 268 µM (±22 µM, n = 5), to the virtually complete emptying of the ER (<20 µM, n = 5). Thirty minutes after the addition of tBuBHQ, cells were treated with 10 µM ceramide, and apoptotic cell death was evaluated after 16 h. Interestingly, for [Ca2+]er reductions comparable to the protective conditions of Figure 2A and B (i.e. 80 µM ± 29 [Ca2+]er obtained with 10 µM tBuBHQ), there was a significant increase in cell survival.
Based on these results, it is expected that alteration of the molecular repertoire responsible for active Ca2+ transport should also modify the susceptibility of the cells to ceramide-induced death. In particular, it has recently been shown that overexpression of the plasma membrane Ca2+ pump (PMCA) causes an ∼20% reduction in [Ca2+]er in CHO cells (Brini et al., 2000). In the experiment of Figure 4A, HeLa cells were transfected with a PMCA expression plasmid and identified by mtGFP co-expression, as in Figure 1C. At this stage, the apoptotic process was initiated by adding increasing ceramide concentrations (from 0 to 20 µM) and evaluated 16 h after ceramide addition, as in previous experiments. Thus, if pump overexpression modifies cell survival, the percentage of living fluorescent cells (which co-express GFP and PMCA) should increase compared with a parallel control transfected with GFP alone. Overexpression of PMCA (Figure 4A) indeed resulted in a significant increase in living GFP-expressing cells, thus indicating that reduction in [Ca2+]er levels under normal values decreases the effect of ceramide.

Fig. 4. PMCA overexpression increases while SERCA overexpression reduces cell viability. HeLa cells were transfected with expression plasmids driving either recombinant PMCA (A) or SERCA (B). Transfected cells were identified by visualizing co-expressed mtGFP as specified in Figure 1C. Increasing ceramide concentrations (from 0 to 20 µM) were added and cell death was evaluated after 16 h. Data are expressed as in Figure 1C. Experiments were repeated at least five times.
Protection against apoptosis depends on the amount of releasable Ca2+, not on the [Ca2+]er
The question then arises as to whether overloading the ER with Ca2+ results in enhanced sensitivity to the apoptotic death induced by ceramide. Brini et al. (2000) showed that overexpression of the ER Ca2+ pump (SERCA) in CHO cells results in an ∼25% increase in the [Ca2+]er. Cells were thus co-transfected with plasmids encoding SERCA2b and mtGFP, and the same protocol employed in Figure 1C was applied. Contrary to PMCA overexpression, a drastic reduction in the percentage of living fluorescent cells was observed (Figure 4B), thus indicating that an increase in [Ca2+]er levels above normal values potentiates the effect of the pro-apoptotic mediator.
The evidence provided so far supports the view that [Ca2+]er is a parameter controlling the susceptibility to apoptotic cell death. As to the mechanism, two routes can be envisaged. In the first, protection could depend on events occurring in the ER lumen (e.g. the sorting or processing of molecules, such as caspases or death receptors), given that [Ca2+]er of the ER environment is known to be important for regulating processes occurring in the organelle (Park et al., 2000). In a second possible mechanism, the lower [Ca2+]er, by reducing the Ca2+ flow across the channels of the ER membrane, could reduce the amplitude of the Ca2+ responses elicited by physiological and/or pathological stimuli. To discriminate between these possibilities, we altered intracellular Ca2+ signaling by overexpressing the main Ca2+ buffering protein of the ER lumen, calreticulin (Krause and Michalak, 1997). By this means, the amplitude and duration of Ca2+ signals can be enhanced (Bastianutto et al., 1995) without increasing [Ca2+]er (Fasolato et al., 1998; Xu et al., 2000). A few data even suggest that [Ca2+]er could be reduced by calreticulin overexpression, due to the direct inhibition of the SERCA mediated by the P-domain of calreticulin (John et al., 1998; Xu et al., 2000). It is thus expected that if protection depends on a signal conveyed to the cytoplasm or to other effector systems (such as mitochondria), calreticulin overexpression should reduce survival upon ceramide treatment, whereas no effect (or even the opposite) would be observed if the [Ca2+]er is the main parameter involved. Figure 5 shows that the former is the case. HeLa cells were co-transfected with a calreticulin and mtGFP expression plasmid and the standard apoptosis protocol was applied as before, by treating cells with different ceramide concentrations. As with SERCA overexpressers, the percentage of fluorescent cells decreased progressively with the increase in ceramide concentration, implying that calreticulin overexpression adversely affects cellular resistance to the apoptotic stimulus.
Effects of ceramide on cellular Ca2+ homeostasis
To support the view that apoptosis is triggered, or enhanced, by the release of Ca2+ from intracellular Ca2+ stores, we investigated whether ceramide had a direct effect on intracellular Ca2+ homeostasis. To this end, HeLa cells were loaded with the Ca2+ indicator Fura-2/AM (Grynkiewicz et al., 1985) and then transferred to a fluorimeter cuvette. [Ca2+]c was calculated from the 340/380 fluorescence ratio, using an algorithm based on the Ca2+ affinity and fluorescence proprieties of Fura-2. Treatment with 10 µM ceramide caused a [Ca2+]c elevation, which gradually increased with time (Figure 6A). This alteration was specific to the apoptogenic lipid, since its analog di-hydroceramide, which does not trigger apoptosis, had no effect on [Ca2+]c (Figure 6B). The simplest explanation for these results is that ceramide causes a progressive release of Ca2+ from intracellular stores, thereby directly causing a [Ca2+]c rise and activating capacitative Ca2+ influx, which in turn is responsible for maintaining a long-lasting [Ca2+]c plateau. Figure 6C and D confirms that this was the case. In these experiments, ceramide, added to cells incubated in EGTA/KRB, caused a slow, transient increase in [Ca2+]c due to the virtually complete release of the Ca2+ content of the intracellular stores. Indeed, the following addition of ionomycin caused a marginal increase in [Ca2+]c (Figure 6C). Conversely, di-hydroceramide had no direct effect on [Ca2+]c, while a large, rapid increase was then observed upon addition of ionomycin, thus confirming that the intracellular Ca2+ stores have not been depleted by the non-apoptogenic lipid (Figure 6D). The same results were obtained when [Ca2+]c was measured in cells transfected with cytosolic aequorin (not shown).

