

Abstract
The Wilms’ tumor gene Wt1 is known for its important functions during genitourinary and mesothelial formation. Here we show that Wt1 is necessary for neuronal development in the vertebrate retina. Mouse embryos with targeted disruption of Wt1 exhibit remarkably thinner retinas than age-matched wild-type animals. A large fraction of retinal ganglion cells is lost by apoptosis, and the growth of optic nerve fibers is severely disturbed. Strikingly, expression of the class IV POU-domain transcription factor Pou4f2 (formerly Brn-3b), which is critical for the survival of most retinal ganglion cells, is lost in Wt1–/– retinas. Forced expression of Wt1 in cultured cells causes an up-regulation of Pou4f2 mRNA. Moreover, the Wt1(–KTS) splice variant can activate a reporter construct carrying 5′-regulatory sequences of the human POU4F2. The lack of Pou4f2 and the ocular defects in Wt1–/– embryos are rescued by transgenic expression of a 280 kb yeast artificial chromosome carrying the human WT1 gene. Taken together, our findings demonstrate a continuous requirement for Wt1 in normal retina formation with a critical role in Pou4f2-dependent ganglion cell differentiation.
Keywords: apoptosis/POU-domain factors/retina development/retinal ganglion cells/Wilms’ tumor gene
Introduction
The Wilms’ tumor gene WT1 (Wt1 in mice) was originally identified on the basis of its mutational inactivation in 10–15% of all Wilms’ tumors (Hastie, 1994 and references therein). Wilms’ tumor of the kidney (nephroblastoma) is the most common solid childhood malignancy, affecting around 1 in 10 000 children (Matsunaga, 1981). Nephroblastoma is a good example of malignant cell growth caused by a failure of the embryonic renal precursor, the metanephric blastema, to differentiate normally (Bennington and Beckwith, 1975). WT1 encodes a zinc finger protein of the C2H2 type, which shares a high degree of structural homology with the early growth response (egr) family of transcription factors (reviewed in Rauscher, 1993). At least 24 different isoforms of WT1 protein exist, which are generated by a combination of alternative splicing, RNA editing and the use of alternative translation start sites (for a recent review see Lee and Haber, 2001). Experimental evidence suggests that the WT1 splice variant which has three amino acids, lysine– threonine–serine (commonly referred as KTS), inserted between zinc fingers three and four of the molecule, plays a role in RNA processing (Englert et al., 1995b; Larsson et al., 1995; Davies et al., 1998; Landomery et al., 1999). The WT1(–KTS) isoform binds with high affinity to GC- and TC-rich DNA consensus sequences and functions as a transcriptional regulator (reviewed in Lee and Haber, 2001). Candidate target genes that are activated by WT1(–KTS) include the cyclin-dependent kinase inhibitor p21Cip1 (Englert et al., 1997), Amphiregulin (Lee et al., 1999), Vitamin D receptor (Maurer et al., 2001; Wagner et al., 2001) and Podocalyxin (Palmer et al., 2001), amongst others. By generating mouse strains in which the Wt1(–KTS) and (+KTS) variants have been specifically removed, it was demonstrated recently that these two isoforms exert distinct functions during sex determination and nephron formation (Hammes et al., 2001).
Besides its tumor suppressor function, Wt1 also exerts important roles in embryonic development. Thus, targeted inactivation of Wt1 in mice caused a lack of formation of the kidneys, gonads, spleen and adrenal glands, in addition to mesothelial defects (Kreidberg et al., 1993; Herzer et al., 1999; Moore et al., 1999). Wt1–/– embryos die in utero, presumably from heart failure due to disturbed cardiac growth (Kreidberg et al., 1993; Moore et al., 1999). Based on its spatio-temporal expression pattern (Pritchard-Jones et al., 1990; Pelletier et al., 1991) and the characteristic phenotype of the Wt1–/– embryos (Kreidberg et al., 1993), Wt1 was proposed to exert its unique role in development by allowing cells to flip back and forward between a mesenchymal and epithelial state (Moore et al., 1999).
It is noteworthy that Wt1 was also detected in populations of neurons in the brainstem and spinal cord (Armstrong et al., 1993; Rackley et al., 1993). These latter observations raise the interesting possibility that Wt1 is involved in the differentiation of ectodermally derived neuronal tissues. However, experimental evidence to support a role for Wt1 in neuron development has not yet been provided. Notably, patients with Wilms’ tumor are at an increased risk for developing ocular disorders including aniridia and, less frequently, optic nerve hypoplasia (Nelson et al., 1984; Bickmore and Hastie, 1989). These eye abnormalities in Wilms’ tumor patients are thought to result from concomitant disruption of the aniridia gene PAX6, which lies telomeric of WT1 on human chromosome 11p13 (Ton et al., 1991). A preliminary report on the expression of Wt1 in the developing eye is remarkable as it points towards a possible role for Wt1 in ocular development (Armstrong et al., 1993).
