

Abstract
Anti-Golgi complex autoantibodies are found primarily in patients with Sjögren's syndrome and systemic lupus erythematosus, although they are not restricted to these diseases. Several Golgi autoantigens have been identified that represent a small family of proteins. Common features of all Golgi autoantigens appear to be their distinct structural organization of multiple α-helical coiled-coil rods in the central domains flanked by non-coiled-coil N-termini and C-termini, and their localization to the cytoplasmic face of Golgi cisternae. Many autoantigens in systemic autoimmune diseases have distinct cleavage products in apoptosis or necrosis and this has raised the possibility that cell death may play a role in the generation of potentially immunostimulatory forms of autoantigens. In the present study, we examined changes in the Golgi complex and associated autoantigens during apoptosis and necrosis. Immunofluorescence analysis showed that the Golgi complex was altered and developed distinctive characteristics during apoptosis and necrosis. In addition, immunoblotting analysis showed the generation of antigenic fragments of each Golgi autoantigen, suggesting that they may play a role in sustaining autoantibody production. Further studies are needed to determine whether the differences observed in the Golgi complex during apoptosis or necrosis may account for the production of anti-Golgi complex autoantibodies.
Keywords: anti-Golgi complex antibody, autoantibody, autoimmunity, cell death
Introduction
The Golgi complex is an elaborate cytoplasmic organelle that has a prominent function in the processing, transporting, and sorting of intracellular proteins. Autoantibodies directed against the Golgi complex were first identified in the serum of a Sjögren's syndrome patient with lymphoma [1]. Subsequent reports described anti-Golgi complex autoantibodies (AGA) in patients with systemic rheumatic diseases such as systemic lupus erythematosus [2], rheumatoid arthritis [3], and Wegener's granulomatosis [4]. Immunoblotting and immunoprecipitation studies have shown that there was heterogeneity of reactivity among AGA [5].
Within the past several years, our laboratories and others have cloned and identified novel Golgi autoantigens (golgins) [6]. This has been achieved primarily by expression cloning using human autoantibody probes. The identified autoantigens referred to are golgin-160/GCP170 [7,8], golgin-95/gm130 [7], golgin-97 [9], golgin-245/ p230 [10-12], giantin/macrogolgin/GCP372 [13,14], and golgin-67 [15]. These proteins are characterized by predominant α-helical coiled-coil domains, except for N-termini and C-termini. It has been reported that some golgins are localized to the cytoplasmic face of Golgi membranes [16]. Functions of the golgins have been proposed in vesicular transport and in maintaining structural integrity of the Golgi complex [17-19]. The potential implication is that these Golgi proteins may have common biochemical characteristics that may make them targets of autoimmune responses in certain disease states.
It is not clear why and how the immune system is able to recognize or target intracellular autoantigens. One possible explanation is that they may be recognized as surface structures on cytoplasmic organelles that are released to the immune system in aberrant disease states associated with unregulated cell death (apoptosis or necrosis) resulting from injury or infection, or defective clearance of dying cells. A variety of autoantigens are cleaved into signature fragments during apoptosis and necrosis [20]. Other cell death post-translational modifications in autoantigens include hyperphosphorylation, (de)ubiquitination, methylation, citrullination, and transglutaminase crosslinking [21,22].
The emerging view is that modified forms of autoantigens generated during cell death might stimulate autoantibody responses if presented to the immune system in a proinflammatory context [23]. Since the fate of the Golgi complex during cell death has not been extensively explored, we investigated whether the Golgi complex and associated autoantigens are altered during apoptosis and necrosis. Using AGA as probes, we observed that the Golgi complex undergoes dynamic changes during apoptosis distinct from those observed in necrosis. Immunoblotting analysis showed distinct cleavage fragments of Golgi autoantigens generated during apoptosis and necrosis. Since the cleaved Golgi autoantigen fragments are antigenic, they may play a role in sustaining autoantibody production in autoimmune disease states.
Materials and methods
Antibodies
Human prototype serum containing highly specific antipolyADP-ribose polymerase (PARP) antibody and AGA sera were obtained from the serum bank of the WM Keck Autoimmune Center, The Scripps Research Institute, CA, USA. Rabbit antibodies to Golgi autoantigens were produced in New Zealand White rabbits [7,9,10]. Briefly, recombinant human Golgi autoantigens were produced using the expression plasmid pET28 system in Escherichia coli (Novagen, Madison, WI, USA). Recombinant golgin-160 (amino acids 787–1348, GenBank accession number BAA23661), giantin (amino acids 851–1496, GenBank accession number NP_004478), gm130 (amino acids 370–990, GenBank accession number AAF65550), and golgin-97 (amino acids 1–767, GenBank accession number AAB81549) proteins were purified by affinity nickel column chromatography. They were then used to immunize one or two rabbits separately by subcutaneous injection of recombinant proteins in an equal volume of Freund's complete adjuvant. After booster immunizations, the immune sera were prepared and stored at -20°C. The appearance and titers of antibodies were monitored by indirect immunofluorescence and immunoblotting analysis.
Induction of cell death
Human Jurkat and HEp-2 cells were obtained from American Type Culture Collection (Rockville, MD, USA) and were cultured in RPMI 1640 and Dulbecco's modified Eagle's medium (Life Technologies, Rockville, MD, USA), respectively, containing 10% fetal bovine serum under standard conditions. Induction of cell death was performed essentially as described elsewhere [20] with some modifications.
Apoptosis was induced in Jurkat T cells (106/ml) by exposure to 1 μM staurosporine (STS) (ALEXIS, San Diego, CA, USA) for up to 4 hours. Apoptosis in HEp-2 cells was induced by exposure to 2 μM STS at 37°C for up to 6 hours. Necrosis was induced in these cells by exposure to 10 μM STS for up to 24 hours or by treatment with 0.1% hydrogen peroxide (H2O2) (Fisher Scientific, Pittsburgh, PA, USA) for 3 hours. Necrosis was quantified using the trypan blue exclusion assay, which measures loss of cytoplasmic membrane integrity, as described previously [20,24].
At least 300 cells were counted in triplicate in three independent experiments. In some experiments, Jurkat cells were incubated for 1 hour in the presence of the pancaspase inhibitor benzylocarbonyl-Val-Ala-Asp-fluromethyl-ketone (zVAD-fmk) (ALEXIS), used at 100 μM, prior to addition of STS. Treated and control cells, and their extracts, were analyzed by indirect immunofluorescence and/or immunoblotting analysis. Spontaneous cell death prior to the experiments was minimized by maintaining exponential cell growth. Cell viability was quantified by trypan blue exclusion analysis at the beginning of every experiment to ensure that cell cultures used in the experiments were healthy (alive cells >95%).
