

Abstract
Reactive oxygen species (ROS) production by NADPH oxidase (NOX) is a key promoter of platelet activation, making NOX inhibition an attractive antiplatelet strategy. This study evaluated the effects of the novel NOX2 inhibitor, GSK2795039, on human platelet functional responses and ROS-mediated signaling pathways. GSK2795039 effectively suppressed both extracellular and intracellular ROS production in collagen-stimulated platelets. Consequently, GSK2795039 treatment significantly inhibited collagen-induced platelet aggregation, dense-granule ATP release, and α-granule P-selectin exposure. Furthermore, GSK2795039 attenuated tyrosine phosphorylation-mediated activation of spleen tyrosine kinase (Syk), linker for the activation of T cells (LAT), vav guanine nucleotide exchange factor 1 (Vav1), and Bruton tyrosine kinase (Btk) within the collagen receptor signaling pathway, leading to decreased phospholipase Cγ2 (PLCγ2) activation and calcium mobilization. GSK2795039 also inhibited collagen-induced integrin αIIbβ3 activation, associated with increased cyclic guanosine monophosphate (cGMP) levels and vasodilator-stimulated phosphoprotein (VASP) phosphorylation. Additionally, the inhibitor reduced collagen-induced p38 mitogen-activated protein kinase (MAPK) activation, which led to lower cytosolic phospholipase A2 (cPLA2) phosphorylation and thromboxane production. GSK2795039 further decreased extracellular signal regulated kinase 5 (ERK5) activation, thereby limiting procoagulant phosphatidylserine exposure. Notably, thrombus formation induced by platelet adhesion to collagen was abolished in the presence of GSK2795039. In vivo, GSK2795039 administration markedly inhibited arterial thrombosis. This is the first study to demonstrate that GSK2795039 suppresses collagen-induced ROS production, platelet activation, and thrombus formation, highlighting its potential as a therapeutic agent for thrombotic and cardiovascular diseases.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-025-20250-z.
Keywords: Platelet, ROS, NOX2 inhibitor, GSK2795039, Antiplatelet, Antithrombotic
Subject terms: Biochemistry, Drug discovery
Introduction
Platelets are essential cells involved in hemostasis and thrombosis, continually monitoring the vascular system to quickly detect damaged or inflamed sites. Upon vascular injury, platelets adhere to the subendothelium by binding von Willebrand factor and glycoprotein (GP) Ib receptors, leading to their firm attachment to subendothelial collagen via collagen receptor GPVI. This interaction initiates a signaling cascade that promotes platelet activation, secretion, and aggregation1,2. Through these processes, platelets function to seal blood vessel injuries, preventing blood loss; however, in certain pathological conditions, uncontrolled platelet activation can lead to thrombus formation. Thrombotic disorders pose significant health risks globally and are major causes of mortality and disability, making effective prevention and treatment strategies essential. Currently, antiplatelet therapies play a central role in reducing the risk of thrombotic events for patients with cardiovascular, cerebrovascular, and peripheral artery disease.
NADPH oxidase (NOX) enzymes, as primary sources of reactive oxygen species (ROS), are pivotal in both physiological and pathological platelet signaling3. A substantial body of research has established the importance of NOX-derived ROS in platelet function and signaling4–7. Furthermore, NOX activity appears to increase platelet sensitivity, contributing to the heightened responsiveness seen in thrombotic conditions5–8. The NOX family comprises seven members-NOX1–5 and the dual oxidases Duox1 and Duox2. Among these, human platelets express NOX1, 2, and 4, with NOX1 and NOX2 playing particularly crucial roles in modulating platelet function, as demonstrated in studies using triple NOX knockout mice6,7. NOX2, in particular, is central to ROS production and platelet activation, as evidenced by various experimental and clinical studies7,9–11. In NOX2-deficient mouse models, platelets exhibit reduced thrombotic activity and platelet-leukocyte interactions, highlighting NOX2’s role in thrombosis and inflammation7,12. The importance of NOX2 in human platelets is further underscored by observations that platelets from individuals with X-linked chronic granulomatous disease produce less ROS and exhibit lower the cluster of differentiation 40 (CD40) ligand expression upon activation7,12. Interestingly, despite NOX2’s contribution to thrombotic processes, patients with this condition11 and NOX2-deficient mice7 do not show increased bleeding, indicating that NOX2’s role may be more specific to thrombosis than hemostasis. Accordingly, NOX2 has emerged as a promising therapeutic target to inhibit platelet function while minimizing bleeding risk.
GSK2795039, a novel NOX2-specific inhibitor, has been shown to inhibit NOX2-derived ROS production in an inflammation model13. Although GSK2795039 is known to inhibit collagen-induced platelet aggregation14, its precise mechanisms in modulating NOX2-mediated ROS pathways that contribute to platelet activation and thrombus formation remain unexplored. This study aims to assess whether GSK2795039 can effectively inhibit platelet activation and thrombus formation by targeting ROS production and interrupting subsequent ROS-mediated signaling pathways.
Results
GSK2795039 attenuates collagen-stimulated platelet aggregation and ROS production
Collagen receptor GPVI is essential for platelet adhesion, activation, and accumulation1. As NOX2 promotes GPVI-stimulated platelet activation7, we investigated the impact of NOX2 inhibitor GSK2795039 on human platelet aggregation after collagen stimulation. GSK2795039 significantly suppressed collagen-induced aggregation in a concentration-dependent manner, with an IC50 value of 22.6 µM (Fig. 1A). To confirm whether this inhibitory effect also occurs in a physiological plasma environment, we evaluated collagen-induced platelet aggregation using platelet-rich plasma (PRP). As shown in Fig. S1, preincubation of PRP with GSK2795039 markedly attenuated collagen-induced platelet aggregation in a dose-dependent manner, consistent with results obtained using washed platelets.
Fig. 1.

GSK2795039 attenuates collagen-stimulated platelet aggregation and ROS production. (A) Washed human platelets (5 × 108/mL) were pretreated with vehicle (0.5% DMSO) or various concentrations of GSK2795039 for 5 min before stimulation with collagen (10 µg/mL). Platelet aggregation was monitored by measuring changes in light transmission, with 100% transmission defined as the buffer control. Data represent the mean ± standard deviation from three independent donors. (B–D) For analyzing the levels of intracellular ROS (B), extracellular ROS (C), and extracellular superoxide anion (D), following CM-H2DCFDA loading (B); in the presence of DCFH2 (C) or L-012 (D), washed human platelets were treated as in A. (B, C) Fluorescence change was measured in a fluorometer cuvette under constant stirring conditions. Upper panels show representative traces. (D) Chemiluminescence was measured in a luminometer. (E) NOX activity assay. Washed human platelets were treated as in A. Cell lysates were prepared and then incubated with NADPH. Superoxide anion production in the lysates was measured by a chemiluminescence assay using L-012. (F) To quantify extracellular H2O2, the supernatant from the reaction mixture was incubated with Amplex Red reagent, and resorufin fluorescence was measured after 30 min. AU, arbitrary units. All data represent the mean ± standard deviation. Statistical significance: * p < 0.05, ** p < 0.01 and *** p < 0.001.
We investigated the effect of GSK2795039 on thrombin- or U46619-induced platelet aggregation, as NOX2-deficient mouse platelets showed partial inhibition7 and GSK2795039 had no inhibitory effect on human platelets14. Despite inhibiting collagen-induced platelet aggregation, GSK2795039 did not influence thrombin- or U46619-induced aggregation at 50 µM concentration (Supplementary Fig. S2).
Next, we examined the effect of GSK2795039 on collagen-induced intracellular ROS levels by evaluating intraplatelet redox status using 5-(and 6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate (CM-H2DCFDA), which is sensitive to oxidation and can detect a wide range of ROS, including peroxides and hydroxyl radicals (Fig. 1B). Similar to earlier research15–17, collagen stimulation significantly increased ROS generation in human platelets. GSK2795039 concentration-dependently reduced collagen-induced intracellular ROS. We examined the impact of GSK2795039 on collagen-induced extracellular ROS generation using 2′,7′-dichlorodihydrofluorescein (DCFH2) a redox-sensitive and membrane-impermeable probe18. Collagen-stimulated platelets significantly increased extracellular ROS production, supporting prior findings15,16. GSK2795039 concentration-dependently reduced collagen-induced extracellular ROS increase (Fig. 1C).
To directly evaluate whether the inhibitory effect of GSK2795039 on ROS generation is attributable to NOX2 suppression, we first quantified extracellular superoxide anion levels in intact platelets using L-012-based real-time chemiluminescence. Collagen stimulation induced a marked increase in superoxide release compared to resting conditions, which was significantly attenuated by GSK2795039 in a concentration-dependent manner (Fig. 1D). Next, to assess enzymatic NOX activity more directly, platelet lysates were incubated with NADPH and L-012 to monitor NADPH oxidase–driven superoxide production. Collagen stimulation markedly enhanced NOX activity relative to resting platelets, whereas pretreatment with GSK2795039 resulted in a dose-dependent suppression of NOX enzymatic activity (Fig. 1E). These findings confirm that GSK2795039 diminishes collagen-induced ROS generation by directly inhibiting NOX2-dependent superoxide production.
The NOX complex generates extracellular superoxide, which spontaneously dismutates to H2O219. Anionic superoxide is not membrane permeable, whereas uncharged H2O2 may diffuse into the cytosol, increasing intracellular ROS. Using the H2O2-specific probe Amplex Red, we found that GSK2795039 significantly inhibits the collagen-induced increase of extraplatelet H2O2 (Fig. 1F), as extraplatelet superoxide does not affect platelet aggregation5. These results suggest that GSK2795039 reduces collagen-stimulated platelet activation by decreasing ROS production.
