

Abstract
Nek2 is a NIMA-related kinase implicated in regulating centrosome structure at the G2/M transition. Two splice variants have been identified that exhibit distinct patterns of expression during cell cycle progression and development. Here we show that Nek2A, but not Nek2B, is destroyed upon entry into mitosis coincident with cyclin A destruction and in the presence of an active spindle assembly checkpoint. Destruction of Nek2A is mediated by the proteasome and is dependent upon the APC/C–Cdc20 ubiquitin ligase. Nek2 activity is not required for APC/C activation. Nek2A destruction in early mitosis is regulated by a motif in its extreme C-terminus which bears a striking resemblance to the extended destruction box (D-box) of cyclin A. Complete stabilization of Nek2A requires deletion of this motif and mutation of a KEN-box. Destruction of Nek2A is not inhibited by the cyclin B-type D-box, but the C-terminal domain of Nek2A inhibits destruction of both cyclins A and B. We propose that recognition of substrates by the APC/C–Cdc20 in early mitosis depends upon possession of an extended D-box motif.
Keywords: APC/Cdc20/centrosome/cyclin A/Nek2
Introduction
Progress through mitosis is governed by a number of regulatory mechanisms that include protein phosphorylation, subcellular distribution and protein degradation (Nigg, 2001; Pines and Rieder, 2001). Of these, protein degradation is perhaps the most effective way to ensure that individual steps cannot be reversed. One key regulator of mitotic destruction is a multisubunit E3 ubiquitin ligase known as the anaphase-promoting complex or cyclosome (APC/C) (Morgan, 1999; Page and Hieter, 1999; Zechariae and Nasmyth, 1999). The APC/C catalyzes the covalent attachment of ubiquitin molecules to a substrate, thereby targeting it for degradation by the multiple peptidase activities of the ATPase-dependent 26S proteasome (Hershko and Ciechanover, 1998).
A number of proteins have now been recognized as APC/C substrates. However, they are not all targeted for destruction at the same time. For example, cyclin A is destroyed in prometaphase (den Elzen and Pines, 2001; Geley et al., 2001), cyclin B1 and securins in metaphase (Clute and Pines, 1999; Wakefield et al., 2000; Zur and Brandeis, 2001) and Ase1 in anaphase (Juang et al., 1997). What regulates this sequential destruction of proteins by the APC/C is not clear. Certainly, activation of the APC/C requires additional events including phosphorylation of APC subunits and association of WD-containing adaptor proteins (Morgan, 1999). Two such adaptors have been identified, Cdc20 and Cdh1. These proteins act as adaptors for the APC/C at different times in mitosis. Phosphorylation of the APC/C in early mitosis promotes association of Cdc20. At the same time, phosphorylation of the other adaptor, Cdh1, by Cdk1 prevents its association with the APC/C (Kotani et al., 1999; Kramer et al., 2000). Destruction of APC/C targets in metaphase and anaphase is therefore regulated through Cdc20. However, in late mitosis, G1 and G0, when Cdk1 has been inactivated, Cdh1 becomes associated with the APC/C and takes over the role of adaptor. This switch from Cdc20 to Cdh1 is completed by the destruction of Cdc20 itself by APC/C–Cdh1 (Pfleger and Kirschner, 2000). Current evidence, however, does not support the notion that Cdc20 and Cdh1 determine the substrate specificity of the APC/C, as most substrates studied so far, including cyclin B1, securin and Cdc6, can be targeted by both Cdc20 and Cdh1 (Petersen et al., 2000; Zur and Brandeis, 2001). The use of both adaptors ensures that destruction begun in mitosis is continued into the subsequent G1. Cdc20 itself is an exception to this rule in that it can be targeted for destruction by APC/C–Cdh1 but not by APC/C–Cdc20.
The APC/C is also negatively regulated by a number of proteins including MAD2, MAD2L2 (also called MAD2B) and Emi1, which inhibit the APC/C via the WD adaptor proteins (Chen and Fang, 2001; Pfleger et al., 2001; Reimann et al., 2001). MAD2 is directly involved in the spindle assembly checkpoint preventing APC/C–Cdc20 from destroying cyclin B and securins in the presence of unattached kinetochores (Shah and Cleveland, 2000). Emi1 also inhibits APC/C–Cdc20 in early mitosis and is itself destroyed later in mitosis through an APC/C-independent pathway (Reimann et al., 2001). Whether Emi1 contributes to the spindle checkpoint is unclear. MAD2L2 can inhibit both Cdc20- and Cdh1-complexed APC/C and it is possible that the different actions of these APC/C inhibitors contribute to the timing of destruction of different APC/C substrates.
Another substrate of the APC/C is the NIMA kinase of Aspergillus nidulans (Ye et al., 1998). NIMA is required for mitotic entry in Aspergillus and its destruction by the APC/C is required for mitotic exit (Pu and Osmani, 1995). The most closely related vertebrate protein to NIMA by sequence is Nek2 (Nigg, 2001). However, whether Nek2 has an equivalent role in regulating mitotic entry remains unclear. Instead, Nek2 has been found to be a core component of the centrosome and, upon overexpression, it can stimulate centrosome splitting (Fry et al., 1998a). Its activity is cell cycle regulated, with peak levels in S and G2 (Fry et al., 1995). However, direct interaction with the catalytic subunit of protein phosphatase 1 may limit the activity of Nek2 to a brief window at the onset of mitosis when PP1 is switched off (Puntoni and Villa-Moruzzi, 1997; Helps et al., 2000). The function of Nek2 may be to facilitate centriole disjunction at G2/M by promoting disassembly of an intercentriolar linkage (Mayor et al., 1999; Fry et al., 2000a; Hinchcliffe and Sluder, 2001). In support of this, a centrosomal substrate of Nek2, called C-Nap1, has properties consistent with holding centrioles together during interphase (Fry et al., 1998b; Mayor et al., 2000). Experiments performed with the Xenopus laevis homolog of Nek2 suggest additional functions for this kinase in assembly and maintenance of centrosome structure (Fry et al., 2000b; Uto and Sagata, 2000).
Two splice variants of Nek2 have been identified, Nek2A and Nek2B, that encode products with distinct C-termini (Uto et al., 1999; Hames and Fry, 2001). In Xenopus, these isoforms display distinct patterns of expression during development, with Nek2B present in oocytes, eggs and early embryos and Nek2A present in late embryos and adults (Uto et al., 1999). Nek2 splice variants also vary in their expression through the cell cycle. In adult human cell cycles, Nek2A is maximal in S/G2 and low in M/G1, whereas Nek2B remains at its peak throughout S, G2 and M, and is low only in G1 (Hames and Fry, 2001). Nek2B expression is invariant during early embryonic cell cycles (Fry et al., 2000b), whereas Nek2A, if added ectopically, is rapidly degraded (Uto and Sagata, 2000). These observations prompted us to consider the possibility that Nek2A is subject to degradation in mitosis as a result of sequences in its C-terminus that are missing from Nek2B. Here, we demonstrate that human Nek2A is an early mitotic target of the APC/C–Cdc20 and present evidence to support a model in which recognition of substrates by the APC/C before inactivation of the spindle assembly checkpoint relies on the presence of a cyclin A-like extended destruction box.
