

Abstract
Eukaryotic initiation factor (eIF) 2B is a heteromeric guanine nucleotide exchange factor that plays an important role in regulating mRNA translation. Here we identify multiple phosphorylation sites in the largest, catalytic, subunit (ε) of mammalian eIF2B. These sites are phosphorylated by four different protein kinases. Two conserved sites (Ser712/713) are phosphorylated by casein kinase 2. They lie at the extreme C-terminus and are required for the interaction of eIF2Bε with its substrate, eIF2, in vivo and for eIF2B activity in vitro. Glycogen synthase kinase 3 (GSK3) is responsible for phosphorylating Ser535. This regulatory phosphorylation event requires both the fourth site (Ser539) and a distal region, which acts to recruit GSK3 to eIF2Bε in vivo. The fifth site, which lies outside the catalytic domain of eIF2Bε, can be phosphorylated by casein kinase 1. All five sites are phosphorylated in the eIF2B complex in vivo.
Keywords: casein kinase/GEF/GSK3/initiation factor/mRNA translation
Introduction
Eukaryotic translation initiation factor eIF2B is composed of five subunits termed α–ε in order of increasing size (Pain, 1996; Hinnebusch, 2000). It is a guanine nucleotide exchange factor and mediates the release of GDP from eIF2 to regenerate active [eIF2⋅GTP] complexes during translation initiation. [eIF2⋅GTP] binds the initiator methionyl (Met)-tRNAi and recruits it to the 40S subunit. The resulting complex, together with other initiation factors, binds to the mRNA, leading to scanning and location of the start codon (probably through its interaction with the anticodon on Met-tRNAi). Late in the initiation process, the GTP bound to eIF2 is hydrolysed to GDP and Pi, yielding the inactive [eIF2⋅GDP] complex, which becomes the substrate for eIF2B (Pain, 1996; Hinnebusch, 2000). The rate at which GDP is released from eIF2 is very slow and eIF2B is required to accelerate the regeneration of active eIF2⋅GTP. eIF2Bε can catalyse guanine nucleotide exchange in the absence of the other subunits showing that it contains the catalytic domain (Fabian et al., 1997; Pavitt et al., 1997; Gomez and Pavitt, 2000; Williams et al., 2001). The roles of the other four subunits remain less well defined. In yeast, eIF2Bγ forms a subcomplex with the ε-subunit, which enhances the activity of the latter (Pavitt et al., 1998). The α-, β- and δ-subunits form a separate regulatory subcomplex (Pavitt et al., 1998) which probably contacts eIF2α. Indeed, eIF2Bα appears to be essential to sensitize the eIF2B complex to inhibition by eIF2(αP) (reviewed in Hinnebusch, 2000).
Formation of eIF2⋅GTP⋅Met-tRNAi complexes appears to be a prerequisite for all translation initiation events, and eIF2B plays a key role in regulating translation initiation under a variety of conditions (Hinnebusch, 2000). Upon amino acid deprivation or viral infection, and under stress conditions, eIF2B activity is decreased (Clemens, 1996; Hinnebusch, 2000), whereas stimulation of cells or tissues by insulin, growth factors or mitogens leads to activation of eIF2B (Kimball and Jefferson, 1988; Welsh and Proud, 1992; Gilligan et al., 1996; Welsh et al., 1996, 1997a; Kleijn et al., 1998).
The activity of eIF2B therefore plays an important role in regulating overall translation initiation: it can be modulated in several ways. Phosphorylation of the α-subunit of eIF2, catalysed by any of several eIF2 kinases, leads to inhibition of eIF2B since eIF2(αP) is a potent competitive inhibitor of eIF2B (reviewed in Clemens, 1996; Hinnebusch, 2000). In response to insulin, mitogens and other agents which activate eIF2B, other mechanisms are involved in activating eIF2B. For example, eIF2Bε from mammals or insects is a substrate for glycogen synthase kinase 3 (GSK3), and this inhibits the activity of eIF2B (Welsh et al., 1998; Williams et al., 2001). GSK3 is inactivated in response to insulin and, concomitantly with this, the corresponding site in eIF2Bε undergoes dephosphorylation concomitantly with the activation of eIF2B (Welsh et al., 1998). eIF2Bε is also phosphorylated in vitro by casein kinases 1 and 2 (CK1 and CK2), although reports of the effects of this on eIF2B activity vary (Dholakia and Wahba, 1988; Oldfield and Proud, 1992; Singh et al., 1994, 1996).
An important gap in our understanding of the control of eIF2B has been the identification of the sites at which eIF2B is phosphorylated in vivo. This study addresses this issue: in this report, we identify the sites at which mammalian eIF2Bε is phosphorylated in vivo. We also study their roles in the function of eIF2B. We show that the phosphorylation sites at the extreme C-terminus of eIF2Bε are required for binding to eIF2 and for full activity of eIF2Bε, while phosphorylation of the GSK3 site requires a serine both at the +4 position and at a region distal to these sites, which may recruit GSK3 to eIF2B, similar to the role of axin in the phosphorylation of β-catenin.
Results
Phosphorylation of eIF2B in vivo
To examine the phosphorylation of eIF2B in vivo, human embryonic kidney (HEK) 293 cells were metabolically labelled with [32P]orthophosphate. Cells were lysed and extracts were subjected to immunoprecipitation using an antiserum that recognizes eIF2Bε. Immunoprecipitates were subjected to SDS–PAGE followed by autoradiography. Radiolabel was clearly present in the position of eIF2Bε (Figure 1A). Four fainter radiolabelled bands were also observed. The positions of two of these corresponded to the α- and β-subunits of eIF2 as judged in comparison with the migration of the subunits of human eIF2 and eIF2B, used as markers (Figure 1A). Both these subunits of eIF2 are known to be phosphoproteins (Proud, 1992) and, since eIF2 interacts tightly with eIF2B and can be co-immunoprecipitated with it (see also below, Figure 9A), we conclude that these radiolabelled bands represent eIF2α and β. The third and faintest band co-migrated with the δ-subunit of eIF2B. However, the amount of radiolabel incorporated was always too low to permit further analysis. The fourth band (marked ‘X’) runs in the position of ABC50, a protein that we recently showed to interact with eIF2, and may correspond to this polypeptide (Tyzack et al., 2000).

Fig. 1. Phosphorylation of eIF2Bε in vivo and in vitro. (A) HEK293 cells transfected with empty vector (lane 1) or a vector encoding HA-tagged rat eIF2Bε (lane 2) were metabolically labelled with [32P]orthophosphate and then extracted as described in Materials and methods. Endogenous eIF2Bε (lane 1, 100 µg of lysate protein) or HA-tagged eIF2Bε (lane 2, equivalent to 5 µg of lysate protein) was immunoprecipitated and analysed by SDS–PAGE followed by autoradiography. Labelled arrows on the left show the migration positions of subunits of human eIF2B, apart from ‘X’ which denotes an additional polypeptide co-immunoprecipitated with eIF2B, probably ABC50. Labelled arrows on the right indicate subunits of eIF2. Arrowheads indicate radiolabelled bands. The position of HA-tagged eIF2Bε is also indicated. (B) Immunoblots of HEK293 cells transfected with empty vector (1) or vector for HA-eIF2Bε (2) developed with an antiserum for eIF2Bε. (C and D) Two-dimensional mapping of peptides from HA-eIF2Bε labelled in vivo. HA-tagged rat eIF2Bε was expressed in HEK293 cells that were metabolically labelled with [32P]orthophosphate. eIF2Bε was isolated by immunoprecipitation and then digested with Asp-N (C) or trypsin (D), and the resulting phosphopeptides were analysed by two-dimensional mapping. The polarity of electrophoresis (+/–) and the direction of chromatography (arrow) are shown. The narrow arrowhead marks the origin and the diamond the position of the dinitrophenyllysine marker. (C) and (D) are annotated indicating the terminology used to refer to the peptides in the text.