GSK2795039 inhibits protein tyrosine phosphorylation in collagen-stimulated platelets
Protein-tyrosine phosphatases (PTPs) are susceptible to ROS-induced oxidative inactivation due to reactive cysteine thiolate in the catalytic region, leading to increased phosphorylation20. GPVI-stimulated platelet signaling involves protein tyrosine phosphorylation cascades21, making ROS-mediated PTP oxidation and deactivation potentially beneficial17,22,23. GSK2795039 reduced ROS rise in collagen-stimulated platelets, thus we evaluated its impact on total protein tyrosine phosphorylation. Figure 2A indicates that collagen-induced protein tyrosine phosphorylation of numerous platelet proteins increased considerably from baseline. In response to collagen stimulation, platelets pretreated with GSK2795039 reduced protein tyrosine phosphorylation.
Fig. 2.

GSK2795039 inhibits protein tyrosine phosphorylation in collagen-stimulated platelets. Washed platelets were treated with vehicle or GSK2795039 at the indicated concentrations for 5 min and stimulated with collagen (10 µg/mL) for 2 min. (A) Total protein tyrosine phosphorylation levels were analyzed by immunoblotting, with β-actin serving as a loading control. (B) After treatment with iodoacetamide and N-ethylmaleimide in a hypoxic chamber, platelet lysates were further incubated with dithiotreitol. The activity of total protein tyrosine phosphatases (PTPs), which were recovered from oxidation, was determined using tyrosine phosphopeptide as the substrate. (C) The phosphorylation of Syk, LAT, Vav1, and Btk at specific tyrosine residues was assessed by immunoblotting with phospho-specific antibodies. Representative immunoblots are shown alongside quantitative analysis of band intensity. Data represent the mean ± standard deviation. Statistical significance: * p < 0.05, ** p < 0.01 and *** p < 0.001.
To explore whether the reduced protein tyrosine phosphorylation was due to preserved PTP activity, we directly assessed the enzymatic function of PTPs following GSK2795039 treatment. Because PTPs can undergo reversible oxidative inactivation by ROS, platelet lysates were pretreated with cysteine-specific alkylating agents under anoxic conditions to prevent further oxidation and to stabilize the thiol redox state. Subsequent reduction of reversibly oxidized cysteines allowed quantification of the active pool of PTPs. As shown in Fig. 2B, collagen stimulation significantly decreased PTP activity, whereas GSK2795039 treatment restored PTP enzymatic activity in a concentration-dependent manner. These findings support the notion that GSK2795039 prevents ROS-dependent PTP oxidation, thereby preserving phosphatase activity and counteracting aberrant phosphorylation during GPVI signal propagation.
We examined the tyrosine phosphorylation-based activation of spleen tyrosine kinase (Syk), linker for the activation of T cells (LAT), vav guanine nucleotide exchange factor 1 (Vav1), and Bruton tyrosine kinase (Btk) to determine whether GSK2795039 affects GPVI signaling cascade activation. In collagen-stimulated platelets, autophosphorylation of Syk-Tyr525/Tyr526 on activation loops implies kinase activity24,25. Phosphorylation of LAT tyrosine residues like Tyr200 by Syk enables src homology 2 (SH2) domain-containing proteins to attach and form a signaling complex21,26,27. Phosphorylations of Vav1-Tyr174 and Btk-Tyr551 in this complex cause phospholipase Cγ2 (PLCγ2)-Tyr753 phosphorylation contributing to its activation28–30. Western blot examination with phospho-specific antibodies showed that collagen stimulation increased the phosphorylation of Tyr525/Tyr526 on Syk, Tyr200 on LAT, Tyr174 on Vav1, and Tyr551 on Btk (Fig. 2C). The tyrosine phosphorylation-based activation of Syk, LAT, Vav1, and Btk in collagen-stimulated platelets was distinctively inhibited by GSK2795039 treatment. Taking into account that phosphatases like low molecular weight-PTP (LMW-PTP), src homology 2-containing inositol phosphatase 1 (SHIP-1), phosphatase and tensin homolog deleted on chromosome 10 (PTEN), and src homology 2-containing PTP2 (SHP-2) can lower GPVI-stimulated platelet activation by dephosphorylating many substrates17,31–33, our results show that GSK2795039 protects PTPs from oxidative inactivation by blocking NOX2-mediated ROS production, which lowers GPVI signaling in platelets.
GSK2795039 suppresses PLCγ2/PKC/Ca2+ pathway in collagen-stimulated platelets
The activation of Vav1 and Btk through tyrosine phosphorylation is associated with the phosphorylation of Tyr753 on the downstream target PLCγ2 following collagen stimulation, thereby enhancing its activity28–30. By inhibiting upstream molecules, GSK2795039 allowed us to study PLCγ2 phosphorylation at Tyr753 in collagen-stimulated platelets. Figure 3A demonstrates that GSK2795039 inhibits collagen-induced PLCγ2 phosphorylation at Tyr753. In collagen-stimulated platelets, PLCγ2-mediated diacylglycerol and inositol-1,4,5-trisphosphate (IP3) production activates protein kinase C (PKC) and increases cytosolic Ca2+ by releasing Ca2+ from intracellular stores21,34. To determine whether GSK2795039 affects PKC signaling, we examined the phosphorylation status of classical PKC substrate motifs35 by immunoblotting. Upon collagen stimulation, a characteristic set of PKC-dependent phosphoprotein bands was observed, indicating pathway activation. Pretreatment with GSK2795039 markedly attenuated these phosphorylation patterns, suggesting inhibition of PKC activation (Fig. 3B).
Fig. 3.

GSK2795039 suppresses PLCγ2/PKC/Ca2+ pathway in collagen-stimulated platelets. (A and B) Washed human platelets were treated with vehicle or GSK2795039 for 5 min and stimulated with collagen (10 µg/mL) for 2 min. Phosphorylation of PLCγ2 at Tyr753 (A) and PKC substrates (B) were analyzed by immunoblotting, respectively. Representative immunoblots are shown alongside quantitative analysis of band intensity. (C) Intracellular calcium mobilization was measured in Fluo-3-AM-loaded washed human platelets stimulated with collagen for 2 min in the presence of either 0.5 mM EGTA (left) or 1 mM extracellular CaCl2 (right). Fluorescence intensity expressed in arbitrary units (AU) was recorded, and maximum intensity values are presented. All data represent the mean ± standard deviation. Statistical significance: * p < 0.05, ** p < 0.01 and *** p < 0.001.
The inhibition of PKC signaling by GSK2795039 carries significant mechanistic implications, given PKC’s established role in promoting the phosphorylation of p47 phagocyte oxidase (p47phox) at Ser304—an essential event for NOX2 activation36. Our data indicate that GSK2795039 attenuates p47phox phosphorylation (Supplementary Fig. S3), potentially through disruption of the PLCγ2–diacylglycerol–PKC axis (Fig. 4A and B), thereby hindering the assembly of the active NOX2 complex. In addition, consistent with a previous report13, GSK2795039 likely interferes with the NADPH-binding site of gp91phox, effectively blocking electron transfer and subsequent ROS generation. These findings suggest that GSK2795039 exerts dual inhibitory actions by impeding both the catalytic function and upstream regulatory mechanisms of NOX2, ultimately reducing the membrane recruitment of cytosolic NOX components.
Fig. 4.

GSK2795039 inhibits collagen-stimulated granule release in platelets. Washed human platelets were treated with vehicle or GSK2795039 for 5 min and stimulated with collagen (10 µg/mL) for 5 min. (A) Platelet dense granule secretion was assessed by measuring ATP release using a luciferin-luciferase assay. Data are presented as representative tracings and maximum luminescence (relative luminescence units, RLU) generated by ATP. (B) Surface expression of P-selectin (CD62P) as a marker of α-granule secretion was measured by flow cytometry after staining with CD62P-PE. Representative histograms and mean fluorescence intensity (MFI) values are shown. All data represent the mean ± standard deviation, with statistical significance indicated: ***p < 0.001.
We used Fluo-3-AM to measure cytosolic Ca2+. When triggered without external Ca2+, platelets release Ca2+ from internal reserves, increasing cytosolic Ca2+. We measured cytosolic Ca2+ in 0.5 mM EGTA (Fig. 3B). GSK2795039 effectively decreased collagen-induced Ca2+ mobilization from internal reserves. GSK2795039 reduced collagen-induced cytosolic Ca2+ rise even in the presence of external Ca2+ (Fig. 3C). These data show that GSK2795039 inhibits Ca2+ mobilization from internal and external sources.
GSK2795039 inhibits collagen-stimulated granule release in platelets
Release of active chemicals from dense and α-granules after activation promotes additional platelet activation. PLCγ2-mediated PKC activation and increased cytosolic Ca2+ promote granule release in collagen-stimulated platelets34,37. GSK2795039 inhibited PLCγ2, thus we investigated its influence on collagen-induced platelet dense-granule release. Results showed considerable inhibition of ATP release from platelets (Fig. 4A). Platelets and leukocytes bind together via P-selectin. Active platelets exhibit it following α-granule degranulation38. Using PE-labelled anti-P-selectin antibody (CD62P-PE), flow cytometry demonstrated that collagen increases platelet surface P-selectin, which GSK2795039 greatly attenuates (Fig. 4B).