Indirect immunofluorescence microscopy
Indirect immunofluorescence was performed as reported previously [7,10,25]. HEp-2 cells were grown on eight-chamber vessel tissue culture slides (Becton Dickinson, Franklin Lakes, NJ, USA) and treated with 2 or 10 μM STS for up to 6 hours. Cells were fixed by methanol and acetone (1:3, -20°C) for 2 min. Sera containing AGA were used in dilutions of 1:200 to 1:10,000. The secondary antibodies were Alexa™ 488 conjugated goat anti-rabbit IgG or anti-human IgG reagents (ALEXIS). Cells were counterstained with 4',6-diamidino-2-phenylindole nuclear stain prior to immunofluorescence microscopy. The estimation of the percentage of cells at each morphological stage described in the following for Golgi staining in apoptotic cells was obtained by scoring 300–500 cells in each experiment.
Immunoblotting analysis of cell lysates
After incubation in the presence of cell-death-inducing reagents, Jurkat cells were centrifuged at 200 × g for 30 min, followed by one wash at 1000 × g for 10 min in PBS containing Complete Protease Inhibitor cocktail (Roche, Mannheim, Germany). Cell pellets (107) were then resuspended directly in lysis buffer containing 150 mM NaCl, 1 mM MgCl2 · 6H2O, 80 mM Tris–HCl and 0.1% NP-40.
The lysates were passed several times sequentially through 18-gauge to 27-gauge needles to shear the DNA, and protein concentrations in the lysates were determined by Protein DC Assay Kit (Bio-Rad, Hercules, CA, USA) to ensure equal loading of proteins in each SDS-PAGE lane. After determination of the protein concentration, lysates were mixed with an equal volume of 2 × gel sample buffer containing 6% sodium dodecyl sulfate, 20% glycerol, 10% β-mercaptoethanol, 0.02% bromphenol blue and Complete Protease Inhibitor cocktail. Lysates were stored at -80°C until use.
The equivalent of 1.5 μg total lysate protein was loaded onto each lane of 10% SDS-PAGE gels, separated by electrophoresis, and transferred to nitrocellulose membranes using a Semi-Dry Trans-Blot apparatus (Bio-Rad). Immunoblotting was performed as described previously [7,10,24] using human anti-PARP specific antibody (1:250) and AGA (1:250). Detection of bound antibodies was achieved using horseradish peroxidase-conjugated goat anti-human or anti-rabbit IgG antibody (CALTAG, San Francisco, CA, USA), used at 1:5000 dilution, in combination with enhanced chemiluminescence (Super Signal; PIERCE Products, Rockford, IL, USA).
Results
Induction of apoptosis and necrosis in HEp-2 cells
HEp-2 cells exposed to 2 and 10 μM STS exhibited apoptotic-like fragmentation into multiple round bodies after 6 hours of treatment (Fig. 1c,1d). Interestingly, cells exposed to 10 μM STS showed a more pronounced loss of cytoplasmic membrane integrity (Fig. 1e), indicating that this STS concentration drove cell death more rapidly into necrosis. As control, cells treated with levels of H2O2 (0.1%) previously shown to provoke massive necrotic cell death [20] exhibited a characteristic necrotic morphology (Fig. 1b) associated with rapid loss of cytoplasmic membrane (Fig. 1e).
Figure 1.
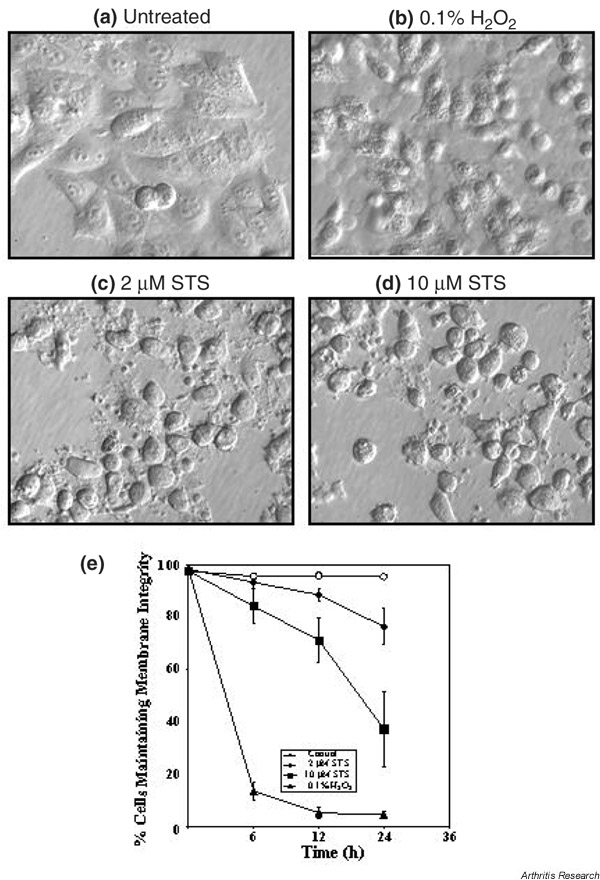
Morphology of HEp-2 cells. (a) Untreated HEp-2 cells, and HEp-2 cells after 6 hours of exposure to (b) hydrogen peroxide (H2O2), (c) 2 μM staurosporine (STS), and (d) 10 μM STS. Images were acquired using an Olympus IX-70 microscope equipped with Hoffman modulation contrast. (e) Time-dependent loss of HEp-2 cytoplasmic membrane integrity in control cells (open circles) and cells exposed to 0.1% H2O2 (triangles), to 2 μM STS (diamonds), and to 10 μM STS (squares). The loss of cytoplasmic membrane integrity, indicative of necrosis, was quantified using the trypan blue exclusion assay. Values are the mean ± standard deviation from three independent experiments.
Changes of the Golgi complex during apoptosis
Changes in the Golgi complex during apoptosis were examined by immunostaining with AGA in HEp-2 cells treated with 2 μM STS. Apoptosis was defined by stereotypic morphological changes, especially evident in the nucleus, where the chromatin condenses and compacts, and assumes a globular, crescent-shaped morphology [26]. Figure 2 shows a composite of four apoptotic stages, provisionally called stages I–IV (Fig. 2b,2c,2d,2e), that are classified on the basis of progression of apoptotic nuclear change (middle panels) and corresponding staining for golgin-97 (left panels).