In this study, we addressed the question of whether the Wilms’ tumor gene Wt1 is required for the differentiation of neurons in the vertebrate retina. Indeed, our findings demonstrate that Wt1 is a critical gene for normal retina formation with impact on the proliferation of retinal progenitors and ganglion cell development.
Results
Wt1 mRNA and protein is expressed in the developing retina
We examined a total of >80 tissue sections, which were obtained from four different mouse retinas, each at the indicated developmental stages. Representative examples of Wt1 expression in the developing retina identified by in situ mRNA hybridization and immunohistochemistry are shown in Figure 1. Wt1 transcripts were initially detected throughout the neural retina and in the developing lens vesicle of 12-day-old (E12) C57BL/6 (B6) mouse embryos (Figure 1). Between E15 and the day after birth (P1), Wt1 expression became restricted to the presumptive retinal ganglion cell layer and was absent from the retinas of adult mice (Figure 1). Wt1 expression in the developing retinas of mice was confirmed by the detection of WT1 immunoreactivity in the future retinal ganglion cell layer of an autopsied 19-week-gestation human embryo (Figure 1).

Fig. 1. Representative micrographs of tissue sections of wild-type (Wt1+/+) eyes at different stages of development. Wt1 transcripts are detected by mRNA in situ hybridization in the lens vesicle and retinal neuroblasts of E12 mice (A), as well as in the developing ganglion cell layer of E15 (C) and P1 eyes (D). No Wt1 mRNA is visible in the retinas of adult mice (E) and in retinal tissue of E12 embryos hybridized with digoxigenin-labeled Wt1 sense RNA strand (B). Note, that WT1 protein is detected by immunohistochemistry in the inner portion of the retina obtained at autopsy of a 19-week-gestation human embryo (F). l, lens; nbl, neuroblast layer; gcl, ganglion cell layer. Scale bars, 100 µm.
Proliferation of the progenitor cells is impaired in Wt1–/– retinas
To examine whether Wt1 is important for normal retinal development, we compared the morphology of wild-type and Wt1–/– eyes at different embryonic stages. Forty retinal sections that were obtained from four different normal and Wt1–/– mutant E12 embryos each [C57BL/6 (B6) mouse strain] were analyzed. Representative tissue sections are shown in Figure 2. Wt1–/– mice at E12 had markedly thinner neuroretinas, which contained significantly fewer cells than those of age-matched Wt1+/+ embryos (Figure 2). Compared with the wild-type animals at E12, immunostaining of proliferating cell nuclear antigen (PCNA) was weaker, and incorporation of 5-bromo-2′-deoxyuridine (BrdU) was decreased in the Wt1–/– retinas (Figure 2). Counting of three retinal sections from BrdU-injected mouse embryos at stage E12 (n = 3 each) revealed that ∼10 times fewer BrdU-positive cells were contained in Wt1–/– than in wild-type retina. Reduced cell numbers were also observed in the E12 retinas of Wt1–/– embryos in the MF1×B6 mouse background (see below). However, the phenotypic changes of the mutant retinas were less severe in MF1×B6 than in C57BL/6 (B6) embryos at stage E12 (data not shown). The retinal morphology of heterozygous (Wt1+/–) embryos at E12 was normal and could not be distinguished from that of wild-type animals (data not shown).

Fig. 2. Representative (immuno)histology of wild-type (+/+) and Wt1 null mutant (–/–) eyes at E12. HE staining reveals notably thinner retinas in Wt1–/– (E and F) than in wild-type embryos (A and B). Immunofluorescent labeling of PCNA is clearly reduced in the mutant (G) versus Wt1+/+ (C) retina. Incorporation of BrdU as an estimate of DNA synthesis is also decreased in Wt1–/– (H) compared with wild-type (D) eyes. l, lens; nbl, neuroblast layer; rpe, retinal pigment epithelium. Scale bars, 100 µm (A, C–E, G and H) and 50 µm (B and F), respectively.
Lack of Wt1 causes defects of retinal ganglion cells and optic nerves
Disruption of the Wt1 gene in mice with a C57BL/6 (B6) genetic background caused embryonic lethality around mid-gestation (Kreidberg et al., 1993). To analyze the role of Wt1 in retina formation at later developmental stages, we made use of the MF1×B6 mouse strain, in which a certain percentage of Wt1–/– embryos survive until birth (Herzer et al., 1999). In comparison with wild-type MF1×B6 mice, the eyes of the Wt1–/– mutants were notably reduced in size at E18 (1403 ± 30 µm maximum diameter in Wt1–/– versus 1735 ± 29 µm in Wt1+/+ embryos, n = 5 each, P <0.0001, Wilcoxon test). The eyes of heterozygotes were phenotypically normal and indistinguishable from those of wild-type embryos (data not shown).