GSK2795039 suppresses the p38 MAPK/cPLA2/TXA2 signaling pathway in collagen-stimulated platelets
Thromboxane A2 (TXA2) is a secondary-wave agonist that enhances collagen-induced platelet aggregation39. Platelets activated by collagen need cytosolic phospholipase A2 (cPLA2) to create TXA2 by releasing arachidonic acid from the plasma membrane40. In collagen-stimulated human platelets, p38 mitogen-activated protein kinase (MAPK) phosphorylates cPLA2 at Ser505, resulting in TXA2 synthesis41. Studies indicate that the absence of p47phox, a regulatory subunit of NOX2, or the selective inhibition of NOX2 hinders the activation of p38 MAPK in GPVI-stimulated platelets9,42. Apoptosis signal-regulating kinase 1 (ASK1), a MAPK kinase kinase family member, regulates p38 MAPK/cPLA2/TXA2 synthesis in mouse platelets upon agonist activation43. In human platelets, H2O2 activates ASK1, phosphorylating MAPK kinases (MKK) 3, 4, and 6, which in turn activating p38 MAPK44. Additionally, removing ROS reduces collagen-induced TXA2 synthesis in human platelets4. Thus, we investigated if GSK2795039 influences ASK1/p38 MAPK signaling axis to activate cPLA2. Collagen stimulation of platelets led to the activation of MKK3/6, which was associated with the phosphorylation of ASK1 at Thr838 (a marker of kinase activity) and p38 MAPK at Thr180/Tyr182 (reflecting activation) (Fig. 5A). This was followed by an increase in cPLA2 phosphorylation at Ser510 (Fig. 5B). Preincubation with GSK2795039 greatly decreased these phosphorylations (Fig. 5A and B). We next examined its effects on the generation of TXB2, a stable TXA2 metabolite. In collagen-stimulated platelets, GSK2795039 reduced TXB2 synthesis (Fig. 5C). These data suggest that the antiplatelet effect of GSK2795039 is linked to the downregulation of the ROS-driven ASK1/p38 MAPK/cPLA2/TXA2 signaling cascade.
Fig. 5.

GSK2795039 suppresses the p38 MAPK/cPLA2/TXA2 signaling pathway in collagen-stimulated platelets. (A, B) Washed human platelets were pretreated with vehicle or GSK2795039 and stimulated with collagen (10 µg/mL) for 2 min. Phosphorylation of ASK1, MKK3/6, p38 MAPK (A), and cPLA2 (B) was analyzed by immunoblotting. Representative immunoblots and quantitative data are presented. (C) Thromboxane B2 (TXB2), a stable metabolite of TXA2, was measured in the supernatant of collagen-stimulated platelets using an ELISA. All data represent the mean ± standard deviation. Statistical significance: *p < 0.05, **p < 0.01, ***p < 0.001.
GSK2795039 reduces ERK5 activation and PS exposure in collagen-stimulated platelets
NOX-derived ROS in stimulated platelets activate extracellular signal regulated kinase 5 (ERK5), a redox-sensitive MAP kinase45,46. ROS-activated ERK5 stimulates caspases, boosting procoagulant phosphatidylserine (PS) externalization and fibrin production while the mechanism is uncertain46. Thus, we investigated whether GSK2795039 influences ERK5’s extracellular PS exposure enhancement. GSK2795039 preincubation inhibited platelet collagen-induced ERK5 phosphorylation at Thr218/Tyr220 (Fig. 6A). Next, using flow cytometry, we examined GSK2795039’s impact on platelet procoagulant reactions by assessing PS exposure marker annexin V-FITC binding to platelets. PS exposure increased after collagen stimulation, whereas GSK2795039 pretreatment decreased annexin V-positive platelets (Fig. 6B). GSK2795039’s capacity to reduce ROS generation may prevent procoagulant states in cardiovascular disease by reducing ROS-sensitive ERK5 activation.
Fig. 6.

GSK2795039 reduces ERK5 activation and PS exposure in collagen-stimulated platelets. (A) Washed human platelets were treated with GSK2795039 or vehicle and stimulated with collagen (10 µg/mL) for 2 min. Phosphorylation of ERK5 at Thr218/Tyr220 was analyzed by immunoblotting. Representative immunoblots and quantitative data are presented. (B) Washed human platelets surface exposure of phosphatidylserine was assessed by annexin V-FITC staining and analyzed using flow cytometry. All data represent the mean ± standard deviation. Statistical significance: ***p < 0.001.
GSK2795039 modulates the cGMP/PKG/VASP/integrin-αIIbβ3 signaling pathway in collagen-stimulated platelets
Activation of integrin-αIIbβ3 leads to high-affinity fibrinogen binding, affecting platelet adhesion and aggregation47. NOX-derived ROS are essential for activating integrin-αIIbβ3 in GPVI-stimulated platelets6. Therefore, we examined the impact of GSK2795039 on this signaling pathway. The FITC-labelled anti-active-integrin αIIbβ3 antibody (PAC1-FITC), which identifies the activated conformation of αIIbβ3, showed that GSK2795039 reduced the rise in active conformational changes of integrin-αIIbβ3 in response to collagen stimulation (Fig. 7A).
Fig. 7.

GSK2795039 modulates the cGMP/PKG/VASP/integrin-αIIbβ3 signaling pathway in collagen-stimulated platelets. Washed human platelets were treated with GSK2795039 (10 or 30 µM) or vehicle for 5 min, then stimulated with collagen (10 µg/mL). (A) Activation of integrin-αIIbβ3 was assessed by PAC1-FITC binding using flow cytometry after 5 min of collagen stimulation. Representative histograms and mean fluorescence intensity (MFI) are shown. (B) Phosphorylation of VASP at Ser239 was analyzed by immunoblotting after 2 min of collagen stimulation. Representative blots and densitometric quantification are presented. (C) Intraplatelet cGMP levels were quantified by ELISA after 2 min of collagen stimulation. All data represent the mean ± standard deviation. Statistical significance: *p < 0.05, **p < 0.01, ***p < 0.001 and ns, p > 0.05 (not significant).
The adaptor molecule vasodilator-stimulated phosphoprotein (VASP) inhibits platelet activation by regulating integrin-αIIbβ3 activation48,49 and links cyclic nucleotide-dependent pathways to actin remodelling, regulating cytoskeletal dynamics50. The function of VASP in cyclic guanosine monophosphate (cGMP)-mediated suppression of platelet integrin-αIIbβ3 activation and aggregation has been demonstrated in VASP-null mouse platelets, albeit the mechanism is unclear48,49. VASP phosphorylation at Ser239 inhibits platelet activation via the cGMP/protein kinase G (PKG) signaling pathway, negatively affecting platelet responsiveness49,51. In collagen-stimulated conditions, triple NOX-deficient mouse platelets showed greater VASP Ser239 phosphorylation compared to the wild type, accompanied by higher intraplatelet cGMP levels in resting conditions6. Research indicates that NOX2-derived ROS in platelets block the cGMP/PKG signaling pathway8.
To examine the effect of GSK2795039 on regulating platelet cGMP signaling, we first examined whether GSK2795039 alone could affect VASP phosphorylation under resting conditions. Treatment of platelets with GSK2795039 significantly increased phosphorylation of VASP at Ser239 in a dose-dependent manner (Fig. 7B, blue bars), accompanied by elevated intracellular cGMP levels (Fig. 7C, blue bars), indicating activation of the NO/cGMP/PKG pathway. The mechanism was confirmed to be PKG-dependent, as the effect was abolished by PKG inhibitor Rp-8-pCPT-cGMPS but not affected by PKA inhibitor H-89 (Supplementary Fig. S4A).
As shown in Fig. 7B and C, collagen stimulation decreased both pVASP and cGMP, and these reductions were significantly reversed by GSK2795039 pretreatment. Supplementary Fig. S4B demonstrates that ROS scavengers Mn(III)TMPyP and Tempol similarly rescued VASP phosphorylation. These findings support a model in which collagen-induced ROS generation decreases NO bioavailability or inhibits soluble guanylate cyclase (sGC), resulting in reduced cGMP synthesis and PKG activation. Consistently, treatment with either an NO donor (DEA-NONOate) or a cGMP analog (8-pCPT-cGMP) in combination with GSK2795039 further augmented pVASP levels (Supplementary Fig. S4C and D), reinforcing the conclusion that ROS suppresses the NO/cGMP/PKG pathway in collagen-stimulated platelets.
Finally, inhibition of PKG by Rp-8-pCPT-cGMPS reversed the anti-aggregatory effect of GSK2795039, whereas PKA inhibition by H-89 had no such effect and instead enhanced aggregation inhibition (Supplementary Fig. S4E). Together, these results show that the modulation of the ROS-mediated cGMP/PKG/VASP/integrin-αIIbβ3 signaling pathway by GSK2795039 contributes to its inhibitory effect on collagen-induced platelet aggregation.
GSK2795039 inhibits thrombus formation under flow and reduces thrombosis in vivo without impairing hemostasis
Subendothelial collagen exposure causes sustained flow-resistant platelet adhesion, aggregation, and thrombus development after endothelial damage1,2. Under shear, NOX2-knockout mouse platelets exhibited decreased adherence and thrombus volume on collagen52. With DiOC6-stained platelets perfused in a collagen-coated flow chamber, we examined platelet adherence and thrombus development under shear. Platelets treated with GSK2795039 had less stable platelet adhesion and smaller platelet thrombi on immobilized collagen under shear (Fig. 8A).
Fig. 8.