Figure 2.
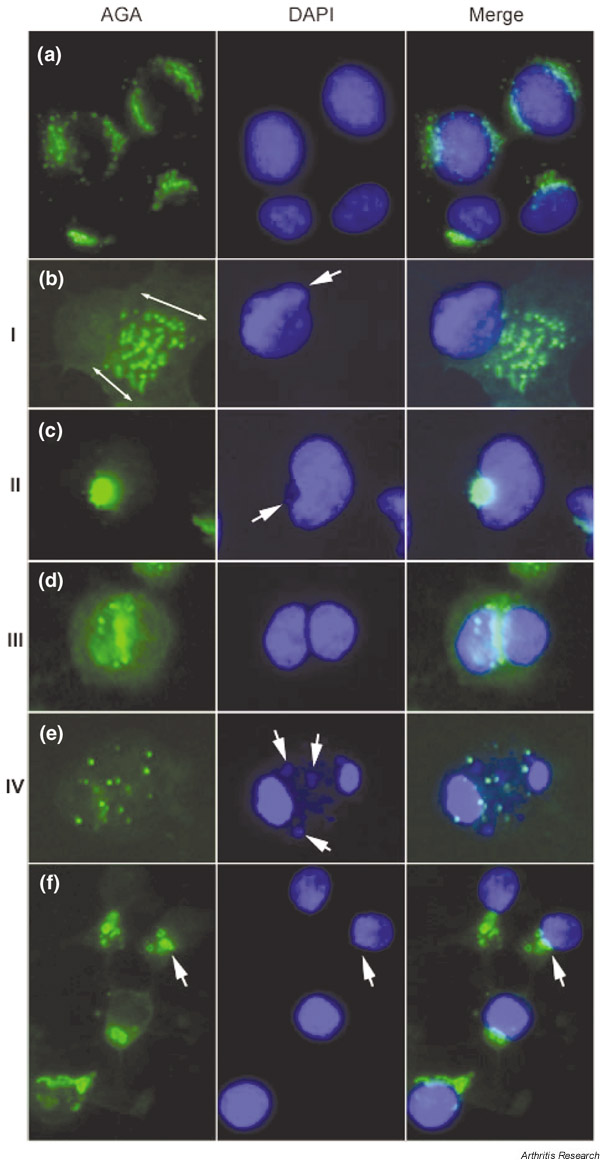
Changes of the Golgi complex during apoptosis and necrosis induced by staurosporine (STS). (a) Untreated HEp-2 cell control, and (b)–(e) cells treated with 2 μM STS for up to 6 hours for the induction of apoptosis. Apoptotic cells can be classified into four basic stages based on the nuclear change and the staining of Golgi antigens. (b) Stage I represents Golgi swelling (double-head arrows) that appears at an early stage when distinctive changes are seen in the shape of the nucleus (arrow). The area occupied by the swelled Golgi can be ~ 5–10 times the normal size for the Golgi complex in HEp-2 cells. (c) Stage II shows the characteristic condensation of the Golgi complex adjacent to the nucleus that was elongated and appeared as a kidney shape or a crescent shape (arrow). (d) Stage III is seen in cells with defined nuclear fragmentation into two approximately equal nuclear fragments, and immunostaining of the Golgi antigen was primarily located at the cleavage site between the two nuclear fragments. (e) In stage IV, nuclei were fragmented into multiple fragments with smaller pieces (middle panel, arrows) and Golgi staining appeared as vesicular structures. (f) HEp-2 cells 6 hours after treatment of 10 μM STS for the induction of necrosis. In contrast to apoptosis, striking fragmentation of the Golgi complex was observed with nuclear condensation rather than nuclear fragmentation. Immunostaining was performed using rabbit anti-golgin-97 and Alexa™ 488 conjugated goat anti-rabbit IgG antibody (left panels), and the nuclei were counterstained by 4',6-diamidino-2-phenylindole (DAPI) (middle panels). Right panels show the merged images. AGA, anti-Golgi complex autoantibodies.
About 50–60% of cells appeared unaffected after 2 hours of incubation in the presence of STS, as evidenced by perinuclear Golgi staining identical to untreated cells (Fig. 2a). Interestingly, 'swelling' of the Golgi complex with vesicular staining (stage I) was observed in 10–20% of cells, and is most strikingly demonstrated in Fig. 2b. It is estimated that the area occupied by the swelled Golgi can be ~ 5–10 times the normal size for Golgi complex in HEp-2 cells. Slight changes in the shape of some nuclei were observed, as shown in the middle panel, but most cells with Golgi swelling exhibited no change in nuclear size and shape.
About 10% of cells appeared to be in stage II (Fig. 2c) where there was elongation of the nucleus, appearing kidney or crescent shaped (middle panel). A characteristic of stage II was that the Golgi complex was condensed at the concave or indented region of the crescentic nucleus (right panel). Stage III (Fig. 2d) was seen in 3–5% of cells with defined nuclear fragmentation into two approximately equal fragments (middle panel) and golgin-97 primarily located at the cleavage zone between the two nuclear fragments (right panel). About 10–15% of the cells were considered to be at stage IV (Fig. 2e), where their nuclei were fragmented into more than two pieces (middle panel) and golgin-97 staining appeared as vesicular structures. This was the final stage when remnants of the Golgi complex could be detected.
Evaluation of cells after 6 hours of incubation with 2 μM STS showed all four stages described (data not shown). At this time, up to 50% of cells were in stage I, 20% were in stage II, 2–3% were in stage III, and 25% of cells were in stage IV. These data suggested that STS-treated cells do not enter the apoptotic pathway at the same time, which is consistent with the notion that apoptosis is a relatively asynchronous process.
Similar staining patterns to those seen with golgin-97 were observed when apoptotic HEp-2 cells were stained with antibodies to golgin-95, golgin-160, golgin-245, and giantin (data not shown). These results were also reproduced in HeLa S3 and mouse J774A.1 cells treated with 2 μM STS (data not shown). The amount of Golgi swelling observed in stage I varied in different experiments, but the characteristics for stages II–IV were highly reproducible in all the experiments we have conducted. Our observations of Golgi swelling during apoptosis are supported by previous studies. For instance, in neurons undergoing apoptosis, morphologic changes were characterized by a highly ordered sequence of organelle abnormalities, with swelling of endoplasmic reticulum and Golgi vesiculation that preceded most nuclear changes or mitochondrial disruption [27,28].
