

Abstract
Rnt1p, the yeast orthologue of RNase III, cleaves rRNAs, snRNAs and snoRNAs at a stem capped with conserved AGNN tetraloop. Here we show that 9 bp long stems ending with AGAA or AGUC tetraloops bind to Rnt1p and direct specific but sequence-independent RNA cleavage when provided with stems longer than 13 bp. The solution structures of these two tetraloops reveal a common fold for the terminal loop stabilized by non-canonical A–A or A–C pairs and extensive base stacking. The conserved nucleotides are stacked at the 5′ side of the loop, exposing their Watson–Crick and Hoogsteen faces for recognition by Rnt1p. These results indicate that yeast RNase III recognizes the fold of a conserved single-stranded tetraloop to direct specific dsRNA cleavage.
Keywords: endoribonuclease/NMR spectroscopy/RNA processing/RNase III/tetraloop
Introduction
Processing of nascent transcripts into mature RNA is an essential step for gene expression and cell growth. In both prokaryotic and eukaryotic cells, many critical RNA processing events are performed by the double-stranded RNA (dsRNA)-specific enzyme, RNase III. In bacteria, this enzyme processes pre-rRNA and several mRNAs (Nicholson, 1999). In eukaryotic cells, at least three orthologues of RNase III were implicated in pre-rRNA processing, including Saccharomyces cerevisiae Rnt1p (Abou Elela et al., 1996), Schizosaccharomyces pombe Pac1 (Zhou et al., 1999) and human RNase III (Wu et al., 2000). In addition, Rnt1p and Pac1 were implicated in the processing of other cellular RNAs including small nuclear RNAs (snRNAs; Chanfreau et al., 1997; Abou Elela and Ares, 1998; Zhou et al., 1999). In vivo, the RNA cleavage by RNase III is highly specific and restricted to a few chemical bonds within a limited number of RNA molecules in the cell. This high specificity indicates an efficient recognition mechanism that allows these enzymes to discriminate between their RNA substrate and other dsRNA molecules in the cell. The basic dsRNA recognition motif of the RNase III family is similar to the conserved dsRNA-binding motif found in many dsRNA-binding proteins such as PKR (RNA-dependent protein kinase) and Staufen (Bycroft et al., 1995; Nanduri et al., 1998). The three-dimensional structures of several dsRNA-binding domains (dsRBDs) and dsRBD–RNA complexes revealed a highly conserved tertiary structure for the protein and the nature of the RNA–protein interaction (Ryter and Schultz, 1998; Ramos et al., 2000). However, the mechanism of RNA recognition by members of the dsRNA-binding RNase III family remains poorly understood and the RNA determinants that confer specificity remain unknown.
Rnt1p shares loose sequence homology with members of the RNase III family. In Rnt1p, the canonical dsRBD motif is located at the C-terminus between positions 372 and 440, with 25% identity to the bacterial RNase III and 31% identity to fission yeast Pac1 (Lamontagne et al., 2000). The Rnt1p 154 amino acid nuclease domain is similar in size to that of RNase III and Pac1 and shares the same charged amino acid clusters. In addition, Rnt1p possesses the characteristic eukaryotic N-terminal domain that spans 191 amino acids. Biochemical and genetic assays suggest that the N-terminal domain influences Rnt1p function by mediating both inter- and intramolecular interactions (Lamontagne et al., 2000). Deletion of the N-terminal domain showed that it is essential for the enzyme function in vivo but not in vitro or when overexpressed using heterologous promoters in yeast cells (Lamontagne et al., 2000; Nagel and Ares, 2000). On the other hand, deletion or alteration of the N-terminal domain does not influence the substrate selectivity or cleavage site selection, indicating that these functions are performed largely by the C-terminal domain that includes the nuclease domain and the dsRBD (Lamontagne et al., 2000; Nagel and Ares, 2000).
The substrate selection by Rnt1p appears to follow a similar but not identical mechanism to that used by other dsRNA-binding proteins. In vitro, Rnt1p recognizes and cleaves long duplex RNAs regardless of their sequence (Abou Elela et al., 1996). In contrast, comparison of Rnt1p natural substrates (Abou Elela and Ares, 1998; Chanfreau et al., 2000; Nagel and Ares, 2000) reveals the presence of a terminal tetraloop with the conserved sequence AGNN at 14–16 bp from the cleavage site (Figure 1). Deletion of this sequence abolishes cleavage, while deletion of nucleotides close to the cleavage site does not affect the substrate recognition or cleavage (Abou Elela and Ares, 1998; Chanfreau et al., 2000; Nagel and Ares, 2000). Mutations that change the conserved AGNN tetraloop sequence to GUNN slow RNA cleavage at low monovalent salt concentrations and block it at concentrations >100 mM (Nagel and Ares, 2000). Changes of the tetraloop sequence to GANN or GAAA also slow the cleavage to various degrees, indicating that the AG sequence plays an important role in substrate recognition (Chanfreau et al., 2000). Indeed, mutations in the AGNN tetraloop that slow the cleavage rate also decrease Rnt1p affinity for the RNA (Chanfreau et al., 2000; Nagel and Ares, 2000). Gel mobility shift assay also demonstrates that the dsRBD of Rnt1p which contains the 74 amino acid canonical RNA-binding motif and an additional non-conserved 54 amino acids is capable of recognizing the AGNN tetraloop (Nagel and Ares, 2000). These experiments prompted the surprising conclusion that Rnt1p uses the apparently single-stranded tetraloop to determine the cleavage site. On the other hand, Rnt1p may not recognize the AGNN sequence itself but may instead recognize a particular structure formed by the AGNN sequence and the adjacent RNA helix. It is also unclear whether the canonical dsRNA-binding motif itself recognizes the tetraloop or if this particular recognition is derived by the adjacent non-conserved amino acid within the C-terminus of Rnt1p (Lamontagne et al., 2000; Nagel and Ares, 2000). A role for the non-conserved amino acids in the recognition of the tetraloop would explain the inability of Pac1 and RNase III to recognize the tetraloop (Rotondo et al., 1997; Nicholson, 1999). In addition to its proposed role as a substrate identity element, the tetraloop appears to assist in determining the site of cleavage. It was shown that Rnt1p cleavage always takes place at a consistent distance from the terminal tetraloop (Chanfreau et al., 2000). These observations suggest that Rnt1p acts as a helical ruler that measures the distance from the tetraloop to the cleavage site (Chanfreau et al., 2000).