Fig. 6. C2 ceramide causes a time-dependent elevation in the [Ca2+]c. HeLa cells were loaded with the Ca2+ indicator Fura-2/AM and [Ca2+]c changes were measured as detailed in Materials and methods. The coverslips with the cells were maintained in 1 mM Ca2+/KRB (A and B) or in Ca2+-free 0.5 mM EGTA/KRB (C and D). Where indicated, the cells were challenged with 10 µM ceramide (+ Cer) (A and C) or 10 µM di-hydroceramide (DH-Cer) (B and D). The traces show the calibrated [Ca2+]c values {Δ[Ca2+] are 212 ± 35 nM (A), 20 ± 15 nM (B), 40 ± 12 nM (C), 6 ± 4 nM (D)}. The experiment shown is representative of at least five similar trials. Ionomycin (Iono).
Downstream of the ER: the effect on mitochondria
The target of the Ca2+-mediated signal must reside outside the ER, either in the cytoplasm or in another subcellular compartment. Mitochondria were obvious candidates to investigate, given their role of modulators of the apoptotic process. Indeed, in a process that might involve the opening of a high-conductance channel, known as the permeability transition pore (PTP) (Bernardi et al., 1998; Crompton et al., 1999; Jacotot et al., 1999), and reflect the apoptotic derangement of physiological stimuli (Szalai et al., 1999) (see Discussion), they can release pro-apoptotic factors acting as co-activators of downstream caspases (Yang et al., 1997; Kluck et al., 1999).
At first we verified whether ceramide induces a mitochondrial [Ca2+] ([Ca2+]m) rise, in parallel with that of the cytosol. For this purpose, HeLa cells were transfected with a mitochondrially targeted aequorin chimera (mtAEQ) (Rizzuto et al., 1992), and analyzed for aequorin luminescence 36 h after transfection, as described in Materials and methods. Figure 7 shows a calibrated [Ca2+]m trace. Ceramide treatment induced a prolonged increase in [Ca2+]m, with a slow kinetic. Interestingly, traces were quite noisy, probably reflecting asynchronous increases of [Ca2+]m in a different group of cells (the aequorin signal is the average of a few thousand cells). Indeed when [Ca2+]c was analyzed at the single cell level, the ceramide-induced increases were somewhat asynchronous (not shown). These results, together with those of Figure 6, suggest that the slow release of ER Ca2+ induced by ceramide, in contrast to agonist-dependent opening of IP3 receptors, allows a relatively modest Ca2+ accumulation via the low affinity mitochondrial uptake systems, which, however, is maintained for tens of minutes, i.e. much longer than a typical physiological challenge.

Fig. 7. C2 ceramide induces a rise in [Ca2+]m. HeLa cells were transfected with a mtAEQ expression plasmid and analyzed 36 h after transfection. Detection of aequorin luminescence and calibration into [Ca2+] values were carried out as described in Materials and methods. The trace shows the calibrated [Ca2+]m values (Δ[Ca2+] is 0.48 ± 0.12 µM). Where indicated, the cells were challenged with 100 µM C2 ceramide (a higher concentration was employed because we observed that perfusion through plastic tubing is very inefficient, and a markedly higher ceramide concentration is needed to elicit the biological effect, as verified by the monitoring of cytosolic [Ca2+] changes and of the apoptotic efficacy of the perfusion effluent). The experiment shown is representative of at least five similar trials.
We then investigated whether the [Ca2+]m changes were paralleled by alterations of mitochondrial morphology, compatible with the opening of PTP (Petit et al., 1998). HeLa cells were thus transfected with mtGFP and organelle structure was evaluated using a high-resolution digital imaging system (Rizzuto et al., 1998a). Figure 8 shows images taken 1 h apart while the coverslip was maintained on the microscope stage. In control cells (A and A′), the interconnected mitochondrial network (Rizzuto et al., 1998b) can be appreciated in both images, despite the occurrence of some motion and structural rearrangements. Conversely, ceramide treatment caused a drastic alteration of mitochondrial morphology (B and B′). Indeed, while changes could already be detected a few minutes after ceramide addition (not shown), the complete rupture of the mitochondrial network can be easily appreciated in the image taken 1 h after the apoptotic challenge (see also the enlargement of the image shown in the inset). We thus verified whether reducing the cytoplasmic Ca2+ increase (i.e. mimicking the Bcl-2 effect) could also prevent the changes in mitochondrial morphology. To this end, two sets of experiments were carried out. In the first (Figure 8C and C′), mtGFP-transfected HeLa cells were transferred to 50 µM Ca2+/KRB, a dose associated with high cell survival (see Figure 2A and B), then ceramide was added. In the second experiment (Figure 8D and D′), cells were maintained in 1 mM Ca2+/KRB and loaded with a Ca2+ chelator (30 min incubation with 5 µM BAPTA), prior to adding ceramide. In both cases, the addition of ceramide caused no evident alteration in mitochondrial morphology.