Changes of the Golgi complex during necrosis
In contrast to the Golgi swelling already described, striking fragmentation of the Golgi complex was observed during necrosis in HEp-2 cells treated with 10 μM STS (Fig. 2f). As shown in Fig. 1b,1e, approximately 50% of cells treated with this STS concentration, while initially showing apoptotic-like fragmentation into multiple bodies, gradually died by necrosis. Figure 2f shows Golgi fragments clearly visible (arrows) in cells with condensed nuclei after 6 hours of treatment with 10 μM STS. Note the absence of nuclear fragmentation in these cells compared with apoptotic cells (Fig. 2e), consistent with previous observations showing that necrotic nuclei exhibit condensation but not fragmentation [20]. In this treatment, designed to drive apoptotic cells more rapidly into necrosis, changes in the Golgi complex similar to the four stages observed in apoptosis were not detected. Similar results were also obtained for other Golgi autoantigens, including golgin-160, giantin, and golgin-95 (data not shown).
No Golgi staining was observed in HEp-2 cells treated with 10 μM STS for 24 hours or with 0.1% H2O2 (data not shown), presumably due to the extensive cytoplasmic damage associated with necrosis. These results suggested that the Golgi complex is affected differently in apoptosis and in necrosis, prompting us to examine whether specific Golgi proteins are targeted for proteolysis in these modes of cell death.
Golgi autoantigens are cleaved during apoptosis and necrosis
It is well established that specific intracellular autoantigens are cleaved into different fragments in Jurkat T cells and other cell types undergoing apoptosis and necrosis [20]. Mancini et al. reported that golgin-160 (native protein, 160 kDa) was cleaved into a 140 kDa fragment during apoptosis [29]. We therefore performed an immunoblotting analysis using extracts from Jurkat cells treated with apoptosis-inducing reagent (1 μM STS) or necrosis-inducing reagent (0.1% H2O2) to investigate whether Golgi autoantigens are cleaved in the two major types of cell death. PARP (110 kDa protein) was selected as a positive control to monitor the cleavage of proteins during apoptosis and necrosis because its cleavage products in Jurkat cells undergoing apoptosis (89 kDa) and undergoing necrosis (60 and 50 kDa) are well established [20,24,30].
Figure 3 shows the results of immunoblotting analysis using antibodies to PARP and four Golgi autoantigens. In cells treated with STS (Fig. 3, left) or H2O2 (Fig. 3, right), PARP was cleaved into an 89 kDa polypeptide (apoptotic fragments) or 60 and 50 kDa polypeptides (necrotic fragments), respectively, with complete degradation of native protein as previously described [24]. Golgin-160 was cleaved into 140 and 80 kDa fragments in apoptotic cells, consistent with published observations [29]. In contrast to the apoptotic fragments, a 70 kDa fragment was detected in necrotic cells. Giantin was cleaved into three major apoptotic fragments (150, 80, and 60 kDa), and these fragments were clearly different from the 75 and 50 kDa fragments derived from necrosis. Interestingly, while apoptotic fragments of golgin-95 and golgin-97 were not detected, necrotic fragments at 50 and 70 kDa were detected for both proteins. A summary of the Golgi protein fragments generated during apoptosis and necrosis is presented in Table 1.
Figure 3.
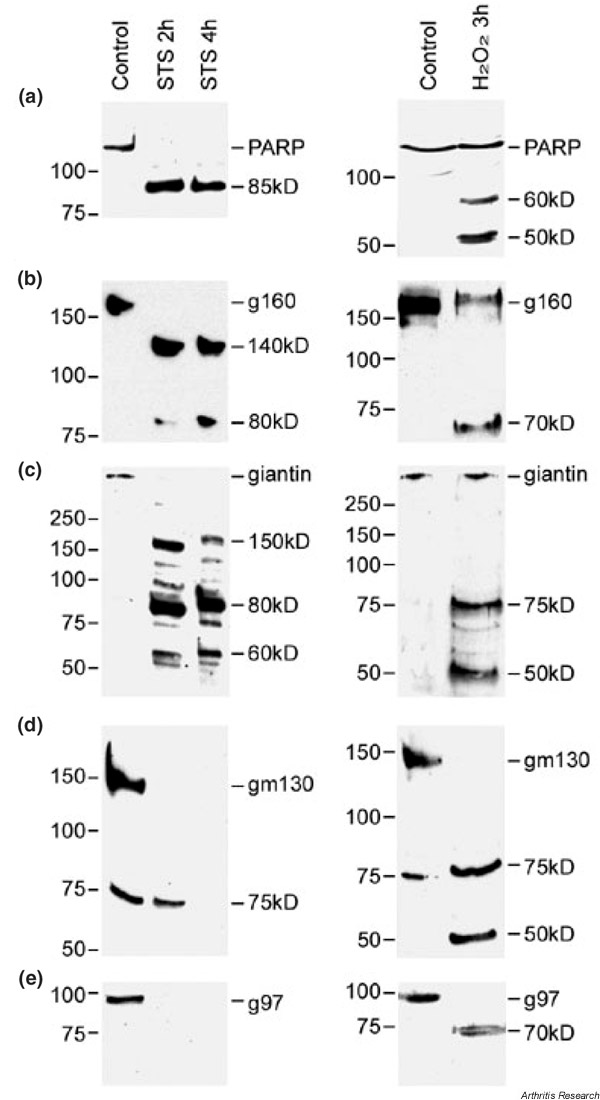
Immunoblot analysis of cleavage fragments of Golgi autoantigens (panels b–e) and polyADP-ribose polymerase (PARP, panel a) during apoptotic and necrotic cell death. Jurkat cells were exposed to either 1μM staurosporine (STS) for 2 or 4 hours (left) or 0.1% hydrogen peroxide (H2O2) for 3 hours (right) for the induction of apoptosis and necrosis, respectively. Intact protein and cleavage fragments are indicated. Numbers to left of each blot represent the relative molecular weight (kDa). Note that the 75 kDa band detected by anti-golgin-95 in the control as well as the sample treated with STS for 2 hours may represent an unrelated protein recognized by the antiserum. Also note that this 75 kDa protein was not detected in HEp-2 cells. g160, golgin-160; g97, golgin-97; gm130, golgin-95.
Table 1.