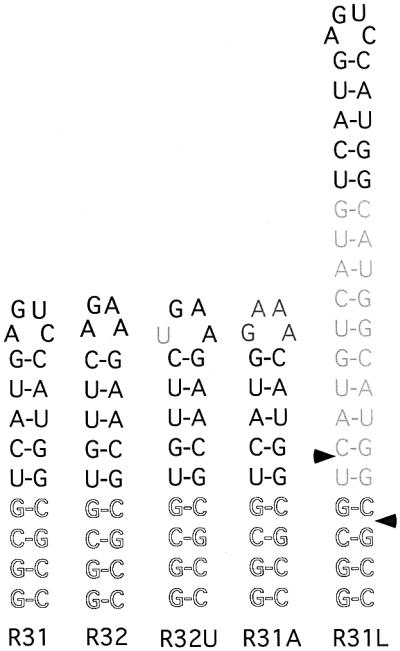
Fig. 1. Illustration of Rnt1p model substrates. The conserved AGNN nucleotides are shown in bold. The mutations introduced in the wild-type sequence are shown in grey. The four heterologous base pairs used to stabilize the structure are shown as an outline. R31 represents the terminal stem–loop of the U5 snRNA. R32 represents the terminal stem–loop of snR47. R31U is R31 with a mutation that changes the conserved A residue in the tetraloop to U. R31A contains the R31 stem sequence but the loop sequence is changed to GAAA. R31L contains the R31 sequence and an insertion of 10 bp that extends the stem size to a total of 19 bp. The sites of cleavages by Rnt1p are indicated by the arrowheads.
To identify the features of the terminal tetraloop recognized by Rnt1p and its role in determining the cleavage site, we have determined the structure of RNA oligonucleotides that contain the conserved AGNN tetraloop (AGAA or AGUC) using nuclear magnetic resonance (NMR) spectroscopy. AGAA and AGUC loops display a similar three-dimensional structure that might represent a conserved fold among AGNN tetraloops. In addition, both tetraloops contain structural features similar to the known UGAA tetraloop (Butcher et al., 1997). A model for how AGNN tetraloops can form a universal signal for Rnt1p recognition and RNA processing is proposed. The structures provide insights into a possible mechanism for specific and efficient RNA processing.
Results
Determination of Rnt1p substrate requirements
Deletion analysis of Rnt1p substrates indicates that the upper stem adjacent to the conserved AGNN tetraloop but not the lower stem is needed for the cleavage (Abou Elela and Ares, 1998; Nagel and Ares, 2000). However, the minimum stem length required for binding or cleavage by Rnt1p remained unknown. As a first step towards determining the structure of Rnt1p substrates, we have searched for short RNA substrates that could bind efficiently to Rnt1p. We have tested two substrates containing the terminal tetraloop and the first 5 bp of Rnt1p substrates at the end of U5 snRNA (R31) and snR47 snoRNA (R32), respectively. An additional four G–C base pairs were added at the end of each substrate to ensure the folding stability needed for subsequent NMR experiments. Each of the two substrates were incubated with recombinant Rnt1p in the absence of Mg2+ and fractionated on a polyacrylamide gel under native conditions. Both RNAs efficiently bound to Rnt1p and caused a band shift that increased by increasing the amount of Rnt1p (Figure 2A and B). Rnt1p bound the substrate that ends with the AGUC tetraloop (R31) with an apparent Kd value of 0.75 µM, while it bound to the substrate that ends with the AGAA tetraloop (R32) with a Kd of 1.5 µM. Substrates that lack the heterologous four G–C base pairs also interacted with Rnt1p but with lower affinity (data not shown). This result indicates that only 9 bp and an AGNN tetraloop are sufficient for recognition by Rnt1p. To determine whether the increase in the stem length enhances binding to Rnt1p, we maintained the presence of the AGUC tetraloop, increased the length of the R31 stem to 19 bp (R31L) and tested it using gel mobility shift assay. As shown in Figure 2C, the increase in the stem size did not enhance the binding to Rnt1p and bound with an apparent Kd of 1.5 µM, suggesting that Rnt1p binds both the short and long substrates with a similar mechanism.

Fig. 2. Gel shift assay of different Rnt1p substrates. Increasing concentrations (µM) of Rnt1p were incubated with 2 fmol of R31 (A), R32 (B), R31L (C), R32U (D) and R31A (E) in 150 mM KCl. RNA incubated without enzyme under the same conditions is included as a control. The positions of the unbound RNA and the shifted RNA are indicated on the left. The protein concentration is indicated on the top.
To examine the specificity of the Rnt1p binding to the R31 and R32 substrates and determine the contribution of the terminal tetraloop, we have introduced two separate mutations that disrupt the conserved tetraloop sequence. As shown in Figure 2D, the first mutant that changes the R32 tetraloop sequence to UGAA bound to Rnt1p poorly, with a calculated Kd of 5 µM, 6- and 3-fold higher than that of R31 and R32, respectively. On the other hand, substitution of the AGNN tetraloop of R31 by a GNRA tetraloop (R31A) completely blocked binding to Rnt1p even at high concentrations (Figure 2E). We conclude that Rnt1p efficiently recognizes RNA substrates with an AGNN tetraloop and a 9 bp stem.
Rnt1p cleaves its substrate 13–15 nucleotides from the terminal tetraloop (Chanfreau et al., 2000). Thus, the substrates R31 and R32 are not expected to be cleaved by Rnt1p, while R31L, which possesses a 19 bp stem, should be cleaved efficiently. To test this hypothesis, we incubated Rnt1p with R31, R32, R32U or R31L in the presence of Mg2+ and separated the resulting RNA fragments on a denaturing gel. As shown in Figure 3, R31, R32 and R32U were not cleaved by Rnt1p. However, R31L was cleaved efficiently, releasing two products of 34 and five nucleotides. The results indicate that the short substrates R31 and R32 bind correctly to Rnt1p and may lead to a productive cleavage reaction when supplied with a stem longer than 13 bp. We conclude that R31 and R32 represent the Rnt1p binding signal and thus may be used to determine the structure of the Rnt1p-binding site.