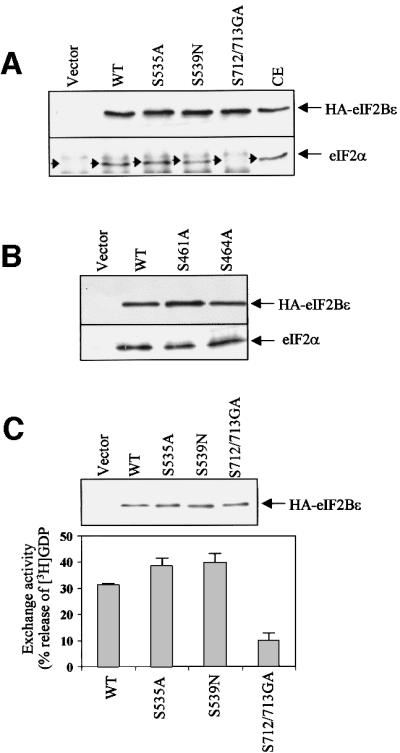
Fig. 9. Effects of point mutations on the interaction of eIF2Bε with eIF2 in vivo and on eIF2Bε activity. (A and B) HEK293 cells were transfected with vectors encoding wild-type eIF2Bε, the indicated point mutants, or vector alone, as a negative control. eIF2Bε was then immunoprecipitated from cell lysates using anti-HA antibodies and analysed by SDS–PAGE followed by immunoblotting with antibodies to either eIF2α or HA (as a loading control). (C) eIF2Bε was immunoprecipitated from transfected HEK293 cells using anti-HA antibodies and then used in eIF2B activity assays, as described in Materials and methods. Immunoprecipitations were carried out in triplicate, two for the exchange assay and one for SDS–PAGE and immunoblotting using anti-HA antibodies to check for equal levels of eIF2Bε (upper panel). The results show the average of two independent experiments ± SD and are expressed as the percentage of [3H]GDP released from complexes with eIF2, corrected for the low activity seen with samples from cells transfected with the empty vector (lower panel). Similar data were obtained in four sets of experiments.
None of the in vivo sites of phosphorylation in eIF2Bε previously has been identified directly. However, eIF2Bε has been shown to be phosphorylated by three different protein kinases in vitro: CK1, CK2 and GSK3 (Dholakia and Wahba, 1988; Oldfield and Proud, 1992; Singh et al., 1994, 1996; Welsh et al., 1998). Thus, the sites in eIF2Bε phosphorylated in vivo were likely to include sites for these three enzymes.
We were unable to achieve sufficient incorporation of radiolabel into endogenous eIF2Bε to allow us to identify the phosphorylation sites. To obtain higher amounts of labelled material, we transfected HEK293 cells with a vector encoding eIF2Bε with a haemagglutinin (HA) tag. Because no full-length cDNA was available for human eIF2Bε, we used the rat cDNA. About 40 h after transfection, cells were extracted, and the overexpressed eIF2Bε was immunoprecipitated and analysed by SDS–PAGE/western blotting. Rat eIF2Bε was expressed efficiently and, indeed, at much higher levels than the endogenous eIF2Bε (Figure 1B). Thus most of this overexpressed protein presumably is present as free eIF2Bε rather than incorporated into eIF2B complexes. Digestion with Asp-N or trypsin was performed and two-dimensional peptide maps were prepared. Several peptides were observed in each case (Figure 1C and D), as well as some material that remained at the origin. This material comprises undigested proteins plus peptides too large to migrate on two-dimensional maps.
Phosphorylation of eIF2Bε in vitro
In order to identify the sites phosphorylated by the known eIF2Bε kinases, rat eIF2Bε was expressed in Escherichia coli as a GST fusion protein and used as a substrate for CK1, CK2 and GSK3. CK2 readily phosphorylated GST–eIF2Bε (Figure 2A). In contrast, CK1 phosphorylated GST–eIF2Bε only weakly (Figure 2A). When phosphorylation of bacterially expressed eIF2Bε by GSK3 was compared with that of His6-eIF2Bε made in insect cells, the latter was found to be a much better substrate (Figure 2B). The reasons for this difference become clear later in this study and are addressed below. Thus, in vitro, GST–eIF2Bε can be phosphorylated efficiently by GSK3 and CK2, but only rather weakly by CK1.
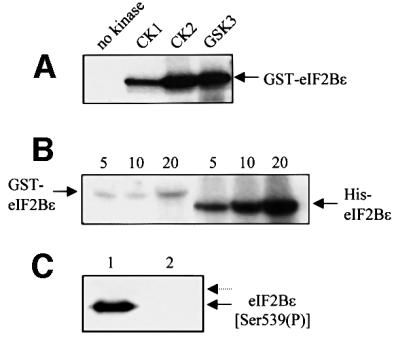
Fig. 2. Phosphorylation of eIF2Bε in vitro. (A) eIF2Bε expressed in E.coli (as a GST fusion protein) was incubated with CK1, CK2 or GSK3 for 20 min in the presence of [γ-32P]ATP, and samples were analysed by SDS–PAGE and autoradiography. (B) eIF2Bε (0.5 µg) expressed in E.coli (as a GST fusion protein) or Sf21 cells (His6 tagged, as indicated) was incubated with GSK3 for the indicated times (min) in the presence of [γ-32P]ATP, and samples were analysed by SDS–PAGE and autoradiography. (C) A 0.5 µg aliquot of eIF2Bε expressed in Sf21 cells (lane 1) or E.coli (as a GST fusion protein;lane 2), as indicated, was analysed by SDS–PAGE and immunoblotting using an antibody which recognizes eIF2Bε when phosphorylated at Ser539 (the arrow shows the position of eIF2Bε). The dotted arrow shows the migration position of GST–eIF2Bε.
Analysis of site(s) phosphorylated by GSK3
In the case of His6-eIF2Bε phosphorylated in vitro by GSK3, Asp-N digestion released two main peptides that eluted close together on HPLC (Figure 3A) and migrated similarly on two-dimensional maps (Figure 3B and C). Mass spectrometry revealed that the peptides in frac tions 65 and 74 corresponded to DSRAGSPQLD and DSRAGSPQL, respectively, each with two attached phosphate groups (masses 1203.52 and 1090.45 Da; theoretical masses 1205.424 and 1090.397 Da). Sequence analysis and detection of released radiolabel indicated that the radiolabelled phosphate was located at the second residue of each of the two peptides, i.e. the equivalent of Ser535 of the rat sequence (Figure 3D and E). This corresponds to the site phosphorylated by GSK3 in vitro (Welsh et al., 1998) in the rabbit eIF2B complex. Judging from the masses, it appears that the second serine in this peptide is already phosphorylated with unlabelled phosphate in the eIF2Bε protein made in insect cells. To study this further, we made use of an antibody that detects eIF2Bε phosphorylated at this position (Ser539). eIF2Bε made in Sf21 cells showed a strong signal with this antibody in an immunoblot (Figure 2C), whereas GST–eIF2Bε from bacteria did not. This suggests that insect cells possess a kinase that phosphorylates this site, whereas E.coli does not. We have recently dentified members of the eukaryotic DYRK family as potential priming kinases as they can phosphorylate Ser539 of eIF2Bε and facilitate subsequent phosphorylation by GSK3 (Woods et al., 2001). Insects possess close relatives of the DYRKs (e.g. minibrain in Drosophila; Tejedor et al., 1995) and it seems likely that its Spodoptera counterpart phosphorylates eIF2Bε expressed in Sf21 cells at the priming site. The fact that only the doubly phosphorylated peptides were observed suggests that, for phosphorylation of Ser535 by GSK3, Ser539 (+4 position) must be phosphorylated. This requirement for a ‘priming’ phosphorylation event is characteristic of the action of GSK3 on several substrates including peptides based on this site in eIF2Bε (Fiol et al., 1990; Welsh et al., 1997b). The role of phosphorylation of Ser539 in priming phosphorylation of Ser535 in vivo is addressed directly in other experiments (see below).