Fragmentation of Golgi autoantigens during apoptosis and necrosis in Jurkat cells.
| Golgi antigens | Molecular weight of intact protein (kDa) | Major apoptotic fragments (kDa) | Major necrotic fragments (kDa) |
| Giantin | 370 | 80, 60 | 75, 50 |
| Golgin-160 | 160 | 140, 80 | 70 |
| Golgin-95 | 130 | None | 50 |
| Golgin-97 | 97 | None | 70 |
Cleavage fragments were detected by immunoblotting of apoptotic and necrotic lysates.
Proteolysis of Golgi autoantigens induced by apoptosis is caspase dependent
The caspase family of proteases plays a central role in apoptosis by participating in a cascade of cleavage events that result in the apoptotic phenotype [31]. We investigated whether the proteolysis of Golgi autoantigens caused by treatment with STS was caspase dependent. Jurkat cells were treated with 1 μM STS for 4 hours after preincubation with the pan-caspase inhibitor 100 μM zVAD-fmk. The generation of proteolytic fragments from Golgi autoantigens was completely blocked in Jurkat cells pretreated with zVAD-fmk (Fig. 4), indicating that cleavage of Golgi autoantigens during apoptosis is mediated by caspases.
Figure 4.
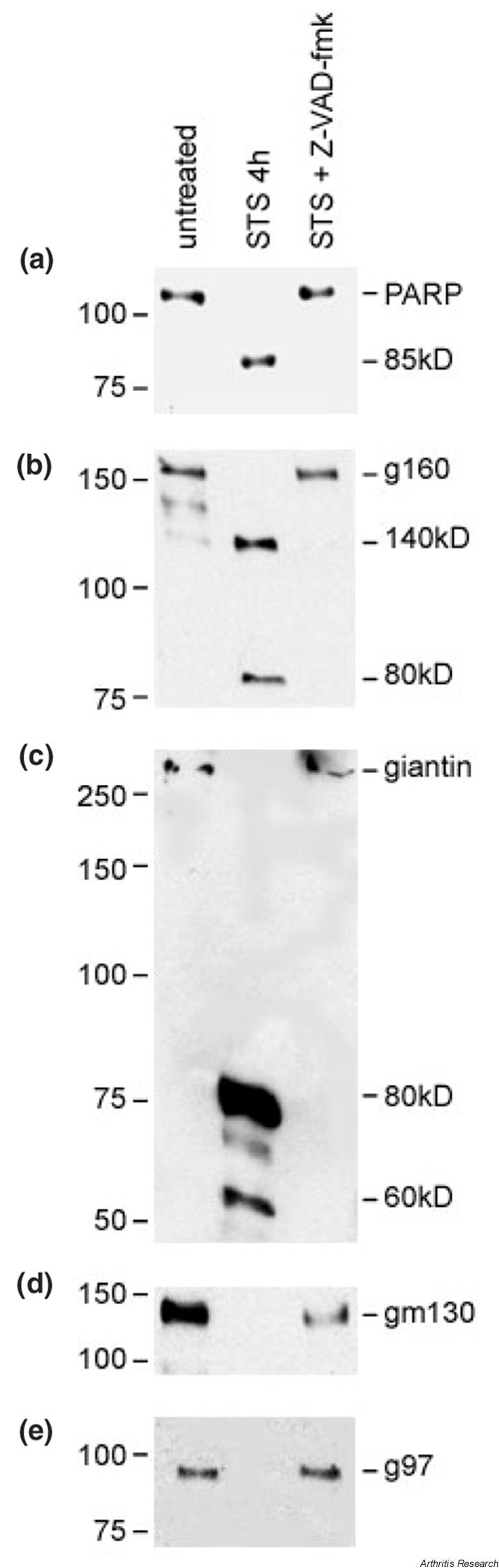
Apoptotic cleavage of Golgi autoantigens is caspase dependent. Benzylocarbonyl-Val-Ala-Asp-fluromethylketone (zVAD-fmk) (100 μM) was added to Jurkat cells 1 hour prior to addition of staurosporine (STS). Cells were then exposed to STS for 4 hours prior to harvest for lysate preparation. Representative blots of lysates from untreated control, 1 μM STS treatment without zVAD-fmk, and 1 μM STS treatment with zVAD-fmk are shown. Intact protein and cleavage fragments are indicated. Numbers to left of each blot represent the relative molecular weight (kDa). g160, golgin-160; g97, golgin-97; gm130, golgin-95; PARP, polyADP-ribose polymerase. See Figure 3 for explanation of a–e.
Discussion
Apoptotic changes of the Golgi complex
Lane et al. reported recently that the Golgi complex in HeLa cells is fragmented into scattered vesicles by caspases due to interference of Golgi reassembly and abnormalities of the 65 kDa stacking protein GRASP65 [32]. In the present report, we describe characteristic changes of the Golgi complex during the progression of apoptosis. It is not known whether the Golgi complex functions as a stress sensor involved in the regulation of apoptosis. Mancini et al. reported that caspase-2 was localized at the Golgi complex and generated a unique cleavage product of golgin-160 [29]. Furthermore, these investigators proposed that the Golgi complex transduces proapoptotic signals [29]. A precedent for this hypothesis was provided by Nakagawa et al., who reported that caspase-12 is localized to the endoplasmic reticulum and mediates an endoplasmic reticulum-specific apoptosis pathway [33]. In the present study, we have found that all Golgi autoantigens examined were cleaved in apoptosis in a caspase-dependent manner, as evidenced by studies showing that the pan-caspase inhibitor zVAD-fmk completely blocked the apoptotic fragmentation of these antigens. Although the requirement for specific caspases was not examined here, it is possible that caspases localized in the Golgi complex (including caspase-2) transduce death signals during the early stages of apoptosis that are associated with fragmentation of specific Golgi proteins.
It has been reported that apoptotic death receptors such as CD95 [34], tumor necrosis factor-related apoptosis inducing ligand receptor 1, tumor necrosis factor-related apoptosis inducing ligand receptor 2 [35], and tumor necrosis factor receptor 1 [36] are enriched in the Golgi complex prior to transport to the plasma membrane, thus suggesting that the Golgi complex may play an important role in apoptotic signalling. We now provide evidence for characteristic alterations in the Golgi complex corresponding to nuclear change during the progression of apoptosis. Swelling of the Golgi complex was particularly detected in the early stages of apoptosis. It could be speculated that apoptotic alterations of the Golgi complex may facilitate the transportation of death receptors from the Golgi complex to the plasma membrane, and may amplify the apoptotic events once apoptosis was initiated. Additional studies are needed to examine the precise function of the Golgi complex during apoptosis.