Fig. 8. C2 ceramide induces early morphological changes in the mitochondrial network, which are inhibited by lowering extracellular Ca2+, or chelating cytosolic Ca2+. HeLa cells were transfected with mtGFP, and treated with 10 µM ceramide for 1 h. Mitochondrial structure was evaluated by visualizing mtGFP with a high-resolution digital imaging system, as specified in Materials and methods. A larger magnification of the images is presented in the insets, to allow a better appreciation of mitochondrial structure. (A and A′) Control; (B and B′) 1 mM Ca2+/KRB + Cer; (C and C′) 40 µM Ca2+/KRB + cer; (D and D′) BAPTA + Cer. (A–D) Time 0; (A′–D′) 1 h after C2 ceramide addition.
Discussion
Understanding the intracellular pathways that commit a cell to (or protect from) apoptosis is currently a topic of major interest in biomedical research. It has long been known that an unchecked increase in [Ca2+]c can trigger apoptosis in various cell types (Li and Yuan, 1999). A protective role against programmed cell death of controlled, small increases in cellular [Ca2+] is also known, particularly for neurons (Ikonomidou et al., 1999).
While these observations point to a direct role of calcium in controlling life and death of cells (for a review see Berridge et al., 2000), in most cases the Ca2+ targets and their mechanism of action have not been identified. Thus, manoeuvres such as cellular Ca2+ overload, ER Ca2+ depletion or heavy cytosolic buffering could affect a number of processes occurring in the cytoplasm (e.g. activation of the protease calpain or the phosphatase calcineurin) (Squier et al., 1999; Wang et al., 1999), or within organelles, e.g. nuclear lamin degradation (Oberhammer et al., 1994; Rao et al., 1996), DNA fragmentation (Pandey et al., 1994; Walker et al., 1994), activation of the transcription factor NFAT (Srivastava et al., 1999), the release of caspase co-factors from the mitochondria (Vander Heiden et al., 1997) and the activation of resident caspases in the ER (Nakagawa et al., 2000). In a previous study, we utilized organelle-targeted aequorin chimeras to demonstrate that Bcl-2 increases the passive Ca2+ leak from the ER, and thus causes a partial emptying of the agonist-sensitive Ca2+ stores (Pinton et al., 2000). As a consequence, Ca2+ release upon cell stimulation is significantly reduced, and thus the [Ca2+] rises occurring in the cytoplasm and in the mitochondria are markedly smaller. Here, we addressed the question of whether these Bcl-2-dependent alterations in cellular Ca2+ handling are causally linked to the anti-apoptotic action of this oncogene.
As apoptotic stimulus we used ceramide, a lipid mediator generated from the hydrolysis of sphingomyelin (Mathias et al., 1998; Hannun and Luberto, 2000), since its apoptotic action is inhibited by Bcl-2 (Rippo et al., 2000). Its mechanism of action is not fully understood, although it is clear that it is not active as such but needs further conversion to the GD3 ganglioside. The whole rationale of the approach is the following: if the reduction in [Ca2+]er caused by Bcl-2 overexpression is part of the anti-apoptotic program set in action by this oncogene, the prediction is that mimicking this effect by completely different approaches should also mimic its protection against ceramide-induced death. A corollary of the hypothesis is that overloading Ca2+ in the ER should exacerbate the ceramide effect. For mimicking the effect of Bcl-2 on Ca2+ homeostasis we followed three conceptually different approaches, i.e. (i) the global reduction in calcium signaling induced by the exposure to sub-physiological extracellular [Ca2+]; (ii) the selective inhibition of the SERCA; and (iii) the recombinant modification of the molecular repertoire to activate Ca2+ extrusion. Independently of the approach, the experimental conditions, which reduced [Ca2+]er through totally independent means, protected cells from the effect of ceramide. This effect appears highly controlled and specific for this Bcl-2-sensitive apoptotic pathway. On the former aspect, it should be noted that massive [Ca2+]er depletions enhance, rather than reduce, the efficacy of ceramide (see Figure 2). On the latter aspect, other stimuli appear either insensitive (e.g. CD95/Fas/APO-1 stimulation; data not shown) or enhanced by large [Ca2+]er depletions (see, for example, the spontaneous apoptosis of primary hepatocytes; Chami et al., 2001).
But where do the ‘sensible’ Ca2+ targets reside? The simplest answer to this question would be the ER lumen, a location where potential targets have recently been described. Indeed, among the executors of apoptotic cell death, caspase-12 has recently been shown to be located in the ER and to be activated by agents that disrupt ER Ca2+ homeostasis, such as the Ca2+ ionophore A23187 or the SERCA inhibitor thapsigargin. Furthermore, loss of caspase-12 was shown to confer selective protection against apoptosis induced by the amyloid-β protein, a factor supposedly targeted to the ER (Nakagawa et al., 2000). However, caspase-12 is activated by a reduction in [Ca2+]er, while here we show not only that reduction in this parameter protects from apoptosis, but also that SERCA overexpression potentiates the apoptotic effect of ceramide. Furthermore, overexpression of calreticulin results in a major increase in total ER Ca2+ content and in an increase of the Ca2+ buffering power of the ER lumen (Bastianutto et al., 1995; Mery et al., 1996). The net effect of calreticulin overexpression is a reduction of the changes in ER free [Ca2+], and an increase in the amount of total releasable Ca2+, while the [Ca2+]er either remains constant or is slightly reduced (John et al., 1998). If the target were caspase-12, activated by Ca2+ depletion, calreticulin overexpression would be expected to increase survival of ceramide-treated cells. Contrary to this anticipation, calreticulin transfectants showed upon ceramide treatment drastically reduced survival. These last data confirm and extend a previous report by Michalak and coworkers, who showed that overexpression of calreticulin enhances the sensitivity to apoptosis, whereas cell lines derived from calreticulin knock-outs are more resistant (Nakamura et al., 2000). The existence of as yet unknown, ER-located anti-apoptotic agents, activated by [Ca2+]er reduction, can not at the moment be excluded, but no evidence for their existence has yet been provided.