Ganglion cells are among the first to appear in the embryonic mouse retina, most of them between E13 and E16 (Sidman, 1961; Cepko et al., 1996). To explore whether Wt1 is critical for normal ganglion cell differentiation, we performed serial sections through the eyes of wild-type and Wt1–/– E18 embryos (n = 5 each). Histological examination of hematoxylin–eosin (HE)-stained tissue sections revealed that ∼40% fewer cells were contained in the future ganglion cell layer of the mutant retinas compared with that of Wt1+/+ mice (Figure 3A and D). TUNEL labeling was performed to explore whether the diminished number of ganglion cells was due to premature death in the Wt1–/– retinas. Indeed, the counts of TUNEL-positive cells in the E18 retinas of Wt1–/– embryos were increased ∼4-fold compared with age-matched Wt1+/+ mice (n = 5 each) (Figure 3I). TUNEL labeling of tissue sections indicated that ∼60% of the apoptotic cells were located in the developing ganglion cell layer of the Wt1–/– retinas (Figure 3H). Apoptosis was not enhanced in the Wt1–/– retinas prior to E18 (Figure 3I).

Fig. 3. Morphology of wild-type (A–C) and Wt1–/– mutant (D–F) retinas at E18. A significant (∼40%) reduction in the number of ganglion cells is detected by HE staining in the E18 retinas of Wt1–/– (D) compared with wild-type (A) embryos. Immunofluorescent labeling of NF200 reveals blind-ending optic nerve fibers exiting from the Wt1–/– eyes (F). NF200 immunoreactivity is absent from cross-sections of optic nerves made at a distance of ∼400 µm beyond the optic disc level (E). For comparison, NF200-positive axon bundles are clearly visible in the optic nerves of wild-type retina (B). Shown are representative results from five different embryos in both groups. gcl, ganglion cell layer; onf, optic nerve fibers. Scale bars, 50 µm (A and D), 100 µm (B and E) and 200 µm (C and F). Representative examples of apoptotic cells identified by TUNEL assay in the E18 retinas of wild-type (G) and Wt1–/– (H) mice. The majority (∼60%) of the TUNEL-positive cells are located in the developing ganglion cell layer of the mutant retina (H). (I) The number of apoptotic cells identified by TUNEL assay in the neural retinas of wild-type (Wt1+/+), null mutant (Wt1–/–) and WT280-YAC transgenic embryos at different stages of development. An ∼4-fold increase in apoptotic cells is counted in mutant versus Wt1+/+ retinas at E18. Note that the average number of apoptotic cells in the developing retina is not significantly different between wild-type and WT280–YAC embryos. Five animals were studied in each group at the indicated time points. Five 10 µm retinal sections were made from each embryo close to the optic disc level. Values shown are means ± SEM. Asterisks indicate statistical significance (P <0.001, ANOVA with Bonferroni as post-hoc test).
Each ganglion cell in the retina forms a single axon, which exits the eye as part of the optic nerve. To establish a second independent criterion of retinal ganglion cell defects, we compared the optic nerves of wild-type and Wt1 null mutant embryos at E18. A polyclonal antibody against neurofilament (NF) 200 was used to mark the retinal ganglion cell axons and optic nerve fibers. Staining of NF200 revealed that outgrowth of axon processes from the retinal ganglion cells was severely disturbed in the Wt1–/– embryos. Indeed, the Wt1 null mutants had abruptly terminating optic nerve fibers that extended no further than some 100 µm beyond the optic disc level (Figure 3F). Disrupted axon growth in the Wt1–/– embryos is also demonstrated by the lack of specific NF200 immunofluorescence on cross-sections made through the optic nerves at a distance of ∼400 µm beyond their exit from the eyes. Normal NF200 staining of the optic nerve fibers was seen in heterozygous embryos at E18 (data not shown). Since Wt1–/– mutants normally die before end-gestation (Kreidberg et al., 1993; Herzer et al., 1999), their retinal morphology could not be analyzed postnatally.