Results
Nek2A is destroyed in early mitosis
In human somatic cells, both Nek2 splice variants are expressed at low levels in G1 and at high levels in S and G2. However, in prometaphase-arrested cells, Nek2A expression is low, whereas that of Nek2B remains high. To determine exactly when Nek2 proteins disappear, extracts were prepared from U2OS cells released from an arrest at the G1/S transition in the presence of nocodazole. Nek2A expression decreased abruptly between 10 and 12 h after release from the block (Figure 1A). This was coincident both with the time of mitotic entry as judged by microscopic examination of cells and with the disappearance of cyclin A, a protein known to be targeted for destruction in prometaphase (den Elzen and Pines, 2001; Geley et al., 2001). The drop in expression of Nek2A was accompanied by the appearance of a weak, higher molecular weight smear suggestive of phosphorylation. However, even taking this into account, the overall level of Nek2A consistently decreased at least 2- to 3-fold with respect to S and G2 phase cells. In contrast, the abundance of Nek2B remained elevated in these cells and, taking into account the appearance of a higher molecular weight smear for Nek2B as well, even increased into mitosis. To observe when Nek2B protein disappears, cells were released from a nocodazole block into G1 (Figure 1B). Nek2B protein decreased between 4 and 8 h after release, whereas cyclin B1 levels disappeared more rapidly. Nek2B protein therefore persists until early G1. In this experiment, Nek2A levels dropped slightly further upon entry into G1 and remained low until the time of entry into the next S phase when there was a sudden 3- to 4-fold increase in its abundance (Figure 1B).

Fig. 1. Nek2A is destroyed in early mitosis. (A) Extracts prepared from exponentially growing U2OS cells (Exp) or cells released from a thymidine–hydroxyurea block into medium containing nocodazole for the times indicated (h) were immunoblotted with antibodies against Nek2, cyclin A, cyclin B1 and α-tubulin. Mitotic entry was observed by phase microscopy to occur between 10 and 12 h after release. The positions of Nek2 splice variants (A and B) are indicated. The abundance of Nek2A and Nek2B proteins present at each time point with respect to the amount in exponential cells was quantified by densitometry and is shown in the histograms on the right. (B) Immunoblots of extracts prepared following release from a nocodazole block for the times indicated (h). Again, histograms on the right show the quantified levels of Nek2A and Nek2B proteins. (C) Protein stability of Nek2A (A) and Nek2B (B) in exponential (Exp) and S phase-arrested (S) cells was measured on immunoblots of cell extracts prepared at the times indicated (h) after addition of cycloheximide. On the right, the amount of Nek2A protein remaining at each time point is plotted with respect to the amount present at time zero (Exp, open triangles; S, closed triangles). (D) In vitro degradation assays were performed by addition of 35S-labeled Nek2A, Nek2B, cyclin B1 or cyclin A to CSF extracts with (anaphase) or without (metaphase) addition of calcium. Samples were collected at the times indicated (min), separated by SDS–PAGE and exposed to autoradiography. The amount of Nek2A protein remaining at each time is plotted with respect to the amount at time zero in metaphase (M, closed squares) and anaphase (A, open squares) extracts. Results are taken from six independent experiments and error bars represent standard deviations.
To determine whether loss of Nek2 proteins was due to increased degradation, their half-lives were measured following treatment with the protein synthesis inhibitor cycloheximide (Figure 1C). In asynchronous cells, the stability of Nek2A was very low, with an estimated half-life of 45 min. The half-life of Nek2B was slightly longer at ∼75 min. However, in S phase-arrested cells, the half-life of Nek2A was extended to >4 h, whereas the half-life of Nek2B was unchanged. In either M or G1 cells, the level of Nek2A was so low that it became undetectable by the first time point (30 min), making calculation of a half-life impossible (data not shown). To test directly whether Nek2 was destroyed in mitosis, recombinant proteins were incubated in cytostatic factor (CSF) (metaphase II-arrested) extracts of Xenopus eggs or in CSF extracts to which calcium had been added to trigger anaphase entry (Figure 1D). In both types of extract, Nek2A was unstable, although its rate of loss was significantly greater in anaphase extracts. The slower degradation that occurred in metaphase extracts was again accompanied by the appearance of a higher molecular weight smear. Nek2B, on the other hand, was completely stable in CSF extracts both before and after addition of calcium (Figure 1D). Both Nek2 isoforms were stable when incubated in interphase extracts, indicating that neither components of the reticulocyte lysate system used for generating recombinant proteins nor those of the interphase egg cytosol were sufficient for Nek2A degradation (data not shown). For comparison, the stability of cyclins A and B1 was measured in these extracts. As previously reported, cyclin B1 was only degraded after calcium addition, whereas cyclin A was degraded both before and after calcium addition although, like Nek2A, degradation was more rapid in anaphase extracts (Glotzer et al., 1991; Geley et al., 2001). Taken together, these results demonstrate that Nek2A is destroyed upon entry into mitosis with very similar timing to cyclin A, whereas Nek2B is stable at least until late mitosis/early G1.
Nek2A is destroyed in mitosis by the proteasome
To determine whether Nek2A is destroyed by the 26S proteasome, its half-life was measured in cells pre-incubated with various protease inhibitors (Lee and Goldberg, 1998). In the presence of either MG132 or lactacystin, strong inhibitors of the proteasome, the half-life of Nek2A was extended to >4 h (Figure 2A and B). In contrast, ALLM, a calpain inhibitor, did not alter the half-life of Nek2A at all. Leupeptin, an inhibitor of trypsin and cysteine proteases, caused a moderate increase in Nek2A half-life, consistent with an inhibition of one of the major peptidase activities (trypsin-like) of the proteasome (Coux et al., 1996). To determine specifically whether the mitotic destruction of Nek2A is due to the proteasome, MG132 was added to cells pre-arrested in either S or M phase (Figure 2C). Whereas addition of MG132 caused no detectable change in the amount of Nek2A present in S phase cells, it led to a significant increase in the level of Nek2A in prometaphase cells. Addition of MG132, but not ALLM, also prevented Nek2A destruction in both metaphase and anaphase egg extracts (Figure 2D and E). These results provide confirmation that the proteasome is responsible for removing Nek2A in mitosis.

Fig. 2. Mitotic destruction of Nek2A is proteasome dependent. (A) Nek2 protein stability was determined as in Figure 1C except following a 1 h pre-incubation with DMSO, 12.5 µM lactacystin, 20 µM MG132, 100 µM ALLM or 200 µg/ml leupeptin. The position of Nek2A is indicated (arrows). (B) The amount of Nek2A in the blots shown in (A) was determined by quantitative densitometry and shown with respect to the amount of protein at time zero (100%); DMSO (open diamonds), lactacystin (open squares), MG132 (filled triangles), ALLM (filled circles), leupeptin (crosses). (C) Cells arrested in either S (hydroxyurea) or M (nocodazole) were treated for 4 h with nothing (–), DMSO or 20 µM MG132, before extraction and immunoblotting with Nek2 antibodies. (D) Degradation assays were performed with cyclin B1 or Nek2A in extracts containing DMSO, 50 µM MG132 or 50 µM ALLM. (E) The amount of Nek2A remaining in metaphase (left graph) and anaphase (right graph) extracts in the presence of DMSO (closed triangles), MG132 (closed squares) and ALLM (open circles) with time was quantified from the autoradiographs shown in (D).
Nek2A is ubiquitylated in mitosis
Proteins bound for destruction by the proteasome generally are covalently modified with multiple chains of ubiquitin (Hershko and Ciechanover, 1998). To determine whether Nek2A can be covalently conjugated with ubiquitin, His-tagged ubiquitin was added to mitotic extracts carrying recombinant proteins before affinity purification with nickel–agarose beads (Figure 3). Some unmodified protein was present in all the precipitates. However, long exposure revealed a ladder of increasing molecular weight products with cyclin B1 in the presence of His6-ubiquitin that was not seen with the control protein, lamin A. Although only destroyed in anaphase extracts, cyclin B1 was ubiquitylated in both metaphase and anaphase extracts, in line with previous reports that ubiquitylation alone is not sufficient for cyclin B1 destruction (Kramer et al., 2000). Despite the intensity of the Nek2A signal being weak, at least two higher molecular weight products were detected in the presence of His6-ubiquitin at intervals of ∼10 kDa, indicative of ubiquitylation. Ubiquitylation of Nek2A occurred in both metaphase and anaphase extracts, with the weaker signal in anaphase most probably due to incomplete inhibition of the proteasome.