Overall, the simplest interpretation of the data is that the relevant parameter is not the ER free [Ca2+], but rather the total ER Ca2+ content, and thus that the ‘sensible’ Ca2+ target may reside outside the ER, i.e. in the cytosol or in another subcellular compartment, i.e. the mitochondria. It is now clear that release into the cytoplasm of pro-apoptotic factors (e.g. cytochrome c, AIF and caspase-2 and -9) located in the intermembrane space of the mitochondria or in their matrix, represents a key step in the progression of apoptosis (Li et al., 1997; Yang et al., 1997; Susin et al., 1999a,b). The still debated mechanism for this release may involve the PTP, a high conductance channel of unknown molecular identity that can be responsible for mitochondrial swelling and consequent rupture of the outer membrane. In this process, Ca2+ is believed to play a key role, given that mitochondrial Ca2+ overload is a potent stimulus for PTP opening (Bernardi et al., 1998) and that mitochondria are strategically located for promptly responding to ER Ca2+ release (Rizzuto et al., 1998b; Csordas et al., 1999). In an elegant series of experiments, Hajnoczky and coworkers demonstrated that the physiological mitochondrial uptake of Ca2+ caused by IP3-producing agonists is turned into an apoptotic signal in the presence of ceramide, possibly via opening of the PTP (Szalai et al., 1999).
All the experiments described above appear thus consistent with a protective role of controlled ER Ca2+ depletion against ceramide-induced apoptosis. An apparent logical contradiction, however, arises from the demonstration that ceramide itself causes a drastic loss of Ca2+ from the ER. We believe that the contradiction is only apparent. In fact all the conditions causing [Ca2+]er depletion act before the apoptotic stimulus is added, i.e. their net effect is to reduce the amount of Ca2+ released by ceramide. The release of Ca2+ induced by ceramide, on the contrary, occurs simultaneously with the initiation of the apoptotic stimulus. A ‘two hits’' model can thus be proposed, similar to that proposed by Hajnoczky and coworkers (Szalai et al., 1999). Ceramide, or better its metabolites, can directly or indirectly damage the mitochondria, but this effect is marginal or totally ineffective if the mitochondria are not contemporaneously exposed to an elevated [Ca2+]. Neither stimulus can affect the mitochondria if applied alone. In other words, as far as ceramide-induced cell death is concerned, mitochondria appear to act as ‘coincidence detectors’, where only the contemporaneous application of both signals can be transduced into an effective triggering signal of apoptosis. Consistent with this interpretation, gross alterations of the mitochondrial structure are observed early in the process of ceramide-induced cell death and are prevented by all the experimental conditions that reduce [Ca2+]er. Similarly, buffering of [Ca2+]c with BAPTA reduces not only the cytoplasmic [Ca2+] increases but also the alterations in mitochondrial morphology and Ca2+ accumulation. We sought direct evidence of the involvement of PTP in this process, but in our experimental conditions cyclosporin A, a known inhibitor of PTP opening, was in itself quite toxic (possibly due to other intracellular effects, such as the inhibition of calcineurin) and it was difficult to evaluate a protective effect on ceramide-induced cell death.
Overall, our results strongly suggest that the ER Ca2+ depletion caused by Bcl-2 overexpression is an integral part of the anti-apoptotic program set in action by this oncoprotein. In fact, mimicking its effect on Ca2+ handling results in reduced efficacy of a classical apoptotic stimulus, ceramide. We certainly can not exclude the existence of other Bcl-2-activated anti-apoptotic pathways, but it is worth mentioning that a mutant of this oncoprotein that does not bind to the ER membrane (and thus presumably does not reduce [Ca2+]er) has a much reduced anti-apoptotic efficacy. Summarizing this work and previously reported data in a comprehensive model, a ‘two hit’ hypothesis can be proposed. On the one hand, ceramide (or a metabolite) drives contemporaneously Ca2+ release from the ER and accumulation in the cytosol and mitochondrial matrix. On the other, ceramide metabolites (i.e. the GD3 ganglioside) interact directly with and perturb the mitochondria, thus modifying their response to an otherwise physiological event, such as Ca2+ uptake. In this context, the efficacy of the Bcl-2-dependent alteration of Ca2+ homeostasis is not surprising. Indeed, not only does it reduce the IP3-dependent release of Ca2+ from the ER, but also, via a long-term adaptive phenomenon, it inhibits capacitative Ca2+ entry, which is largely responsible for the prolonged elevations of [Ca2+]c. While much still needs to be learnt about the molecular mechanisms involved, we believe that the clarification of the signaling pathway utilized by a key endogenous regulator, such as Bcl-2, may provide new insight and potential pharmacological approaches to modulate this major pathophysiological event.