Fig. 3. Analysis of peptides from eIF2Bε phosphorylated by GSK3. eIF2Bε made in insect cells was radiolabelled in vitro using GSK3. (A) This was then subjected to digestion by Asp-N and the resulting peptides were analysed by reverse-phase (C18) HPLC. The dotted line indicates the percentage (v/v) of CH3CN. (B and C) The material in fractions 65 (B) or 74 (C) from the HPLC was analysed by two-dimensional mapping. (D and E) These fractions (as indicated) were subjected to solid-phase sequencing, and release of radioactivity was determined at each cycle. The sequences of the peptides are also displayed (as shown by the release of radioactivity and the masses of the peptides determined by mass spectrometry, see text: Sp denotes phosphoserine). (F and G) Two-dimensional analysis of tryptic peptides from either bacterially expressed rat eIF2Bε phosphorylated in vitro by DYRK2 (F) or rat eIF2Bε expressed in insect cells and phosphorylated by GSK3 (G). Peptides t1 and t3 (see text) are indicated.
The peptides obtained from Asp-N digests of rat eIF2Bε phosphorylated by GSK3 migrate on two-dimensional maps in identical positions to peptides a2 and a3 from recombinant eIF2Bε labelled in vivo (Figure 3B and C; cf. Figure 1C). Analysis of peptide a2 from HA-eIF2Bε labelled in 293 cells (Figure 1C) showed that the associated radioactivity was released at cycles 2 and 6 of the Edman degradation, consistent with it being the doubly phosphorylated peptide containing Ser535 and Ser539 as deduced above (not shown).
Tryptic two-dimensional maps for rat eIF2Bε phosphorylated in vivo showed two main peptides (Figure 1D). To ascertain whether any of these contained Ser535 or Ser539, GST–eIF2Bε was phosphorylated in vitro by DYRK (Figure 3F). Tryptic digestion released a peptide corresponding to t1 (Figure 3F). When rat eIF2Bε expressed in insect cells was phosphorylated by GSK3 in vitro, tryptic digestion gave peptide t3 and a small amount of t1 (due to the presence in the substrate of a priming kinase from the Sf21 cells; Woods et al., 2001; data not shown; Figure 3G). Peptide t3, seen in maps from rat eIF2Bε phosphorylated in 293 cells, thus contains the GSK3 site, Ser535. It is not clear why the yield of t3 in Figure 1C is so low compared with t1.
To confirm the identification of peptides a2 and a3 for eIF2Bε phosphorylated in vivo, and to test the role of Ser539 in priming phosphorylation of Ser535 in vivo, HEK293 cells were transfected with vectors encoding a mutant of eIF2Bε in which Ser535 or Ser539 had been changed to alanine or asparagine, respectively. Asparagine was chosen in preference to alanine at position 539 since, unlike alanine, it preserves the ability to form hydrogen bonds, which might be important. Indeed, there are examples, such as Ser53 in eIF4E, of an amino acid residue which is not a phosphorylation site but where mutation to alanine nevertheless exhibits marked phenotypes (Joshi-Barve et al., 1990; Lazaris-Karatzas et al., 1990). Western blotting revealed that eIF2Bε(S535A) was no longer phosphorylated at that site but was still phosphorylated at S539, while no signal with either antibody was seen for the S539N mutant (Figure 4A). This shows that mutation of Ser539 to a non-phosphorylatable residue does indeed eliminate phosphorylation at Ser535, as predicted for a target of GSK3.
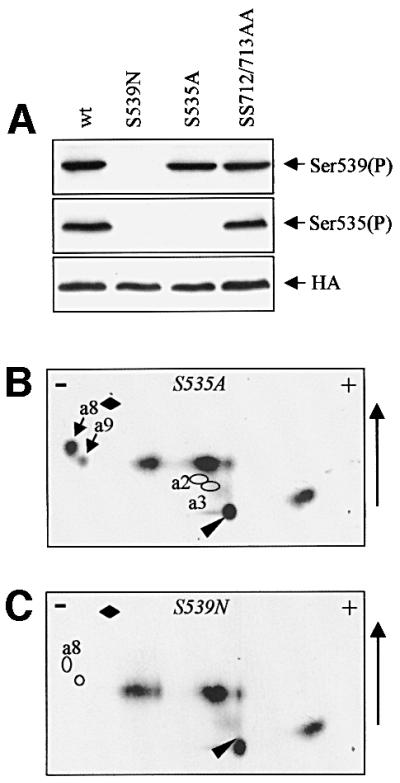
Fig. 4. Phosphorylation of eIF2Bε(S535A) and eIF2Bε(S539N) in vivo. HEK293 cells were transfected with vectors encoding each of these mutant proteins or wild-type (wt) eIF2Bε. (A) Cells were extracted and analysed by SDS–PAGE and immunoblotting using phosphospecific antisera or anti-HA (loading control). (B and C) Transfected cells were metabolically labelled with [32P]orthophosphate and eIF2Bε proteins were isolated by immunoprecipitation/SDS–PAGE. They were then digested with Asp-N and peptides displayed on two-dimensional maps. Labelled arrows in (B) mark the additional peptides termed a8/9 seen in this map, and the labelled rings show the expected positions of the absent peptides a2/3. In (C), the positions of the absent peptides a8/9 are indicated by rings.
To verify this directly, cells were transfected with vectors encoding either HA-eIF2Bε(S535A) or HA-eIF2Bε(S539N), labelled with [32P]orthophosphate, and the eIF2Bε was isolated and digested with Asp-N. Peptide maps revealed that the S535A mutation resulted in loss of peptides a2 and a3, and led to the appearance of ‘new’ peptides a8 and a9 (Figure 4B). For the S539N mutant peptides, a2, a3, a8 and a9 were all absent (Figure 4C). These findings confirm the identification of a2 and a3 as doubly phosphorylated variants of the peptide DSRAGSPQL(D). a8 and a9 presumably represent singly phosphorylated forms of the peptide (with asparagine in place of Ser539). Since two-dimensional maps from wild-type eIF2Bε do not contain peptides close to a8/a9, HA-eIF2Bε appears to be entirely doubly phosphorylated in HEK293 cells.
Analysis of site(s) phosphorylated by CK2
For GST–eIF2Bε labelled in vitro by CK2, Asp-N digestion failed to release a peptide that could be resolved by HPLC analysis (data not shown), so tryptic digestion was employed as an alternative. This gave two major peptides eluting in fractions 43 and 46 on HPLC and migrating close to the origin on two-dimensional maps (Figure 5A and B). Gas-phase sequence analysis revealed that fractions 43 and 46 contained the peptides EAEEE(S)(S)D and EAEEE(S)(S)DDD, respectively, which are variants of the extreme C-terminus of eIF2Bε (i.e. peptide 43 is generated by loss of the final two aspartate residues). Fraction 46 contained sufficient radioactivity for reliable solid-phase Edman degradation to identify the sites of phosphorylation. After one round of gas-phase sequencing to remove the first glutamic acid (which cyclizes and blocks the Edman degradation), solid-phase sequencing revealed that radioactivity was released at positions 6 and 7, and the pattern of release indicated that both serines were phosphorylated (Figure 5C). This accounts for the failure to observe serine at these two sequencing cycles (since phosphoserine undergoes β-elimination) and the sequencing data thus also imply that both peptides are doubly phosphorylated. As shown in Figure 5D, mutants of eIF2Bε lacking the extreme C-terminus were not substrates for CK2 in vitro. Thus, it appeared that CK2 phosphorylates a site(s) in the extreme C-terminus of eIF2B. To study this further, vectors encoding truncation mutants lacking the final 33 (Δ33) or 63 (Δ63) residues of eIF2Bε were transfected into HEK293 cells, which were then radiolabelled. Tryptic peptide maps revealed that for each truncated mutant, peptide t2 was absent, consistent with the presence in the C-terminus of eIF2Bε of significant sites of phosphorylation (data not shown). For final confirmation of these sites, we transfected cells with a vector encoding a mutant of eIF2Bε in which the codons for Ser712 and Ser713 are altered to encode residues that cannot undergo phosphorylation, i.e. glycine and alanine, respectively. After in vivo radiolabelling, the mutant eIF2Bε polypeptide was subjected to tryptic digestion. Two-dimensional mapping revealed that peptide t2 was absent, showing that it does indeed correspond to this C-terminal region of eIF2Bε (Figure 5E). The more acidic tryptic peptide obtained from eIF2Bε phosphorylated by CK2 in vitro corresponds to peptide t2 from tryptic maps of eIF2Bε phosphorylated in vivo (Figure 1D), indicating that in the overexpressed protein in vivo the final two aspartates are retained. All the available sequences of eIF2Bε contain a potential CK2 site(s) in this region (i.e. one or more serines followed by several acidic residues) although the distance from the highly conserved WL motif varies (Figure 5F).