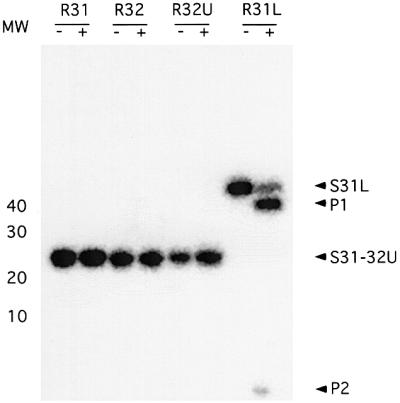
Fig. 3. Cleavage assay of Rnt1p substrates. The different RNA substrates were incubated with recombinant Rnt1p in the presence of Mg2+ and 150 mM KCl. After completion of the cleavage reactions, the RNA was fractionated using 20% denaturing PAGE and the bands visualized using Instant Imager. The positions of the different substrates (S31L–S32U) and the cleavage products (P1 and P2) are indicated on the right. The position of the 10 bp molecular weight marker is shown on the left. The (+) and (–) indicate the presence and absence of Rnt1p, respectively.
NMR spectroscopy and structure determination
To determine the feature of the tetraloop recognized by Rnt1p and verify whether Rnt1p indeed uses a single-stranded tetraloop region to identify its substrate, we set out to determine the solution structure of two different conserved AGNN tetraloops. The solution structures of AGAA and AGUC tetraloops were characterized using the 22 nucleotide RNA molecules R31 and R32 (Figure 1) that contain a fragment of the stem–loop recognized by Rnt1p (Figure 2A and B). Both RNA constructs contain a four nucleotide loop closing a 9 bp A-form helix and were prepared by in vitro transcription from an oligonucleotide DNA template.
Milligram quantities of unlabelled and 13C-15N-labelled RNAs were prepared. Complete 1H assignments were obtained using standard heteronuclear (1H, 15N, 13C, 31P) multidimensional NMR experiments and through-bond assignment strategies. All base-paired imino proton resonances in the stem regions were assigned via sequential nuclear Overhauser effects (NOEs) observed in two-dimensional NOESY spectra and with 1H–15N HSQC spectra. Watson–Crick N–H–N hydrogen bonds were observed directly in heteronuclear J (N,N)–HNN-COSY experiments performed in H2O (Dingley and Grzesiek, 1998). In both AGAA and AGUC RNAs, the G11 imino proton is partially protected from solvent exchange as observed in the imino spectra and the 1H–15N HSQC spectra at 5°C (Figure 4). The chemical shifts and NOE patterns observed for the two loops were not affected significantly by addition of 3 mM Mg2+ (data not shown), indicating that the structural data are relevant to conditions that more closely resemble the environment in vivo.

Fig. 4. Imino proton region of the one-dimensional spectrum (A) and the HSQC spectrum (B) of the AGUC tetraloop RNA. The imino proton region of the one-dimensional spectrum (C) and the HSQC spectrum (D) of the AGAA tetraloop RNA. All one-dimensional imino and HSQC spectra were recorded at 5°C.
Complete assignments for all the non-exchangeable proton resonances and their directly bound carbons were obtained using standard homonuclear two-dimensional and heteronuclear three-dimensional experiments. All H2 resonances of both RNA molecules could be assigned with a 1H–13C constant time HSQC experiment (Santoro and King, 1992) and a HCCH-TOCSY experiment for adenine H2–H8 correlation (Legault et al., 1994; Marino et al., 1994).
Structures were calculated using restrained molecular dynamics in a simulated annealing protocol. A total of 293 NOE distance restraints and 68 dihedral torsion restraints obtained from the NMR data were used to determine the structure of the AGUC tetraloop RNA. For the AGAA tetraloop RNA, a total of 295 NOE distance restraints and 73 dihedral torsion restraints were used. The overall structures are well defined by the NMR data (Table I), with a heavy atom root-mean-square deviation (r.m.s.d.) of 0.83 and 0.88 Å, respectively, for AGUC and AGAA RNAs (Table I; Figure 5A and B) for the 20 structures.
Table I. Structural statistics and atomic r.m.s.ds.
| AGUC tetraloop <SA>a | AGAA tetraloop <SA>a | |
|---|---|---|
| Total number of experimental restraints | ||
| distance restraints | 293 | 295 |
| dihedral restraints | 68 | 73 |
| Final distance and dihedral | 2.2 ± 0.7 | 1.0 ± 0.3 |
| restraint violation energies (kcal/mol) | ||
| R.m.s.d. from experimental restraints | ||
| distance restraints (Å) | 0.007 ± 0.003 | 0.007 ± 0.002 |
| dihedral restraints (°) | 0.003 ± 0.001 | 0.003 ± 0.001 |
| Deviations from idealized geometry | ||
| bonds (Å) | 0.0336 ± 0.0001 | 0.0336 ± 0.0001 |
| angle (°) | 0.0756 ± 0.0003 | 0.0753 ± 0.0003 |
| impropers (°) | 0.039 ± 0.004 | 0.039 ± 0.004 |
| Heavy-atoms r.m.s.d. (Å) | <SA> versus SA | <SA> versus SA |
| All RNA | 0.83 ± 0.23 | 0.88 ± 0.26 |
a<SA> refers to the final 20 simulated annealing structures, SA to the average structure obtained by taking the average coordinates of the 20 simulated annealing structures best-fitted to one another. The 20 final structures did not contain distance violations of >0.15 Å or dihedral violations of >10°.

Fig. 5. Best-fit superposition of the 20 final simulated annealing structures of AGUC (A) and AGAA (B) tetraloop RNAs. The heavy atoms of the RNA have been superimposed. Bases are shown in dark blue and the backbone in light blue.
AGAA and AGUC tetraloop structures
The AGUC tetraloop RNA folds as a compact and stable structure, with the loop capping an A-form helix containing a U5–G18 base pair (Figure 5A). The loop is stabilized by an A10–C13 non-canonical base pair where the N4 amino group of C13 is hydrogen bonded to the N3 of A10 (average distance of 3.1 ± 0.1 Å in the ensemble of 20 structures). In the loop, a sharp turn in the phosphodiester backbone occurs between G11 and U12. The turn results from non-standard values for torsion angles (U12γ was defined by the NMR data as gauche–). Conserved nucleotides A10 and G11 of the AGNN tetraloop sequence are stacked continuously at the 5′ side of the loop in the major groove, while U12 and C13 are stacked at the 3′ side in the minor groove. Base G11 was found in a syn conformation (Figure 6A) based on the intensity of the G11–H8/G11–H1′ NOE (as strong as a H5/H6) and additional NOEs. An intraresidue hydrogen bond is formed between the N2 amino of G11 and its phosphate (average distance of 2.1 ± 0.2 Å within the 20 lowest energy structures). This hydrogen bond and a possible contact mediated by an H2O molecule between G11 imino and the phosphate of A10 may contribute to stabilize the guanine in a syn conformation.