Fig. 3. Ubiquitylation of Nek2A. 35S-labeled cyclin B1, lamin A or Nek2A were added to CSF extracts either with or without Ca2+ and His6-ubiquitin as indicated. After 30 min incubation, samples were precipitated using Ni2+-NTA–agarose beads and analyzed by SDS–PAGE and autoradiography. In all samples, some unmodified protein (*) was precipitated irrespective of the presence of His6-ubiquitin. However, high molecular weight products (Ub) were only detected with cyclin B1 and Nek2A in the presence of His6-ubiquitin.
Nek2A destruction is APC/C dependent
The major E3 ubiquitin ligase that is active in mitosis is the APC/C. However, other ubiquitin ligases are active in mitosis including the SCF complex (Jackson et al., 2000). To determine whether Nek2A is a target of the APC/C, monoclonal antibodies directed against the core APC/C subunit Cdc27 (Yamano et al., 1998) were used to remove the APC/C from egg extracts (Figure 4A). In these extracts, the destruction of cyclin B1, cyclin A and Nek2A was significantly delayed as compared with mock-depleted extracts (Figure 4B). In a different approach, antibodies raised against the Xenopus APC/C adaptor protein Cdc20 (also called Fizzy), that block APC/C– Cdc20-dependent degradation, were added to extracts (Lorca et al., 1998). These antibodies blocked the degradation not only of cyclin B1 but also of Nek2A in both metaphase and anaphase extracts, providing additional strong evidence for the role of the APC/C–Cdc20 in the mitotic destruction of Nek2A (Figure 4C).
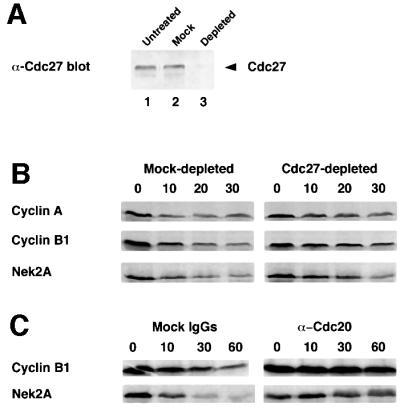
Fig. 4. Mitotic destruction of Nek2A is APC/C-Cdc20 dependent. (A) Egg extracts, which had been untreated (lane 1) or depleted with non-specific mouse IgGs (lane 2) or anti-Cdc27 mAb AF3 (lane 3), were immunoblotted with commercial anti-Cdc27 antibody. (B) Degradation assays were performed on cyclin A, cyclin B1 and Nek2A in anaphase extracts that had been either mock-depleted (left panels) or Cdc27-depleted (right panels). (C) Degradation assays were performed on cyclin B1 and Nek2A in anaphase extracts in the presence of either mock IgGs (left panels) or anti-Cdc20 antibodies (right panels).
Nek2 activity is not required for APC/C activation or Nek2A destruction
Phosphorylation of APC/C subunits is necessary for its activation (Charles et al., 1998; Descombes and Nigg, 1998; Shirayama et al., 1998; Kotani et al., 1999). We therefore tested whether Nek2 kinase activity was required for APC/C activation by removing the predominant splice variant present in egg extracts, X-Nek2B, by immunodepletion. Despite removal of >95% of X-Nek2B (Figure 5A), depleted extracts remained as efficient as mock-treated extracts in their ability to destroy cyclin B1, cyclin A and Nek2A (Figure 5B). It therefore seems highly unlikely that Nek2 kinase activity is required for APC/C activity. In metaphase extracts, the slower destruction of Nek2A is accompanied by the transient appearance of a higher molecular weight smear suggestive of phosphorylation. Nek2A has been shown previously to dimerize and trans-autophosphorylate on its C-terminal domain (Fry et al., 1999). To determine whether the higher molecular weight products are the result of autophosphorylation, catalytically inactive Nek2 (K37R) was incubated in X-Nek2B-depleted extracts. To our surprise, this smear was still apparent, albeit a little weaker than with the wild-type kinase, indicating that Nek2A must also be phosphorylated by a distinct cytoplasmic kinase (Figure 5B). Similar results were obtained using inactive Xenopus X-Nek2A in depleted extracts (data not shown). As the inactive kinase was still destroyed, these experiments demonstrate that Nek2 activity is not required for its own destruction.
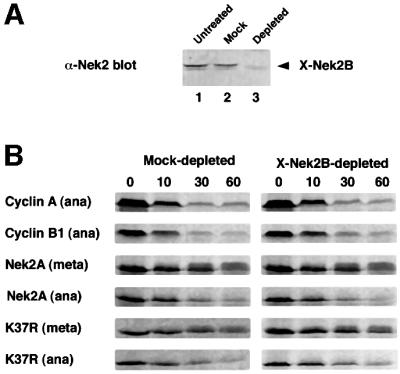
Fig. 5. Nek2 kinase activity is not required to activate the APC/C. (A) Egg extracts that were untreated (lane 1) or depleted with non-specific rabbit IgGs (lane 2) or anti-Xenopus Nek2B antibodies (lane 3) were immunoblotted with anti-Xenopus Nek2B antibodies. (B) Degradation assays were performed on cyclin A, cyclin B1, Nek2A or catalytically inactive Nek2A (K37R) in mock-depleted (left panels) or Nek2-depleted (right panels) anaphase (ana) or metaphase (meta) extracts.
Nek2A destruction depends upon a cyclin A-like destruction box in its extreme C-terminus
Two sequence motifs have been described that target proteins to the APC/C: a nine amino acid destruction box (D-box), first identified in cyclin B, and a KEN-box, initially found in the APC/C regulator, Cdc20 (Glotzer et al., 1991; Pfleger and Kirschner, 2000). Human Nek2A contains putative versions of both these motifs: a D-box (RKFLSLASN) at amino acids 361–369 and a KEN-box (KENIMRSENS) at amino acids 391–400. The positions of these motifs with respect to the splice site are such that both Nek2A and Nek2B contain the D-box, whereas only Nek2A contains the KEN-box (Figure 6A). During the initial description of the KEN-box, it was shown that human Nek2A was unstable in interphase egg extracts supplemented with recombinant Cdh1 but could be stabilized by mutation of its KEN-box (Pfleger and Kirschner, 2000). To test the importance of the D- and KEN-box motifs for Nek2A destruction, they were mutated individually or in combination and the stability of the resulting constructs assessed in egg extracts (Figure 6B). Mutation of the D-box had no effect on Nek2 destruction, while mutation of the KEN-box slowed the destruction of Nek2A in metaphase, but not anaphase, extracts. Mutation of the D- and KEN-box in the same construct provided no additional stabilization over mutation of the KEN-box alone. The stability of these proteins was also tested in vivo by analyzing the abundance of transfected protein (Figure 6C and D). All ectopically expressed proteins were detected in S phase but were significantly reduced in prometaphase-arrested cells. Hence, the D- and KEN-box alone or in combination cannot account for the destruction of Nek2A that occurs in early mitotic cells. In line with this, ubiquitylation was still observed with the KEN-box mutant (data not shown).