Fig. 5. Phosphorylation of eIF2Bε by CK2 and CK1. (A and B) GST–eIF2Bε made in E.coli was phosphorylated in vitro by CK2 and then subjected to cleavage with trypsin. The resulting peptides were analysed by reverse-phase chromatography (A) or two-dimensional mapping (B) (additional mapping data, not shown, identified the individual peptides with either the first or second peak observed on HPLC, as indicated). (C) The material in fraction 46 was subjected to one cycle of gas-phase Edman degradation followed by 10 cycles of solid-phase sequencing, release of radioactivity being monitored during the latter. (D) Full-length eIF2Bε (FL) and truncation mutants lacking the last 33 (Δ33) or 63 (Δ63) residues were expressed in E.coli and purified. A 0.5 µg aliquot of each was then incubated with [γ-32P]ATP and CK2, as indicated. Samples were then analysed by SDS–PAGE and autoradiography. (E) HEK293 cells were transfected with vectors encoding wild-type eIF2Bε or a double point mutant, S712/713GA, each with HA tags. After metabolic labelling of cells with [32P]orthophosphate, immunoprecipitation, SDS–PAGE and tryptic digestion, peptides were analysed by two-dimensional mapping. Only the region around the origin and including peptide t2 is shown. (F) Alignment of the extreme C-termini of eIF2Bε for a range of species (Dm, Drosophila melanogaster; Rn, Rattus norvegicus; Sc, Saccharomyces cerevisiae; At, Arabidopsis thaliana; Ce, Caenorhabditis elegans) showing the conservation of potential CK2 sites (S in bold) and the highly conserved WL motif (bold). Numbers are those of the final residue in the polypeptide. (G) GST–eIF2Bε was phosphorylated by CK1 in vitro and digested with Asp-N. Resulting peptides were displayed by two-dimensional mapping.
Analysis of site(s) phosphorylated by CK1
As described above (Figure 2A), CK1 phosphorylated GST–eIF2Bε only weakly. Proteolytic digestion followed by HPLC (data not shown) or two-dimensional maps (Figure 5G) revealed numerous minor peptides mainly in the same positions as those seen for eIF2Bε phosphorylated in vitro by GSK3 and CK2. CK1 thus seems to phosphorylate eIF2Bε weakly at several sites that are better substrates for CK2 or GSK3. The Asp-N-derived map for eIF2Bε phosphorylated in vitro by CK1 also showed spots in the positions of peptides a4/5 seen in the digests of eIF2Bε phosphorylated in vivo (Figure 1C), suggesting that these sites might be phosphorylated by CK1 in vivo. Most of the peptides from eIF2Bε phosphorylated by CK1 and digested by Asp-N failed to give a signal on mass spectrometric analysis (most probably because they were negatively charged in the instrument used, operating on positive-ion mode), but one peptide with m/z = 1437.27 was clearly identified. This mass corresponds to the sequence DEDDGQFSDDSGA (residues 454–466; miscleavage product) with one phosphate.
Analysis revealed that peptides a4/5 from HA-eIF2Bε phosphorylated in vivo contained phosphoserine. Its negative charge at pH 1.9 showed that it contained no basic amino acids (arginine, histidine or lysine) and inspection of the sequence of eIF2Bε revealed only three possible candidate peptides, i.e. those containing Ser46, Ser461 and Ser464. These three residues were each mutated individually to alanine. Vectors encoding these three mutant polypeptides were each transfected into HEK293 cells, which were radiolabelled. The labelled mutant polypeptides were immunoprecipitated and subjected to digestion with Asp-N. Two-dimensional peptide mapping revealed that peptides a4/5 were still present in maps from eIF2Bε(S46A) but were absent from maps of eIF2Bε(S461A) or (S464A) (Figure 6). These data indicate that peptides a4/5 correspond to phosphorylation at one or other of these residues. Why should mutation of either site result in loss of the peptide? One possibility is that altering the local sequence (changing one serine) precludes phosphorylation at the other adjacent site. A phosphopeptide corresponding to this region was identified in Asp-N digests of eIF2Bε phosphorylated in vitro by CK1, suggesting that Ser461 or Ser464 may be phosphorylated by this kinase in vivo. With two acidic residues N-terminal to it, Ser464 lies in a sequence context similar to that required for phosphorylation by CK1 whereas Ser461 does not, suggesting that Ser464 is the true site of phosphorylation. This site and most of the adjacent residues are conserved in the known mammalian eIF2Bε sequences, but this region is not well conserved in other species and the sites appear to be absent.

Fig. 6. Analysis of in vivo phosphorylation of S46A, S461A and S464A mutants of eIF2Bε. HEK293 cells were transfected with vectors encoding wild-type eIF2Bε or point mutants as indicated. After in vivo labelling, isolation of eIF2Bε by immunoprecipitation (anti-HA) and SDS–PAGE, eIF2Bε polypeptides were digested with Asp-N and the resulting peptides were analysed on two-dimensional maps.
Sites phosphorylated in endogenous eIF2Bε
The above analysis enabled us to identify a number of sites phosphorylated in recombinant eIF2Bε in vitro and in 293 cells. An important objective was to identify the sites phosphorylated in endogenous eIF2Bε. Figure 7A and B shows two-dimensional maps for endogenous eIF2Bε radiolabelled in vivo and subsequently digested with Asp-N or trypsin. Two factors complicated our study: (i) the low level of incorporation of radiolabel which precluded a number of direct analyses and (ii) the fact that there are some sequence differences between the rat and human sequences, in particular around Ser535/539. However, the sequences around Ser461/464 and Ser712/713 are identical. Peptide A2 on the map from endogenous eIF2Bε thus corresponds to peptide a4/5 from the overexpressed protein, with which it co-migrates, and represents the S461/464 region, which is probably phosphorylated by CK1 in vivo. Peptide T2 migrates exactly as the C-terminal peptide seen for eIF2Bε containing the CK2 sites (Figure 5B), suggesting that these sites are phosphorylated in eIF2Bε in vivo.

Fig. 7. Analysis of phosphorylation of endogenous eIF2Bε. (A and B) Endogenous eIF2Bε from metabolically labelled HEK293 cells was digested with Asp-N or trypsin, and peptides were resolved on two-dimensional maps. (C) Schematic summary of the peptides in (A) and (B). (D) Peptide A1 was digested with trypsin and the products were analysed by two-dimensional mapping. (E and F) Peptide a2 (Figure 1C) was digested with trypsin. Two-dimensional maps of the starting peptide (E) and the tryptic products (F) are shown. For details of the annotation see text.
The Asp-N map for endogenous eIF2Bε lacked the major peptides a1–3 seen for the overexpressed rat protein, but contained a ‘new’ peptide, A1, which was almost neutral in charge and relatively hydrophilic (Figure 7A). As mentioned above, the sequence around the GSK3 and priming sites does differ between rat and human (A537G and L542M, and other differences in more distal positions), which could explain the lack of peptides a2/3. It was clearly possible that A1 represented the peptide containing the GSK3 and DYRK sites from human eIF2Bε (predicted sequence DSpRGGSpPQM), although the very small amounts of material and low level of radioactive peptide precluded direct analysis of this. As an alternative, A1, and also a2 from the rat eIF2Bε labelled in vivo (cf. Figure 1), were each digested with trypsin, and the products were analysed on two-dimensional maps (Figure 7D and F). Peptide a2 (rat) yielded two products; one (labelled ‘X’ in Figure 7F) was positively charged (indicating that it contained the N-terminal part of a2) while the other migrated in a similar position to the parent peptide (Yr). Similarly, tryptic digestion of A1 gave two products. One migrated identically to X in Figure 7D: this peptide presumably corresponds to the tripeptide DSpR, which is the predicted N-terminal product from rat or human eIF2Bε. The other product (‘Yh’, Figure 7D) again migrated in a similar position to the starting peptide, i.e. A1. These data strongly suggest that the peptide A1 from the endogenous eIF2Bε is the one containing the GSK3 and priming sites, which migrates differently on chromatography relative to peptide a2 from rat eIF2Bε due to the replacement of two residues by less hydrophobic ones (A→G, L→M). To confirm this, a peptide corresponding to the predicted sequence of the human protein in this region was synthesized, with a phosphate in the priming position. Phosphorylation of this peptide by GSK3 in vitro gave a product which behaved exactly like A1 on two-dimensional maps (data not shown).