Role of apoptotic or necrotic fragments in autoantibody production
Analysis of post-translational modifications in intracellular autoantigens associated with apoptosis and necrosis should provide important insights into the possible role of cell death in the generation of potentially autoantigenic forms of cellular antigens. Casciola-Rosen and coworkers have proposed that modifications of autoantigens during cell death, particularly proteolytic cleavage, might be crucial for the generation of autoantibodies in autoimmune diseases [23,37]. Unique proteolytic fragments of α-fodrin and type 3 muscarinic acetylcholine receptor have been implicated in the generation of autoantibody responses in primary Sjögren's syndrome [38,39].
Although direct evidence that autoantigen cleavage fragments produced during cell death elicit autoantibody responses is still scarce, Pollard et al. reported that immunization of B10.S mice (H-2s) with a 19 kDa proteolytic cleavage fragment of fibrillarin (derived from nonapoptotic cell death) elicited antibodies that are comparable with human anti-fibrillarin autoantibodies and those derived from mice exposed to mercury [40]. In a study using human sera, Greidinger et al. reported that the recognition of apoptosis-derived and oxidatively modified forms of the 70 kDa subunit of U1 small nuclear ribonucleoprotein autoantigen was associated with distinct disease manifestations [41]. Furthermore, Oriss et al. demonstrated that a combination of antigen-processing cells and a fragment of DNA topoisomerase I efficiently elicited autoreactive T-cell proliferation, whereas the full-length topoisomerase I required additional stimulus of exogenous interleukin-2 [42].
These data point to a crucial role for fragments of autoantigens in the generation of autoantibody responses. In the present study, several Golgi autoantigens were detected cleaved into distinctive fragments during apoptosis and necrosis. It has been speculated that these modified forms of autoantigens may have enhanced immunogenicity through exposure of immunocryptic epitopes that are not generated during antigen processing [21,23,37,43]. These epitopes could trigger autoimmune responses if presented to the immune system under proinflammatory conditions [44], and they may be recognized as surface structures on cytoplasmic organelles that are released to the immune system in aberrant disease states.
Autoantibody responses could be amplified and maintained on repeated stimulation if the exposure of intracellular antigens to the immune system is associated with defective clearance of apoptotic cells, prolonged necrosis (primary or secondary), T-cell cytotoxicity associated with chronic infection, or even antigen mutation or overexpression. It would be important not only to assess the immunogenic potential of subcellular particles and proteolytic fragments released during cell death, but also to continue investigating possible defects leading to aberrant apoptosis or phagocyte function and/or aberrant antigen expression in systemic autoimmune diseases.
Kinetics of degradation of golgins and generation of apoptotic fragments
Although only well-defined apoptotic fragments of golgin-160 and giantin, not of golgin-95 and golgin-97, were detected after 2 or 4 hours of incubation with STS, total disappearance of intact proteins was detected with all Golgi autoantigens and complete blockage was observed in the presence of z-VAD-fmk. These results suggested that there might be differences in the kinetics of degradation among the golgins, with golgin-95 and golgin-97 perhaps being more sensitive to proteolysis and being targeted very early during apoptosis. While the absence of apoptotic fragments for golgin-95 and golgin-97 suggests that total degradation of these proteins occurred without generation of intermediate protease-resistant fragments, it cannot be ruled out that the intact proteins were released from apoptotic cells or that their apoptotic fragments were not recognized by autoantibodies.
In the present study, Jurkat cells were relatively sensitive to the treatment with STS such that the kinetics of degradation of different golgins relative to that of PARP could not be accurately differentiated. Since HEp-2 cells grown as a monolayer were observed to be more resistant to apoptosis compared with Jurkat cells, we used HEp-2 cells to investigate more precisely the relative kinetics of degradation for the native golgins during apoptosis.
An identical pattern of fragmentation was essentially observed for all the Golgi autoantigens examined except that the overall kinetics of native protein degradation was slower (data not shown). The integrity of native golgin-95 and golgin-97, in which apoptotic fragments were not detected, was almost completely maintained at 12 hours after the addition of STS (data not shown). In contrast, native golgin-160 and giantin were degraded into their respective fragments 12 hours after the addition of STS. The processing of golgin-160 and giantin occurred as early as that for PARP, suggesting that these proteins are cleaved during the onset of apoptosis (data not shown). Cleavage of golgin-160 and giantin may thus play a potentially important role in the early stages of apoptosis.
Although we did not determine which golgins are substrates for specific caspases, further studies aimed at defining the caspases responsible for golgin cleavage, the cleavage sites, and the biological consequences of these cleavages should provide important insights into the possible role of some of the Golgi autoantigens in apoptosis.
Conclusion
The present study has shown overall morphologic changes of the Golgi complex during apoptosis and necrosis, with distinct fragmentation patterns of its associated autoantigens. We propose that cleavage of golgin-160 and giantin may play a role in early events in apoptosis. We also speculate that Golgi fragmentation in cell death might participate in sustaining autoantibody response to the Golgi complex in aberrant disease states characterized by unregulated cell death resulting from injury or infection or defective clearance of dying cells.
Abbreviations
AGA = anti-Golgi complex autoantibodies; H2O2 = hydrogen peroxide; PARP = polyADP-ribose polymerase; PBS = phosphate-buffered saline; STS = staurosporine; zVAD-fmk = benzylocarbonyl-Val-Ala-Asp-fluromethylketone.
Acknowledgments
Acknowledgments
The present study is publication 14756-MEM from The Scripps Research Institute. This work was supported in part by National Institutes of Health Grants AI39645 and AI47859 (EKLC), AI44088 (CAC), Canadian Institutes for Health Research Grant MOP-38034 (MJF), and the Sam and Rose Stein Charitable Trust.