Fig. 6. Stereo views of single representative structures of AGUC (A) and AGAA (B) tetraloops. All heavy atoms are displayed. Bases are coloured in light blue, with nitrogen and oxygen atoms in dark blue and red, respectively. Ribose–phosphate backbones are coloured in yellow, and phosphate oxygen in red.
The AGAA tetraloop RNA folds into a very similar structure (Figures 5B and 6B). The non-canonical A–C base pair closing the AGUC tetraloop is replaced by a non-canonical A–A base pair. The geometry of the A–A base pair was defined by the NMR data; the N3 amino group of A10 is hydrogen bonded to the N6 amino group of A13 (average distance of 2.85 ± 0.05 Å in the ensemble of 20 structures). As in the AGUC tetraloop, a sharp turn in the phosphodiester backbone occurs between nucleotides 11 and 12 (G11γ and A12γ were found to be gauche– from the NMR data). The conserved A10 and G11 residues are also stacked at the 5′ side of the loop in the major groove, while A12 and A13 are stacked at the 3′ side in the minor groove (Figure 6B). Like the AGUC tetraloop, G11 is in a syn conformation and stabilized with the same contacts between G11–N2 amino and G11 phosphate (average distance of 3.1 ± 0.8 Å within the 20 lowest energy structures). G11 imino is partially protected from solvent exchange (Figure 4C and D) and may also form a hydrogen bond with the A10 phosphate via an H2O molecule (average distance of 4.1 ± 0.6 Å within the 20 lowest energy structures). The structure qualitatively explains the unusual chemical shifts that are observed in the tetraloop. G11 H4′ and H5′ protons experience upfield shifts in the vicinity of the A12 base moiety (Figure 6B).
Discussion
The AGNN tetraloop is essential for the selection of Rnt1p substrates
In this study, we show binding between Rnt1p and two different AGNN tetraloops supported with a 9 bp stem (Figure 2A and B). Extension of the stem length and changes of the lower stem sequence did not significantly alter the interaction with Rnt1p (Figure 2C). In contrast, a single mutation that changes the conserved AGNN tetraloop to the closely related UGNN tetraloop reduced its affinity for Rnt1p (Figure 2D), indicating a critical role for AG residues in the interaction with Rnt1p. This result agrees with previous studies on longer RNA substrates where substitution of A10 or G11 by other nucleotides was found to be deleterious to Rnt1p binding (Chanfreau et al., 2000; Nagel and Ares, 2000). Furthermore, in our short model RNA substrates, change of the tetraloop to the GAAA sequence blocked Rnt1p binding completely (Figure 2E), indicating that Rnt1p cannot bind to a short stem in the absence of the conserved AGNN tetraloop. In contrast, the in vitro cleavage assay indicates that although Rnt1p may bind a short stem–loop structure, it can only cleave stems longer than 13 nucleotides (Figure 3).
The AGUC and AGAA tetraloops contain a common structural motif
The AGUC and AGAA tetraloops fold into very similar structures. This observation strongly suggests that the conformation of the AGNN tetraloop is important for Rnt1p recognition, rather than the sequence of the loop. Conserved nucleotides A10 and G11 are stacked at the 5′ side of the loop. The Hoogsteen face of A10 and G11 and the Watson–Crick face of A10 are accessible in the major groove, allowing Rnt1p to contact the loop. In both AGNN tetraloops, the residue G11 was found in a syn conformation stabilized by contacts with phosphate groups. The AGUC tetraloop is stabilized by a non-canonical A–C pair, which is substituted by an A–A base pair in the AGAA sequence.
Compilation of Rnt1p cleavage sites (Chanfreau et al., 2000) allowed the identification of five tetraloop sequences (AGUA, AGGA, AGUU, AGUG and UGGU) in addition to the AGUC or AGAA tetraloops used in this study. Based on the NMR structures of the AGUC and AGAA tetraloops presented here, the other five tetraloops are predicted to have a similar conformation for the conserved 5′ AG nucleotides, permitting a common mode of recognition of AGNN tetraloops by Rnt1p. This structural similarity that also extends to the UGNN tetraloops (Butcher et al., 1997) explains Rnt1p’s ability to recognize the UGGU capped stem–loop structures at the U1 snRNA 3′ end (Seipelt et al., 1999). The AGUC and AGAA structures (Figures 5 and 6) reveal a base stacking on the 3′ side of both loops, which maximizes the thermodynamic stability of the structure and suggests that the nucleotides at the 3′ side are necessary for stability but not protein recognition. This may explain the sequence variability of the 3′ side nucleotides in AGNN tetraloops. AGNN loops may fold in a common structure, and further structure determination will be required to rationalize this model.
Structural similarity of AGNN and UGAA hairpins
The structures of AGUC and AGAA tetraloops are similar to that of a UGAA tetraloop (Butcher et al., 1997). All three structures have the G residue stacked in syn conformation at the 5′ side of the loop above the uracil in the UGAA sequence or the adenine in the AGNN sequence. In addition, all three tetraloops share a sharp turn in the backbone that occurs at the phosphate of the third nucleotide and stacking on the 3′ side of the loop. However, the position of the syn G relative to the backbone is slightly different in the UGAA loop and its two adenines stacked at the 3′ side are directed to the inside of the loop, resulting in a more compact structure. The r.m.s.d. of the phosphodiester backbones of the AGUC nucleotides in the AGUC RNA and the segment UGAA is 2.7 Å. The same comparison with the AGAA hairpin gives an r.m.s.d. of 3.1 Å. Interestingly, a UGNN tetraloop is found in the U1 snRNA substrate, and the structural similarity between a AGNN loop and a UGAA tetraloop may explain why a UGNN tetraloop can be recognized by Rnt1p. However, the AGNN tetraloop appears to bind more efficiently to Rnt1p than the UGNN tetraloop. Changing the first adenine of the AGAA tetraloop to uracil reduced the binding to Rnt1p by 3-fold (Figure 2D). The reduction in binding does not translate into a reduced cleavage rate of the U1 3′ end in vivo because no unprocessed U1 is observed in normal cells (Seipelt et al., 1999). Therefore, U1 may employ other features in the lower stem to enhance binding or cleavage, or use other factors in vivo to stimulate binding and cleavage by Rnt1p. In any case, the observed reduction in the interaction with the UGNN tetraloop explains why the AGNN is the predominant motif of the Rnt1p substrate.
