

Abstract
The catalytic determinants for the cleavage and ligation reactions mediated by the hairpin ribozyme are integral to the polyribonucleotide chain. We describe experiments that place G8, a critical guanosine, at the active site, and point to an essential role in catalysis. Cross-linking and modeling show that formation of a catalytic complex is accompanied by a conformational change in which N1 and O6 of G8 become closely apposed to the scissile phosphodiester. UV cross-linking, hydroxyl-radical footprinting and native gel electrophoresis indicate that G8 variants inhibit the reaction at a step following domain association, and that the tertiary structure of the inactive complex is not measurably altered. Rate–pH profiles and fluorescence spectroscopy show that protonation at the N1 position of G8 is required for catalysis, and that modification of O6 can inhibit the reaction. Kinetic solvent isotope analysis suggests that two protons are transferred during the rate-limiting step, consistent with rate-limiting cleavage chemistry involving concerted deprotonation of the attacking 2′-OH and protonation of the 5′-O leaving group. We propose mechanistic models that are consistent with these data, including some that invoke a novel keto–enol tautomerization.
Keywords: cleavage/hairpin ribozyme/mechanism
Introduction
The hairpin ribozyme is a small catalytic motif that catalyzes a reversible, site-specific RNA cleavage reaction essential for the propagation of plant RNA replicons, and is an important model system for understanding biological catalysis by RNA (Figure 1A; Fedor, 2000). Cleavage requires deprotonation of the 2′-OH group of substrate nucleotide A–1 and a nucleophilic attack on the phosphorus within the A–1·G+1 linkage, yielding cleavage products with 5′-OH and 2′-3′-cyclic phosphate termini (Buzayan et al., 1986). The reaction mechanism of the hairpin ribozyme is of particular interest, in that metal ions are not direct participants in the reaction chemistry, indicating that the RNA itself is likely to play a direct role in acid–base catalysis (Hampel and Cowan, 1997; Nesbitt et al., 1997; Young et al., 1997; Murray et al., 1998). However, the pKa values of nucleobases G, C, A and U, which are <4.5 or >9, appear to be inappropriate for efficient acid–base catalysis at neutral pH. These pKa values may be shifted to values closer to neutrality in the tertiary structure of catalytic RNA molecules (Muth et al., 2000). Here we have examined the role of guanosine 8 (G8) in the cleavage reaction (Pinard et al., 2001; Rupert and Ferré-D’Amaré, 2001). Our results strongly suggest that N1, and possibly O6, of G8 participate in the reaction chemistry and lead us to propose specific mechanistic models for the hairpin ribozyme.


Fig. 1. (A) Ribozyme constructs used in this study. Constructs that are used for structural assays have 2′-deoxy substitutions of the A–1 nucleotide in order to prevent ribozyme-mediated cleavage. For selected structure probing experiments, the two-way junction construct was modified to connect A50 with the 5′ end of the substrate through a C5 linker. (B) Docking of the two-way junction ribozyme showing the key inter-domain base pair between G+1 of the substrate (lower case) and C25 of the ribozyme (upper case). UV-cross-linking shows that, upon docking, G8 stacks on A–1 (double-headed arrow). The heavy arrow indicates the cleavage site. Nucleotides surrounding the cleavage site where the backbone is strongly protected from hydroxyl-radical cleavage are indicated by gray shading. (C) Model of the hairpin ribozyme active site showing the proposed catalytic base, G8 (light blue), stacked on A–1 (red), and the Watson–Crick base pair between G+1 (red) and C25 (green). Distances between N1 of G8 and the 5′-O leaving group of G+1 (2.42 Å), and between O6 of G8 and the attacking nucleophile, 2′-OH of A–1 (3.16 Å), are shown. The SN2 angle measured is 154°. The model was developed using MC-SYM software, and subjected to energy minimization. With the exception of A–1, the bases shown are essentially invariant in both natural and artificial phylogenies of the hairpin ribozyme.
Results and discussion
The importance of G8 for hairpin ribozyme activity
To form a catalytic complex, the two domains (A and B) of the hairpin–ribozyme complex must interact with one another during a step termed docking (Figure 1; Butcher et al., 1995; Murchie et al., 1998; Walter et al., 1998). The hairpin ribozyme reactions can be inhibited by base substitutions, backbone modifications, denaturation, and chelation of divalent cations (Walter and Burke, 1998; Fedor, 2000). Fluorescence resonance energy transfer (FRET) and hydroxyl-radical footprinting each showed that inhibition of the reaction by any of these methods is almost always accompanied by a loss of docking ability, leading to the hypothesis that inhibition resulted from disruption of interdomain tertiary contacts rather than from the loss of essential catalytic determinants (Hampel et al., 1998; Walter and Burke, 1998; Walter et al., 1998). Therefore, we developed ribozyme constructs with improved folding properties and higher docking efficiencies, including a two-way helical junction with modified sequences, and a native-like four-way junction (Figure 1A) to re-examine those bases that were found to be important for the activity of earlier folding-deficient ribozymes (Esteban et al., 1998; Walter et al., 1999). Strikingly, these constructs strongly suppress the loss of activity induced by base substitutions (for A9, A10, G21, A22, A23, A24, A40, U41) (Hampel et al., 1990; Berzal-Herranz et al., 1993; Schmidt et al., 1996; Hampel et al., 1998; Walter and Burke, 1998) that have strong effects in the folding-deficient ribozyme constructs (J.Han, R.Pinard, J.E.Heckman, K.J.Hampel and J.M.Burke, unpublished data). Except for the replacement of A–1 with 2′-deoxy or 2′-O-methyl adenosine, G8 is the only nucleotide in the ribozyme–substrate complex where substitutions have been found to inhibit cleavage dramatically at a step that follows docking. Moreover, it has been shown recently using FRET that the ion-induced folding of a G8U variant, which strongly inhibits catalysis, is virtually identical to that of the wild-type sequence ribozyme (Wilson et al., 2001). The only other bases where substitutions strongly inhibit catalysis by stably docked versions of the hairpin ribozyme that we have used (two- and four-way junction constructs) without severely affecting docking are G+1 and C25, which form an essential but interchangeable interdomain Watson–Crick base pair (Berzal-Herranz et al., 1993; Pinard et al., 1999a; Rupert and Ferré-D’Amaré, 2001; Wilson et al., 2001).