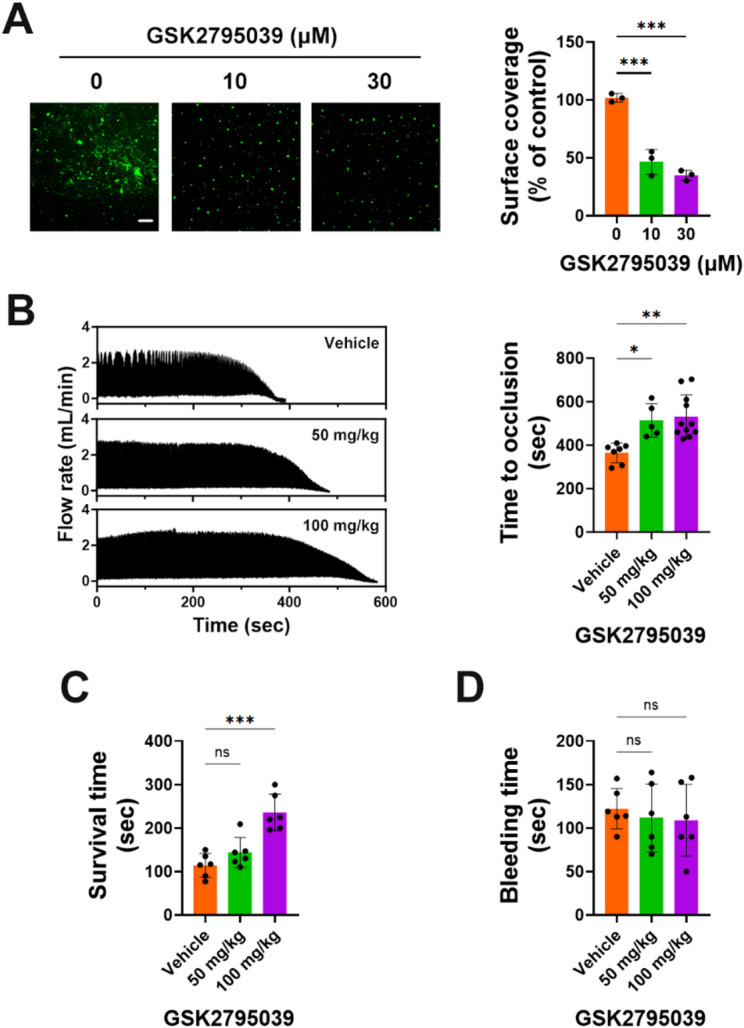
GSK2795039 inhibits thrombus formation under flow and reduces thrombosis in vivo without impairing hemostasis. (A) Washed human platelets labeled with DiOC6 were perfused over a collagen-coated surface at a shear rate, and platelet adhesion and thrombus formation were visualized. Representative images and quantification of surface coverage are presented. Scale bar represents 100 μm. (B–D) Mice were pretreated with vehicle or GSK2795039 (50 or 100 mg/kg). (B) Mice were subjected to ferric chloride-induced carotid artery injury. Occlusion times were recorded using a flowmeter. The left panel shows representative tracings of blood flow. (C) Pulmonary thromboembolism was induced by retro-orbital injection of collagen and epinephrine. Time for thromboembolic death was measured and plotted. (D) Bleeding time upon tail-tip transection is shown. All data represent the mean ± standard deviation. Statistical significance: *p < 0.05, **p < 0.01, ***p < 0.001 and ns, p > 0.05 (not significant).
We next tested the antithrombotic efficacy of GSK2795039 in vivo using two mouse models of thrombosis. Topical FeCl3 application to the carotid artery is a proven animal model for thrombosis and arterial injury53. Injuring the vascular endothelium with FeCl3 exposes collagen to platelet adhesion and thrombus, leading to endothelial denudation from ferric ion transendothelial migration54. Figure 8B shows that GSK2795039-administered animals protected against carotid occlusion better than vehicle controls, suggesting that GSK2795039 may delay thrombus formation in vivo. To further examine platelet-specific thrombotic potential, we utilized a pulmonary thromboembolism model induced by intravenous injection of collagen and epinephrine. Mice pretreated with GSK2795039 showed a dose-dependent increase in survival time compared to controls (Fig. 8C), supporting its protective role against platelet-driven thromboembolism.
Importantly, to assess whether GSK2795039 interferes with physiological hemostasis, we performed a tail bleeding time assay. Mice treated with GSK2795039 at either dose did not show a significant difference in bleeding time relative to vehicle-treated animals (Fig. 8D), indicating that the compound does not impair primary hemostasis at therapeutically effective doses.
These findings collectively demonstrate that GSK2795039 suppresses pathological thrombus formation under flow and in vivo, without compromising hemostatic function.
Discussion
Our data, along with previous studies14, indicate that GSK2795039 does not affect thrombin- or U46619-induced human platelet aggregation but can inhibit collagen-induced platelet aggregation. This discrepancy may be due to the essential role of NOX2-derived ROS in collagen-mediated platelet activation and aggregation, whereas its impact on thrombin- or U46619-mediated responses is limited7. Studies demonstrating that GPVI stimulation significantly increases ROS production in human platelets, compared to much lower ROS levels induced by thrombin or U4661915–17, further highlight the critical role of NOX2-mediated ROS in platelet activation. However, in contrast, the studies by Vara et al.6,55 reported no significant inhibition of collagen-induced aggregation in NOX2−/− platelets. These findings suggest that the properties of NOX may differ between human and mouse platelets, and further research is needed to clarify these differences.
Our findings demonstrate that GSK2795039 effectively suppresses platelet activation by attenuating NOX2-derived ROS generation and maintaining PTP activity. Although NOX2 was not genetically or immunologically isolated in this study, we employed two complementary biochemical approaches to assess NADPH oxidase activity in human platelets. The first assay measured extracellular superoxide production using L-012 chemiluminescence, while the second directly quantified NADPH oxidase enzymatic activity in platelet lysates. Both assays showed significant inhibition in response to GSK2795039, supporting its functional suppression of NADPH oxidase activity in human platelets (Fig. 1D and F). Given that GSK2795039 selectively inhibits NOX2 via competitive binding to its NADPH-binding site13, and that no effect was observed on aggregation induced by non-GPVI agonists (thrombin, U46619), the observed inhibition is most likely attributed to NOX2. Given that PTPs serve as key regulators of phosphorylation-dependent signaling and are vulnerable to redox-dependent inactivation20,56, preserving their function is essential for modulating platelet responses. Treatment with GSK2795039 led to a marked reduction in intracellular ROS levels in response to collagen (Fig. 1), and notably restored PTP enzymatic activity under oxidative conditions (Fig. 2B). As a result, the phosphorylation status of critical signaling mediators, including Syk, LAT, Vav1, Btk, and PLCγ2, was diminished, indicating impaired GPVI-mediated signal transduction (Figs. 2C and 3A). These data collectively suggest that GSK2795039 mitigates aberrant platelet activation by sustaining PTP function through redox regulation.
GPVI-PLCγ2 signaling activates IP3 receptor (IP3R) to release Ca2+ from the dense tubular system, like the endoplasmic reticulum in platelets. The sarcoendoplasmic reticulum and plasma membrane Ca2+-ATPases (SERCA and PMCA) recover cytosolic Ca2+ to near-resting levels after stimulation57. Collagen-stimulated platelets can produce H2O2 at 1 mM15. H2O2-induced cytosolic Ca2+ elevations in platelets are linked to sulfhydryl oxidation-dependent IP3R activation, SERCA inhibition, and PMCA inhibition58. Our findings suggest a model in which GSK2795039 inhibits collagen-induced elevation of extracellular H2O2 (Fig. 1), which can freely diffuse into the cytosol and increase cytosolic Ca2+ by oxidizing IP3R and SERCA and inhibiting PMCA’s ability to extrude Ca2+. Further research is needed to identify which of the various ROS generated by NOX2 in activated platelets regulates the modulator of intracellular Ca2+ levels in a redox-dependent manner.
Both plasma fibrinogen and fibrinogen released from α-granules bind to the activated integrin-αIIbβ3 on the platelet membrane, which is essential for platelet aggregate formation47. P-selectin, an α-granule membrane protein, moves to platelet membrane and interacts with P-selectin glycoprotein ligand-1 on the surface of the leukocytes or endothelial cells to generate thrombus in vivo38. Dense-granule factors, like ADP and epinephrine, stimulate platelets autocrinely and paracrinely. Consistent with its inhibitory effect on the GPVI-PLCγ2 signaling pathway, GSK2795039 also inhibits collagen-stimulated granule release. These findings suggest that the inhibitory effect of GSK2795039 on platelet-mediated thrombosis in vivo is largely attributed to its suppression of granule release, in addition to its inhibition of the GPVI-PLCγ2 signaling pathway.
Another mechanism contributing to the antiplatelet activity of GSK2795039 is its modulation of the NO/cGMP/PKG/VASP signaling axis. This pathway plays a crucial inhibitory role in platelet activation by suppressing integrin αIIbβ3 activation and aggregation through PKG-mediated phosphorylation of VASP at Ser23949,59. However, NOX2-derived ROS can scavenge NO to form peroxynitrite, thereby reducing NO bioavailability and impairing sGC activation and downstream cGMP signaling60,61. Our results show that GSK2795039 restores VASP phosphorylation and intracellular cGMP levels in collagen-stimulated platelets, indicating preservation of NO/sGC/PKG signaling by suppressing NOX2-derived ROS. These effects were PKG-dependent, unaffected by PKA inhibition, and mimicked by antioxidants, highlighting a redox-sensitive regulatory mechanism. Notably, reduced integrin αIIbβ3 activation and aggregation were observed in parallel, consistent with enhanced VASP phosphorylation. Thus, GSK2795039 appears to inhibit platelet aggregation not only by disrupting GPVI signaling but also by preserving the endogenous NO/cGMP/PKG inhibitory pathway through redox modulation. Lastly, we note that all experiments were conducted under continuous stirring to reflect physiological shear. This experimental design was chosen to recapitulate physiological shear environments necessary for ROS production by NOX2, as established by Xu et al.52. Consistent with previous reports showing that stirring alone can enhance VASP Ser239 phosphorylation in human platelets62, this condition likely accounts for the elevated baseline levels of VASP phosphorylation observed in our study.