Maps from overexpressed Asp-N-digested eIF2Bε contained three peptides which we had not been able to identify (a1, a major peptide; a6 and a7, minor ones; Figure 1C). However, they were absent from maps of the endogenous protein. This suggested that these peptides were perhaps ‘artefactually’ phosphorylated in the overexpressed recombinant protein, perhaps because most of this protein is not incorporated into eIF2B complexes so that residues that are normally occluded are exposed and available for phosphorylation. We nevertheless made strenuous attempts to identify these peptides: however, they could not be detected on reversed-phase HPLC analyses, and samples derived from the two-dimensional maps were refractory to analysis by Edman degradation or MALDI-TOF mass spectrometry. As they are absent in maps from endogenous eIF2Bε, we decided not to pursue their identification. Similarly, t4 and t5, seen for the HA-eIF2Bε (Figure 1D) but not for the endogenous protein, may also be sites only phosphorylated in the overexpressed polypeptide. In the case of human eIF2Bε, the tryptic peptide containing the GSK3 site is much larger (23 residues) than that from rat eIF2Bε (10 residues), explaining why no peptide corresponding to t3 is seen in Figure 7B. This large peptide is likely to migrate close to the origin.
The C-terminus of eIF2Bε is required for efficient phosphorylation at the GSK3 site in vivo
HEK293 cells were transfected with vectors encoding each of the two truncation mutants of eIF2Bε [eIF2Bε(Δ33) and eIF2Bε(Δ63)]. Analysis of these proteins using the appropriate phosphospecific antibody revealed that phosphorylation of Ser535 was substantially decreased in the Δ63 truncation mutant relative to the full-length or Δ33 polypeptides (Figure 8A). These findings were con firmed in experiments in which the Δ33 or Δ63 mutants were expressed in 293 cells, radiolabelled in vivo and analysed by peptide mapping after Asp-N digestion. Again, decreased phosphorylation of the GSK3 site was observed for the Δ63 truncated protein, as shown by a marked decrease in the amount of peptides a2 and a3, and increases in peptides migrating close to a8 and a9 (see Figure 4B), peptides in which only Ser539 is phosphorylated (data not shown).

Fig. 8. A region near the C-terminus of eIF2Bε modulates phosphorylation of Ser535 by GSK3. (A) HEK293 cells were transfected with vectors as indicated (‘vector’ = empty vector). Cell lysates were analysed by SDS–PAGE and immunoblotting using phosphospecific antisera or anti-HA (loading control) as indicated. (B and C) Full-length (FL) GST–eIF2Bε and the two truncation mutants (Δ33 and Δ63) were expressed in E.coli and purified. They were incubated with DYRK2 and ATP-MgCl2 for the times indicated before analysis by SDS–PAGE and either autoradiography (B; incubations included [γ-32P]ATP) or immunoblotting using anti-(P)Ser539 (C). (D) FL and truncated GST–eIF2Bε proteins were pre-incubated with DYRK and unlabelled ATP for 30 min. GSK3 and[γ-32P]ATP were then added and incubations continued for the times shown, after which samples were analysed by SDS–PAGE and autoradiography. (E) Alignment of sequences of the GSK3-interacting region of axin and part of the region between residues 665 and 679 from mammalian eIF2Bε. Identical residues are shown in bold while similar ones are underlined. Hs, Homo sapiens, Mm, Mus musculus, Rn, Rattus norvegicus. (F) HEK293 cells were transfected with vectors encoding FL HA-eIF2Bε, the Δ63 mutant or empty vector and, after immunoprecipitation of GSK3α and β, samples were subjected to SDS–PAGE followed by immunoblotting with either anti-HA or an antiserum which recognizes GSK3α and β (as a loading control).
This suggested that the Δ63 truncated proteins might be phosphorylated less well by GSK3 than full-length eIF2Bε. To test this, we studied the phosphorylation in vitro of eIF2Bε(Δ63) and eIF2Bε(Δ33) expressed in E.coli as GST fusion proteins. Since they are not phosphorylated at Ser539, the priming site for GSK3, we first incubated them in vitro with DYRK2. Each of the three recombinant eIF2Bε polypeptides was phosphorylated with similar efficiency (Figure 8B and C). Similar levels of phosphorylation of Ser539 were also seen in vivo for full-length eIF2Bε and both the truncation mutants (Figure 8A and peptide mapping data not shown). In contrast, the truncated proteins, especially the Δ63 mutant, were phosphorylated much less well by GSK3 in vitro (Figure 8D). These data suggest that the region between residues 655 and 685 contains a motif necessary for the efficient action of GSK3 in vivo and in vitro. There is a precedent for this in the role of axin in promoting phosphorylation of β-catenin by GSK3 (Hart et al., 1998; Hedgepeth et al., 1999). It is therefore of considerable interest that the sequence of this region of eIF2Bε shows similarity to the region of axin that promotes GSK3-mediated phosphorylation of β-catenin (Figure 8E). To test this idea further, we studied whether we could detect an interaction between GSK3 and eIF2Bε and whether this was affected by removal of the C-terminus of eIF2Bε. GSK3 was immunoprecipitated from extracts of HEK293 cells that had been transfected with full-length eIF2Bε or the Δ63 mutant, and the samples were probed for GSK3 (as a loading control) or HA-eIF2Bε. The data (Figure 8F) show that HA-eIF2Bε co-immunoprecipitates with GSK3, indicating a rather stable interaction between them. However, markedly less of the truncated Δ63 mutant associated with GSK3. The C-terminus of eIF2Bε does therefore seem to be important for the interaction of GSK3 with eIF2Bε, offering an explanation for the reduced phosphorylation of the Δ63 mutant.
Analysis of the effects of mutations at phosphorylation sites on the function of eIF2Bε
To test whether any of the point mutations affected the interaction of eIF2Bε with eIF2, mutant proteins were expressed in HEK293 cells and immunoprecipitated from cell extracts using the anti-HA antibody. Immuno precipitates were analysed by SDS–PAGE and immunoblotting using an antibody to eIF2α. As shown in Figure 9A, similar amounts of eIF2 were found to associate with the S535A and S539N mutants, and with wild-type eIF2Bε, showing that these mutations do not affect the stable association of eIF2 with eIF2Bε. However, association between eIF2 and the S712/713GA mutant was almost undetectable, indicating that loss of the CK2 sites greatly decreases this interaction. This is especially significant as they lie in the region of eIF2Bε previously identified as playing a key role in binding eIF2 (Asano et al., 1999) and indicates that phosphorylation of these sites is involved in the interaction of eIF2Bε with eIF2. The effect of mutating the CK1 phosphorylation site at residues 461 or 464 on the interaction of eIF2Bε with eIF2 was also analysed in a similar way. In four separate experiments, no significant effect of mutating these residues was observed on the interaction between eIF2 and eIF2Bε (Figure 9B).
To assess the effects of the mutations at Ser540, 544, 712 and 713 on eIF2B activity, the mutant proteins were again expressed in 293 cells. Recombinant eIF2Bε was immunoprecipitated from cell extracts using anti-HA, and eIF2B activity was then determined using the standard assay (i.e. with eIF2⋅[3H]GDP complexes as substrate; Figure 9C). Immunoprecipitates were also analysed by SDS–PAGE/western blotting using anti-HA, to verify that the amounts of HA-eIF2Bε were equal. The S535A and S539N mutant proteins did not consistently show a change in activity relative to the wild-type protein. In contrast, the S712/713GA mutant displayed substantially decreased activity relative to wild-type eIF2Bε (∼25% of the activity seen with HA-eIF2Bε, Figure 9C). This presumably reflects the very marked impairment of the ability of this mutant to bind stably to eIF2 in vivo (Figure 9A). The fact that it still shows some exchange activity suggests that, although these sites are important for the interaction with eIF2, they are not the only sites of interaction. For example, the catalytic region itself must presumably also interact directly with eIF2, and there are extensive data from studies with yeast eIF2B suggesting that the α-, β- and δ-subunits also contact eIF2 (see, for example, Pavitt et al., 1997, 1998). Since mutation of Ser461 or Ser464 to alanine did not affect the ability of eIF2Bε to bind eIF2, and as these residues lie outside the catalytic domain of eIF2Bε defined by Gomez and Pavitt (2000) and are relatively minor sites of phosphorylation in the endogenous eIF2B (Figure 7A), we have not examined the effects of these mutations on eIF2B activity.