Photo-cross-linking analysis of unmodified RNA demonstrates that G8 undergoes a conformational change during docking of the two domains. G8 is unreactive to UV-induced cross-linking when domain B is absent, when chelators are used to inhibit docking and when mutations that inhibit docking are introduced into the ribozyme or substrate. However, upon docking in cobalt(III) hexammine, G8 can be cross-linked to A–1 (Pinard et al., 2001), the nucleotide immediately 5′ to the cleavage site, indicating cross-strand stacking of bases (Figure 1B). G8·A–1 stacking occurs at the same rate constant, within experimental error, as docking of the two domains, indicating that the two processes are concerted or occur in rapid sequence (Pinard et al., 2001).
Molecular modeling with MC-SYM (Major et al., 1991) has been conducted using G8·A–1 stacking as a structural constraint, together with available biochemical data. Previously, we successfully employed this approach to predict, and then experimentally demonstrate, a C25·G+1 tertiary base pair that is a key interdomain contact and a significant factor in stabilizing loop–loop interactions (Pinard et al., 1999a). An energy-minimized model fulfilling these constraints indicates that G8 stacks over A–1, and predicts that the Watson–Crick face of G8 is closely apposed to the scissile bond (Figure 1B), physically suggesting the involvement of this nucleotide in reaction chemistry. This model is part of a three-dimensional representation of domain A (Pinard et al., 2001) in the docked complex and of a complete model of the hairpin ribozyme currently being built and refined in our laboratories using chemical protections and inter-domain as well as intra-domain cross-links (D.Lambert, R.Pinard, J.E.Heckman, K.J.Hampel, J.M.Burke and F.Major, manuscript in preparation). The recent 2.4 Å resolution crystal structure of the hairpin ribozyme confirms the presence of the inter-domain G+1·C25 base pair and, strikingly, indicates that G8 is located in very close proximity to the cleavage site, as predicted by our model (Rupert and Ferré-D’Amaré, 2001). With respect to the positioning of G8 at the active site, the MC-SYM model predicts that the N1 proton is within 3 Å of the 5′-O of G+1, while in the crystal structure this proton is within hydrogen bonding distance of the 2′-O of A–1, the nucleophile for the cleavage reaction.
Effect of G8 variants on tertiary structure formation
The observations that G8 is essential for a step that follows docking and moves to a position closely apposed to the reactive linkage upon docking lead us to suggest that G8 is an essential component of the active site and directly participates in the reaction chemistry. To test this hypothesis, we examined the effect of a wide range of base substitutions at position 8 (Figure 2), and found that only a subset of these variants retains catalytic activity. Since we have shown that several mutations perturb cleavage activity by impairing the formation of a stable docked complex, it was imperative to study how G8 variants affect the docking process and the tertiary structure of the docked complex. Three complementary approaches were used. In order to arrest the normal cleavage reaction at a step after docking, the substrate strand in these experiments contains a 2′-deoxy-substitution at A–1. This substitution inhibits the cleavage reaction by disabling the intramolecular nucleophile, but does not affect docking of the A and B domains (Walter et al., 1998).

Fig. 2. Activity of ribozymes with G8 modifications. Single-turnover cleavage rate constants (single or double exponential) were determined for the two-way junction trans-cleavage construct shown in Figure 1A.
Native gel electrophoresis at pH 7.5 reveals the fraction of docked molecules, and shows that all variants at position 8, except 7-deaza 2′-deoxyxanthosine and cytosine, dock efficiently, including those that strongly inhibit the reaction (Figure 3A). Hydroxyl-radical footprinting reflects the tertiary structure of the docked molecules at single-nucleotide resolution; in all variants examined at pH 7.0, identical ribose moieties were protected and no additional protections were observed (Figure 3B; data not shown), indicating that, to the limit of the method’s resolution, the final docked conformation of all G8 variants appears to be identical to that of the unmodified RNA. For the 2′-deoxypurine variant, a decrease in protection at all sites was observed, but the identity of the protected sites and their relative intensity did not change (data not shown). This result is consistent with the slight decrease in docking efficiency observed by native gel analysis (Figure 3A, lane 11).



Fig. 3. The structural phenotypes of G8 mutants. In all three experiments cleavage was inhibited by a 2′-deoxy substitution of the A–1 nucleotide. This modification does not impair docking efficiency (Walter et al., 1998). (A) Non-denaturing polyacrylamide gel analysis of the two-way junction ribozyme variants at pH 7.5. The construct used is shown at the top left. The linker between the 3′ portion of helix three and the 5′ end of the substrate is required in order to observe the gel shift upon inter-domain docking (K.J.Hampel, unpublished data). C25A and G+1A mutants served as undocked controls (Walter et al., 1998; Pinard et al., 1999a). Docking requires multivalent cations; thus, the minus Mg2+ gel served as a control for the cation dependence of the formation of a docked complex. (B) Hydroxyl-radical footprinting of the wild-type and G8–2-aminopurine-variant ribozyme–substrate complexes at pH 7.0. The constructs used are shown above the respective gels. The formation of a solvent-inaccessible core structure requires assembly of the complete secondary structure and Mg2+. Protected regions are indicated by black bars to the right of each set of lanes. The assignment of the identities of protected residues is shifted one nucleotide below the corresponding band on partial alkali hydrolysis (OH) and RNase T1 digest (T1) lanes. (C) UV-cross-linking analysis of wild-type and G8 variants at pH 7.5. Wild-type and variant two-way junction trans-cleavage ribozyme–substrate complexes (Figure 1A) were incubated in the absence (lane 1) or presence (lanes 2–7) of 1 mM Co(NH3)63+. The docking-dependent cross-links between G8 and A–1, and between G36 and U+2, are indicated by arrows to the right of the autoradiogram. X indicates an intra-molecular cross-link involving substrate alone.