Our findings indicate that the downregulation of the ROS-driven ASK1/p38 MAPK/cPLA2/TXA2 signaling pathway is associated with the antiplatelet action of GSK2795039. Additionally, GSK2795039 seems to inhibit TXA2 production independently of the ASK1/p38 axis. Activated by cytosolic Ca2+, cPLA2 translocates to membranes via a Ca2+-dependent phospholipid‐binding domain63. Thus, it is plausible that GSK2795039’s impact on TXA2 production may partially stem from a reduction in Ca2+-dependent cPLA2 activation, as it suppressed the rise in cytosolic Ca2+ in platelets stimulated with collagen. Notably, Sledz et al.44 showed that although ASK1 initiates p38 MAPK phosphorylation in human platelets, continued phosphorylation is driven by autophosphorylation through Syk. Given this, and GSK2795039’s inhibitory effect on Syk activation (Fig. 2), it is likely that GSK2795039 also contributes to the suppression of p38 MAPK/cPLA2/TXA2 signaling by inhibiting Syk-mediated autophosphorylation of p38 MAPK.
Moreover, we acknowledge that the inhibition of serine/threonine phosphorylation observed in Fig. 5 suggests broader redox-sensitive signaling modulation beyond tyrosine phosphatases. Collagen stimulation increases intracellular ROS, which activates ASK1 (MAP3K) via autophosphorylation at Thr838. Activated ASK1, in turn, phosphorylates and activates MKK3 (at Ser189) and MKK6 (at Ser207), both of which are dual-specificity MAPK kinases. These kinases subsequently phosphorylate p38 MAPK at Thr180/Tyr182, promoting its activation. Activated p38 MAPK then phosphorylates cPLA2 at Ser510, enhancing its enzymatic activity. The stepwise inhibition of these phosphorylation events by GSK2795039 strongly suggests suppression of the ROS–ASK1–MKK–MAPK signaling axis. Therefore, while PTP preservation may contribute to inhibition of tyrosine phosphorylation, GSK2795039 also appears to modulate serine/threonine kinase cascades downstream of NOX2-dependent ROS production.
The sustained and continuous adherence of platelets to the extracellular matrix is crucial for thrombus formation at sites of arterial injury1,2. Both intracellular and extracellular ROS are key factors that facilitate platelet adhesion and enhance platelet activation in response to shear stress, thereby promoting thrombus formation6,7,52. Given that GSK2795039 significantly reduces intracellular and extracellular ROS levels triggered by collagen (Fig. 1) and inhibits thrombus formation as well as platelet adhesion to collagen under shear conditions (Fig. 8A), it suggests that GSK2795039 may hinder stable platelet adhesion and thrombus progression within the vasculature of cardiovascular disease patients. This hypothesis is further supported by our in vivo results showing that GSK2795039 delays thrombus formation in a ferric chloride-induced carotid artery injury model and significantly improves survival in a pulmonary thromboembolism model (Fig. 8B and C). These data indicate that NOX2 inhibition by GSK2795039 confers protection against platelet-mediated arterial and pulmonary thrombosis. Notably, despite its antithrombotic efficacy, GSK2795039 did not prolong bleeding time in the tail transection assay (Fig. 8D), suggesting that it does not compromise primary hemostasis. This distinguishes GSK2795039 from conventional antiplatelet agents, which often increase bleeding risk by interfering with essential hemostatic mechanisms. The apparent dissociation between antithrombotic activity and hemostatic function may arise from the preferential targeting of NOX2-derived ROS, which are more critical in pathological thrombus propagation than in physiological platelet plug formation. These findings reinforce the potential of GSK2795039 as a novel therapeutic with a favorable safety profile.
However, the present study has several limitations. First, while GSK2795039 is a promising selective NOX2 inhibitor, its therapeutic development must focus on specificity and off-target effects. GSK2795039 lowers ROS production by competitively binding to NOX2’s NADPH binding site13. The selective inhibition of NOX2 over other NADPH oxidase isoforms is due to differences in the NADPH binding site, making GSK2795039 more specific. NOX inhibitors, such as GSK2795039, can have unintended side effects on other NADPH-dependent enzymes, making their development difficult. Because NADPH is required for antioxidant defence and metabolism, inhibitors may have an unintended effect on enzymes. Second, patients with X-linked chronic granulomatous disease are vulnerable to serious infections and immune disorders due to their genetic NOX2 deficiency. This implies that GSK2795039’s inhibition of NOX2 could potentially lead to unfavourable immunological adverse effects. Although the current study does not entirely eliminate this possibility, we do not expect that transient NOX2 inhibition will produce the same clinical outcomes as a permanent NOX2 absence, since the temporary and dose-dependent effects of GSK2795039 are likely to differ from those caused by a genetic NOX2 deficiency. Further studies are needed to evaluate the long-term safety of GSK2795039, particularly with respect to its impact on the immune system, to confirm its viability as a safe and effective antiplatelet therapy.
In conclusion, our work demonstrates that NOX2 inhibitor GSK2795039 is capable of inhibiting collagen-induced ROS generation and subsequent signaling events mediated by ROS, ultimately leading to attenuation of collagen-induced platelet aggregation and platelet-dependent thrombosis. These observations suggest that GSK2795039 may be potentially beneficial in the prevention of platelet-mediated thrombotic disorder.
Materials and methods
Ethics statements
All animal experiments were approved by the Institutional Animal Care and Use Committee of Seoul National University (Approval No. SNU-230731-2-3), and all methods were performed in accordance with relevant guidelines and regulations. The study is reported in accordance with the ARRIVE guidelines (https://arriveguidelines.org).
The collection and use of human blood samples from healthy volunteers were approved by the Seoul National University Institutional Review Board under Protocol No. 2206/001–006, and all methods involving human participants were conducted in accordance with the relevant guidelines and regulations. Informed consent was obtained from all participants.
Reagents and antibodies
The study utilized several reagents, including GSK2795039, DCFH2, polyethylene glycol 300, Rp-8-pCPT-cGMPS, H-89, 8-pCPT-cGMP, (all from MedChem Express, Princeton, NJ, USA), CM-H2DCFDA, 3,3’-dihexyloxacarbocyanine Iodide (DiOC6), Fluo-3-AM (all from Molecular Probes, Eugene, OR, USA), 4-(2-aminoethyl)benzenesulfonyl fluoride (AEBSF) (Gold Biotechnology, St. Louis, MO, USA), aprotinin, NaCl, NaHCO3, Nonidet P-40, Tris (all from Amresco, Solon, OH, USA), CaCl2, catalase, (both from Calbiochem, San Diego, CA, USA), DMSO, EGTA, Evans blue, FeCl3, glucose, iodoacetamide, KH2PO4, Na2HPO4, Na3VO4, Na4P2O7•10H2O, NaF, N-ethylmaleimide, paraformaldehyde, prostaglandin E1, sodium citrate, thrombin, Triton X-100, β-glycerophosphate, Tween 80, U46619, NADPH, L-012, Tempol (all from Sigma-Aldrich, St. Louis, MO, USA), collagen (Chrono-Log, Havertown, PA, USA), citric acid (Duksan, Seoul, Korea), EDTA, KCl, leupeptin, MgCl2 (all from USB, Cleveland, OH, USA), dithiothreitol (Duchefa Biochemie, Haarlem, Netherlands), FITC-labelled annexin V (BD Biosciences, San Jose, CA, USA), HEPES (Thermo Fisher Scientific, Waltham, MA, USA), DEA-NONOate (Enzo Biochem, Farmingdale, NY, USA), Mn(III)TMPyP (Cayman, Ann Arbor, MI, USA).
All antibodies used in this study were used are summarized in Supplementary Table S1.
Human platelet preparation
Blood samples from healthy, medication-free individuals were collected into tubes containing acid/citrate/dextrose (22.0 g sodium citrate, 24.5 g dextrose, and 7.3 g citric acid per 1 L) (Becton Dickson, Franklin, NJ, USA). Platelet-rich plasma (PRP) was separated by centrifugation using low speed centrifuge (Hanil Scientific Inc., Gimpo, Republic of Korea) at 150×g for 15 min, followed by an additional centrifugation at 300×g for 10 min at 25 °C using to concentrate the platelets. The pellet was resuspended in Tyrode’s-HEPES buffer (10 mM HEPES [pH 7.4], 129 mM NaCl, 0.8 mM KH2PO4, 8.9 mM NaHCO3, 2.8 mM KCl, 0.8 mM MgCl2, and 5.6 mM glucose) supplemented with 2 mM EDTA, 10% of citric acid/citrate/dextrose solution, and 1 µM prostaglandin E1, and washed once more. Washed platelets were resuspended in Tyrode’s-HEPES buffer at the appropriate concentration. Unless specified, washed platelets were treated with 1 mM CaCl2 for 2 min prior to stimulation.
Light transmission aggregometry
Platelet aggregation was assessed by measuring changes in light transmission using a 4-channel aggregometer (Chrono-Log, Havertown, PA, USA) at 37 °C under continuous stirring (1000 rpm). Data were recorded using Aggrolink software, with 100% transmission calibrated using buffer and washed platelets for the respective controls.