Materials and methods
Reagents and solutions
Ionomycin, histamine, digitonin, tBuBHQ and N-acetyl-d-sphingosine (C2 ceramide) were purchased from Sigma (Sigma-Aldrich, Milan, Italy), and coelenterazine from Molecular Probes (The Netherlands). KRB contained: 125 mM NaCl, 5 mM KCl, 1 mM MgSO4, 1 mM Na2HPO4, 5.5 mM glucose, 20 mM NaHCO3, 2 mM l-glutamine and 20 mM HEPES pH 7.4, and was supplemented with CaCl2 as indicated in the text.
Cell culture and transfection
HeLa cells were grown in DMEM supplemented with 10% FCS, in 75 cm2 Falcon flasks (Becton-Dickinson, NJ). For the aequorin measurements, cells were seeded onto 13 mm coverslips (BDH, Milan, Italy) and transfected with 4 µg of mtAEQ, cytAEQ or erAEQmut using the Ca2+-phosphate technique; experiments were performed 36 h after transfection, as previously described (Rizzuto et al., 1995a). For microscopic analysis of GFP-expressing cells, HeLa cells were seeded onto 24 mm coverslips and transfected with 8 µg of DNA using the Ca2+-phosphate technique [4 µg of mtGFP + 4 µg of pcDNAI (Figure 2B); 4 µg of mtGFP + 4 µg of Bcl-2 (Figure 2A); 4 µg of mtGFP + 4 µg of PMCA (Figure 4A); 4 µg of mtGFP + 4 µg of SERCA (Figure 4B); 4 µg of mtGFP + 4 µg of calreticulin (Figure 5)].
Measurement of caspase activity
Caspase-3-like activity was evaluated by using the EnzChek Caspase-3 Assay Kit #2 (Molecular Probes, The Netherlands). Enzymatic activity was determined spectrofluorimetrically (LS 50 B Perkin Elmer spectrometer, Perkin Elmer Italia, Italy) by measuring the kinetics of fluorescence increase at excitation/emission wavelengths of 496/520 nm.
Staining of nuclei with propidium iodide
HeLa cells were incubated for 15 min with 1 µM propidium iodide in the presence of 100 µM digitonin and then examined by using an inverted Nikon Eclipse TE300 microscope (Nikon, Tokyo, Japan) equipped with epifluorescence and a piezoelectric motorization of the objective (Physik Instrumente, GmbH & Co., Germany) (see Microscopic analyses for details).
Aequorin measurements
For mtAEQ and cytAEQ, 36 h after transfection the coverslips with the cells were incubated with 5 µM coelenterazine for 1–2 h in DMEM supplemented with 1% FCS, and then transferred to the perfusion chamber. For reconstituting with high efficiency the aequorin chimera targeted to the ER (erAEQ) the luminal [Ca2+] of this compartment must first be reduced. This was obtained by incubating the cells for 1 h at 4°C in KRB supplemented with 5 µM coelenterazine, the Ca2+ ionophore ionomycin and 600 µM EGTA. After this incubation, cells were extensively washed with KRB supplemented with 2% bovine serum albumin (BSA) (Pinton et al., 1998) before the luminescence measurement.
All aequorin measurements were carried out in KRB, supplemented with either 1 mM Ca2+ or the indicated [Ca2+]. Agonists and other drugs were added to the same medium, as specified in the figure legends. The experiments were terminated by lysing the cells with 100 µM digitonin in a hypotonic Ca2+-rich solution (10 mM CaCl2 in H2O), thus discharging the remaining aequorin pool. The light signal was collected and calibrated into [Ca2+] values, as previously described (Brini et al., 1995; Rizzuto et al., 1995a). In brief, a 13 mm-round coverslip with the transfected cells was placed in a perfused, thermostatted chamber located in close proximity to a low-noise photomultiplier, with built-in amplifier-discriminator. The output of the discriminator was captured by a Thorn-EMI photon counting board and stored in an IBM-compatible computer for further analyses. The aequorin luminescence data were calibrated off-line into [Ca2+] values, using a computer algorithm based on the Ca2+ response curve of wild-type and mutant aequorins, as previously described (Brini et al., 1995; Barrero et al., 1997).
Microscopic analyses
The 24 mm-round coverslips with the cells were placed in a thermostatted Leyden chamber, (model TC-202A; Medical Systems Corp., NY) on the stage of an inverted Nikon Eclipse TE300 microscope (Nikon) equipped with epifluorescence and a piezoelectric motorization of the objective (Physik Instrumente). The light field or fluorescence images were captured by a back-illuminated CCD camera (Pricenton Instruments, AZ) using the Metamorph software (Universal Imaging Corporation, PA). In the computationally deblurred images (Figure 8) a stack of images through the z plane was acquired (200 ms/image; 20 plans 0.5 µm apart) and processed using the EPR software developed by the Biomedical Imaging group of the University of Massachusetts Medical School (Worcester, MA).
Fura-2/AM measurements
Changes in [Ca2+]c were measured with the fluorescent indicator Fura-2/AM (Molecular Probes) using an LS50 Perkin Elmer fluorometer (Perkin Elmer Ltd, Beaconsfield, UK), as previously described (Falzoni et al., 1995). For Fura-2/AM loading, cells (1 × 107/ml) were resuspended in 1 mM Ca2+/KRB, in the presence of 4 µM Fura-2/AM and 250 µM sulfinpyrazone (Sigma). Incubation was performed at 37°C for 15 min. Cells were then washed in the same solution and [Ca2+]c changes were determined in a thermostatted, magnetically stirred cuvette, with the 340/380 excitation ratio at an emission wavelength of 505 nm.