To probe local tertiary structure at the active site, we examined two docking-dependent photo-cross-links at pH 7.5: the intradomain A–1·G8 cross-link described above, and an inter-domain cross-link between U+2 in the substrate and G36 in domain B (Figure 3C; J.E.Heckman and J.M.Burke, unpublished data). Both of the highly unreactive variants that were examined, 2-aminopurine and O6-methylguanosine, formed identical cross-links with efficiencies similar to G8, indicating that they preserve the local tertiary structure required to generate these specific docking-dependent cross-links. When G8 was replaced with 2,6-diaminopurine or with 6-thioguanosine, two nucleobases that are known to have enhanced photo-cross-linking activity, UV-irradiation yielded cross-linked products identical in mobility to those obtained with G, and the efficiency of the A–1 position 8 cross-link formation was higher than with G8, as expected. In the case of inosine, and other variants at position 8 that lack significant absorbance of UV-light at the wavelength used (312 nm), we were not able to detect the A–1 position 8 cross-link, although the U+2·G36 cross-link was still detected.
This careful analysis of the G8 variants clearly indicates that the two domains interact with one another and that the tertiary structure of the native sequence is not detectably perturbed by inactivating mutations. We conclude that the loss of activity by the G8 variants cannot be attributed to a loss of docking or to changes in the tertiary structure of the docked complex.
G8 is involved in the reaction chemistry
The rate-limiting step of the hairpin ribozyme reaction has been shown to follow docking (Walter et al., 1998), but its nature has been unclear. The rate of the reaction is virtually independent of solvent pH over a broad range (Nesbitt et al., 1997), in sharp contrast to the well-characterized hammerhead ribozyme, where the reaction rate is directly proportional to the concentration of hydroxide ions, an effect attributed to the participation of metal hydroxides or hydroxide ions in the mechanistic chemistry (Dahm and Uhlenbeck, 1991; Slim and Gait, 1991; Dahm et al., 1993; McKay, 1996). The relative pH independence of the hairpin ribozyme reaction initially suggested that a slow, pH-independent conformational change could mask a rapid, pH-dependent chemical step (Nesbitt et al., 1997; Walter et al., 1998). An alternative explanation is that the reaction is pH independent simply because hydroxide ions do not participate in the reaction chemistry. Deuterium isotope substitution results in a 3- to 4-fold decrease in reaction rate over a wide range of pH values, consistent with a rate-limiting chemical step (see below).
The reaction catalyzed by the hairpin ribozyme is metal ion independent, indicating that the RNA itself may function in acid–base catalysis (Hampel and Cowan, 1997; Nesbitt et al., 1997; Young et al., 1997; Murray et al., 1998; Walter and Burke, 1998). In the hairpin ribozyme, metal ions or metal-bound water would function to organize the tertiary structure of the ribozyme–substrate complex in a manner that facilitates catalysis by the RNA where a specific nucleotide (e.g. G8) would assume the role of the general acid and/or base. Metal-binding sites have been identified, but are located well away from G8 and the reactive phosphodiester (Walter et al., 2000; Rupert and Ferré-D’Amaré, 2001).
In our energy-minimized model, the attacking 2′-OH is positioned for an in-line SN2 attack on the scissile phosphorous, while N1, and potentially O6, of G8 are located at positions that are highly suggestive of participation in mechanistic chemistry (Figure 1C), functioning as proton donor and acceptor, respectively. To examine this hypothesis, we incorporated several guanine analogs at position 8, including those with altered N1 pKa values, and examined their cleavage activity as a function of pH (Figure 4B). These experiments were carried out using a construct with a shortened, 10 nucleotide substrate such that the helix 1 pairing has been reduced from 6 to 2 bp. This construct has been shown to prohibit religation of cleavage products after product release (K.J.Hampel and J.M.Burke, unpublished observations). Variants with acidic N1 pKa values (adenine, 2-aminopurine and 2,6-diaminopurine) were severely impaired at elevated pH (>8.0), but activity could be rescued at lower pH. Bases with alkaline N1 pKa values, guanosine and inosine, did not show a loss of activity at high pH values. These results support the proposal that protonation of N1 specifically at position 8 is required for cleavage. Moreover, a careful comparative analysis of the rate–pH profiles for the trans-cleavage reaction reveals a reaction pKa for 2-aminopurine that is 0.4 pH units lower than that for 2,6-diaminopurine, in agreement with the direction of the pKa difference between the two base analogs in solution, although the magnitude of the pKa shift between these two mutants is less than the value of 1.5 pH units for the free base analogs (Brown, 1971). The loss of catalytic activity at low pH for all variants can be suppressed by increasing the concentration of Mg2+ from 12 to 48 mM; this effect appears to be structural rather than a reflection of changes in the ionization of active site constituents (Figure 4B).



Fig. 4. (A) Purine base analogs and their free base N1 pKa values (Brown, 1971). (B) Rate–pH profiles for the trans-cleavage reaction with guanosine (triangles), inosine (squares), 2,6-diaminopurine (circles) and 2-aminopurine (diamonds) at position 8. Single-turnover cleavage assays were carried out on the SV5 trans-cleavage construct in the presence of 12 (open symbols) or 48 mM Mg2+ (closed symbols). The resulting rate data were plotted as a function of pH. Individual rates represent the average of at least three determinations in various buffers. The data from pH 5 to 9 for 2,6-diaminopurine 8 and from pH 5.5 to 8 for 2-aminopurine in 48 mM Mg2+ were fitted to the equation kobs = kmax/[1 + 10(pH – pKa)]. The calculated cleavage reaction pKa values for the 2,6-diaminopurine 8 and 2-aminopurine 8 were 6.94 ± 0.04 and 6.50 ± 0.04, respectively. (C) Relative fluorescence intensity of 2-aminopurine at position 8 as a function of pH in 50 mM NaCl or 10 mM MgCl2. The four-way junction construct with 2′-deoxy A–1 substitution was used for these experiments. The wavelength of excitation was 319 nm and the emission spectra were monitored at 360 nm. The pKa values of 5.0 and 5.7 in the presence of 50 mM NaCl and 12 mM MgCl2, respectively, were derived by fitting the data to the equation Fobs = [Fa + Fb × 10(pH – pKa)]/[1 + 10(pH – pKa)].