Specific contacts between the AGNN hairpin and the C-terminal domain of Rnt1p
The structures of the AGUC and AGAA tetraloops reveal that the AG nucleotides critical for recognition by Rnt1p are found in the major groove of the loop structure (Figure 6A and B), indicating that Rnt1p may establish specific contacts with the RNA major groove. In support of this idea, modification interference experiments (Chanfreau et al., 2000) revealed that the N7 atoms of residues G11 and A15, found in the major groove of the AGAA hairpin, are important for Rnt1p recognition. In addition, substitution of A12 Rp phosphate oxygen by sulfur inhibits binding of Rnt1p (Chanfreau et al., 2000). In the AGAA hairpin structure presented here, the phosphate of A12 is located at the sharp turn in the backbone, as in the AGUC tetraloop, suggesting that the conformation of the turn is also important for protein recognition. Changes of the terminal tetraloop structure upon binding of the dsRBD were not observed in the case of the Staufen–RNA complex (Ramos et al., 2000), suggesting that Rnt1p dsRBD indeed contacts the A12 residue in the conformation observed in this study (Figures 5 and 6). However, we cannot rule out the possibility that the loop structure of Rnt1p may undergo a conformational change upon Rnt1p binding. Our structural data suggest that Rnt1p recognizes the unique fold adopted by conserved nucleotides of AGNN tetraloops.
In vitro binding assays demonstrate that a 130–142 amino acid fragment of the C-terminal domain of Rnt1p that contains the 74 amino acid dsRNA-binding motif selectively binds to RNA stems capped with terminal AGNN tetraloops (Lamontagne et al., 2000; Nagel and Ares, 2000). This suggests that the dsRBD of Rnt1p is responsible for the AGNN tetraloop specificity. The dsRBD motif is predicted to form an αβββα structure like its bacterial orthologue RNase III and other dsRNA-binding proteins (Bycroft et al., 1995; Kharrat et al., 1995; Nanduri et al., 1998). Two structures of the conserved dsRBD in complex with RNA (Ryter and Schultz, 1998; Ramos et al., 2000) revealed the nature of the RNA– protein interaction and explained the general dsRBD specificity for dsRNA (over ssRNA or dsDNA). In both structures, the N-terminal α-helix and loop 2 interact with 2′-OH groups of the minor groove of the RNA, while loop 4 interacts with the phosphodiester backbone across the major groove. Interaction between the two loops and 2′-OH groups in the RNA minor groove in addition to the spacing between the loops, which fits well the groove distances of A-type helix RNA (but not B-type dsDNA), explain dsRNA versus dsDNA discrimination. Addition ally, large variations in the N-terminal α-helix sequence and structural difference in the α-helix–RNA interaction suggested that specific interaction between helix α1 and single-stranded RNA loops could provide selectivity in the recognition of RNA substrates (Ramos et al., 2000). In the NMR-derived structure of the Staufen dsRBD3–RNA complex (Ramos et al., 2000), a conserved lysine at the C-terminal end of helix α1 makes electrostatic interactions with the phosphate of the third residue of a UUCG tetraloop where a sharp turn in the backbone occurs. In the AGNN tetraloop structure (Figures 5 and 6), the phosphate of the third residue also makes a sharp turn in the backbone and was found to be important for Rnt1p binding (Chanfreau et al., 2000). The requirements for the stem length in the substrates of Rnt1p and Staufen are very similar, 9 and 9–12 bp, respectively. Our study is consistent with the idea that in the case of Rnt1p, loop 2 and loop 4 continue to provide a general specificity to dsRNA leaving helix α1 of the Rnt1p dsRBD to interact with the AGNN tetraloop and provide the additional specificity (Ramos et al., 2000). Other regions within the C-terminal domain of Rnt1p such as the non-conserved 54 amino acid extension may also play a crucial role in the specific recognition of AGNN hairpins. Previously, it was suggested that Rnt1p’s specificity for the conserved AGNN tetraloop is due to the amino acid sequence of loop 2, which differs from the conserved GXXH motif found in both Staufen and bacterial RNase III dsRBDs (Nagel and Ares, 2000). It remains possible, however, that interaction of loop 2 with the AGNN tetraloop requires binding of the Rnt1p dsRBD in the opposite direction to that of Staufen. Such a drastic change in the mode of interaction cannot be explained at the moment by the structure of the AGNN tetraloop.
Variation in the stem length of the Rnt1p substrate from 9 to 19 bp did not significantly alter the binding affinity for Rnt1p (Figure 2), despite the fact that the 19 bp substrate is cleaved by Rnt1p while the 9 bp substrate is not (Figure 3). This indicates that the inability of Rnt1p to cleave substrates shorter than 13 nucleotides is not due to the absence of binding determinants in the lower stem but to the inability of the nuclease domain to contact RNA closer than 13 bp away from the tetraloop. This also indicates that in stem–loops that vary between 9 and 19 bp, the AGNN hairpin accounts for most of the affinity. However, additional but independent contacts between Rnt1p and the lower stem of duplexes longer than 25 bp cannot be ruled out. Rnt1p efficiently binds long duplex RNA without tetraloops and it binds with higher affinity to long stem–loop structures (Abou Elela et al., 1996). This is consistent with the fact that changes in the conserved tetraloop sequence of long stems reduce binding to Rnt1p only 4-fold (Chanfreau et al., 2000; Nagel and Ares, 2000) while the same change in a short stem–loop structure completely abolishes binding (Figure 2).
The solution structure of the conserved AGNN loop suggests that the proposed additional contacts between Rnt1p and the major groove of the AGNN tetraloop may account for the strong selectivity of the RNA substrates by Rnt1p. The presence of the AGNN tetraloop allows selective recruitment of Rnt1p, which then would cleave the substrate with the help of weaker dsRNA interactions. Crystallographic data (Ryter and Schultz, 1998) and NMR data (Ramos et al., 2000) on dsRBD–RNA complexes revealed that the protein–RNA interface is dynamic, resulting in weak affinity and low sequence specificity. A similar flexible interface may be seen in the complex between the dsRBD of Rnt1p and the stem region of the RNA substrate. Single mutation within the conserved AGNN tetraloop severely inhibits Rnt1p binding and RNA processing. These observations suggest a dual mode of recognition of the RNA substrates by Rnt1p, specific interaction with the AGNN hairpin and weak sequence-independent binding of the RNA stem. This should be investigated further by biochemical and structural experiments and emphasis should now be put on determining the amino acid residues in Rnt1p responsible for the AGNN selectivity.