Fig. 6. Examination of putative destruction boxes in Nek2A. (A) Schematic diagram of the human Nek2 splice variants indicating the catalytic domain (CAT), leucine zipper (LZ), coiled-coil domain (CC), splice site, D-box and KEN-box. (B) In vitro degradation assays were performed in metaphase and anaphase extracts with Nek2A, Nek2A-KEN, Nek2A-R361L and Nek2A-R361L/KEN. (C) HeLa cells were transfected with myc-tagged wild-type Nek2A, Nek2A-KEN, Nek2A-R361L or Nek2A-R361L/KEN before arrest in either S phase (S) or prometaphase (M). Cell extracts were immunoblotted with Nek2 antibodies to detect transfected myc-Nek2A and endogenous Nek2A proteins as indicated. (D) The mitotic degradation of each myc-tagged Nek2A construct is represented by the fraction (%) remaining in M phase-arrested cells, where the amount in S phase-arrested cells is considered as 100%. For comparison, the change in endogenous Nek2A is also shown.
Destruction of cyclin A by the APC/C in early mitosis is dependent upon a novel extended form of D-box that contains both the core cyclin B-type D-box motif plus an additional short sequence downstream (amino acids 45–72 in human cyclin A2; Geley et al., 2001). As Nek2A is destroyed at the same time as cyclin A, we compared the sequence of the cyclin A extended D-box with the region of Nek2A that is absent from Nek2B. We found that the extreme C-terminal 23 amino acids of Nek2A have startling similarity to the cyclin A D-box, possessing both the core B-type D-box motif and a 10 amino acid sequence downstream (LKSRQILGMR) that is highly conserved with the cyclin A extension (Figure 7A). Expression of a Nek2A construct lacking the C-terminal 25 amino acids (Nek2A-Δ25) produced a protein that was no longer degraded in prometaphase-arrested cells (Figure 7B and 6D) and had a half-life of significantly more than 4 h in asynchronous cells (Figure 7C and D). Hence, it is this A-type D-box that is key to Nek2A destruction in prometaphase cells. Nek2A-Δ25 was stable in metaphase extracts but still slowly degraded in anaphase extracts (Figure 7E). However, a double mutant lacking both the KEN-box and C-terminal 25 amino acids was completely stable (Figure 7E).

Fig. 7. Identification of a cyclin A-type D-box in the C-terminus of Nek2A. (A) Comparison of the C-terminal 25 amino acids of human (Hs), mouse (Mm) and frog (Xl) Nek2A with the extended D-box of human and mouse cyclin A2. The three key residues of the classical D-box are shaded, together with the residues downstream that are either identical or have only conservative changes. (B) Extracts of HeLa cells transfected with myc-Nek2A (left panel) or myc-Nek2A-Δ25 (right panel) and arrested in S phase (S) or prometaphase (M) were immunoblotted with Nek2 antibodies and the position of transfected and endogenous Nek2 proteins indicated. (C) HeLa cells transfected with myc-Nek2A (top panel), myc-Nek2A-R361L/KEN (middle panel) and myc-Nek2A-Δ25 (bottom panel) were pre-treated with DMSO or MG132 before addition of cycloheximide for either 0 or 4 h as indicated. (D) The fraction (%) of each expressed protein remaining at 4 h in the absence of MG132 is shown with respect to the amount at 0 h. (E) Destruction assays were carried out in metaphase or anaphase extracts with Nek2A, Nek2A-Δ25 and Nek2A-KEN/Δ25.
To test whether the Nek2A C-terminal D-box could compete with cyclin B destruction, we purified the entire C-terminal non-catalytic domain as a GST fusion protein (Figure 8A). At low concentrations, this protein was itself destroyed in egg extracts in a proteosomal-dependent manner, indicating that it carries motifs sufficient for mitotic destruction (Figure 8B). When added in excess, it delayed the destruction of both cyclin A and cyclin B1, emphasizing that Nek2A is a target of the same destruction machinery as the cyclins (Figure 8C). When an excess of an N-terminal fragment of Schizosaccharomyes pombe cyclin B that contains the B-type D-box (N70; Yamano et al., 1998) was added, the destruction of cyclin B1 was strongly inhibited, but that of cyclin A and Nek2A was only weakly inhibited (Figure 8D). These intriguing results suggest that the cyclin A/Nek2A-type D-box contains additional sequences, presumably in the downstream extension, that allow recognition by the APC/C independently of its core B-type D-box.
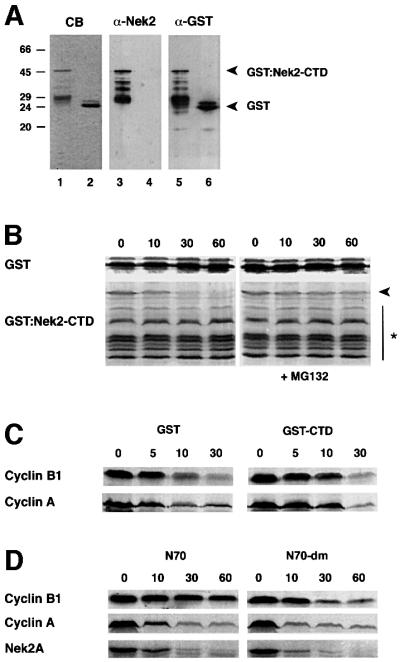
Fig. 8. The Nek2 C-terminal domain interferes with destruction of cyclins A and B. (A) Purified GST–Nek2-CTD (lanes 1, 3 and 5) and GST alone (lanes 2, 4 and 6) separated on 15% SDS–polyacrylamide gels and stained with Coomassie Blue (lanes 1 and 2), or immunoblotted with antibodies against Nek2 (lanes 3 and 4) or GST (lanes 5 and 6). Molecular weights (kDa) are indicated on the left. (B) Degradation assays were performed with 200 ng of GST or GST–Nek2-CTD in 10 µl of anaphase extracts in the absence (left panels) or presence (right panels) of 50 µM MG132. Samples taken at the times indicated (min) were analyzed by immunoblotting with anti-GST antibodies. Note that only the full-length GST–Nek2-CTD protein (arrowhead), and not the lower molecular weight truncated forms (asterisk), is degraded in the absence of MG132. (C) Degradation assays were performed with cyclin B1 and cyclin A in anaphase extracts in the presence of 1 µg GST (left panels) or GST–Nek2-CTD (right panels). (D) Degradation assays were performed in anaphase extracts with cyclin B1, cyclin A and Nek2A in the presence of 1 µg of an N-terminal 70 amino acid fragment of S.pombe Cdc13 (N70, left panels) or an identical fragment but with mutations in the destruction box (N70-dm right panels).
Discussion
Previous work has demonstrated that Nek2A disappears in mitotically arrested cells (Schultz et al., 1994; Fry et al., 1995; Hames and Fry, 2001), is unstable in interphase egg extracts supplemented with recombinant Cdh1 (Pfleger and Kirschner, 2000) and is degraded when injected into dividing embryos (Uto and Sagata, 2000). This prompted us to investigate if, when and how human Nek2 proteins are destroyed during mitosis. The results presented here show that Nek2A is destroyed by the proteasome following ubiquitylation by the E3 ubiquitin ligase APC/C– Cdc20. Furthermore, degradation of Nek2A first occurs in early mitosis, coincident with cyclin A destruction, and depends upon a motif in its extreme C-terminus that is almost identical to the extended D-box present in cyclin A (Geley et al., 2001).
Nek2A is an early mitotic target of the APC/C
In synchronized cells, Nek2A is destroyed early in mitosis coincident, within the limits of the experiment, with the destruction of cyclin A. Moreover, Nek2A is absent in cells arrested in prometaphase with the microtubule poison nocodazole. Hence, the destruction of Nek2A, like that of cyclin A but not cyclin B, is independent of the spindle assembly checkpoint. Proteasome inhibitors added to cells arrested in prometaphase allowed the re-accumulation of Nek2A, indicating that its loss is due to continual turnover by the 26S proteasome rather than to inhibition of transcription or translation. Also in common with cyclin A, Nek2A is unstable in egg extracts arrested in metaphase II of meiosis but is destroyed more rapidly in CSF extracts triggered to enter anaphase. Only these latter extracts can destroy cyclin B. Hence, the destruction mechanism that is active in prometaphase of an adult cell cycle seems also to be active in metaphase-II of meiosis.