Acknowledgments
Acknowledgements
We thank Dr Marisa Brini for the SERCA2b and PMCA expression plasmids and Drs Condo, Fogarty, Rippo and Testi for helpful discussion and experimental advice. This work was supported by grants from the Italian Association for Cancer Research (AIRC), the Armenise Harvard Foundation, the National Research Council of Italy (target project on Biotechnology), the Ministry of Scientific Research (MURST), the Human Frontier Science Program (HFSP), the Italian Space Agency (ASI) and Telethon of Italy (grants #1285, 1226, 961).
References
- Barrero M.J., Montero,M. and Alvarez,J. (1997) Dynamics of [Ca2+] in the endoplasmic reticulum and cytoplasm of intact HeLa cells. A comparative study. J. Biol. Chem., 272, 27694–27699. [DOI] [PubMed] [Google Scholar]
- Bastianutto C., Clementi,E., Codazzi,F., Podini,P., De Giorgi,F., Rizzuto,R., Meldolesi,J. and Pozzan,T. (1995) Overexpression of calreticulin increases the Ca2+ capacity of rapidly exchanging Ca2+ stores and reveals aspects of their lumenal microenvironment and function. J. Cell Biol., 130, 847–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardi P. et al. (1998) The mitochondrial permeability transition. Biofactors, 8, 273–281. [DOI] [PubMed] [Google Scholar]
- Berridge M.J., Lipp,P. and Bootman,M.D. (2000) The versatility and universality of calcium signalling. Nature, 1, 11–21. [DOI] [PubMed] [Google Scholar]
- Boise L.H., Gottschalk,A.R., Quintans,J. and Thompson,C.B. (1995) Bcl-2 and Bcl-2-related proteins in apoptosis regulation. Curr. Top. Microbiol. Immunol., 200, 107–121. [DOI] [PubMed] [Google Scholar]
- Brini M., Marsault,R., Bastianutto,C., Alvarez,J., Pozzan,T. and Rizzuto,R. (1995) Transfected aequorin in the measurement of cytosolic Ca2+ concentration ([Ca2+]c). A critical evaluation. J. Biol. Chem., 270, 9896–9903. [DOI] [PubMed] [Google Scholar]
- Brini M., Bano,D., Manni,S., Rizzuto,R. and Carafoli,E. (2000) Effects of PMCA and SERCA pump overexpression on the kinetics of cell Ca2+ signalling. EMBO J., 19, 4926–4935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chami M. et al. (2001) SERCA1 splice variants encoding C-terminally truncated proteins reduce ER calcium content and induce apoptosis. J. Cell Biol., in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao D.T. and Korsmeyer,S.J. (1998) BCL-2 family: regulators of cell death. Annu. Rev. Immunol., 16, 395–419. [DOI] [PubMed] [Google Scholar]
- Crompton M., Virji,S., Doyle,V., Johnson,N. and Ward,J.M. (1999) The mitochondrial permeability transition pore. Biochem. Soc. Symp., 66, 167–179. [DOI] [PubMed] [Google Scholar]
- Csordas G., Thomas,A.P. and Hajnoczky,G. (1999) Quasi-synaptic calcium signal transmission between endoplasmic reticulum and mitochondria. EMBO J., 18, 96–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchen M.R. (1999) Contributions of mitochondria to animal physiology: from homeostatic sensor to calcium signalling and cell death. J. Physiol., 16, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis R.E., Yuan,J.Y. and Horvitz,H.R. (1991) Mechanisms and functions of cell death. Annu. Rev. Cell Biol., 7, 663–698. [DOI] [PubMed] [Google Scholar]
- Falzoni S., Munerati,M., Ferrari,D., Spisani,S., Moretti,S. and Di Virgilio,F. (1995) The purinergic P2Z receptor of human macrophage cells: characterization and possible physiological role. J. Clin. Invest., 95, 1207–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fasolato C., Pizzo,P. and Pozzan,T. (1998) Delayed activation of the store-operated calcium current induced by calreticulin overexpression in RBL-1 cells. Mol. Biol. Cell, 9, 1513–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foyouzi-Youssefi R., Arnaudeau,S., Borner,C., Kelley,W.L., Tschopp,J., Lew,D.P., Demaurex,N. and Krause,K.H. (2000) Bcl-2 decreases the free Ca2+ concentration within the endoplasmic reticulum. Proc. Natl Acad. Sci. USA, 97, 5723–5728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grynkiewicz G., Poenie,M. and Tsien,R.Y. (1985) A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem., 260, 3440–3450. [PubMed] [Google Scholar]
- Hannun Y.A. and Luberto,C. (2000) Ceramide in the eukaryotic stress response. Trends Cell Biol., 10, 73–80. [DOI] [PubMed] [Google Scholar]
- He H., Lam,M., McCormick,T.S. and Distelhorst,C.W. (1997) Mainten ance of calcium homeostasis in the endoplasmic reticulum by Bcl-2. J. Cell Biol., 138, 1219–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hengartner M.O. and Horvitz,H.R. (1994) Programmed cell death in Caenorhabditis elegans. Curr. Opin. Genet. Dev., 4, 581–586. [DOI] [PubMed] [Google Scholar]
- Hetts S.W. (1998) To die or not to die: an overview of apoptosis and its role in disease. J. Am. Med. Assoc., 279, 300–307. [DOI] [PubMed] [Google Scholar]