NOX activity assay
NOX activity was determined as previously described64. Briefly, washed platelet lysates (10 µg protein) were incubated with 100 µM L-012 and 0.5 mM NADPH at 37 °C in the dark, and luminescence was detected using a luminometer (Berthold Technologies, Oak Ridge, TN, USA). NOX activity was first calculated as relative luminescence units (RLU) per µg protein, and the results were presented as relative fold changes compared to the control group.
Assessment of intracellular ROS and cytosolic Ca2+ levels
For the measurement of intracellular ROS and Ca2+ levels, washed platelets (5 × 108/mL) were loaded with CM-H2DCFDA (5 µM) or Fluo-3-AM (1 µM) by incubation at 37 °C for 30 min in the dark. At 37 °C, a fluorometer cuvette was used to stimulate platelets (5 × 108/mL) in Tyrode’s-HEPES buffer with constant stirring at 1000 rpm. A spectrofluorophotometer (Jasco, Tokyo, Japan) measured the fluorescence of CM-DCF (495 nm excitation, 525 nm emission) or Fluo-3 (488 nm excitation, 525 nm emission).
Assessment of extracellular ROS levels
Washed platelets (5 × 108/mL) in Tyrode’s-HEPES buffer with 1 µM DCFH2 were stimulated with 10 µg/mL collagen in a fluorometer cuvette at 37 °C with continual stirring at 1000 rpm. The fluorescence of DCF (488 nm excitation, 525 nm emission) was measured using a spectrofluorophotometer as described above.
Assessment of extracellular superoxide anion levels
Extracellular superoxide anion levels were assessed using L-012, a chemiluminescent probe specific for superoxide65. After washed platelets (5 × 108/mL) in Tyrode’s-HEPES buffer containing 100 µM L-012 were stimulated, luminescence was read immediately 30-second intervals over 5 min using a luminometer (Berthold Technologies, Oak Ridge, TN, USA).
Assessment of extracellular H2O2 levels
The Amplex Red hydrogen peroxide/peroxidase assay kit (Invitrogen, New York, NY, USA) was used. Washed platelets (5 × 108/mL) were treated at 1000 rpm at 37 °C in a Thermomixer (Eppendorf, Hamburg, Germany). The supernatant (50 µL) obtained by centrifugation using micro centrifuge (Hanil Scientific Inc., Gimpo, Republic of Korea) at 12,000×g for 30 s at 25 °C was added to each black 96-well containing a reaction mixture (50 µL) containing horseradish peroxidase (0.2 U/mL) and Amplex Red reagent (100 µM) to start the reaction. After 30 min of incubation, a Cytation3 microplate reader (BioTek, Burlington, VT, USA) detected fluorescence (520 nm excitation, 590 nm emission).
PTP activity assay
PTP activity was measured as described previously66. Washed platelets were stimulated under experimental conditions and lysed in an oxygen-deprived environment (< 1% O₂; Mbraun glovebox workstation, Garching, Germany) using a buffer (50 mM Tris-HCl, pH 6.5, 150 mM NaCl, 1% Nonidet P-40) supplemented with 1 µg/mL leupeptin, 1 µg/mL aprotinin, 1 mM AEBSF, 10 mM iodoacetamide, and 10 mM N-ethylmaleimide to preserve the reduced state of catalytic cysteines. Samples were centrifuged using micro centrifuge (Hanil Scientific Inc., Gimpo, Republic of Korea) at 12,500×g for 10 min at 4 °C to remove cellular debris. The supernatant was incubated at room temperature for 30 min in the dark to allow complete alkylation of free thiol groups. Residual alkylating agents were quenched by the addition of 50 mM dithiothreitol. Protein concentration was then determined using the Bradford assay (Bio-Rad, Hercules, CA, USA). PTP enzymatic activity was measured using a commercially available colorimetric PTP assay kit (EMD Millipore, Billerica, MA, USA) according to the manufacturer’s instructions. Liberated inorganic phosphate was quantified by absorbance measurement, and PTP activity was normalized and expressed as nanomoles of phosphate released per minute per milligram of total protein.
Western blotting
After stimulation, washed platelets were lysed in cell extraction buffer (20 mM HEPES [pH 7.0], 150 mM NaCl, 1% Triton X-100, 10% glycerol, 1 mM EDTA, 2 mM EGTA, 20 mM β-glycerophosphate, 1 mM Na3VO4, 1 µg/mL leupeptin, 1 µg/mL aprotinin, and 1 mM AEBSF). Lysates were centrifuged using micro centrifuge (Hanil Scientific Inc., Gimpo, Republic of Korea) at 12,000×g for 10 min at 4 °C to remove debris, and total protein concentrations were determined using the Bio-Rad protein assay dye reagent (Bio-Rad Laboratories, Richmond, CA). Equal amounts of protein (20–40 µg) were loaded per lane and subjected to SDS-PAGE. Western blot analysis was performed using specific antibodies, and band intensity was quantified with ImageJ software (NIH, Bethesda, MD, USA).
TXB2 ELISA
Washed platelets (5 × 10⁸/mL) were stimulated and the reaction was halted by adding EDTA (5 mM) and indomethacin 200 µM). The supernatant was collected, and TXB₂ levels were measured using an ELISA kit (Cayman, Ann Arbor, MI, USA) and a Cytation3 microplate reader.
Flow cytometry and analysis
Flow cytometry was performed to assess P-selectin exposure, integrin activation, and PS exposure using CD62P-PE (0.5 µg/mL) and PAC1-FITC (5 µg/mL) antibodies and annexin V-FITC (0.1 mg/mL), respectively. Washed platelets were fixed in 1% paraformaldehyde, diluted with PBS, and analyzed using a FACSCalibur flow cytometer, with data processed in FlowJo software (FlowJo LLC, Ashland, OR, USA).
ATP release assay
Collagen-stimulated washed platelets were analyzed for ATP release using a ATP bioluminescent assay kit (Sigma-Aldrich, St. Louis, MO, USA) in a white 96-well plate. Luminescence was monitored at 30-second intervals over 5 min using an LB 960 Centro microplate luminometer (Berthold, Bad Wildbad, Germany).
cGMP ELISA
Following centrifugation using micro centrifuge (Hanil Scientific Inc., Gimpo, Republic of Korea) at 12,000×g for 30 s at 4 °C, washed platelets were lysed in 0.1 M HCl with 1% Triton X-100. After additional centrifugation using micro centrifuge (Hanil Scientific Inc., Gimpo, Republic of Korea) at 12,000×g for 10 min at 4 °C, cGMP levels in the supernatant were quantified using an ELISA kit (Enzo Life Sciences, Exeter, UK) and a Cytation3 microplate reader.
Determination of platelet adhesion and thrombus formation under flow conditions
As previously described64, DiOC6-labeled washed platelets were perfused over a collagen-coated surface in a flow chamber (Chamlide CF) at 1000 s⁻¹. Adherent platelets were fixed with paraformaldehyde and visualized under a fluorescence microscope. Image analysis for surface coverage was performed with ImageJ software.
Animal Preparation and drug administration
C57BL/6J male mice (10–11 weeks old) were obtained from the Jackson Laboratory (Bar Harbor, ME, USA) and maintained in a specific-pathogen-free animal facility at Seoul National University. Mice were housed under controlled environmental conditions and fed a standard chow diet (Harlan Teklad, Madison, WI, USA) ad libitum. Mice were randomly divided into three experimental groups. GSK2795039 was orally administered at a dose of 10 mL/kg body weight via oral gavage in a vehicle composed of 20% DMSO, 20% Tween 80, and 60% polyethylene glycol 300, as previously described13. Control animals received an equivalent volume of the vehicle alone. One hour after GSK2795039 administration, mice were anesthetized by intraperitoneal injection of alfaxalone (100 mg/kg; Jurox, New South Wales, Australia) and xylazine (10 mg/kg; Rompun, Bayer Korea, Seoul, Korea).
Pulmonary thromboembolism model
To induce pulmonary thromboembolism, a mixture of collagen (0.8 mg/kg; Type I, bovine, Sigma-Aldrich) and epinephrine (60 µg/kg; Sigma-Aldrich) dissolved in sterile saline was retro-orbitally injected in a final volume of 100 µL using a 30G insulin syringe. After injection, mice were returned to a warmed cage and continuously monitored for respiratory distress, paralysis, or death for up to 15 min. Mice exhibiting severe respiratory symptoms or sudden death were recorded as thromboembolism-positive. Time to death by pulmonary embolism was analyzed and utilized as a measure of susceptibility to thrombosis.
Carotid occlusion model
To assess arterial thrombosis the right common carotid artery was surgically exposed. A Perivascular Flowprobe (model MA0.5PSB, Transonic Systems Inc., Ithaca, NY, USA) connected to a Perivascular Flowmeter (model TS420) was positioned around the isolated artery and calibrated to baseline flow (0.3–0.9 mL/s). Vascular injury was induced by applying a filter paper (1 × 1 mm) soaked in 20% FeCl₃ to the surface of the artery for 3 min. The filter paper was then removed, and blood flow was continuously recorded every 0.5 s. Stable occlusion was defined as complete cessation of flow (0 mL/second) sustained for at least 30 s.
Tail bleeding assay
Hemostatic function was assessed using a tail bleeding time assay. The tail was transected at a fixed diameter of 1.5 mm from the tip using a sterile scalpel, and the remaining portion was immediately immersed in prewarmed isotonic saline at 37 °C. Bleeding was monitored visually, and the time to complete cessation of bleeding was recorded. Cessation was defined as no rebleeding for at least 1 min following the initial stop.