Certain adenine analogs such as 2-aminopurine are fluorophores that can be selectively excited under physiological conditions, even in the presence of normal bases. The fluorescence of 2-aminopurine can be measured in oligonucleotides with excitation at 319 nm and emission at 360 nm. Upon lowering the solution pH, the relative fluorescence intensity of 2-aminopurine is reduced as a result of N1 protonation (Ward et al., 1969; Sowers et al., 2000). We took advantage of this feature and monitored the protonation status of the N1 of 2-aminopurine when introduced at position 8 by measuring the fluorescence intensity as a function of pH. Fluorescence emission by 2-aminopurine is also quenched by stacking of the base (Ward et al., 1969). In our case we expect that this stacking effect would oppose the effect of base protonation on the fluorescence emission since 2-aminopurine at position 8 probably becomes unstacked from A–1 at very low pH, resulting in a loss of catalytic activity. The plot of fluorescence intensity versus pH, however, appears to be dominated by the effect of the ionization of the base. From the available data we cannot unambiguously determine what role, if any, base stacking plays in the exact shape of the curve. Using the sigmoidal dependencies of the pH titration curves (Figure 4C), we determined an apparent N1 pKa for 2-aminopurine within the hairpin ribozyme– substrate complex with a 2′-deoxy substitution of the A–1 nucleotide. The native four-way junction construct was employed for these experiments since the structural equilibrium of this form in Mg2+ strongly favors the docked form (Walter et al., 1999). The reported pKa of monomeric 2-aminopurine is 3.8 (Brown, 1971). This value increases to 4.5 in the RzA oligonucleotide (data not shown). Interestingly, in conditions that favor secondary structure formation (50 mM NaCl), the apparent pKa value that we have measured by fluorescence is ∼5, and increases further to 5.7 upon formation of the docked complex (Figure 4C). These results confirm that docking of the A and B domains shifts the apparent N1 pKa of 2-aminopurine at position 8 toward neutrality, most likely due to charge stabilization and stacking interactions as indicated by previous NMR studies of adenine protonation (Ravindranathan et al., 2000). The pKa of 2-aminopurine 8 as determined by fluorescence spectroscopy is lower than that determined for the cleavage reaction by ∼1 pH unit. It was, however, not possible to obtain a good fit to the rate–pH data in Figure 4B for 2-aminopurine with a pKa of 5.7. The clearest interpretation of these data is that the presence of a 2′-OH group at the cleavage site shifts the pKa of 2-aminopurine by 1 pH unit relative to the uncleavable 2′-deoxy variant.
In the recent crystal structure of the hairpin ribozyme, the N1 proton of G8 is within hydrogen bonding distance of the 2′-O of A–1 of the 2′-methoxy-substituted substrate strand (Rupert and Ferré-D’Amaré, 2001). Such an interaction is expected to increase the pKa of the N1, a hypothesis that is testable since a methoxy-substituted substrate can be used for the fluorescence experiments as well. Our data are also consistent with a shift in the pKa of 2-aminopurine at position 8 during the rate-limiting step of the reaction. The transition state intermediate contains one additional negative charge relative to that of the ground state and, therefore, could contribute to a basic shift in the pKa of a neighboring functional group such as the N1 of 2-aminopurine at position 8.
Support for the involvement of the carbonyl oxygen of G8 in catalysis comes from variants in which O6 is deleted or modified (Figure 2). Variants in which O6 is deleted (2-aminopurine, ribopurine) significantly inhibit cleavage. The cleavage rate penalty for losing the carbonyl group at position 6 can be estimated by comparing the wild-type G8 with the 2-aminopurine 8 variant. The kmax values for these ribozymes are 0.236 and 0.00413 min–1 respectively, indicating >50-fold loss in activity upon loss of the 6-keto group. Methylation of O6 is also strongly inhibitory (Figure 2). These results are consistent with the potential involvement of O6 in catalysis, although other interpretations may be possible.
Mechanistic models involving G8
Several potential reaction mechanisms are consistent with these findings. One is a novel mechanistic model in which G8 functions as both the proton donor and acceptor through the concerted transfer of two protons, from the attacking hydroxyl to O6 and from N1 to the leaving group (Figure 5A), yielding the reaction products and converting G8 from its keto form to the enol tautomer. N1 therefore serves as both the initial proton donor and ultimate proton acceptor in this mechanistic model. This model rationalizes both the strong inhibition by O6-methylguanosine, which is unable to tautomerize, and the significant activity of 2,6-diaminopurine, which can tautomerize in a manner analogous to guanosine, through conversion of the 6-amino group to a 6-imino group (Figure 5B). Tautomerization of G8 may be facilitated through formation of a hydrogen bond between the nucleobase and the backbone.