- Ikonomidou C. et al. (1999) Blockade of NMDA receptors and apoptotic neurodegeneration in the developing brain. Science, 283, 70–74. [DOI] [PubMed] [Google Scholar]
- Jacotot E., Costantini,P., Laboureau,E., Zamzami,N., Susin,S.A. and Kroemer,G. (1999) Mitochondrial membrane permeabilization during the apoptotic process. Ann. NY Acad. Sci., 887, 18–30. [DOI] [PubMed] [Google Scholar]
- John L.M., Lechleiter,J.D. and Camacho,P. (1998) Differential modulation of SERCA2 isoforms by calreticulin. J. Cell Biol., 142, 963–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kass G.E., Duddy,S.K., Moore,G.A. and Orrenius,S. (1989) 2,5-Di-(tert-butyl)-1,4-benzohydroquinone rapidly elevates cytosolic Ca2+ concentration by mobilizing the inositol 1,4,5-trisphosphate-sensitive Ca2+ pool. J. Biol. Chem., 264, 15192–15198. [PubMed] [Google Scholar]
- Kluck R.M. et al. (1999) The pro-apoptotic proteins, Bid and Bax, cause a limited permeabilization of the mitochondrial outer membrane that is enhanced by cytosol. J. Cell Biol., 147, 809–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause K.H. and Michalak,M. (1997) Calreticulin. Cell, 88, 439–443. [DOI] [PubMed] [Google Scholar]
- Kuo T.H., Kim,H.R., Zhu,L., Yu,Y., Lin,H.M. and Tsang,W. (1998) Modulation of endoplasmic reticulum calcium pump by Bcl-2. Oncogene, 17, 1903–1910. [DOI] [PubMed] [Google Scholar]
- Lam M., Dubyak,G., Chen,L., Nunez,G., Miesfeld,R.L. and Distelhorst,C.W. (1994) Evidence that BCL-2 represses apoptosis by regulating endoplasmic reticulum-associated Ca2+ fluxes. Proc. Natl Acad. Sci. USA, 91, 6569–6573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. and Yuan,J. (1999) Deciphering the pathways of life and death. Curr. Opin. Cell Biol., 11, 261–266. [DOI] [PubMed] [Google Scholar]
- Li P., Nijhawan,D., Budihardjo,I., Srinivasula,S.M., Ahmad,M., Alnemri,E.S. and Wang,X. (1997) Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell, 91, 479–489. [DOI] [PubMed] [Google Scholar]
- Lithgow T., van Driel,R., Bertram,J.F. and Strasser,A. (1994) The protein product of the oncogene bcl-2 is a component of the nuclear envelope, the endoplasmic reticulum and the outer mitochondrial membrane. Cell Growth Differ., 5, 411–417. [PubMed] [Google Scholar]
- Mathias S., Pena,L.A. and Kolesnick,R.N. (1998) Signal transduction of stress via ceramide. Biochem. J., 335, 465–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson M.P. (2000) Apoptotic and anti-apoptotic synaptic signaling mechanisms. Brain Pathol., 10, 300–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mery L., Mesaeli,N., Michalak,M., Opas,M., Lew,D.P. and Krause,K.H. (1996) Overexpression of calreticulin increases intracellular Ca2+ storage and decreases store-operated Ca2+ influx. J. Biol. Chem., 271, 9332–9339. [DOI] [PubMed] [Google Scholar]
- Minn A.J., Velez,P., Schendel,S.L., Liang,H., Muchmore,S.W., Fesik,S.W., Fill,M. and Thompson,C.B. (1997) Bcl-xL forms an ion channel in synthetic lipid membranes. Nature, 385, 353–357. [DOI] [PubMed] [Google Scholar]
- Nagata S. (1997) Apoptosis by death factor. Cell, 88, 355–365. [DOI] [PubMed] [Google Scholar]
- Nakagawa T., Zhu,H., Morishima,N., Li,E., Xu,J., Yankner,B.A. and Yuan,J. (2000) Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-β. Nature, 403, 98–103. [DOI] [PubMed] [Google Scholar]
- Nakamura K. et al. (2000) Changes in endoplasmic reticulum luminal environment affect cell sensitivity to apoptosis. J. Cell Biol., 150, 7317–7340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson D.W. (1999) Caspase structure, proteolytic substrates and function during apoptotic cell death. Cell Death Differ., 6, 1028–1042. [DOI] [PubMed] [Google Scholar]
- Nicotera P., Leist,M. and Manzo,L. (1999) Neuronal cell death: a demise with different shapes. Trends Pharmacol. Sci., 20, 46–51. [DOI] [PubMed] [Google Scholar]
- Oberhammer F.A., Hochegger,K., Froschl,G., Tiefenbacher,R. and Pavelka,M. (1994) Chromatin condensation during apoptosis is accompanied by degradation of lamin A+B, without enhanced activation of cdc2 kinase. J. Cell Biol., 126, 827–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey S., Walker,P.R. and Sikorska,M. (1994) Separate pools of endonuclease activity are responsible for internucleosomal and high molecular mass DNA fragmentation during apoptosis. Biochem. Cell Biol., 72, 625–629. [DOI] [PubMed] [Google Scholar]
- Park M.K., Petersen,O.H. and Tepikin,A.V. (2000) The endoplasmic reticulum as one continuous Ca2+ pool: visualization of rapid Ca2+ movements and equilibration. EMBO J., 19, 5729–5739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry D.K. (1999) Ceramide and apoptosis. Biochem. Soc. Trans., 27, 399–404. [DOI] [PubMed] [Google Scholar]