Materials and methods
Enzymatic assays
Rnt1p was expressed in bacteria and purified as described previously (Lamontagne and Abou Elela, 2001). The radiolabelled RNA used as a substrate in the enzymatic assays was generated by T7 RNA polymerase in the presence of [α-32P]UTP (Lamontagne and Abou Elela, 2001). The DNA oligos used to generate the RNA substrates are (5′→3′): R31, GGCGCCATGGACTCATGACGCCTATAGTGAGTCGTATTA; R32, GGCGCGTTCTTCTGACACCGCCTATAGTGAGTCGTATTA; R32/A-U, GGCGCGTTCTTCAGAACACGCCTATAGTGAGTCGTAT; R31L, GGCGCCATGCCATGCCATGGACTCATGACATGACATGACGCCTATAGTGAGTCGTAT; and R31A, GGCGCCATGTTTCCATGACGCCTATAGTGAGTCGTAT.
For the in vitro cleavage, between 0.40 and 2.8 pmol of substrate were incubated in the presence of 0.2 pmol of Rnt1p for 20 min at 30°C in 20 µl of reaction buffer [30 mM Tris pH 7.5, 5 mM spermidine, 20 mM MgCl2, 0.1 mM dithiothreitol (DTT), 0.1 mM EDTA pH 7.5]. The reactions were stopped by addition of a stop buffer (20 mM EDTA pH 7.5 and 0.1% bromophenol blue in formamide) and directly loaded on a denaturing 20% polyacrylamide gel. The cleavage rate was quantified using the Instant Imager (Packard, Meriden, CT).
Gel mobility shift assay
RNA-binding experiments were performed using 2 fmol of radiolabelled RNA in 20 µl of binding buffer (20% glycerol, 30 mM Tris pH 7.5, 150 mM KCl, 5 mM spermidine, 0.1 mM DTT and 0.1 mM EDTA pH 7.5) for 10 min on ice (Lamontagne and Abou Elela, 2001). The amount of protein used is indicated for each experiment. The reactions were fractionated on 4% non-denaturing polyacrylamide gels using 0.5 V/cm2 at 4°C.
Preparation of the RNAs for NMR
Milligram quantities of the RNAs (22 nucleotides) were prepared unlabelled and 13C–15N labelled by in vitro transcription from an oligonucleotide template containing a 2′OmeG in position 2 (Kao et al., 1999) and purified as described previously (Puglisi and Wyatt, 1995). After electroelution and ethanol precipitation, the resuspended RNA was then dialysed for 48 h against the buffer used for the NMR experiment in a microdialysis apparatus with a 3500 Da molecular weight cut-off membrane. Labelled NTPs were isolated from Escherichia coli strain MRE600 grown in rich 13C–15N medium (Martek CN9). Labelled NMPs were purified and converted to NTPs as described previously (Batey et al., 1992; Nikonowicz et al., 1992).
Proton and heteronuclear NMR
NMR experiments were recorded at 800 MHz on a Bruker DRX spectrometer equipped with triple resonance, z-gradient probes. NMR experiments involving 31P were recorded on a Bruker DRX spectrometer at 600 MHz. NMR data were processed using XWINNMR, Aurelia (Bruker) and Gif A (Pons et al., 1996) software packages. NMR experiments were performed in 10 mM sodium phosphate (pH 6.4). The concentrations of unlabelled and 13C–15N-labelled RNA ranged from 1 to 2 mM. Sample volumes were 280 µl in Shigemi NMR tubes. 15N and 13C chemical shifts were referenced with the known chemical shifts of the ribose carbon or base nitrogen from the 5′ and 3′ ends region (Yoshizawa et al., 1998).
1H, 13C, 15N and 31P assignments were obtained using standard homonuclear and heteronuclear methods. NMR data were acquired at either 20 or 30°C, and data for exchangeable protons were collected at either 2, 5 or 15°C. Solvent suppression for samples in 90% H2O/10% D2O was achieved using the WATERGATE sequence (Sklenar et al., 1993; Piotto et al., 1992). The residual HDO resonance in D2O was suppressed using low power pre-saturation. Two-dimensional NOESY spectra in 95% H2O/5% D2O were acquired with mixing times of 50, 75, 150 and 300 ms. The hydrogen bonding patterns of the base pairs were determined from analysis of NOESY spectra in H2O at different mixing times and directly observed from heteronuclear J(N,N)–HNN COSY experiments performed in 90% H2O/10% D2O (Dingley and Grzesiek, 1998). NOESY spectra with mixing times of 50, 150, 250 and 400 ms in D2O were measured at 20 and 30°C for the AGUC and AGAA tetraloop RNA.
Heteronuclear NMR spectra were measured at 20°C in 99.9% D2O, with the exception of 1H–15N HSQC experiments which were acquired at 2, 5 and 15°C. 13C and/or 15N decoupling during the acquisition time was achieved using the GARP composite pulse sequence. The assignment of the non-exchangeable protons of the labelled RNA was completed using constant-time HSQC (Santoro and King, 1992), two-dimensional HCCH-TOCSY and three-dimensional NOESY-HMQC (100 and 200 ms mixing times) (Clore et al., 1990). Sequential connectivities between nucleotides in the AGAA and AGUC tetraloop RNAs were obtained by HP-COSY experiments (Sklenar et al., 1986) at 20°C and HCP experiments (Marino et al., 1995). The H2 protons of adenines were assigned unambiguously for the labelled RNAs by correlation of the H2–H8 resonances in two-dimensional HCCH-TOCSY experiments (Legault et al., 1994; Marino et al., 1994).
Distance and dihedral restraints for structure calculation
Distance restraints involving non-exchangeable RNA protons were derived from visual inspection of cross-peak intensities in 50, 150, 250 and 400 ms NOESY experiments and three-dimensional NOESY-HMQC spectra. The H5–H6 cross-peak of pyrimidines was used as an internal standard. NOEs were classified into four distance bound ranges: strong 0–3 Å, medium 0–4 Å, weak 0–5 Å and very weak 0–6.0 Å. In several instances, very weak NOE restraints observed in the loop region were loosened to 0–7 Å. The appropriate pseudoatom distance corrections were used.