References
- Rodriguez JL, Gelpi C, Thomson TM, Real FJ, Fernandez J. Anti-Golgi complex autoantibodies in a patient with Sjögren syndrome and lymphoma. Clin Exp Immunol. 1982;49:579–586. [PMC free article] [PubMed] [Google Scholar]
- Fritzler MJ, Etherington J, Sokoluk C, Kinsella TD, Valencia DW. Antibodies from patients with autoimmune disease react with a cytoplasmic antigen in the Golgi apparatus. J Immunol. 1984;132:2904–2908. [PubMed] [Google Scholar]
- Hong HS, Morshed SA, Tanaka S, Fujiwara T, Ikehara Y, Nishioka M. Anti-Golgi antibody in rheumatoid arthritis patients recognizes a novel antigen of 79 kDa (doublet) by western blot. Scand J Immunol. 1992;36:785–792. doi: 10.1111/j.1365-3083.1992.tb03140.x. [DOI] [PubMed] [Google Scholar]
- Mayet WJ, Hermann E, Csernok E, Knuth A, Poralla T, Gross WL, Meyer zum Buschenfelde KH. A human renal cancer line as a new antigen source for the detection of antibodies to cytoplasmic and nuclear antigens in sera of patients with Wegener's granulomatosis. J Immunol Methods. 1991;143:57–68. doi: 10.1016/0022-1759(91)90272-h. [DOI] [PubMed] [Google Scholar]
- Kooy J, Toh BH, Gleeson PA. Heterogeneity of human anti-Golgi autoantibodies: reactivity with components from 35 to 260 kDa. Immunol Cell Biol. 1994;72:123–127. doi: 10.1038/icb.1994.19. [DOI] [PubMed] [Google Scholar]
- Chan EKL, Fritzler MJ. Golgins: coiled-coil proteins associated with the Golgi Complex. Electron J Biotechnol. 1998;1:1–10. [Google Scholar]
- Fritzler MJ, Hamel JC, Ochs RL, Chan EKL. Molecular characterization of two human autoantigens: Unique cDNAs encoding 95- and 160-kD proteins of a putative family in the Golgi complex. J Exp Med. 1993;178:49–62. doi: 10.1084/jem.178.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misumi Y, Sohda M, Yano A, Fujiwara T, Ikehara Y. Molecular characterization of GCP170, a 170-kDa protein associated with the cytoplasmic face of the Golgi membrane. J Biol Chem. 1997;272:23851–23858. doi: 10.1074/jbc.272.38.23851. [DOI] [PubMed] [Google Scholar]
- Griffith KJ, Chan EKL, Lung CC, Hamel JC, Guo X, Miyachi K, Fritzler MJ. Molecular cloning of a novel 97-kd Golgi complex autoantigen associated with Sjögren's syndrome. Arthritis Rheum. 1997;40:1693–1702. doi: 10.1002/art.1780400920. [DOI] [PubMed] [Google Scholar]
- Fritzler MJ, Lung CC, Hamel JC, Griffith K, Chan EKL. Molecular characterization of golgin-245: a novel Golgi complex protein containing a granin signature. J Biol Chem. 1995;270:31262–31268. doi: 10.1074/jbc.270.52.31262. [DOI] [PubMed] [Google Scholar]
- Erlich R, Gleeson PA, Campbell P, Dietzsch E, Toh BH. Molecular characterization of trans-Golgi p230. A human peripheral membrane protein encoded by a gene on chromosome 6p12-22 contains extensive coiled-coil alpha-helical domains and a granin motif. J Biol Chem. 1996;271:8328–8337. doi: 10.1074/jbc.271.14.8328. [DOI] [PubMed] [Google Scholar]
- Kooy J, Toh BH, Pettitt JM, Erlich R, Gleeson PA. Human autoantibodies as reagents to conserved Golgi components. Characterization of a peripheral, 230-kDa compartment-specific Golgi protein. J Biol Chem. 1992;267:20255–20263. [PubMed] [Google Scholar]
- Sohda M, Misumi Y, Fujiwara T, Nishioka M, Ikehara Y. Molecular cloning and sequence analysis of a human 372-kDA protein localized in the Golgi complex. Biochem Biophys Res Commun. 1994;205:1399–1408. doi: 10.1006/bbrc.1994.2821. [DOI] [PubMed] [Google Scholar]
- Seelig HP, Schranz P, Schroter H, Wiemann C, Renz M. Macro-golgin–a new 376 kD Golgi complex outer membrane protein as target of antibodies in patients with rheumatic diseases and HIV infections. J Autoimmun. 1994;7:67–91. doi: 10.1006/jaut.1994.1006. [DOI] [PubMed] [Google Scholar]
- Eystathioy T, Jakymiw A, Fujita DJ, Fritzler MJ, Chan EKL. Human autoantibodies to a novel Golgi protein golgin-67: high similarity with golgin-95/gm 130 autoantigen. J Autoimmun. 2000;14:179–187. doi: 10.1006/jaut.1999.0359. [DOI] [PubMed] [Google Scholar]
- Munro S, Nichols BJ. The GRIP domain–a novel Golgi-targeting domain found in several coiled-coil proteins. Curr Biol. 1999;9:377–380. doi: 10.1016/s0960-9822(99)80166-3. [DOI] [PubMed] [Google Scholar]
- Orci L, Stamnes M, Ravazzola M, Amherdt M, Perrelet A, Sollner TH, Rothman JE. Bidirectional transport by distinct populations of COPI-coated vesicles. Cell. 1997;90:335–349. doi: 10.1016/s0092-8674(00)80341-4. [DOI] [PubMed] [Google Scholar]
- Orci L, Perrelet A, Rothman JE. Vesicles on strings: morphological evidence for processive transport within the golgi stack. Proc Natl Acad Sci USA. 1998;95:2279–2283. doi: 10.1073/pnas.95.5.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnichsen B, Lowe M, Levine T, Jamsa E, Dirac-Svejstrup B, Warren G. A role for giantin in docking COPI vesicles to Golgi membranes. J Cell Biol. 1998;140:1013–1021. doi: 10.1083/jcb.140.5.1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casiano CA, Ochs RL, Tan EM. Distinct cleavage products of nuclear proteins in apoptosis and necrosis revealed by autoantibody probes. Cell Death Differ. 1998;5:183–190. doi: 10.1038/sj/cdd/4400336. [DOI] [PubMed] [Google Scholar]