Fig. 5. Reaction mechanisms for phosphodiester bond cleavage involving G8. (A) RNA cleavage catalyzed by the keto–enol tautomerization of G8 is achieved by abstraction of a proton from the 2′-OH by the 6-keto group of G and donation of a proton from the N1 to the 5′-O leaving group. (B) Imino–amino tautomerization cleavage mechanism involving 2,6-diaminopurine at position 8. (C) The imino proton of G8 facilitates nucleophilic attack of the phosphorus by donating a hydrogen bond to a non-bridging phosphate oxygen, indicated by a dashed line, thereby rendering the phosphorus more electropositive. (D) Hydrogen bonding between O6 and 2′-H. The imino proton of G8 and 2′-O polarizes and destabilizes the 2′-O–H bond: base abstraction of the 2′-H by O6 or the action of an unidentified general base results in bond cleavage. (E) General base and general acid catalysis by the N1 of G8.
Other mechanistic models may be compatible with the requirement for N1 protonation of G8 (Figure 5). In one such model, the N1 proton of G8 could form a hydrogen bond with the pro-Rp oxygen of the scissile phosphate and in doing so make the phosphorus more electropositive, a better candidate for nucleophilic attack by the neighboring 2′-O (Figure 5B). In a second model, suggested by the crystal structure, the imino proton of G8 could form a hydrogen bond to the 2′-O nucleophile (Rupert and Ferré-D’Amaré, 2001; Figure 5C). This interaction could assist in deprotonation of the nucleophile in conjunction with the action of the general base, by polarizing the H–O bond and/or by stabilizing the developing negative charge on the nucleophile. An analogous role has been suggested for an essential protonated His40 in the cleavage mechanism for ribonuclease T1 (Martinez-Oyanedel et al., 1991; Zegers et al., 1994). Interestingly, in the case of ribonuclease T1, a glutamate, Glu58, has been implicated as the general base in the cleavage reaction (reviewed by Steyaert, 1997). A similar role may be played by the O6 of G8. The very basic pKa of G8 is ideally suited to serve in one of these roles. In Figure 5C, the 6-keto group of G8 is depicted as the general base, but we cannot rule out the possibility that another base or solvent fulfills this role. Finally, the N1 of G8 may serve a role as a general acid or base in the RNA cleavage reaction with an as yet unidentified base or solvent molecule functioning as the conjugate base or acid (Figure 5D). We are not able to rule out any of these models based on our current data. The rate–pH profiles for 2-aminopurine and 2,6-diaminopurine, while consistent with a role for the functional form of a general acid, are equally consistent with a general base role for N1 of G8. This ambiguity is a general problem with interpreting rate–pH profiles in terms of function (Jencks, 1987).
Kinetic solvent isotope effects
Each of the above models predicts that two protons should be transferred during the rate-limiting step, as expected for general acid–base catalysis. To test this possibility, we examined effects of deuterium isotope substitution on the reaction rates. The pKa of an acid or a base varies as deuterium is introduced into an aqueous medium and, therefore, the interpretation of the kinetic solvent isotope effect (KSIE) can be ambiguous. There is no absolute, general or reliable relationship to predict the variation of pKa with deuterium substitution. From a practical standpoint, it is fortunate that large deviations from the simplest possible result, ΔpKa = 0.5, do not seem to be common (Schowen and Schowen, 1982). In order to avoid the complexity due to the ΔpKa, KSIE usually focuses on the plateau region of rate–pH profile (Schowen and Schowen, 1982). In the case of the hairpin ribozyme, we are fortunate that the reaction is virtually independent of solvent pH over a broad range. We found that upon substitution of the solvent with deuterium oxide, the rate of the reaction is diminished by a factor of 3–4 throughout the pH range (Figure 6A). A similar effect was observed with 2,6-diaminopurine at position 8. Photo-cross-linking and docking assays show that neither the local tertiary structure nor the ability of the complex to dock is impaired by deuterium substitution (Figure 6B; data not shown).

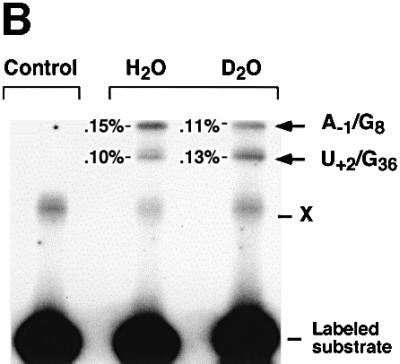

Fig. 6. Deuterium isotope effect. (A) Cleavage rates measured in H2O or D2O for a wide range of pH and pD (pL). The native sequence and 2,6-diaminopurine at position 8 were used; native sequence in H2O (filled diamonds); native sequence in D2O (filled squares); 2,6-diamino purine at position 8 in H2O (open circles); 2,6-diaminopurine at position 8 in D2O (open triangles). Mean values of kH/kD over the tested range were 3.35 for the native sequence and 5.26 for 2,6-diaminopurine. (B) Cross-linking assays were performed with the native complex containing a 5′-end-labeled, non-cleavable, 2′-deoxy A–1 substrate analog. Lane 1 contains hairpin ribozyme–substrate complex incubated and irradiated in the absence of Co(NH3)63+. Lane 2 contains native complex in H2O. Lane 3 contains native complex in D2O. Cross-linked products were analyzed by electrophoresis through denaturing polyacrylamide gels as described (Pinard et al., 1999a,b). (C) Proton inventory of the wild-type cleavage reaction. The square-root (top) and non-squared (bottom) data for the rate constants in a given medium relative to that in water (kH/kD) were plotted against the mole fraction of deuterium oxide (nD2O). The average of the data from four independent experiments performed in the plateau region of the rate–pH curve, pL = 7.5, was plotted. Solid and dashed lines indicate the expected shape of the curves for one and two proton inventories, respectively.