It was important to establish whether the eIF2B activity measured in anti-HA immunoprecipitates from whole lysates reflected the properties of eIF2B complexes con taining the mutant ε-subunit or whether free HA-eIF2Bε, which is expressed at considerably higher levels than the endogenous polypeptide, also contributed to exchange activity. To address this, we used ion-exchange chromatography on Mono-Q (Figure 10A) to resolve complexes containing recombinant (wild-type or mutant) HA-eIF2Bε from the excess free HA-eIF2Bε. Samples of each fraction were subjected to SDS–PAGE and western blotting. As shown in Figure 10B, although the overall protein resolution achieved was very good, as judged by the sharp peaks in the A280 profile, HA-eIF2Bε emerged from the column across a broad range of salt concentrations. This effect could be due to partial misfolding of the free subunit or perhaps interactions with other proteins, e.g. chaperones. Western blotting with antisera for the α-, γ- and δ-subunits of eIF2B showed that these polypeptides emerged together as a sharp peak around fractions 52–54 (Figure 10C). (We did not test for eIF2Bβ due to lack of a suitable antibody, and the endogenous eIF2Bε ran too close to the HA-tagged band to be resolved reliably from it on this western blot analysis.)

Fig. 10. eIF2Bε only displays significant activity when incorporated into eIF2B complexes. Extracts from HEK293 cells transfected with the vector encoding HA-tagged eIF2Bε (wild-type) were subjected to ion-exchange chromatography on Mono-Q as described in the Supplementary material. (A) The absorbance and conductivity traces from a typical chromatogram (representative of >15 runs performed). The absorbance (A280, AU = absorbance units) and conductivity (salt gradient) traces are shown. (B) Selected fractions (numbers shown) were subjected to SDS–PAGE and western blotting using the indicated antisera. The elution positions of eIF2, eIF2B complexes and HA-eIF2Bε free of other eIF2B subunits are shown. (C) Selected fractions were subjected to immunoprecipitation with anti-HA and immunoprecipitates were analysed by SDS–PAGE and western blotting using anti-HA, anti-eIF2Bγ and anti-eIF2Bδ antibodies as indicated. Note that these data are from a different run from those in (A) and (B), so fraction numbers are shifted ‘rightwards’ by about half of one fraction. (D) Selected fractions were subjected to immunoprecipitation with anti-HA followed by a standard eIF2B activity assay using eIF2⋅[3H]GDP as substrate. Data are shown as the percentage of total bound [3H]GDP released during the 15 min assay and are typical of six sets of data for 293 cells expressing wild-type HA-eIF2Bε.
To assess whether the HA-eIF2Bε in fractions around 52–54 was incorporated into complexes containing other eIF2B subunits, samples were subjected to immunopre-cipitation with anti-HA antibody and analysed by SDS–PAGE/western blotting. This revealed that the HA-eIF2Bε in these fractions does co-immunoprecipitate with other eIF2B subunits, indicating that it is indeed incorporated into eIF2B complexes (Figure 10C). The most important point was to explore whether only eIF2B complexes have exchange activity or whether free HA-eIF2Bε is also active. Fractions from across the column profile were therefore subjected to immunoprecipitation with anti-HA and the precipitates were assayed for eIF2B activity. As shown in Figure 10D, only material in the fractions containing eIF2B complexes displayed significant exchange activity, even when fractions contained similar amounts of HA-eIF2Bε as assessed by western blotting (compare fractions 52–54 with 57–59). Most importantly, the fractions eluting at higher salt concentrations that contain HA-eIF2Bε, but not other subunits of eIF2B, displayed very low, or in some cases negligible, activity compared with those containing eIF2B complexes. Similar data were obtained in >10 separate experiments. The observation that HA-eIF2Bε only has significant activity as part of eIF2B complexes is in agreement with previous findings that incorporation into eIF2B complexes greatly enhances the activity of yeast or mammalian eIF2Bε (Fabian et al., 1997; Pavitt et al., 1998; Gomez and Pavitt, 2000).
The above experiments imply that the exchange activity measured in HA immunoprecipitates from cell lysates (Figure 9C) overwhelmingly reflects the activity of HA-eIF2Bε that has been incorporated into eIF2B complexes. Thus, within the context of such complexes, the S712/713GA mutation markedly impairs eIF2B activity while S535A has little effect on activity.
To study this further, we performed similar analyses to that shown in Figure 10 for the S712/713GA and S535A mutant proteins. Four separate experiments were conducted for both mutants, each analysed in each case in parallel with the wild-type protein. However, given the nature of the experiments and their complexity, direct quantitative comparisons proved difficult. Nevertheless, it was found consistently that the S712/713GA mutant displayed only low activity as part of eIF2B complexes while the activity of complexes containing the S535A mutant did not differ significantly from that of complexes containing wild-type eIF2B.
Discussion
This study identifies five sites in the catalytic subunit of eIF2B that are phosphorylated in intact cells and shows that all of them are targets for protein kinases known to phosphorylate eIF2Bε in vitro. Four of the sites lie in the region identified by Gomez and Pavitt (2000) as the catalytic region of eIF2Bε, including two in the extreme C-terminus which previously has been shown to be involved in the interaction between eIF2Bε and eIF2β (Asano et al., 1999). The fifth site, a relatively minor one in vivo, is in the central part outside the conserved catalytic domain and, unlike the other sites, is not found in any of the known eIF2Bε sequences from non-mammalian species.
The only previous study on the in vivo phosphorylation of eIF2Bε employed labelling in reticulocytes followed by CNBr digestion, resolution of the resulting fragments on acrylamide gels and N-terminal sequence analysis of the radiolabelled fragments (Aroor et al., 1994). This approach, unlike ours, did not allow the identification of any specific sites of phosphorylation. Consistent with our studies, only serine residues were found to be labelled, but several labelled CNBr fragments were detected. This type of analysis was made more difficult by the occurrence of incomplete cleavage products, adding to the complexity of the picture. Their analysis suggested that several regions of eIF2Bε contained sites that are phosphorylated in vivo. These included the C-terminal region (which we show contains sites for CK2), the regions between residues 500 and 600 (which contains the GSK3 and priming sites) and the central region (which contains Ser461/464).
In our study, we did not find any sites that are not substrates for kinases already known to phosphorylate eIF2Bε in vitro, i.e. CK1, CK2, GSK3 and DYRK. The material remaining at the origin of two-dimensional maps probably reflects the fact that some of these sites are contained within very large peptides which migrate poorly on maps. The CK2 site is in a 27 residue Asp-N peptide, while the CK1 site is in a tryptic peptide of 49 amino acids. Mutation of each of the sites identified on the basis of in vitro experiments resulted in loss of the expected phosphopeptides (or signal with the relevant phospho-specific antibody) in vivo. Furthermore, mutation of these phosphorylation sites gave three further important pieces of information.
First, mutation of the CK2 sites at the extreme C-terminus resulted in a marked loss of activity when eIF2Bε was expressed in 293 cells. This also greatly diminished the binding of eIF2Bε to eIF2 in vivo. These data strongly suggest that phosphorylation of one or both of these sites is required for efficient interaction between eIF2Bε and its substrate, eIF2. Previous data from Wahba and colleagues (Dholakia and Wahba, 1988) suggested that phosphorylation of eIF2Bε by CK2 increased its activity in nucleotide exchange, and our findings thus provide an explanation of this effect. We previously were unable to reproduce this effect (Oldfield and Proud, 1992): a likely explanation for this is that our preparations of eIF2B from reticulocytes are only weakly phosphorylated by CK2 (Oldfield and Proud, 1992), presumably because phosphate is already present at these sites. Thus, low levels of phosphorylation were seen and this probably explains the absence of an effect on activity in our study. Hormones and growth factors that activate eIF2B have little, if any, effect upon the activity of CK2 (Litchfield et al., 1994; our unpublished data), so that phosphorylation by CK2 is unlikely to make an input to the control of eIF2Bε activity but instead provides a permanent modification of eIF2Bε that facilitates substrate binding.