Statistical analysis
Data were statistically analyzed using GraphPad PRISM software 10 (GraphPad Software Inc., La Jolla, CA, USA). The Shapiro–Wilk test was used for normality testing. Statistical significance was calculated using the t-test for two-group comparisons and one-way or two-way ANOVA tests for multiple comparisons with Bonferroni correction, respectively. P values less than 0.05 were considered statistically significant.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
This work was supported by the Basic Science Research Program through the National Research Foundation of Korea funded by the Ministry of Education (NRF-2022R1A6A1A03046247), and the Brain Korea 21 Plus Program funded by the Korean government (MOE) in the Republic of Korea.
Author contributions
Conceptualization: TSC. Data curation: EBO, YJK, and TK. Formal analysis: EBO, YJK, TK, JJ, HY, JWP, SK, TK, and JS. Methodology: EBO, YJK, TK, JJ, HY, JWP, SK, TK, JS, and TSC. Software: EBO, YJK, TK, JJ, and HY. Visualization: EBO, YJK, and TK. Writing—original draft preparation: EBO, YJK, and TK. Writing—review and editing: EBO and TSC. Supervision: TSC. Project administration: TSC. Funding acquisition: TSC. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Basic Science Research Program through the National Research Foundation of Korea funded by the Ministry of Education (NRF-2022R1A6A1A03046247), and the Brain Korea 21 Plus Program funded by the Korean government (MOE) in the Republic of Korea.
Data availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
Declarations
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Eun Bee Oha, Yun Jeong Kong and Taeil Kim contributed equally to this work.
References
- 1.Nieswandt, B. et al. Glycoprotein VI but not alpha2beta1 integrin is essential for platelet interaction with collagen. EMBO J.20, 2120–2130. 10.1093/emboj/20.9.2120 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nieswandt, B., Pleines, I. & Bender, M. Platelet adhesion and activation mechanisms in arterial thrombosis and ischaemic stroke. J Thromb. Haemost 9 Suppl. (1), 92–104. 10.1111/j.1538-7836.2011.04361.x (2011). [DOI] [PubMed]
- 3.Lambeth, J. D. Nox enzymes, ROS, and chronic disease: An example of antagonistic Pleiotropy. Free Radic Biol. Med.43, 332–347. 10.1016/j.freeradbiomed.2007.03.027 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chlopicki, S. et al. Functional role of NADPH oxidase in activation of platelets. Antioxid. Redox Signal.6, 691–698. 10.1089/1523086041361640 (2004). [DOI] [PubMed] [Google Scholar]
- 5.Vara, D., Cifuentes-Pagano, E., Pagano, P. J. & Pula, G. A novel combinatorial technique for simultaneous quantification of oxygen radicals and aggregation reveals unexpected redox patterns in the activation of platelets by different physiopathological stimuli. Haematologica104, 1879–1891. 10.3324/haematol.2018.208819 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vara, D. et al. NADPH oxidases are required for full platelet activation in vitro and thrombosis in vivo but dispensable for plasma coagulation and hemostasis. Arterioscler. Thromb. Vasc Biol.41, 683–697. 10.1161/ATVBAHA.120.315565 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Delaney, M. K. et al. Differential roles of the NADPH-Oxidase 1 and 2 in platelet activation and thrombosis. Arterioscler. Thromb. Vasc Biol.36, 846–854. 10.1161/ATVBAHA.116.307308 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Magwenzi, S. et al. Oxidized LDL activates blood platelets through CD36/NOX2-mediated Inhibition of the cGMP/protein kinase G signaling cascade. Blood125, 2693–2703. 10.1182/blood-2014-05-574491 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Akbar, H. et al. Small molecule targeting the Rac1-NOX2 interaction prevents collagen-related peptide and thrombin-induced reactive oxygen species generation and platelet activation. J. Thromb. Haemost. 16, 2083–2096. 10.1111/jth.14240 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu, Y. et al. PI3K positively regulates platelet activation and thrombosis via PI(3)P-Directed function of NADPH oxidase. Arterioscler. Thromb. Vasc Biol.37, 2075–2086. 10.1161/ATVBAHA.117.309751 (2017). [DOI] [PubMed] [Google Scholar]
- 11.Pignatelli, P. et al. gp91phox-dependent expression of platelet CD40 ligand. Circulation110, 1326–1329. 10.1161/01.cir.0000134963.77201.55 (2004). [DOI] [PubMed] [Google Scholar]
- 12.Kim, K., Li, J., Tseng, A., Andrews, R. K. & Cho, J. NOX2 is critical for heterotypic neutrophil-platelet interactions during vascular inflammation. Blood126, 1952–1964. 10.1182/blood-2014-10-605261 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hirano, K. et al. Discovery of GSK2795039, a novel small molecule NADPH oxidase 2 inhibitor. Antioxid. Redox Signal.23, 358–374. 10.1089/ars.2014.6202 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lu, W. J. et al. VAS2870 and VAS3947 attenuate platelet activation and thrombus formation via a NOX-independent pathway downstream of PKC. Sci. Rep.9, 18852. 10.1038/s41598-019-55189-5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pignatelli, P., Pulcinelli, F. M., Lenti, L., Gazzaniga, P. P. & Violi, F. Hydrogen peroxide is involved in collagen-induced platelet activation. Blood91, 484–490. 10.1182/blood.V91.2.484 (1998). [PubMed] [Google Scholar]
- 16.Krotz, F. et al. NAD(P)H oxidase-dependent platelet superoxide anion release increases platelet recruitment. Blood100, 917–924. 10.1182/blood.v100.3.917 (2002). [DOI] [PubMed] [Google Scholar]
- 17.Jang, J. Y. et al. Reactive oxygen species play a critical role in collagen-induced platelet activation via SHP-2 oxidation. Antioxid. Redox Signal.20, 2528–2540. 10.1089/ars.2013.5337 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reiniers, M. J. et al. Preparation and practical applications of 2’,7’-Dichlorodihydrofluorescein in redox assays. Anal. Chem.89, 3853–3857. 10.1021/acs.analchem.7b00043 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Holmstrom, K. M. & Finkel, T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat. Rev. Mol. Cell. Biol.15, 411–421. 10.1038/nrm3801 (2014). [DOI] [PubMed] [Google Scholar]
- 20.Tonks, N. K. Redox redux: Revisiting PTPs and the control of cell signaling. Cell121, 667–670. 10.1016/j.cell.2005.05.016 (2005). [DOI] [PubMed] [Google Scholar]
- 21.Watson, S. P., Auger, J. M., McCarty, O. J. & Pearce, A. C. GPVI and integrin αIIbβ3 signaling in platelets. J. Thromb. Haemost. 3, 1752–1762. 10.1111/j.1538-7836.2005.01429.x (2005). [DOI] [PubMed] [Google Scholar]
- 22.Salmeen, A. & Barford, D. Functions and mechanisms of redox regulation of cysteine-based phosphatases. Antioxid. Redox Signal.7, 560–577. 10.1089/ars.2005.7.560 (2005). [DOI] [PubMed] [Google Scholar]
- 23.Karisch, R. et al. Global proteomic assessment of the classical protein-tyrosine phosphatome and redoxome. Cell146, 826–840. 10.1016/j.cell.2011.07.020 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fujii, C. et al. Involvement of protein-tyrosine kinase p72syk in collagen-induced signal transduction in platelets. Eur. J. Biochem.226, 243–248. 10.1111/j.1432-1033.1994.tb20047.x (1994). [DOI] [PubMed] [Google Scholar]
- 25.Furlong, M. T. et al. Identification of the major sites of autophosphorylation of the murine protein-tyrosine kinase Syk. Biochim. Biophys. Acta1355, 177–190. 10.1016/s0167-4889(96)00131-0 (1997). [DOI] [PubMed] [Google Scholar]
- 26.Donner, L. et al. The collagen receptor glycoprotein VI promotes platelet-mediated aggregation of beta-amyloid. Sci. Signal.1310.1126/scisignal.aba9872 (2020). [DOI] [PubMed]
- 27.Pasquet, J. M. et al. LAT is required for tyrosine phosphorylation of phospholipase cγ2 and platelet activation by the collagen receptor GPVI. Mol. Cell. Biol.19, 8326–8334 (1999). http://www.ncbi.nlm.nih.gov/pubmed/10567557 [DOI] [PMC free article] [PubMed]
- 28.Pearce, A. C. et al. Vav1, but not Vav2, contributes to platelet aggregation by CRP and thrombin, but neither is required for regulation of phospholipase C. Blood100, 3561–3569. 10.1182/blood.V100.10.3561 (2002). [DOI] [PubMed] [Google Scholar]
- 29.Atkinson, B. T., Ellmeier, W. & Watson, S. P. Tec regulates platelet activation by GPVI in the absence of Btk. Blood102, 3592–3599. 10.1182/blood-2003-04-1142 (2003). [DOI] [PubMed] [Google Scholar]