The partial KSIE (proton inventory analysis) was determined for the cleavage reaction catalyzed by the unmodified ribozyme (Figure 6C). The lack of significant curvature in the plot of (kH/kD)1/2 as a function of solvent isotopic composition strongly supports the hypothesis that the rate-limiting step involves the transfer of two protons or deuterons. Interestingly, in the case of ribonuclease A, the proton inventory data indicate that two protons are exchanged in one transition state and an overall isotope effect of 3.07 was measured in the plateau region of the rate–pH profile (Matta and Vo, 1986). While our results do not exclude the possibility that the transfer of two protons within the RNA drives a rate-limiting conformational change, they are fully consistent with a rate-limiting transfer of two protons in the course of the reaction catalyzed by the hairpin ribozyme. These results appear to be analogous to those obtained with ribonuclease A, in which His12 and His119 act as a base and an acid in the reaction and two protons are transferred in a concerted mechanism (Roberts et al., 1969). Our analysis of the plot of the non-square root data against the solvent isotope composition, however, does not allow us to rule out the possibility of a one proton inventory, as has been found recently by Shih and Been (2001) for the HDV ribozyme.
Together, these experiments provide strong support for the participation of G8 in active site chemistry within the hairpin ribozyme–substrate complex. They point to a possible novel catalytic mechanism, in which RNA employs keto–enol tautomerization, a process that has been proposed to contribute to mis-incorporation of bases by DNA and RNA polymerases. The demonstration that the metal-independent reaction mechanism for the hairpin ribozyme, which involves acid–base catalysis, is mediated in part, if not in its entirety, by a key ribonucleotide residue (G8) demonstrates how RNA can be endowed with catalytic activity that does not depend on either metal ions or solvent-borne acids or bases. These results have particular relevance to studies of the RNA-catalyzed mechanism of peptide synthesis, where the peptidyl transferase center of the ribosome has been shown to be comprised of RNA and where, like the hairpin ribozyme, proteins and metal ions are absent (Ban et al., 2000; Nissen et al., 2000).
Materials and methods
Preparation of RNA samples
All RNAs used in this study were generated by solid-phase synthesis and deprotected and purified as described previously (Walter et al., 2000), using reagents from Chemgenes and Glen Research. Deprotection of RNAs containing specific modified bases (6-methyl 2′-deoxyguanosine, 6-thio 2′-deoxyguanosine) followed a protocol provided by Glen Research.
Ribozyme cleavage assays
Activities of ribozymes with G8 modifications were determined for a two-way junction hairpin ribozyme cleaving a 32P-5′-end-labeled, 14 nucleotide substrate as depicted in Figure 1A. Cleavage reactions were carried out in a buffer containing 60 mM Tris–HCl pH 7.5 and 12 mM MgCl2, as described previously (Pinard et al., 1999a). Cleavage rates in D2O were determined as described above. The buffers used were MES (pH 5–6), MOPS (pH 6.5–7.5) and HEPES (pH 7.5–8.5). The pH of buffers prepared in D2O was adjusted by adding 0.4 to the pH meter reading as described (Schowen and Schowen, 1982; Shim and Benkovic, 1999). For the pH dependency reactions, the trans-acting ribozyme depicted in Figure 1A and a 32P-5′-end-labeled, 10 nucleotide substrate strand (5′-UCGCA↓GUCCU-3′) were employed. The ribozyme mix, consisting of 500 nM unlabeled ribozyme components, 12 or 48 mM MgCl2 and 50 mM buffer and substrate mix, consisting of 1 nM 32P-5′-end-labeled substrate in 50 mM buffer, were incubated separately for 10 min at 37°C. The solutions were equilibrated at 25°C for 5 min and then equal volumes of the substrate and ribozyme mixes were combined and incubated at 25°C. At least 10 reaction aliquots were removed at various times and quenched on ice in 10 vol. of 95% formamide and dye markers. The buffers were sodium acetate (pH 4.5–5.0), MES (pH 5.5–6), MOPS (pH 6.0–7.0), HEPES (pH 7.0–8.0) and Tris (pH 8.0–9.0). Buffer-specific effects were not observed using two different buffers at pH 6, 7 and 8. Adding 50 mM NaCl to reaction mixes, in order to correct for fluctuations in ionic strength, did not affect the relative cleavage rates of 2,6-diaminopurine mutants at pH 7–9. Cleavage reaction products were separated on 20% polyacrylamide 8 M urea gels and quantified using a Bio-Rad Molecular Imager System GS-525. Cleavage rate values >0.001 min–1 were calculated by curve-fitting to the single-exponential equation y = yo + A(1 – e–t/τ). Rates <0.001 min–1 were calculated by fitting the data to a linear equation using the initial 25% of the reaction.
Native gel electrophoresis
A saturating concentration of RzA (250 nM) and 1 nM 32P-5′-end-labeled 3′-LB-S were incubated in a folding buffer containing 40 mM Tris–acetate pH 7.5, 15 mM Mg2+ acetate and 10% (v/v) glycerol for 10 min at 25°C, loaded onto a running 10% polyacrylamide gel, and electrophoresed for 18 h at 4°C. The gel buffers consisted of 40 mM Tris–acetate pH 7.5, 15 mM Mg2+ acetate (plus Mg2+ gel) or 40 mM Tris–acetate pH 7.5, 1 mM EDTA (minus Mg2+ gel).
Hydroxyl-radical footprinting
A mixture of a cold RNA (250 nM) and 200 × 105 d.p.m. of 32P-5′-end-labeled RNA was incubated for 10 min in a buffer containing 20 mM Na-cacodylate pH 7, with or without 15 mM Mg2+. Hydroxyl-radicals were generated and the reactions were quenched as described previously (Hampel et al., 1998). Products were separated on a 15% polyacrylamide 8 M urea gel.
UV cross-linking
Hairpin ribozyme–substrate complexes were assembled in 60 mM Tris–HCl pH 7.5, with or without 1 mM Co(NH3)63+, and irradiated for 10 min at room temperature with a 312 nm UV-light. The complex prepared in H2O or D2O was assembled in 50 mM HEPES pH 7.5 and 1 mM Co(NH3)63+, then irradiated with 312 nm UV-light as above. Purification and characterization of all of the cross-linked RNA species were performed as described previously (Pinard et al., 1999b).