Based on mutational analysis, it has been shown that this region of eIF2Bε is required for its interaction with eIF2 and that binding of eIF2Bε to eIF2 involves lysine blocks in eIF2β (Das et al., 1997; Asano et al., 1999). A likely explanation of the importance of the CK2 phosphorylation sites is that the negatively charged phosphoserines interact with (part of) these lysine blocks. The earlier data showed that acidic residues in this region were important for this interaction: since mutation of these two serine residues alone abolishes binding to eIF2 in vivo, they also seem to play an important role in this interaction, probably by virtue of their phosphorylation. The finding that the S712/713GA mutant also shows markedly reduced activity against eIF2⋅GDP supports this conclusion.
Secondly, mutation of Ser539 led to the loss of phosphorylation of Ser535. This strongly supports the idea, previously based on the use of synthetic peptides (Welsh et al., 1997b), that phosphorylation of Ser539 primes the phosphorylation of Ser535 by GSK3. eIF2Bε thus resembles a number of other substrates for GSK3 in requiring phosphorylation of a serine residue at the +4 position in order to become an efficient substrate for GSK3. We have shown recently that members of the DYRK group of kinases can phosphorylate Ser539 in vitro (Woods et al., 2001). Further studies will be required to assess whether DYRKs are the kinases acting on this site in vivo and whether the activities of these poorly understood kinases are subject to regulation. All known mammalian eIF2Bε sequences (Welsh et al., 1998) and that from Drosophila (Williams et al., 2001) contain serine residues at the positions corresponding to Ser535 in rat, and a Ser/Pro or Thr/Pro pair at the priming position.
Thirdly, mutation of the GSK3 site (Ser535) to alanine did not have a marked effect on the activity of eIF2Bε expressed in 293 cells. This is surprising given that phosphorylation of the mammalian (Welsh et al., 1998) or insect (Williams et al., 2001) eIF2B complex by GSK3 in vitro inhibits its activity by ∼50%. This site is heavily phosphorylated in HEK293 cells (as indicated by the fact that only the doubly phosphorylated peptide containing Ser535 and 539 is observed) so this mutation might have been expected to enhance activity. The absence of a marked effect of this mutation on eIF2B activity may reflect other, counteracting, effects of replacement of Ser535 by alanine on eIF2B activity.
A further intriguing finding is that a region towards the C-terminus of eIF2Bε is required for its efficient phosphorylation at Ser535, both in vitro and in vivo. Deletion of this region results in a marked decrease in the association of GSK3 with eIF2Bε. It shows similarity to the region of axin required for its ability to promote phosphorylation of β-catenin by GSK3 (Hedgepeth et al., 1999) and may therefore represent a common docking sequence for GSK3. However, the situation differs from that for axin/β-catenin as the recognition element lies within the substrate, eIF2Bε, rather than in a separate polypeptide. This is, to our knowledge, the first example of such a GSK3-binding element operating within the same polypeptide (‘in cis’) rather than in trans, as for axin.
Materials and methods
Materials
CK1 and CK2 were obtained from Promega. GSK3 and DYRK2 were kindly provided by Drs A.Paterson and Y.Woods, respectively (both University of Dundee). Details of the antisera used are given in the Supplementary data available at The EMBO Journal Online.
Vectors
Details of vectors and the expression of eIF2Bε in E.coli appear in the Supplementary data.
Cell culture
HEK293 cells were routinely grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal calf serum (FCS), 2 mM glutamine and a commercial mixture of antibiotics and antimycotics (Gibco-BRL Life Technologies). Cells were transfected using a calcium phosphate transfection procedure (Hall-Jackson et al., 1999). Cells were harvested into standard extraction buffer [100 mM Tris–HCl pH 7.6; 50 mM β-glycerophosphate; 0.5 mM sodium orthovanadate; 1.5 mM EGTA; 0.1% (v/v) Triton X-100] unless otherwise indicated. In some cases, where indicated, an alternative extraction buffer (buffer B) containing no detergent [20 mM HEPES–KOH pH 7.6; 50 mM KCl; 25 mM β-glycerophosphate; 0.1 mM EDTA; 10% (v/v) glycerol] was used.
Immunoprecipitation and western blotting
Cells were harvested into extraction buffer B and immediately snap frozen. The cells were then subjected to two freeze–thaw cycles to lyse the cells and centrifuged at 14 000 r.p.m. for 10 min at 4°C to pellet the debris. Supernatants were removed to fresh tubes and then rotated at 4°C in the presence of protein G–Sepharose pre-bound with anti-HA or anti-eIF2Bε pre-equilibrated in buffer B. Immunocomplexes were washed twice with this buffer and then resuspended in SDS–PAGE sample buffer. Samples were then run on SDS–polyacrylamide gels (12.5% acrylamide), transferred to Immobilon (Millipore) and probed with the appropriate antibody. Blots were visualized by enhanced chemiluminescence (ECL).
eIF2B assays
Cells were harvested in extraction buffer and centrifuged at 14 000 r.p.m. for 10 min at 4°C to pellet the cell debris. Lysates were rotated at 4°C in the presence of protein G–Sepharose for 30–45 min. This supernatant was then removed and varying amounts of extract (to compensate for any differences in the expression levels of eIF2Bε) were added to protein G–Sepharose bound with anti-HA in 100 µl of buffer B. Immunocomplexes were washed twice with this buffer and then 10 µl of extraction buffer was added to resuspend the pellets. eIF2B assays were essentially as described earlier (Kleijn et al., 1998) with constant shaking (1000 r.p.m.).
Supplementary data
Details of in vitro phosphorylation, protein chemistry and in vivo phosphorylation site mapping procedures are given in the Supplementary data available at The EMBO Journal Online.
Acknowledgments
Acknowledgements
We are grateful to Drs Gert Scheper and Chris Armstrong (Dundee) for their help with protein chemistry and baculovirus expression, respectively. This work was supported by a Programme Grant (046110) from the Wellcome Trust.
References
- Aroor A.R., Denslow,N.D., Singh,L.P., O’Brien,T.W. and Wahba,A.J. (1994) Phosphorylation of rabbit reticulocyte guanine nucleotide exchange factor in vivo. Identification of putative casein kinase II phosphorylation sites. Biochemistry, 33, 3350–3357. [DOI] [PubMed] [Google Scholar]
- Asano K., Krishnamoorthy,T., Phan,L., Pavitt,G.D. and Hinnebusch, A.G. (1999) Conserved bipartite motifs in yeast eIF5 and eIF2Bε, GTPase-activating and GDP–GTP exchange factors in translation initiation, mediate binding to their common substrate eIF2. EMBO J., 18, 1673–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemens M.J. (1996). Protein kinases that phosphorylate eIF2 and eIF2B, and their role in eukaryotic cell translational control. In Hershey,J.W.B., Mathews,M.B. and Sonenberg,N. (eds), Translational Control. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 139–172.