The destruction of Nek2A, again in common with cyclin A, is directed by the APC/C ubiquitin ligase. In this study, it is shown that Nek2A can be ubiquitylated during mitosis. More specifically, depletion of the APC/C via anti-Cdc27 antibodies significantly reduced the rate of Nek2A destruction, while inhibitory Cdc20 antibodies blocked it completely. Nek2A can also be destroyed in interphase extracts, which possess APC/C proteins, simply by addition of the other APC/C activator Cdh1 (Pfleger and Kirschner, 2000). The identification of its C-terminal destruction motifs provides final confirmation that Nek2A is an APC/C substrate. Hence, Nek2A shares another common property with its fungal homolog NIMA. Although their destruction motifs appear to be different, both are targeted by the APC/C through their non-catalytic C-terminal domains.
Early embryos of frogs and flies possess only one APC/C adaptor protein, Cdc20, as Cdh1 is only expressed later in development when cell cycles introduce the first G1 phase (Sigrist and Lehner, 1997; Lorca et al., 1998). Nek2A must therefore be targeted for destruction, at least in meiotic egg extracts, by the APC/C–Cdc20 complex, explaining the prevention of Nek2A destruction by inhibitory anti-Cdc20 antibodies. However, Nek2A can also be destroyed by APC/C–Cdh1 (Pfleger and Kirschner, 2000). Thus, Nek2A, like other APC/C substrates (e.g. securin, cyclin B and Cdc6), can be recognized by the APC/C in complex with either Cdc20 or Cdh1. Nek2A abundance remains at very low levels throughout G1 and, although not formally proven here, it seems reasonable to predict that destruction begun in early mitosis by APC/C– Cdc20 is maintained into G1 (and G0) by APC/C–Cdh1. Nek2B, the shorter splice variant, is not destroyed in prometaphase cells or metaphase II extracts. Neither is it destroyed in anaphase or interphase extracts, in line with results showing that Xenopus Nek2B is stable throughout the first embryonic cell cycle (Fry et al., 2000b). However, Nek2B does decrease in abundance in cells upon re-entry into G1. The half-life of Nek2B did not change significantly between asynchronous and S phase-arrested cells, although the proteasome inhibitor MG132 did cause a moderate increase in its stability. It therefore remains unclear whether the decrease of Nek2B in G1 is due to degradation or some other mechanism such as reduced transcription.
What regulates the timing of mitotic APC/C substrates?
Nek2A and cyclin A are both destroyed via the APC/C early in mitosis when the APC/C is unable to target other substrates, such as cyclin B or securin, for destruction. Inhibition of the APC/C towards these latter substrates is regulated by the spindle checkpoint protein Mad2 which, in the presence of unattached kinetochores, binds directly to Cdc20 (Shah and Cleveland, 2000). The mechanism by which the APC/C is prevented from destroying cyclin B in metaphase II-arrested eggs is not known, because at this cell cycle stage, not only are all kinetochores attached, but also the spindle assembly checkpoint is not functional (Minshull et al., 1994).
The finding that Nek2A and cyclin A share a similar extended destruction box suggests that this may be key to recognition of early mitotic substrates by the APC/C. Deletion of this motif stabilized Nek2A in both cells and extracts. Moreover, this motif is highly conserved among vertebrate Nek2 kinases, unlike the other potential D-box (amino acids 361–369) that is present in human Nek2A and Nek2B, but not in the Nek2 sequences of other vertebrates. Indeed, mutation of this latter D-box had no stabilizing effect whatsoever, suggesting that it has no function in regulating Nek2 destruction. The KEN-box, on the other hand, is conserved in mammalian and frog Nek2A and its mutation partially stabilized Nek2A in extracts, although not in mitotic cells. It seems plausible that the extended destruction box present in the extreme C-terminus is targeted by APC/C–Cdc20 in early mitosis, whereas the KEN-box is targeted by APC/C–Cdh1 in late mitosis and G1. This fits with the notion that Cdc20 acts most effectively on D-box-type sequences, but can also recognize KEN-boxes, whereas Cdh1 is more selective for the KEN-box (Pfleger and Kirschner, 2000).
The destruction of cyclin B1 was strongly inhibited by addition of a protein fragment containing a B-type D-box. However, this fragment had only a mild inhibitory effect on the destruction of cyclin A or Nek2A. In contrast, the destruction of both cyclins was significantly delayed by a GST fusion protein containing the degradation motifs of Nek2A. This fusion protein could itself be destroyed, indicating that the Nek2A C-terminus could act as a transferable degradation signal. These competition experiments could reflect a quantitative difference in the extent of ubiquitylation required to destroy these proteins, with cyclin B requiring more ubiquitylation than cyclin A or Nek2A. Alternatively, they could indicate that, as a result of the extended nature of the cyclin A-type D-box, the presence of the smaller B-type D-box could not entirely prevent the APC/C from recognizing cyclin A or Nek2A. If Mad2 acts by competing for the same site as the B-type D-box, then this could equally explain why the spindle assembly checkpoint does not prevent destruction of proteins with the extended A-type D-box. Exactly which residues are critical within this A-type D-box will require further investigation, but one possibility is that a phosphorylation site is involved in a manner reminiscent of substrate recognition by the SCF ubiquitin ligase (Jackson et al., 2000). Nek2A is transiently phosphorylated before its destruction and this appears not to be solely via autophosphorylation. It is attractive to speculate that there may be a Nek2 kinase kinase that is somehow involved in targeting Nek2A, and perhaps cyclin A, to the APC/C.
What is the purpose of Nek2A destruction?
The timing of Nek2 kinase activation, its interaction with PP1 and now the timing of its destruction all support a role for Nek2A immediately prior to the onset of mitosis. Taken together with its localization to the centrosome, its ability to stimulate centrosome splitting upon overexpression and its interaction with C-Nap1, a protein implicated in centrosome cohesion, it seems likely that Nek2A plays a critical role in centrosome disjunction at the G2/M transition (Hinchcliffe and Sluder, 2001). If, as has been proposed, Nek2A stimulates loss of cohesion between centrosomes by triggering disassembly of an intercentriolar linkage (Mayor et al., 1999), then Nek2A destruction may somehow be necessary to allow re-establishment of the intercentriolar linkage in late mitosis (Figure 9). C-Nap1, which disappears from centrosomes in late G2, reappears around telophase (Mayor et al., 2000), at the same time as a flexible linker is re-established between the new centriole pairs of the future daughter cells (Piel et al., 2000). The persistence of Nek2B at this time may seem to pose a problem for this hypothesis. However, Nek2B does not bind PP1, nor does it stimulate centrosome splitting with the same efficiency as Nek2A (Hames and Fry, 2001). Hence, Nek2B may not have an equivalent function in regulating the intercentriolar linkage. Instead, Nek2B protein may contribute to the stability of mitotic spindle poles as inhibition of Nek2B leads to centrosome fragmentation in early Xenopus embryos (Uto and Sagata, 2000). Another important question is whether the destruction of Nek2A is necessary for mitotic exit. So far, we have not seen any evidence for substantial delays in mitosis upon expression of mutant Nek2A constructs (data not shown). Overexpression of a stabilized NIMA mutant did cause a mitotic arrest in Aspergillus cells without preventing destruction of cyclin B, suggesting that there is a genuine need to down-regulate NIMA activity (Pu and Osmani, 1995). Cyclin A overexpression also delays progression through mitosis, although this could be due to either competition for the APC/C or abnormal maintenance of Cdk1 activity (den Elzen and Pines, 2001; Geley et al., 2001). The challenge now is to demonstrate the role of Nek2A destruction in both centrosome dynamics and mitotic progression in living cells.