Fluorescence assays
Fluorescence spectra and intensities were recorded on an Aminco-Bowman Series 2 (AB2) spectrophotofluorometer. Samples (150 µl) containing 400 nM 2-aminopurine oligonucleotide were incubated at 25°C for at least 15 min in 50 mM NaCl and different pH buffers as mentioned previously. The 2-aminopurine was excited at a wavelength of 319 nm and the emission spectrum was monitored from 340 to 500 nm. The intensity of the 360 nm peak was recorded for all pH values. These experiments were repeated in the presence of oligonucleotides (excess concentrations of 1.2 µm each) to complete the assembly of a four-way junction ribozyme containing a 2′-deoxy-A–1-modified substrate, and the addition of MgCl2 to achieve a final concentration of 10 mM. Data were normalized by dividing by the highest fluorescence reading in each case and plotted as a function of pH. The pKa values were derived by fitting the data to the equation Fobs = [Fa + Fb × 10(pH – pKa)]/[1 + 10(pH – pKa)]. The observed fluorescence change at a given pH value can be expressed by the equation Fobs = Fapa + Fbpb where pa represents the fraction of unprotonated species and Fa and Fb represent the fluorescence intensities corresponding to the protonated and unprotonated states of the 2-aminopurine, respectively. Experiments were repeated three times and error bars fitted as ±1 standard deviation of the data.
Acknowledgments
Acknowledgements
We thank Bill Scott for helpful discussions, David Pecchia for synthesis of oligonucleotides, insightful comments and suggestions of reviewers, and Anne MacLeod for manuscript preparation. This work was supported by a grant from the National Institutes of Health (AI 44186) to J.M.B. and from the Medical Research Council of Canada (MT 14604) to F.M. R.P. was supported by a postdoctoral fellowship from the Medical Research Council of Canada.
References
- Ban N., Nissen,P., Hansen,J., Moore,P.B. and Steitz,T.A. (2000) The complete atomic structure of the large ribosomal subunit at 2.4 Å resolution. Science, 289, 905–920. [DOI] [PubMed] [Google Scholar]
- Berzal-Herranz A., Joseph,S., Chowrira,B.M., Butcher,S.E. and Burke,J.M. (1993) Essential nucleotide sequences and secondary structure elements of the hairpin ribozyme. EMBO J., 12, 2567–2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown D.J. (1971) Fused Pyrimidines Part II: Purines. Wiley, New York, NY.
- Butcher S.E., Heckman,J.E. and Burke,J.M. (1995) Reconstitution of hairpin ribozyme activity following separation of functional domains. J. Biol. Chem., 270, 29648–29651. [DOI] [PubMed] [Google Scholar]
- Buzayan J.M., Gerlach,W.L. and Bruening,G. (1986) Nonenzymatic cleavage and ligation of RNAs complementary to a plant virus satellite RNA. Nature, 323, 349–353. [Google Scholar]
- Dahm S. and Uhlenbeck,O.C. (1991) Role of divalent metal ions in the hammerhead RNA cleavage reaction. Biochemistry, 30, 9464–9469. [DOI] [PubMed] [Google Scholar]
- Dahm S., Derrick,W.B. and Uhlenbeck,O.C. (1993) Evidence for the role of solvated metal hydroxide in the hammerhead cleavage mechanism. Biochemistry, 32, 13040–13045. [DOI] [PubMed] [Google Scholar]
- Esteban J.A., Walter,N.G., Kotzorek,G., Heckman,J.E. and Burke,J.M. (1998) Structural basis for heterogeneous kinetics: re-engineering the hairpin ribozyme. Proc. Natl Acad. Sci. USA, 95, 6091–6096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedor M.J. (2000) Structure and function of the hairpin ribozyme. J. Mol. Biol., 297, 269–291. [DOI] [PubMed] [Google Scholar]
- Hampel A. and Cowan,J.A. (1997) A unique mechanism for RNA catalysis: the role of metal co-factors in hairpin ribozyme cleavage. Chem. Biol., 4, 513–517. [DOI] [PubMed] [Google Scholar]
- Hampel A., Tritz,R., Hicks,M. and Cruz,P. (1990) Hairpin catalytic RNA model: evidence for helices and sequence requirement for substrate RNA. Nucleic Acids Res., 18, 299–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampel K.J., Walter,N.G. and Burke,J.M. (1998) The solvent-protected core of the hairpin ribozyme–substrate complex. Biochemistry, 37, 14672–14682. [DOI] [PubMed] [Google Scholar]
- Jencks W.P. (1987) Catalysis in Chemistry and Enzymology. Dover Publications Inc., New York, NY.