- Das S., Maiti,T., Das,K. and Maitra,U. (1997) Specific interaction of eukaryotic translation initiation factor 5 (eIF5) with the β-subunit of eIF2. J. Biol. Chem., 272, 31712–31718. [DOI] [PubMed] [Google Scholar]
- Dholakia J.N. and Wahba,A.J. (1988) Phosphorylation of the guanine nucleotide exchange factor from rabbit reticulocytes regulates its activity in polypeptide chain initiation. Proc. Natl Acad. Sci. USA, 85, 51–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabian J.R., Kimball,S.R., Heinzinger,N.K. and Jefferson,L.S. (1997) Subunit assembly and guanine nucleotide exchange factor activity of eukaryotic initiation factor eIF2B subunits expressed in Sf9 cells. J. Biol. Chem., 272, 12359–12365. [DOI] [PubMed] [Google Scholar]
- Fiol C.J., Wang,A., Roeske,R.W. and Roach,P.J. (1990) Ordered multisite protein phosphorylation. Analysis of glycogen synthase kinase 3 action using model peptide substrates. J. Biol. Chem., 265, 6061–6065. [PubMed] [Google Scholar]
- Gilligan M., Welsh,G.I., Flynn,A., Bujalska,I., Proud,C.G. and Docherty,K. (1996) Glucose stimulates the activity of the guanine nucleotide-exchange factor eIF2B in isolated rat islets of Langerhans. J. Biol. Chem., 271, 2121–2125. [DOI] [PubMed] [Google Scholar]
- Gomez E. and Pavitt,G.D. (2000) Identification of domains and residues within translation initiation factor eIF2Bε required for guanine nucleotide-exchange reveals a novel activation function promoted by eIF2B complex formation. Mol. Cell. Biol., 20, 3965–3976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall-Jackson C.A., Cross,D.A., Morrice,N. and Smythe,C. (1999) ATR is a caffeine-sensitive, DNA-activated protein kinase with a substrate specificity distinct from DNA-PK. Oncogene, 18, 6707–6713. [DOI] [PubMed] [Google Scholar]
- Hart M.J., de los Santos,R., Albert,I.N., Rubinfeld,P. and Polakis,P. (1998) Downregulation of β-catenin by human axin and its association with the APC tumor suppressor, β-catenin and GSK3β. Curr. Biol., 8, 573–581. [DOI] [PubMed] [Google Scholar]
- Hedgepeth C.M., Deardorff,M.A., Rankin,K. and Klein,P.S. (1999) Regulation of glycogen synthase kinase 3β and downstream Wnt signaling by axin. Mol. Cell. Biol., 19, 7147–7157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinnebusch A.G. (2000). Mechanism and regulation of methionyl-tRNA binding to ribosomes. In Sonenberg,N., Hershey,J.W.B. and Mathews,M.B. (eds), Translational Control of Gene Expression. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 185–243.
- Joshi-Barve S., Rychlik,W. and Rhoads,R.E. (1990) Alteration of the major phosphorylation site of eukaryotic protein synthesis initiation factor 4E prevents its association with the 48S initiation complex. J. Biol. Chem., 265, 2979–2983. [PubMed] [Google Scholar]
- Kimball S.R. and Jefferson,L.S. (1988) Effect of diabetes on guanine nucleotide exchange factor activity in skeletal muscle and heart. Biochem. Biophys. Res. Commun., 156, 706–711. [DOI] [PubMed] [Google Scholar]
- Kleijn M., Welsh,G.I., Scheper,G.C., Voorma,H.O., Proud,C.G. and Thomas,A.A.M. (1998) Nerve and epidermal growth factors induce protein synthesis and eIF2B activation in PC12 cells. J. Biol. Chem., 273, 5536–5541. [DOI] [PubMed] [Google Scholar]
- Lazaris-Karatzas A., Montine,K.S. and Sonenberg,N. (1990) Malignant transformation by a eukaryotic initiation factor subunit that binds to mRNA 5′ cap. Nature, 345, 544–547. [DOI] [PubMed] [Google Scholar]
- Litchfield D.W., Dobrowolska,G. and Krebs,E.G. (1994) Regulation of casein kinase-II by growth factors: a re-evaluation. Cell. Mol. Biol. Res., 40, 373–381. [PubMed] [Google Scholar]
- Oldfield S. and Proud,C.G. (1992) Purification, phosphorylation and control of the guanine-nucleotide-exchange factor from rabbit reticulocyte lysates. Eur. J. Biochem., 208, 73–81. [DOI] [PubMed] [Google Scholar]
- Pain V.M. (1996) Initiation of protein synthesis in eukaryotic cells. Eur. J. Biochem., 236, 747–771. [DOI] [PubMed] [Google Scholar]
- Pavitt G.D., Yang,W. and Hinnebusch,A.G. (1997) Homologous segments in three subunits of the guanine nucleotide exchange factor eIF2B mediate translational regulation by phosphorylation of eIF2. Mol. Cell. Biol., 17, 1298–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavitt G.D., Ramaiah,K.V.A., Kimball,S.R. and Hinnebusch,A.G. (1998) eIF2 independently binds two distinct eIF2B subcomplexes that catalyse and regulate guanine-nucleotide exchange. Genes Dev., 12, 514–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proud C.G. (1992) Protein phosphorylation in translational control. Curr. Top. Cell Regul., 32, 243–369. [DOI] [PubMed] [Google Scholar]
- Singh L.P., Aroor,A.R. and Wahba,A.J. (1994) Phosphorylation of the guanine nucleotide exchange factor and eukaryotic initiation factor 2 by casein kinase 2 regulates guanine nucleotide binding and GDP/GTP exchange. Biochemistry, 33, 9152–9157. [DOI] [PubMed] [Google Scholar]
- Singh L.P., Denslow,N.D. and Wahba,A.J. (1996) Modulation of rabbit reticulocyte guanine nucleotide exchange factor activity by casein kinases 1 and 2 and glycogen synthase kinase 3. Biochemistry, 35, 3206–3212. [DOI] [PubMed] [Google Scholar]
- Tejedor F., Zhu,X.R., Kaltenbach,E., Ackermann,A., Baumann,A., Canal,I., Heisenberg,M., Fischbach,K.F. and Pongs,O. (1995) Minibrain: a new protein kinase family involved in postembryonic neurogenesis in Drosophila. Neuron, 14, 287–301. [DOI] [PubMed] [Google Scholar]
- Tyzack J.K., Belsham,G.J. and Proud,C.G. (2000) ABC50 interacts with eukaryotic initiation factor (eIF) 2 and binds to ribosomes in an ATP-dependent manner. J. Biol. Chem., 275, 34131–34139. [DOI] [PubMed] [Google Scholar]
- Welsh G.I. and Proud,C.G. (1992) Regulation of protein synthesis in Swiss 3T3 fibroblasts. Rapid activation of the guanine-nucleotide-exchange factor by insulin and growth factors. Biochem. J., 284, 19–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh G.I., Miyamoto,S., Proud,C.G. and Safer,B. (1996) T-cell activation leads to rapid stimulation of translation initiation factor eIF2B and inactivation of glycogen synthase kinase-3. J. Biol. Chem., 271, 11410–11413. [DOI] [PubMed] [Google Scholar]
- Welsh G.I., Stokes,C.M., Wang,X., Sakaue,H., Ogawa,W., Kasuga,M. and Proud,C.G. (1997a) Activation of translation initiation factor eIF2B by insulin requires phosphatidylinositol 3-kinase. FEBS Lett., 410, 418–422. [DOI] [PubMed] [Google Scholar]
- Welsh G.I., Patel,J.C. and Proud,C.G. (1997b) Peptide substrates suitable for assaying glycogen synthase kinase-3 (GSK-3) in crude cell extracts. Anal. Biochem., 244, 16–21. [DOI] [PubMed] [Google Scholar]
- Welsh G.I., Miller,C.M., Loughlin,A.J., Price,N.T. and Proud,C.G. (1998) Regulation of eukaryotic initiation factor eIF2B: glycogen synthase kinase-3 phosphorylates a conserved serine which undergoes dephosphorylation in response to insulin. FEBS Lett., 421, 125–130. [DOI] [PubMed] [Google Scholar]
- Williams D.D., Pavitt,G.D. and Proud,C.G. (2001) Characterisation of the initiation factor eIF2B and its regulation in Drosophila melanogaster. J. Biol. Chem., 276, 3733–3742. [DOI] [PubMed] [Google Scholar]
- Woods Y., Cohen,P., Becker,W., Jakes,R., Goedert,M., Wang,X. and Proud,C.G. (2001) DYRK phosphorylates protein synthesis initiation factor eIF2Bε at Ser539 and the microtubule-associated protein τ at Thr212; potential role for DYRK as a GSK3 priming kinase. Biochem. J., 355, 609–615. [DOI] [PMC free article] [PubMed] [Google Scholar]