Fig. 9. The role of Nek2A destruction in mitosis. Current data, presented here and elsewhere, predict that a complex would exist in G2 composed of Nek2A, C-Nap1 and PP1. This complex, present at the proximal ends of centrioles, may form an anchoring structure for the intercentriolar linkage. Nek2A expression is high in G2, but its association with active PP1 keeps it and C-Nap1 in a dephosphorylated state. Upon phosphorylation and inactivation of PP1 at the G2/M transition, Nek2 and C-Nap1 become hyperphosphorylated, leading to displacement or degradation of C-Nap1 and disjunction of centrosomes. Centrosomes can then separate to opposite ends of the cell as a bipolar spindle is formed. Upon progress into mitosis, the APC/C becomes active towards Nek2A, causing its ubiquitylation and destruction. With Nek2A no longer present, C-Nap1 can be dephosphorylated at some point later in mitosis, allowing it to reassociate with disoriented centrioles and establish a new intercentriolar linkage.
Materials and methods
Cell culture, synchronization and transfection
HeLa and U2OS cells were grown at 37°C in a 5% CO2 atmosphere in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal calf serum and penicillin–streptomycin (100 IU/ml and 100 µg/ml, respectively). The following protocols were used to obtain U2OS cells arrested at specific stages of the cell cycle. G1/S: cells were pre-synchronized in S phase by the addition of 2 mM thymidine for 12 h, released by transferring to fresh medium for 8 h and then re-synchronized at the G1/S transition by incubation for 16 h with 1 mM hydroxyurea. S phase: cells were incubated with 1 mM hydroxyurea for 16 h. M phase: cells were treated with 500 ng/ml nocodazole for 16 h before collecting the mitotic population by gentle pipetting. Release of cells from drug-induced cell cycle blocks was performed by three washes in 1× phosphate-buffered saline (PBS) and replacement into fresh medium. Cell cycle distributions were confirmed by flow cytometry. To measure protein stability in cells, cycloheximide was added to 50 µg/ml and cells were harvested at given time intervals after cycloheximide addition. To test the role of different proteases, cells were first pre-incubated for 1 h with dimethylsulfoxide (DMSO), 12.5 µM clasto-lactacystin, 20 µM MG132, 100 µM ALLM (all from Calbiochem) or 200 µg/ml leupeptin (Sigma) before addition of cycloheximide. Transient transfections were carried out using Lipofectamine 2000 reagent (Invitrogen Life Technologies) according to the manufacturer’s instructions.
Cell extraction and immunoblotting
Cell extractions and immunoblotting were performed as described previously (Fry et al., 1998a). The following antibodies were used for immunoblots: anti-Nek2 (Zymed anti-peptide antibody; Fry et al., 1999), 1.0 µg/ml; anti-human cyclin B1 (UBI), 0.8 µg/ml; anti-human cyclin A (UBI), 1.0 µg/ml; anti-α-tubulin (Amersham Pharmacia), 0.15 µg/ml; anti-Cdc27 (Transduction Laboratories), 1.0 µg/ml; and anti-GST (Molecular Probes), 0.3 µg/ml.
Plasmid construction and mutagenesis
pGEM:Nek2A-KEN was generated by PCR-based mutagenesis of the pGEM:Nek2 plasmid (Schultz et al., 1994) using the QuikChange Site-directed Mutagenesis Kit (Stratagene). This changes amino acids 391–393, KEN to AAA, and 399, N to A. pCMVmyc:Nek2A-KEN was made by excising an NheI C-terminal fragment containing the KEN-box motif from pCMVmyc:Nek2A (Fry et al., 1998a) and replacing it with the corresponding fragment containing the KEN-box mutation from pGEM:Nek2A-KEN. pGEM:Nek2A-R361L was made by mutagenizing the pGEM-Nek2 plasmid using the Transformer Site-directed Mutagenesis Kit (Clontech). To make pCMVmyc:Nek2A-R361L, the Nek2A-R361L fragment was excised from pGEM:Nek2A-R361L on a NaeI–XbaI fragment and subcloned into the SmaI–XbaI sites of a pBlueScript-myc vector (Schmidt-Zachmann and Nigg, 1993), creating pBS:myc-Nek2A-R361L. The myc-Nek2A-R361L insert was then excised from this plasmid on a SalI (blunted)–XbaI fragment and subcloned into pRcCMV (Invitrogen) cut with HindIII (blunted)– XbaI. pGEM:Nek2A-R361L/KEN was generated by performing site-directed mutagenesis on the pGEM:Nek2A-R361L plasmid using the QuikChange Site-directed Mutagenesis Kit (Stratagene), and to make pCMVmyc:Nek2A-R361L/KEN site-directed mutagenesis was performed on the pCMVmyc:Nek2A-R361L plasmid. To make pGEM: Nek2A-Δ25, a C-terminal fragment of Nek2A was amplified by PCR using a 3′ PCR primer that introduced a stop and BamHI site immediately after amino acid 420. This was digested at an internal EcoRV site and the BamHI site. An EcoRV–BamHI fragment was then excised from pGEM:Nek2 and replaced with the corresponding amplified fragment. pCMVmyc:Nek2A-Δ25 was made by excising the N-terminal 420 amino acids of Nek2A together with an N-terminal myc tag from pBSmyc-Nek2 (Fry et al., 1998a) on a SalI–PvuII blunted fragment and subcloning into pRcCMV (Invitrogen) cut with HindIII–XbaI and blunted. Finally, to make pGEM:Nek2A-KEN/Δ25, a DraII fragment containing most of the Nek2A coding sequence and encompassing the KEN-box was excised from pGEM:Nek2A-KEN and used to replace the corresponding DraII fragment from pGEM:Nek2A-Δ25. All constructs were confirmed by DNA sequencing on an ABI sequencer (Perkin Elmer) using BigDye technology at the University of Leicester Protein and Nucleic Acid Laboratory.
In vitro degradation assays and immunodepletions
CSF (metaphase II-arrested) or interphase extracts were prepared as described (Desai et al., 1999). To generate anaphase extracts, calcium chloride was added to 0.5 mM. Substrates were prepared by coupled in vitro transcription–translation in reticulocyte lysates (TNT, Promega), in the presence of [35S]cysteine/methionine (NEN), using the appropriate plasmids. Egg extracts (10 µl) were supplemented with 1.5 µl of in vitro translation mix and 0.3 µl of cycloheximide (10 mg/ml) before incubation at 22°C. Aliquots (2.5 µl) were taken at the times indicated, mixed with protein sample buffer and separated on 12% SDS–polyacrylamide gels. Gels were fixed and stained with Coomassie Blue, soaked in Amplify (Amersham), dried and exposed either to X-ray film (Fuji) or to phosphoimaging screens. Immunodepletions were carried out as described (Descombes and Nigg, 1998), with the exception of using two 30 min rounds of depletion with antibody coated to protein A– dynabeads (Dynal).