RNA dihedral restraints were assigned following the general strategy of Varani and co-workers (Allain and Varani, 1995). β dihedral angles were restrained from estimates of the 3JP–H5′, 3JP–H5′′ and 3JP–C4′ coupling constants from the HP-COSY experiment on the unlabelled RNA. ε was also restrained from estimates of 3JH3′–P, 3JC2′–P and 3JC4′–P from the HP-COSY and HCP experiments. The ε dihedral angles were constrained to 210 ± 30° (trans), 260 ± 20° (gauche–) or 235 ± 55° (when the trans or gauche– conformations could not be distinguished).
The γ dihedral angles were constrained where possible using estimates of 3JH4′–H5′ and 3JH4′–H5′′ coupling constants from the 31P decoupled DQF-COSY. For a gauche+ conformation, γ was constrained to 55 ± 20°. The ribose sugar pucker was estimated from analysis of the H1′–H2′ coupling constants in the 31P decoupled DQF-COSY spectrum. Nucleotides with a H1′–H2′ coupling constant of > 8 Hz in the COSY spectrum were classified as C2′-endo (δ = 150 ± 30°). Nucleotides with no COSY and TOCSY cross-peaks between the H1′ and H2′ protons (J <3 Hz) were classified as C3′-endo (δ = 85 ± 20°). Some nucleotides had weak H1′–H2′ cross-peaks in the TOCSY spectrum, but not in the COSY spectrum. When a mixed population of C2′/C3′-endo conformations was observed, the ribose puckers for these nucleotides were restrained in the range of the C2′ + C3′-endo conformations during molecular dynamics. The glycosidic torsion angle χ was determined by the intensity of the intranucleotide H8–H1′ NOE and additional intra- and internucleotide NOEs. Only one torsion angle χ (the syn G residue from the tetraloop) was restrained explicitly to 45 ± 45°.
Structure determination
Structures were calculated using a simulated annealing protocol (AMBER force field) within the Insight II package (Biosym Technologies, San Diego, CA) as described previously (Puglisi and Puglisi, 1998). A randomized array of atoms corresponding to RNA was heated to 1000 K. Bonding, distance and dihedral restraints were gradually increased to full value over 15 ps of molecular dynamics, while maintaining low values for interatomic repulsion (repulsive quartic potential), which subsequently were increased to full value during another 15 ps of dynamics. The molecules were then cooled during 10 ps to 300 K and subjected to a final energy minimization step that includes an attractive Lennard–Jones potential. No electrostatic term was included in the target function. Using this protocol, 15% of the structures converged, as based on restraint violation energies, and 30 of them were collected to be refined further with the final set of restraints. The lowest energy non-converged structures had total energies >4 SDs for the converged structures. During refinement, molecules were heated to 1000 K and subjected to 30 ps of molecular dynamics following the same protocol as above. The molecules were then cooled during 10 ps to 300 K and subjected to a final energy minimization step that included an attractive Lennard–Jones potential and electrostatic terms with a dielectric constant of 7. The 20 final structures shown in Figure 5 had the lowest total energy and restraint violation energies. For the AGUC tetraloop RNA, a total of 293 distance restraints were used including 96 intranucleotide RNA restraints, 147 internucleotide RNA restraints and 50 base pair hydrogen bonding restraints; no hydrogen bonding restraints were used for non-canonical base pairs. A total of 68 experimental dihedral restraints were used. For the AGAA tetraloop RNA, a total of 295 distance restraints were used including 99 intranucleotide RNA restraints, 146 internucleotide RNA restraints and 50 base pair hydrogen bonding restraints; no hydrogen bonding restraints were used for non-canonical base pairs. A total of 73 experimental dihedral restraints were used. The final force constants for distance restraints were 40 kcal/mol. Final force constants for base pairing hydrogen bond and dihedral restraints were set to 250 and 60 kcal/mol, respectively. All colour figures were generated with the program InsightII (Biosym Technologies, San Diego, CA).
Coordinates
The coordinates for the ensemble of 20 structures for the AGUC and AGAA tetraloop RNAs have been deposited in the Protein Data Bank with accession Nos for R31 and R32 of 1K6G and 1K6H, respectively.
Acknowledgments
Acknowledgements
We thank Philip Johnson for critical reading of the manuscript. This work was supported by grant MOP-14305 from the Canadian Institute for Health Research (CIHR) to S.A.E. Support for the RNA group core was provided by CIHR grant MGC-48372. S.A.E. is a Chercheur-Boursier Junior II of the Fonds de la Recherche en Santé du Québec. S.Y. was supported by a grant from the HFSP (Human Frontier Science Program).
References
- Abou Elela S. and Ares,M.,Jr (1998) Depletion of yeast RNase III blocks correct U2 3′ end formation and results in polyadenylated but functional U2 snRNA. EMBO J., 17, 3738–3746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abou Elela S., Igel,H. and Ares,M.,Jr (1996) RNase III cleaves eukaryotic preribosomal RNA at a U3 snoRNP-dependent site. Cell, 85, 115–124. [DOI] [PubMed] [Google Scholar]
- Allain F.H. and Varani,G. (1995) Structure of the P1 helix from group I self-splicing introns. J. Mol. Biol., 250, 333–353. [DOI] [PubMed] [Google Scholar]
- Batey R.T., Inada,M., Kujawinski,E., Puglisi,J.D. and Williamson,J.R. (1992) Preparation of isotopically labeled ribonucleotides for multidimensional NMR spectroscopy of RNA. Nucleic Acids Res., 20, 4515–4523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butcher S.E., Dieckmann,T. and Feigon,J. (1997) Solution structure of a GAAA tetraloop receptor RNA. EMBO J., 16, 7490–7499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bycroft M., Grunert,S., Murzin,A.G., Proctor,M. and St Johnston,D. (1995) NMR solution structure of a dsRNA binding domain from Drosophila staufen protein reveals homology to the N-terminal domain of ribosomal protein S5. EMBO J., 14, 3563–3571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chanfreau G., Elela,S.A., Ares,M.,Jr and Guthrie,C. (1997) Alternative 3′-end processing of U5 snRNA by RNase III. Genes Dev., 11, 2741–2751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chanfreau G., Buckle,M. and Jacquier,A. (2000) Recognition of a conserved class of RNA tetraloops by Saccharomyces cerevisiae RNase III. Proc. Natl Acad. Sci. USA, 97, 3142–3147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clore G.M., Bax,A., Driscoll,P.C., Wingfield,P.T. and Gronenborn,A.M. (1990) Assignment of the side-chain 1H and 13C resonances of interleukin-1β using double- and triple-resonance heteronuclear three-dimensional NMR spectroscopy. Biochemistry, 29, 8172–8184. [DOI] [PubMed] [Google Scholar]