- Major F., Turcotte,M., Gautheret,D., Lapalme,G., Fillion,E. and Cedergren,R. (1991) The combination of symbolic and numerical computation for three-dimensional modeling of RNA. Science, 253, 1255–1260. [DOI] [PubMed] [Google Scholar]
- Martinez-Oyanedel J., Choe,H.W., Heinemann,U. and Saenger,W. (1991) Ribonuclease T1 with free recognition and catalytic site: crystal structure analysis at 1.5 Å resolution. J. Mol. Biol., 222, 335–352. [DOI] [PubMed] [Google Scholar]
- Matta M.S. and Vo,D.T. (1986) Proton inventory of the second step of ribonuclease catalysis. J. Am. Chem. Soc., 108, 5318–5328. [Google Scholar]
- McKay D.B. (1996) Structure and function of the hammerhead ribozyme: an unfinished story. RNA, 2, 395–403. [PMC free article] [PubMed] [Google Scholar]
- Murchie A.I., Thomson,J.B., Walter,F. and Lilley,D.M. (1998) Folding of the hairpin ribozyme in its natural conformation achieves close physical proximity of the loops. Mol. Cell, 1, 873–881. [DOI] [PubMed] [Google Scholar]
- Murray J.B., Seyhan,A.A., Walter,N.G., Burke,J.M. and Scott,W.G. (1998) The hammerhead, hairpin and VS ribozymes are catalytically proficient in monovalent cations alone. Chem. Biol., 5, 587–595. [DOI] [PubMed] [Google Scholar]
- Muth G.W., Ortoleva-Donnelly,L. and Strobel,S.A. (2000) A single adenosine with a neutral pKa in the ribosomal peptidyl transferase center. Science, 289, 947–950. [DOI] [PubMed] [Google Scholar]
- Nesbitt S., Hegg,L.A. and Fedor,M.J. (1997) An unusual pH-independent and metal-ion-independent mechanism for hairpin ribozyme catalysis. Chem. Biol., 4, 619–630. [DOI] [PubMed] [Google Scholar]
- Nissen P., Hansen,J., Ban,N., Moore,P.B. and Steitz,T.A. (2000) The structural basis of ribosome activity in peptide bond synthesis. Science, 289, 920–930. [DOI] [PubMed] [Google Scholar]
- Pinard R., Lambert,D., Walter,N.G., Heckman,J.E., Major,F. and Burke,J.M. (1999a) Structural basis for the guanosine requirement of the hairpin ribozyme. Biochemistry, 38, 16035–16039. [DOI] [PubMed] [Google Scholar]
- Pinard R., Heckman,J.E. and Burke,J.M. (1999b) Alignment of the two domains of the hairpin ribozyme–substrate complex defined by inter-domain photoaffinity cross-linking. J. Mol. Biol., 287, 239–251. [DOI] [PubMed] [Google Scholar]
- Pinard R. et al. (2001) The hairpin ribozyme substrate binding-domain: a highly constrained D-shaped conformation. J. Mol. Biol., 307, 51–65. [DOI] [PubMed] [Google Scholar]
- Ravindranathan S., Butcher,S.E. and Feigon,J. (2000) Adenine protonation in domain B of the hairpin ribozyme. Biochemistry, 39, 16026–16032. [DOI] [PubMed] [Google Scholar]
- Roberts G.C.K., Dennis,E.A., Meadows,D.H., Cohen,J.S. and Jardetzky,O. (1969) The mechanism of action of ribonuclease. Proc. Natl Acad. Sci. USA, 62, 1151–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rupert P.B. and Ferré-D’Amaré,A.R. (2001) Crystal structure of a hairpin ribozyme–inhibitor complex with implications for catalysis. Nature, 410, 780–786. [DOI] [PubMed] [Google Scholar]
- Schmidt S., Beigelman,L., Karpeisky,A., Usman,N., Sorensen,U.S. and Gait,M.J. (1996) Base and sugar requirements for RNA cleavage of essential nucleoside residues in internal loop B in the hairpin ribozyme: implication for secondary structure. Nucleic Acids Res., 24, 573–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schowen K.B. and Schowen,R.L. (1982) Solvent isotope effects of enzyme systems. Methods Enzymol., 87, 551–606. [PubMed] [Google Scholar]
- Shih I. and Been,M.D. (2001) Involvement of a cytosine side chain in proton transfer in the rate-determining step of ribozyme self-cleavage. Proc. Natl Acad. Sci. USA, 98, 1489–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim J.H. and Benkovic,S.J. (1999) Catalytic mechanism of Escherichia coli glycinamide ribonucleotide transformylase probed by site-directed mutagenesis and pH-dependent studies. Biochemistry, 38, 10024–10031. [DOI] [PubMed] [Google Scholar]
- Slim G. and Gait,M.J. (1991) Configurationally defined phosphorothioate-containing oligoribonucleotides in the study of the mechanism of cleavage of hammerhead ribozymes. Nucleic Acids Res., 19, 1183–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sowers L.C., Boulard,Y. and Fazakerley,G.V. (2000) Multiple struc tures for the 2-aminopurine–cytosine mispair. Biochemistry, 39, 7613–7620. [DOI] [PubMed] [Google Scholar]
- Steyaert J. (1997) A decade of protein engineering on ribonuclease T1: atomic dissection of the enzyme–substrate interactions. Eur. J. Biochem., 247, 1–11. [DOI] [PubMed] [Google Scholar]
- Walter N.G. and Burke,J.M. (1998) The hairpin ribozyme: structure, assembly and catalysis. Curr. Opin. Chem. Biol., 2, 24–30. [DOI] [PubMed] [Google Scholar]
- Walter N.G., Hampel,K.J., Brown,K.M. and Burke,J.M. (1998) Tertiary structure formation in the hairpin ribozyme monitored by fluorescence resonance energy transfer. EMBO J., 17, 2378–2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter N.G., Burke,J.M. and Millar,D.P. (1999) Stability of hairpin ribozyme tertiary structure is governed by the interdomain junction. Nature Struct. Biol., 6, 544–549. [DOI] [PubMed] [Google Scholar]
- Walter N.G., Yang,N. and Burke,J.M. (2000) Probing non-selective cation binding in the hairpin ribozyme with Tb(III). J. Mol. Biol., 298, 539–555. [DOI] [PubMed] [Google Scholar]
- Ward D.C., Reich,E. and Stryer,L. (1969) Fluorescence studies of nucleotides and polynucleotides. J. Biol. Chem., 244, 1228–1237. [PubMed] [Google Scholar]
- Wilson T.J., Zhao,Z.Y., Maxwell,K., Kontogiannis,L. and Lilley,D.M.J. (2001) Importance of specific nucleotides in the folding of the natural form of the hairpin ribozyme. Biochemistry, 40, 2291–2302. [DOI] [PubMed] [Google Scholar]
- Young K.J., Gill,F. and Grasby,J.A. (1997) Metal ions play a passive role in the hairpin ribozyme-catalyzed reaction. Nucleic Acids Res., 25, 3760–3766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zegers I., Haikal,A.F., Palmer,R. and Wyns,L. (1994) Crystal structure of RNase T1 with 3′-guanylic acid and guanosine. J. Biol. Chem., 269, 127–133. [PubMed] [Google Scholar]