Ubiquitylation assay
Ubiquitylation assays were essentially as described in Funabiki and Murray (2000). Specifically, CSF extract (20 µl) was pre-incubated for 15 min at 22°C with MG132 (50 µM final concentration) before addition of cycloheximide (250 µg/ml final concentration) and 3 µl of in vitro translation mix containing 35S-labeled recombinant protein. To this was added 0.2 µl of either dH2O or CaCl2 (0.5 mM final concentration) and 1.3 µl of either His6-ubiquitin (0.5 mg/ml final concentration; Calbiochem) or His6-ubiquitin buffer [20 mM Tris–HCl pH 8, 20 mM NaCl, 1 mM dithiothreitol (DTT)]. Samples were incubated for 30 min at 22°C before removing 2.5 µl for direct analysis by SDS–PAGE. To the remainder, 1 ml of buffer I [20 mM Tris–HCl pH 7.5, 500 mM NaCl, 10 mM N-ethylmaleimide, 5 mM imidazole, 1 mM phenylmethylsulfonyl fluoride (PMSF)] was added, mixed and spun at 14 K for 5 min at 4°C to remove insoluble material. Ni2+-NTA–agarose beads (Qiagen) were prepared by washing three times in buffer I, incubating with rabbit reticulocyte lysate for 30 min at 4°C on a rotating wheel, and washing again three times with buffer I. The supernatant from the ubiquitylation reaction was added to 10 µl of beads and the sample rotated for 30 min at 4°C. Beads were pelleted and washed three times with 1 ml of buffer II (20 mM Tris–HCl pH 8.0, 500 mM NaCl, 25 mM imidazole, 10 mM N-ethylmaleimide, 1 mM PMSF, 0.5% Triton X-100, 0.5% Tween-20) and once with buffer I, before finally resuspending in 30 µl of protein sample buffer. Samples were separated on 8% SDS–polyacrylamide gels, dried and exposed to X-ray film or phosphoimager screens.
Recombinant protein expression in Escherichia coli
The C-terminal domain (CTD) of human Nek2A (amino acids 265–445) was excised from pGEM-Nek2 on an XmnI fragment and inserted into the SmaI site of the pGEX-KG vector to generate pGEX:Nek2-CTD. This was transformed into BL21 E.coli and induced for 4 h with 0.4 mM isopropyl-β-d-thiogalactopyranoside before bacterial lysis and protein purification using standard procedures.
Acknowledgments
Acknowledgements
We thank Tim Hunt (South Mimms) and Jon Pines (Cambridge) for Xenopus cyclin B1 and human cyclin A plasmids, respectively, Sue Shackleton (Leicester) for human lamin A plasmid, Thierry Lorca (Montpellier) for anti-Cdc20 (Fizzy) antibodies, and Eilis Byrne (Leicester) for purified GST protein. We are grateful to Erich Nigg (Munich) for helpful comments on the manuscript and to all members of the laboratory for useful discussion. This work was supported by grants to A.M.F. from The Wellcome Trust and Cancer Research Campaign. A.M.F. is a Lister Institute Research Fellow. R.S.H. is supported by a studentship from the BBSRC. H.Y. is supported by a grant from PRESTO of the Japan Science and Technology Corporation.
References
- Charles J.F., Jaspersen,S.L., Tinker-Kulberg,R.L., Hwang,L., Szidon,A. and Morgan,D.O. (1998) The Polo-related kinase Cdc5 activates and is destroyed by the mitotic cyclin destruction machinery in S.cerevisiae. Curr. Biol., 8, 497–507. [DOI] [PubMed] [Google Scholar]
- Chen J. and Fang,G. (2001) MAD2B is an inhibitor of the anaphase-promoting complex. Genes Dev., 15, 1765–1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clute P. and Pines,J. (1999) Temporal and spatial control of cyclin B1 destruction in metaphase. Nature Cell Biol., 1, 82–87. [DOI] [PubMed] [Google Scholar]
- Coux O., Tanaka,K. and Goldberg,A.L. (1996) Structure and functions of the 20S and 26S proteasomes. Annu. Rev. Biochem., 65, 801–847. [DOI] [PubMed] [Google Scholar]
- den Elzen N. and Pines,J. (2001) Cyclin A is destroyed in prometaphase and can delay chromosome alignment and anaphase. J. Cell Biol., 153, 121–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai A., Murray,A., Mitchison,T.J. and Walczak,C.E. (1999) The use of Xenopus egg extracts to study mitotic spindle assembly and function in vitro. In Rieder,C.L. (ed.), Mitosis and Meiosis. Academic Press, San Diego, CA, pp. 385–412. [DOI] [PubMed]
- Descombes P. and Nigg,E.A. (1998) The polo-like kinase Plx1 is required for M phase exit and destruction of mitotic regulators in Xenopus egg extracts. EMBO J., 17, 1328–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fry A.M., Schultz,S.J., Bartek,J. and Nigg,E.A. (1995) Substrate specificity and cell cycle regulation of the Nek2 protein kinase, a potential human homolog of the mitotic regulator NIMA of Aspergillus nidulans. J. Biol. Chem., 270, 12899–12905. [DOI] [PubMed] [Google Scholar]
- Fry A.M., Meraldi,P. and Nigg,E.A. (1998a) A centrosomal function for the human Nek2 protein kinase, a member of the NIMA-family of cell cycle regulators. EMBO J., 17, 470–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fry A.M., Mayor,T., Meraldi,P., Stierhof,Y.-D., Tanaka,K. and Nigg,E.A. (1998b) C-Nap1, a novel centrosomal coiled-coil protein and candidate substrate of the cell cycle-regulated protein kinase Nek2. J. Cell Biol., 141, 1563–1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fry A.M., Arnaud,L. and Nigg,E.A. (1999) Activity of the human centrosomal kinase, Nek2, depends upon an unusual leucine zipper dimerization motif. J. Biol. Chem., 274, 16304–16310. [DOI] [PubMed] [Google Scholar]
- Fry A.M., Mayor,T. and Nigg,E.A. (2000a) Regulating centrosomes by protein phosphorylation. Curr. Top. Dev. Biol., 49, 291–312. [DOI] [PubMed] [Google Scholar]
- Fry A.M., Descombes,P., Twomey,C., Bacchieri,R. and Nigg,E.A. (2000b) The NIMA-related kinase X-Nek2B is required for efficient assembly of the zygotic centrosome in Xenopus laevis. J. Cell Sci., 113, 1973–1984. [DOI] [PubMed] [Google Scholar]
- Funabiki H. and Murray,A.W. (2000) The Xenopus chromokinesin Xkid is essential for metaphase chromosomes alignment and must be degraded to allow anaphase chromosomes movement. Cell, 102, 411–424. [DOI] [PubMed] [Google Scholar]
- Geley S., Kramer,E., Gieffers,C., Gannon,J., Peters,J.-M. and Hunt,T. (2001) Anaphase-promoting complex/cyclosome-dependent proteolysis of human cyclin A starts at the beginning of mitosis and is not subject to the spindle assembly checkpoint. J. Cell Biol., 153, 137–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glotzer M., Murray,A.W. and Kirschner,M.W. (1991) Cyclin is degraded by the ubiquitin pathway. Nature, 349, 132–138. [DOI] [PubMed] [Google Scholar]
- Hames R.S. and Fry,A.M. (2002) Alternative splice variants of the human centrosome kinase Nek2 exhibit distinct patterns of expression in mitosis. Biochem. J., 361, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helps N.R., Luo,X., Barker,H.M. and Cohen,P.T.W. (2000) NIMA-related kinase 2 (Nek2), a cell cycle-regulated protein kinase localized to centrosomes, is complexed to protein phosphatase 1. Biochem. J., 349, 509–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershko A. and Ciechanover,A. (1998) The ubiquitin system. Annu. Rev. Biochem., 67, 425–479. [DOI] [PubMed] [Google Scholar]