- 30.Suzuki-Inoue, K. et al. Glycoproteins VI and Ib-IX-V stimulate tyrosine phosphorylation of tyrosine kinase Syk and phospholipase Cγ2 at distinct sites. Biochem. J.378, 1023–1029. 10.1042/BJ20031430 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mancini, F., Rigacci, S., Berti, A., Balduini, C. & Torti, M. The low-molecular-weight phosphotyrosine phosphatase is a negative regulator of FcgammaRIIA-mediated cell activation. Blood110, 1871–1878. http://dx.doi.org/blood-2007-03-081414 (2007). [DOI] [PubMed] [Google Scholar]
- 32.Chari, R. et al. PKC-delta, SHIP-1 interactions regulate GPVI-mediated platelet-dense granule secretion. Blood114, 3056–3063. 10.1182/blood-2008-11-188516 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Weng, Z. et al. PTEN regulates collagen-induced platelet activation. Blood116, 2579–2581. 10.1182/blood-2010-03-277236 (2010). [DOI] [PubMed] [Google Scholar]
- 34.van der Meijden, P. E. J. & Heemskerk, J. W. M. Platelet biology and functions: New concepts and clinical perspectives. Nat. Rev. Cardiol.16, 166–179. 10.1038/s41569-018-0110-0 (2019). [DOI] [PubMed] [Google Scholar]
- 35.Nishikawa, K., Toker, A., Johannes, F. J., Songyang, Z. & Cantley, L. C. Determination of the specific substrate sequence motifs of protein kinase C isozymes. J. Biol. Chem.272, 952–960. 10.1074/jbc.272.2.952 (1997). [DOI] [PubMed] [Google Scholar]
- 36.Fontayne, A., Dang, P. M., Gougerot-Pocidalo, M. A. & El-Benna, J. Phosphorylation of p47phox sites by PKC alpha, beta II, delta, and zeta: Effect on binding to p22phox and on NADPH oxidase activation. Biochemistry41, 7743–7750. 10.1021/bi011953s (2002). [DOI] [PubMed] [Google Scholar]
- 37.Durrant, T. N., van den Bosch, M. T. & Hers, I. Integrin alpha(IIb)beta(3) outside-in signaling. Blood130, 1607–1619. 10.1182/blood-2017-03-773614 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Frenette, P. S. et al. D. P-Selectin glycoprotein ligand 1 (PSGL-1) is expressed on platelets and can mediate platelet-endothelial interactions in vivo. J. Exp. Med.191, 1413–1422. 10.1084/jem.191.8.1413 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Atkinson, B. T., Stafford, M. J., Pears, C. J. & Watson, S. P. Signalling events underlying platelet aggregation induced by the glycoprotein VI agonist convulxin. Eur. J. Biochem.268, 5242–5248. 10.1046/j.0014-2956.2001.02448.x (2001). [DOI] [PubMed] [Google Scholar]
- 40.Wong, D. A., Kita, Y., Uozumi, N. & Shimizu, T. Discrete role for cytosolic phospholipase A(2)alpha in platelets: Studies using single and double mutant mice of cytosolic and group IIA secretory phospholipase A(2). J. Exp. Med.196, 349–357. 10.1084/jem.20011443 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Borsch-Haubold, A. G., Kramer, R. M. & Watson, S. P. Phosphorylation and activation of cytosolic phospholipase A2 by 38-kDa mitogen-activated protein kinase in collagen-stimulated human platelets. Eur. J. Biochem.245, 751–759. 10.1111/j.1432-1033.1997.t01-1-00751.x (1997). [DOI] [PubMed] [Google Scholar]
- 42.Wang, X. et al. p47phox deficiency impairs platelet function and protects mice against arterial and venous thrombosis. Redox Biol.34, 101569. 10.1016/j.redox.2020.101569 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Naik, M. U. et al. Ask1 regulates murine platelet granule secretion, thromboxane A(2) generation, and thrombus formation. Blood129, 1197–1209. 10.1182/blood-2016-07-729780 (2017). [DOI] [PMC free article] [PubMed]
- 44.Sledz, K. M. et al. Redundant role of ASK1-mediated p38MAPK activation in human platelet function. Cell. Signal.68, 109528. 10.1016/j.cellsig.2020.109528 (2020). [DOI] [PubMed] [Google Scholar]
- 45.Cameron, S. J. et al. Platelet extracellular regulated protein kinase 5 is a redox switch and triggers maladaptive platelet responses and myocardial infarct expansion. Circulation132, 47–58. 10.1161/CIRCULATIONAHA.115.015656 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang, M. et al. Platelet CD36 signaling through ERK5 promotes caspase-dependent procoagulant activity and fibrin deposition in vivo. Blood Adv.2, 2848–2861. 10.1182/bloodadvances.2018025411 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bennett, J. S. Structure and function of the platelet integrin αIIbβ3. J. Clin. Invest.115, 3363–3369. 10.1172/JCI26989 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hauser, W. et al. Megakaryocyte hyperplasia and enhanced agonist-induced platelet activation in vasodilator-stimulated phosphoprotein knockout mice. Proc. Natl. Acad. Sci. U S A. 96, 8120–8125. 10.1073/pnas.96.14.8120 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Aszodi, A. et al. The vasodilator-stimulated phosphoprotein (VASP) is involved in cGMP- and cAMP-mediated Inhibition of agonist-induced platelet aggregation, but is dispensable for smooth muscle function. EMBO J.18, 37–48. 10.1093/emboj/18.1.37 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Reinhard, M., Jarchau, T. & Walter, U. Actin-based motility: Stop and go with ena/vasp proteins. Trends Biochem. Sci.26, 243–249. 10.1016/s0968-0004(00)01785-0 (2001). [DOI] [PubMed] [Google Scholar]
- 51.Smolenski, A. et al. Analysis and regulation of vasodilator-stimulated phosphoprotein Serine 239 phosphorylation in vitro and in intact cells using a phosphospecific monoclonal antibody. J. Biol. Chem.273, 20029–20035. 10.1074/jbc.273.32.20029 (1998). [DOI] [PubMed] [Google Scholar]
- 52.Xu, Z. et al. Shear and integrin outside-in signaling activate NADPH-Oxidase 2 to promote platelet activation. Arterioscler. Thromb. Vasc Biol.41, 1638–1653. 10.1161/ATVBAHA.120.315773 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bonnard, T. & Hagemeyer, C. E. Ferric Chloride-induced thrombosis mouse model on carotid artery and mesentery vessel. J. Vis. Exp. e52838. 10.3791/52838 (2015). [DOI] [PMC free article] [PubMed]
- 54.Tseng, M. T., Dozier, A., Haribabu, B. & Graham, U. M. Transendothelial migration of ferric ion in FeCl3 injured murine common carotid artery. Thromb. Res.118, 275–280. 10.1016/j.thromres.2005.09.004 (2006). [DOI] [PubMed] [Google Scholar]
- 55.Vara, D. et al. NADPH oxidase 1 is a novel Pharmacological target for the development of an antiplatelet drug without bleeding side effects. FASEB J.34, 13959–13977. 10.1096/fj.202001086RRR (2020). [DOI] [PubMed] [Google Scholar]
- 56.Rhee, S. G. H2O2, a necessary evil for cell signaling. Science312, 1882–1883 (2006). [DOI] [PubMed] [Google Scholar]
- 57.Varga-Szabo, D., Braun, A. & Nieswandt, B. Calcium signaling in platelets. J. Thromb. Haemost. 7, 1057–1066. 10.1111/j.1538-7836.2009.03455.x (2009). [DOI] [PubMed] [Google Scholar]
- 58.Redondo, P. C., Salido, G. M., Rosado, J. A. & Pariente, J. A. Effect of hydrogen peroxide on Ca2+ mobilisation in human platelets through sulphydryl oxidation dependent and independent mechanisms. Biochem. Pharmacol.67, 491–502. 10.1016/j.bcp.2003.09.031 (2004). [DOI] [PubMed] [Google Scholar]
- 59.Massberg, S. et al. Enhanced in vivo platelet adhesion in vasodilator-stimulated phosphoprotein (VASP)-deficient mice. Blood103, 136–142. 10.1182/blood-2002-11-3417 (2004). [DOI] [PubMed] [Google Scholar]
- 60.Clutton, P., Miermont, A. & Freedman, J. E. Regulation of endogenous reactive oxygen species in platelets can reverse aggregation. Arterioscler. Thromb. Vasc Biol.24, 187–192. 10.1161/01.ATV.0000105889.29687.CC (2004). [DOI] [PubMed] [Google Scholar]
- 61.Chakrabarti, S. et al. Glycoprotein iib/iiia Inhibition enhances platelet nitric oxide release. Thromb. Res.113, 225–233. 10.1016/j.thromres.2004.02.018 (2004). [DOI] [PubMed] [Google Scholar]
- 62.Gambaryan, S. et al. Potent Inhibition of human platelets by cGMP analogs independent of cGMP-dependent protein kinase. Blood103, 2593–2600. 10.1182/blood-2003-09-3349 (2004). [DOI] [PubMed] [Google Scholar]
- 63.Clark, J. D. et al. A novel arachidonic acid-selective cytosolic PLA2 contains a Ca(2+)-dependent translocation domain with homology to PKC and GAP. Cell65, 1043–1051. 10.1016/0092-8674(91)90556-e (1991). [DOI] [PubMed]
- 64.Jang, J. Y. et al. Resveratrol inhibits collagen-induced platelet stimulation through suppressing NADPH oxidase and oxidative inactivation of SH2 domain-containing protein tyrosine phosphatase-2. Free Radic Biol. Med.89, 842–851. 10.1016/j.freeradbiomed.2015.10.413 (2015). [DOI] [PubMed] [Google Scholar]
- 65.Daiber, A. et al. Measurement of NAD(P)H oxidase-derived superoxide with the luminol analogue L-012. Free Radic. Biol. Med.36, 101–111. 10.1016/j.freeradbiomed.2003.10.012 (2004). [DOI] [PubMed] [Google Scholar]
- 66.Jang, J. et al. Targeting NADPH oxidase with APX-115: Suppression of platelet activation and thrombotic response. Antioxid. Redox Signal.10.1089/ars.2024.0695 (2025). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.