- Utz PJ, Anderson P. Posttranslational protein modifications, apoptosis, and the bypass of tolerance to autoantigens. Arthritis Rheum. 1998;41:1152–1160. doi: 10.1002/1529-0131(199807)41:7<1152::AID-ART3>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- van Venrooij WJ, Pruijn GJ. Citrullination: a small change for a protein with great consequences for rheumatoid arthritis. Arthritis Res. 2000;2:249–251. doi: 10.1186/ar95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casciola-Rosen L, Andrade F, Ulanet D, Wong WB, Rosen A. Cleavage by granzyme B is strongly predictive of autoantigen status: implications for initiation of autoimmunity. J Exp Med. 1999;190:815–826. doi: 10.1084/jem.190.6.815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Molinaro C, Johnson N, Casiano CA. Secondary necrosis is a source of proteolytically modified forms of specific intracellular autoantigens: implications for systemic autoimmunity. Arthritis Rheum. 2001;44:2642–2652. doi: 10.1002/1529-0131(200111)44:11<2642::aid-art444>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- Andrade LEC, Chan EKL, Peebles CL, Tan EM. Two major autoantigen-antibody systems of the mitotic spindle apparatus. Arthritis Rheum. 1996;39:1643–1653. doi: 10.1002/art.1780391006. [DOI] [PubMed] [Google Scholar]
- Leist M, Jaattela M. Four deaths and a funeral: from caspases to alternative mechanisms. Nat Rev Mol Cell Biol. 2001;2:589–598. doi: 10.1038/35085008. [DOI] [PubMed] [Google Scholar]
- Smith RG, Siklos L, Alexianu ME, Engelhardt JI, Mosier DR, Colom L, Habib Mohamed A, Appel SH. Autoimmunity and ALS. Neurology. 1996;47:40–45. doi: 10.1212/wnl.47.4_suppl_2.40s. [DOI] [PubMed] [Google Scholar]
- Portera-Cailliau C, Price DL, Martin LJ. Excitotoxic neuronal death in the immature brain is an apoptosis–necrosis morphological continuum. J Comp Neurol. 1997;378:70–87. [PubMed] [Google Scholar]
- Mancini M, Machamer CE, Roy S, Nicholson DW, Thornberry NA, Casciola-Rosen LA, Rosen A. Caspase-2 is localized at the Golgi complex and cleaves golgin-160 during apoptosis. J Cell Biol. 2000;149:603–612. doi: 10.1083/jcb.149.3.603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gobeil S, Boucher CC, Nadeau D, Poirier GG. Characterization of the necrotic cleavage of poly(ADP-ribose) polymerase (PARP-1): implication of lysosomal proteases. Cell Death Differ. 2001;8:588–594. doi: 10.1038/sj.cdd.4400851. [DOI] [PubMed] [Google Scholar]
- Creagh EM, Martin SJ. Caspases: cellular demolition experts. Biochem Soc Trans. 2001;29:696–702. doi: 10.1042/0300-5127:0290696. [DOI] [PubMed] [Google Scholar]
- Lane JD, Lucocq J, Pryde J, Barr FA, Woodman PG, Allan VJ, Lowe M. Caspase-mediated cleavage of the stacking protein GRASP65 is required for Golgi fragmentation during apoptosis. J Cell Biol. 2002;156:495–509. doi: 10.1083/jcb.200110007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BA, Yuan J. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature. 2000;403:98–103. doi: 10.1016/S0168-9002(97)01086-3. [DOI] [PubMed] [Google Scholar]
- Bennett M, Macdonald K, Chan SW, Luzio JP, Simari R, Weissberg P. Cell surface trafficking of Fas: a rapid mechanism of p53-mediated apoptosis. Science. 1998;282:290–293. doi: 10.1002/(SICI)1097-010X(19981015)282:3<290::AID-JEZ2>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Zhang XD, Franco AV, Nguyen T, Gray CP, Hersey P. Differential localization and regulation of death and decoy receptors for TNF-related apoptosis-inducing ligand (TRAIL) in human melanoma cells. J Immunol. 2000;164:3961–3970. doi: 10.4049/jimmunol.164.8.3961. [DOI] [PubMed] [Google Scholar]
- Jones SJ, Ledgerwood EC, Prins JB, Galbraith J, Johnson DR, Pober JS, Bradley JR. TNF recruits TRADD to the plasma membrane but not the trans-Golgi network, the principal subcellular location of TNF-R1. J Immunol. 1999;162:1042–1048. [PubMed] [Google Scholar]
- Andrade F, Roy S, Nicholson D, Thornberry N, Rosen A, Casciola-Rosen L. Granzyme B directly and efficiently cleaves several downstream caspase substrates: implications for CTL-induced apoptosis. Immunity. 1998;8:451–460. doi: 10.1016/s1074-7613(00)80550-6. [DOI] [PubMed] [Google Scholar]
- Haneji N, Nakamura T, Takio K, Yanagi K, Higashiyama H, Saito I, Noji S, Sugino H, Hayashi Y. Identification of alpha-fodrin as a candidate autoantigen in primary Sjögren's syndrome. Science. 1997;276:604–607. doi: 10.1126/science.276.5312.604. [DOI] [PubMed] [Google Scholar]
- Nagaraju K, Cox A, Casciola-Rosen L, Rosen A. Novel fragments of the Sjogren's syndrome autoantigens alpha-fodrin and type 3 muscarinic acetylcholine receptor generated during cytotoxic lymphocyte granule-induced cell death. Arthritis Rheum. 2001;44:2376–2386. doi: 10.1002/1529-0131(200110)44:10<2376::aid-art402>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- Pollard KM, Pearson DL, Bluthner M, Tan EM. Proteolytic cleavage of a self-antigen following xenobiotic-induced cell death produces a fragment with novel immunogenic properties. J Immunol. 2000;165:2263–2270. doi: 10.4049/jimmunol.165.4.2263. [DOI] [PubMed] [Google Scholar]
- Greidinger EL, Casciola-Rosen L, Morris SM, Hoffman RW, Rosen A. Autoantibody recognition of distinctly modified forms of the U1-70-kd antigen is associated with different clinical disease manifestations. Arthritis Rheum. 2000;43:881–888. doi: 10.1002/1529-0131(200004)43:4<881::AID-ANR20>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Oriss TB, Hu PQ, Wright TM. Distinct autoreactive T cell responses to native and fragmented DNA topoisomerase I: influence of APC type and IL-2. J Immunol. 2001;166:5456–5463. doi: 10.4049/jimmunol.166.9.5456. [DOI] [PubMed] [Google Scholar]
- Casciola-Rosen LA, Anhalt GJ, Rosen A. DNA-dependent protein kinase is one of a subset of autoantigens specifically cleaved early during apoptosis. J Exp Med. 1995;182:1625–1634. doi: 10.1084/jem.182.6.1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rovere P, Sabbadini MG, Fazzini F, Bondanza A, Zimmermann VS, Rugarli C, Manfredi AA. Remnants of suicidal cells fostering systemic autoaggression. Apoptosis in the origin and maintenance of autoimmunity. Arthritis Rheum. 2000;43:1663–1672. doi: 10.1002/1529-0131(200008)43:8<1663::AID-ANR1>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]