- Dingley A.J. and Grzesiek,S. (1998) Direct observation of hydrogen bonds in nucleic acid base pairs by internucleotide 2JNN couplings. J. Am. Chem. Soc., 120, 8293–8297. [Google Scholar]
- Kao C., Zheng,M. and Rudisser,S. (1999) A simple and efficient method to reduce nontemplated nucleotide addition at the 3′ terminus of RNAs transcribed by T7 RNA polymerase. RNA, 5, 1268–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharrat A., Macias,M.J., Gibson,T.J., Nilges,M. and Pastore,A. (1995) Structure of the dsRNA binding domain of E.coli RNase III. EMBO J., 14, 3572–3584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamontagne B. and Abou Elela,S. (2001) Purification and character ization of Saccharomyces cerevisiae Rnt1p nuclease. Methods Enzymol., 342, 159–167. [DOI] [PubMed] [Google Scholar]
- Lamontagne B., Tremblay,A. and Abou Elela,S. (2000) The N-terminal domain that distinguishes yeast from bacterial RNase III contains a dimerization signal required for efficient double-stranded RNA cleavage. Mol. Cell. Biol., 20, 1104–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legault P., Farmer,B.T.,II, Mueller,L. and Pardi,A. (1994) Through-bond correlation of adenine protons in a 13C-labeled ribozyme. J. Am. Chem. Soc., 116, 2203–2204. [Google Scholar]
- Marino J.P., Prestegard,J.H. and Crothers,D.M. (1994) Correlation of adenine H2/H8 resonances in uniformly 13C labeled RNAs by 2D HCCH-TOCSY: a new tool for 1H assignment. J. Am. Chem. Soc., 116, 2205–2206. [Google Scholar]
- Marino J.P., Schwalbe,H., Anklin,C., Bermel,W., Crothers,D.M. and Griesinger,C. (1995) Sequential correlation of anomeric ribose protons and intervening phosphorus in RNA oligonucleotides by a 1H, 13C, 31P triple resonance experiment: HCP-CCH-TOCSY. J. Biomol. NMR, 5, 87–92. [DOI] [PubMed] [Google Scholar]
- Nagel R. and Ares,M.,Jr (2000) Substrate recognition by a eukaryotic RNase III: the double-stranded RNA-binding domain of Rnt1p selectively binds RNA containing a 5′-AGNN-3′ tetraloop. RNA, 6, 1142–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanduri S., Carpick,B.W., Yang,Y., Williams,B.R. and Qin,J. (1998) Structure of the double-stranded RNA-binding domain of the protein kinase PKR reveals the molecular basis of its dsRNA-mediated activation. EMBO J., 17, 5458–5465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson A.W. (1999) Function, mechanism and regulation of bacterial ribonucleases. FEMS Microbiol. Rev., 23, 371–390. [DOI] [PubMed] [Google Scholar]
- Nikonowicz E.P., Sirr,A., Legault,P., Jucker,F.M., Baer,L.M. and Pardi,A. (1992) Preparation of 13C and 15N labelled RNAs for heteronuclear multi-dimensional NMR studies. Nucleic Acids Res., 20, 4507–4513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piotto M., Saudek,V. and Sklenar,V. (1992) Gradient-tailored excitation for single-quantum NMR spectroscopy of aqueous solutions. J. Biomol. NMR, 2, 661–665. [DOI] [PubMed] [Google Scholar]
- Pons J.L., Malliavin,T. and Delsuc,M.A. (1996) A complete package for NMR data set processing. J. Biomol. NMR, 8, 445–452. [DOI] [PubMed] [Google Scholar]
- Puglisi E.V. and Puglisi,J.D. (1998) HIV-1 A-rich RNA loop mimics the tRNA anticodon structure. Nature Struct. Biol., 5, 1033–1036. [DOI] [PubMed] [Google Scholar]
- Puglisi J.D. and Wyatt,J.R. (1995) Biochemical and NMR studies of RNA conformation with an emphasis on RNA pseudoknots. Methods Enzymol., 261, 323–350. [DOI] [PubMed] [Google Scholar]
- Ramos A., Grunert,S., Adams,J., Micklem,D.R., Proctor,M.R., Freund,S., Bycroft,M., St Johnston,D. and Varani,G. (2000) RNA recognition by a Staufen double-stranded RNA-binding domain. EMBO J., 19, 997–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotondo G., Huang,J.Y. and Frendewey,D. (1997) Substrate structure requirements of the Pac1 ribonuclease from Schizosaccharomyces pombe. RNA, 3, 1182–1193. [PMC free article] [PubMed] [Google Scholar]
- Ryter J.M. and Schultz,S.C. (1998) Molecular basis of double-stranded RNA–protein interactions: structure of a dsRNA-binding domain complexed with dsRNA. EMBO J., 17, 7505–7513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro J. and King,G. (1992) A constant-time 2D overbodenhausen experiment for inverse correlation of isotopically enriched species. J. Magn. Reson., 97, 202–207. [Google Scholar]
- Seipelt R.L., Zheng,B., Asuru,A. and Rymond,B.C. (1999) U1 snRNA is cleaved by RNase III and processed through an Sm site-dependent pathway. Nucleic Acids Res., 27, 587–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sklenar V., Miyashiro,H., Zon,G., Miles,H.T. and Bax,A. (1986) Assignment of the 31P and 1H resonances in oligonucleotides by two-dimensional NMR spectroscopy. FEBS Lett., 208, 94–98. [DOI] [PubMed] [Google Scholar]
- Sklenar V., Piotto,M., Leppik and Saudek,V. (1993) Gradient-tailored water suppression for 1H–15N experiments optimized to retain full sensitivity. J. Magn. Reson., 102, 241–245. [Google Scholar]
- Wu H., Xu,H., Miraglia,L.J. and Crooke,S.T. (2000) Human RNase III is a 160-kDa protein involved in preribosomal RNA processing. J. Biol. Chem., 275, 36957–36965. [DOI] [PubMed] [Google Scholar]
- Yoshizawa S., Fourmy,D. and Puglisi,J.D. (1998) Structural origins of gentamicin antibiotic action. EMBO J., 17, 6437–6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou D., Frendewey,D. and Lobo Ruppert,S.M. (1999) Pac1p, an RNase III homolog, is required for formation of the 3′ end of U2 snRNA in Schizosaccharomyces pombe. RNA, 5, 1083–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]