- Hinchcliffe E.H. and Sluder,G. (2001) ‘It takes two to tango’: understanding how centrosome duplication is regulated throughout the cell cycle. Genes Dev., 15, 1167–1181. [DOI] [PubMed] [Google Scholar]
- Jackson P.K., Eldridge,A.G., Freed,E., Furstenthal,L., Hsu,J.Y., Kaiser,B.K. and Reimann,J.D.R. (2000) The lore of the RINGs: substrate recognition and catalysis by ubiquitin ligases. Trends Cell Biol., 10, 429–439. [DOI] [PubMed] [Google Scholar]
- Juang Y.L., Huang,J.-Y., Peters,J.-M., McLaughlin,M.E., Tai,C.Y. and Pellman,D. (1997) APC-mediated proteolysis of Ase1 and the morphogenesis of the mitotic spindle. Science, 275, 1311–1314. [DOI] [PubMed] [Google Scholar]
- Kotani S., Tanaka,H., Yasuda,H. and Todokoro,K. (1999) Regulation of APC activity by phosphorylation and regulatory factors. J. Cell Biol., 146, 791–800. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Kramer E.R., Scheuringer,N., Podtelejnikov,V., Mann,M. and Peters, J.-M. (2000) Mitotic regulation of the APC activator proteins CDC20 and CDH1. Mol. Biol. Cell, 11, 1555–1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee D.H. and Goldberg,A.L. (1998) Proteasome inhibitors: valuable new tools for cell biologists. Trends Cell Biol., 8, 397–403. [DOI] [PubMed] [Google Scholar]
- Lorca T., Castro,A., Martinez,A.-M., Morin,N., Sigrist,S., Lehner,C., Doree,M. and Labbe,J.-C. (1998) Fizzy is required for activation of the APC-cyclosome in Xenopus egg extracts. EMBO J., 17, 3565–3575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayor T., Meraldi,P., Stierhof,Y.-D., Nigg,E.A. and Fry,A.M. (1999) Protein kinases in control of the centrosome cycle. FEBS Lett., 452, 92–95. [DOI] [PubMed] [Google Scholar]
- Mayor T., Tanaka,K., Stierhof,Y.-D., Fry,A.M. and Nigg,E.A. (2000) The centrosomal protein C-Nap1 displays properties supporting a role in cell cycle-regulated centrosome cohesion. J. Cell Biol., 151, 837–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minshull J., Sun,H., Tonks,N.K. and Murray,A.W. (1994) A MAP kinase-dependent spindle assembly checkpoint in Xenopus egg extracts. Cell, 79, 475–486. [DOI] [PubMed] [Google Scholar]
- Morgan D.O. (1999) Regulation of the APC and the exit from mitosis. Nature Cell Biol., 1, E47–E53. [DOI] [PubMed] [Google Scholar]
- Nigg E.A. (2001) Mitotic kinases as regulators of cell division and its checkpoints. Nature Rev. Mol. Cell Biol., 2, 21–32. [DOI] [PubMed] [Google Scholar]
- Page A.M. and Hieter,P. (1999) The anaphase-promoting complex: new subunits and regulators. Annu. Rev. Biochem., 68, 583–609. [DOI] [PubMed] [Google Scholar]
- Petersen B.O. et al. (2000) Cell cycle- and cell growth-regulated proteolysis of mammalian CDC6 is dependent on APC–CDH1. Genes Dev., 14, 2330–2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfleger C.M. and Kirschner,M.W. (2000) The KEN box: an APC recognition signal distinct from the D box targeted by Cdh1. Genes Dev., 14, 655–665. [PMC free article] [PubMed] [Google Scholar]
- Pfleger C.M., Salic,A., Lee,E. and Kirschner,M. (2001) Inhibition of Cdh1–APC by the MAD2-related protein MAD2L2: a novel mechanism for regulating Cdh1. Genes Dev., 15, 1759–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piel M., Meyer,P., Khodjakov,A., Rieder,C.L. and Bornens,M. (2000) The respective contributions of the mother and daughter centrioles to centrosome activity and behaviour in vertebrate cells. J. Cell Biol., 149, 317–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pines J. and Rieder,C.L. (2001) Re-staging mitosis: a contemporary view of mitotic progression. Nature Cell Biol., 3, E3–E6. [DOI] [PubMed] [Google Scholar]
- Pu R.T. and Osmani,S.A. (1995) Mitotic destruction of the cell cycle regulated NIMA protein kinase of Aspergillus nidulans is required for mitotic exit. EMBO J., 14, 995–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puntoni F. and Villa-Moruzzi,E. (1997) Protein phosphatase-1α, γ1 and δ: changes in phosphorylation and activity in mitotic HeLa cells and in cells released from the mitotic block. Arch. Biochem. Biophys., 340, 177–184. [DOI] [PubMed] [Google Scholar]
- Reimann J.D.R., Freed,E., Hsu,J.Y., Kramer,E.R., Peters,J.-M. and Jackson,P.K. (2001) Emi1 is a mitotic regulator that interacts with Cdc20 and inhibits the anaphase promoting complex. Cell, 105, 645–655. [DOI] [PubMed] [Google Scholar]
- Schmidt-Zachmann M.S. and Nigg,E.A. (1993) Protein localization to the nucleolus: a search for targeting domains in nucleolin. J. Cell. Sci., 105, 799–806. [DOI] [PubMed] [Google Scholar]
- Schultz S.J., Fry,A.M., Sütterlin,C., Ried,T. and Nigg,E.A. (1994) Cell cycle-dependent expression of Nek2, a novel human protein kinase related to the NIMA mitotic regulator of Aspergillus nidulans. Cell Growth Differ., 5, 625–635. [PubMed] [Google Scholar]
- Shah J.V. and Cleveland,D.W. (2000) Waiting for anaphase: Mad2 and the spindle assembly checkpoint. Cell, 103, 997–1000. [DOI] [PubMed] [Google Scholar]
- Shirayama M., Zachariae,W., Ciosk,R. and Nasmyth,K. (1998) The polo-like kinase Cdc5p and the WD-repeat protein Cdc20p/fizzy are regulators and substrates of the anaphase promoting complex in Saccharomyces cerevisiae. EMBO J., 17, 1336–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigrist S.J. and Lehner,C.F. (1997) Drosophila fizzy-related downregulates mitotic cyclins and is required for cell proliferation arrest and entry into endocycles. Cell, 90, 671–681. [DOI] [PubMed] [Google Scholar]
- Uto K. and Sagata,N. (2000) Nek2B, a novel maternal form of Nek2 kinase, is essential for the assembly or maintenance of centrosomes in early Xenopus embryos. EMBO J., 19, 1816–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uto K., Nakajo,N. and Sagata,N. (1999) Two structural variants of Nek2 kinase, termed Nek2A and Nek2B, are differentially expressed in Xenopus tissues and development. Dev. Biol., 208, 456–464. [DOI] [PubMed] [Google Scholar]
- Wakefield J.G., Huang,J.-Y. and Raff,J.W. (2000) Centrosomes have a role in regulating the destruction of cyclin B in early Drosophila embryos. Curr. Biol., 10, 1367–1370. [DOI] [PubMed] [Google Scholar]
- Yamano H., Tsurumi,C., Gannon,J. and Hunt,T. (1998) The role of the destruction box and its neighbouring lysine residues in cyclin B for anaphase ubiquitin-dependent proteolysis in fission yeast: defining the D-box receptor. EMBO J., 17, 5670–5678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye X.S., Fincher,R.R., Tang,A., Osmani,A.H. and Osmani,S.A. (1998) Regulation of the anaphase-promoting complex/cyclosome by bimAAPC3 and proteolysis of NIMA. Mol. Biol. Cell, 9, 3019–3030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zechariae W. and Nasmyth,K. (1999) Whose end is destruction: cell division and the anaphase-promoting complex. Genes Dev., 13, 2039–2058. [DOI] [PubMed] [Google Scholar]
- Zur A. and Brandeis,M. (2001) Securin degradation is mediated by fzy and fzr and is required for complete chromatid separation but not for cytokinesis. EMBO J., 20, 792–801. [DOI] [PMC free article] [PubMed] [Google Scholar]