- Petit P.X., Goubern,M., Diolez,P., Susin,S.A., Zamzami,N. and Kroemer,G. (1998) Disruption of the outer mitochondrial membrane as a result of large amplitude swelling: the impact of irreversible permeability transition. FEBS Lett., 426, 111–116. [DOI] [PubMed] [Google Scholar]
- Pinton P., Pozzan,T. and Rizzuto,R. (1998) The Golgi apparatus is an inositol 1,4,5-trisphosphate-sensitive Ca2+ store, with functional properties distinct from those of the endoplasmic reticulum. EMBO J., 17, 5298–5308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinton P., Ferrari,D., Magalhaes,P., Schulze-Osthoff,K., Di Virgilio,F., Pozzan,T. and Rizzuto,R. (2000) Reduced loading of intracellular Ca2+ stores and downregulation of capacitative Ca2+ influx in Bcl-2-overexpressing cells. J. Cell Biol., 148, 857–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao L., Perez,D. and White,E. (1996) Lamin proteolysis facilitates nuclear events during apoptosis. J. Cell Biol., 135, 1441–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rippo M.R. et al. (2000) GD3 ganglioside directly targets mitochondria in a bcl-2-controlled fashion. FASEB J., 14, 2047–2054. [DOI] [PubMed] [Google Scholar]
- Rizzuto R., Simpson,A.W., Brini,M. and Pozzan,T. (1992) Rapid changes of mitochondrial Ca2+ revealed by specifically targeted recombinant aequorin. Nature, 358, 325–327. [DOI] [PubMed] [Google Scholar]
- Rizzuto R., Brini,M., Bastianutto,C., Marsault,R. and Pozzan,T. (1995a) Photoprotein mediated measurement of [Ca2+] in mitochondria of living cells. Methods Enzymol., 260, 417–428. [DOI] [PubMed] [Google Scholar]
- Rizzuto R., Brini,M., Pizzo,P., Murgia,M. and Pozzan,T. (1995b) Chimeric green fluorescent protein as a tool for visualizing subcellular organelles in living cells. Curr. Biol., 5, 635–642. [DOI] [PubMed] [Google Scholar]
- Rizzuto R., Carrington,W. and Tuft,R.A. (1998a) Digital imaging microscopy of living cells. Trends Cell Biol., 8, 288–292. [DOI] [PubMed] [Google Scholar]
- Rizzuto R., Pinton,P., Carrington,W., Fay,F.S., Fogarty,K.E., Lifshitz,L.M., Tuft,R.A. and Pozzan,T. (1998b) Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science, 280, 1763–1766. [DOI] [PubMed] [Google Scholar]
- Schendel S.L., Xie,Z., Montal,M.O., Matsuyama,S., Montal,M. and Reed,J.C. (1997) Channel formation by antiapoptotic protein Bcl-2. Proc. Natl Acad. Sci. USA, 94, 5113–5118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Squier M.K., Sehnert,A.J., Sellins,K.S., Malkinson,A.M., Takano,E. and Cohen,J.J. (1999) Calpain and calpastatin regulate neutrophil apoptosis. J. Cell Physiol., 178, 311–319. [DOI] [PubMed] [Google Scholar]
- Srivastava R.K., Sasaki,C.Y., Hardwick,J.M. and Longo,D.L. (1999) Bcl-2-mediated drug resistance: inhibition of apoptosis by blocking nuclear factor of activated T lymphocytes (NFAT)-induced Fas ligand transcription. J. Exp. Med., 190, 253–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susin S.A. et al. (1999a) Molecular characterization of mitochondrial apoptosis-inducing factor. Nature, 397, 441–446. [DOI] [PubMed] [Google Scholar]
- Susin S.A., Lorenzo,H.K., Zamzami,N., Marzo,I., Brenner,C., Larochette,N., Prevost,M.C., Alzari,P.M. and Kroemer,G. (1999b) Mitochondrial release of caspase-2 and -9 during the apoptotic process. J. Exp. Med., 189, 381–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szalai G., Krishnamurthy,R. and Hajnoczky,G. (1999) Apoptosis driven by IP3-linked mitochondrial calcium signals. EMBO J., 18, 6349–6361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden M.G., Chandel,N.S., Williamson,E.K., Schumacker,P.T. and Thompson,C.B. (1997) Bcl-xL regulates the membrane potential and volume homeostasis of mitochondria. Cell, 91, 627–637. [DOI] [PubMed] [Google Scholar]
- Walker P.R., Weaver,V.M., Lach,B., LeBlanc,J. and Sikorska,M. (1994) Endonuclease activities associated with high molecular weight and internucleosomal DNA fragmentation in apoptosis. Exp. Cell Res., 213, 100–106. [DOI] [PubMed] [Google Scholar]
- Wang H.G. et al. (1999) Ca2+-induced apoptosis through calcineurin dephosphorylation of BAD. Science, 284, 339–343. [DOI] [PubMed] [Google Scholar]
- Xu W., Longo,F.J., Wintermantel,M.R., Jiang,X., Clark,R.A. and DeLisle,S. (2000) Calreticulin modulates capacitative Ca2+ influx by controlling the extent of inositol 1,4,5-trisphosphate-induced Ca2+ store depletion. J. Biol. Chem., 275, 36676–36682. [DOI] [PubMed] [Google Scholar]
- Yang J., Liu,X., Bhalla,K., Kim,C.N., Ibrado,A.M., Cai,J., Peng,T.I., Jones,D.P. and Wang,X. (1997) Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science, 275, 1129–1132. [DOI] [PubMed] [Google Scholar]
- Zhang J., Alter,N., Reed,J.C., Borner,C., Obeid,L.M. and Hannun,Y.A. (1996) Bcl-2 interrupts the ceramide-mediated pathway of cell death. Proc. Natl Acad. Sci. USA, 93, 5325–5328. [DOI] [PMC free article] [PubMed] [Google Scholar]