The class IV POU-domain factor Pou4f2 is absent from the Wt1–/– retinas
The three closely related class IV POU-domain genes Pou4f1, Pou4f2 and Pou4f3 (formerly Brn-3a, Brn-3b and Brn-3c) are thought to exert distinct functions in the cell-fate specification of sensory neurons (Xiang et al., 1998). Selective loss of ∼70% of retinal ganglion cells and reduced optic nerve diameters were reported in mice that lack the transcription factor Pou4f2 (Erkman et al., 1996; Gan et al., 1996). To explore whether Wt1 is necessary for Pou4f2 expression in the developing retina, we performed immunohistochemistry on tissue sections from wild-type and Wt1–/– retinas at E18 (n = 5 each). A representative immunostaining of Pou4f2 is shown in Figure 4. In agreement with previous studies (Xiang et al., 1993; Gan et al., 1996; Xiang, 1998), Pou4f2 immunoreactivity was detected in the developing ganglion cell layer of normal E18 mice (Figure 4B). However, Pou4f2 was clearly absent from the Wt1–/– retinas at E18 (Figure 4E), and also could not be detected at earlier embryonic stages in the Wt1 mutant retinas (our unpublished observation). In situ labeling of Pou4f2 protein in the developing retinal ganglion cell layer was confirmed at the mRNA level. Thus, Pou4f2 transcripts were identified by RT–PCR in the E18 retinas of normal mice, but could not be amplified in retinal tissue obtained from Wt1–/– embryos at E18 (n = 5 each) (Figure 4H). For comparison, expression of two other POU-domain family members, Pou4f1 and Pou4f3, seemed normal in the Wt1 null mutant E18 retinas (Figure 4D and F). These observations suggest that Wt1 is required for the expression of Pou4f2 in the embryonic retina.

Fig. 4. Expression of Pou4f genes in retinal ganglion cells of wild-type (+/+) and Wt1 null mutant (–/–) retinas at E18. Negative immuno fluorescent staining of the Wt1–/– retina (E) indicates lack of expression of the class IV POU-domain transcription factor Pou4f2. RT–PCR demonstrates absence of Pou4f2 mRNA from the E18 retinas of Wt1–/– embryos (H). No differences are seen in Pou4f1 and Pou4f3 expression (mRNA and protein) between wild-type and Wt1–/– retinas. Shown are representative data obtained from five different embryos each. gcl, ganglion cell layer. Scale bars, 100 µm. A 100 bp DNA ladder was used to estimate the sizes of the expected PCR products. The predicted lengths of the amplified targets are 274 bp (Pou4f1), 300 bp (Pou4f2), 229 bp (Pou4f3) and 642 bp (β-actin), respectively.
WT1 activates the POU4F2 gene
Next we investigated whether Wt1 can directly activate the POU4F2 gene. For this purpose, we examined whether forced expression of Wt1 stimulates expression of the endogenous POU4F2 gene in cultured cells. Furthermore, it was tested whether the activity of the POU4F2 promoter is enhanced by co-expression of Wt1. A native Wt1-expressing cell line, which is derived from the neural retina, does not exist to our knowledge. We therefore made use of the previously established human embryonic kidney (HEK) 293 cell line to compare POU4F2 mRNA levels in cells that were stably transfected either with a Wt1(–KTS) expression construct or with an empty pCB6+ vector (Wagner et al., 2001). Real-time RT–PCR (Roche Molecular Biochemicals) was performed with total RNA isolated from four independent clones each of Wt1-expressing and non-expressing HEK293 cells. POU4F2 transcripts were normalized to GAPDH mRNA in each sample. Notably, HEK293 cells that stably expressed the Wt1(–KTS) isoform had on average 8-fold higher POU4F2 mRNA levels than the pCB6+-transfected cells (Figure 5A). We isolated ∼3.8 kb of the 5′-regulatory region of the human POU4F2 gene from a BAC clone (RZPD clone ID RPCIB753D12667Q2) and ligated it into the pGL2basic reporter plasmid (Figure 5B). Sequence analysis of this construct, which was designated phBrn3bUS3.8, revealed a high GC content and several predicted WT1 consensus binding sites in the 5′-upstream region of the human POU4F2 gene. Plasmid phBrn3bUS3.8 was transiently introduced into U2OS human osteosarcoma cells along with expression constructs encoding either of the Wt1(+KTS) and Wt1(–KTS) isoforms (Haber et al., 1991). The U2OS cell line has previously been used as a tool for identifying other candidate target genes of Wt1 (Lee et al., 1999). Basal activity of phBrn3bUS3.8 was enhanced >4-fold by the Wt1(–KTS) variant (Figure 5B). However, the Wt1(+KTS) isoform, which is thought to act at the post-transcriptional level (Englert et al., 1995; Larsson et al., 1995; Davies et al., 1998; Landomery et al., 1999), had no significant effect on reporter gene activity (Figure 5B). These findings show that the Wt1(–KTS) isoform can stimulate gene transcription from the putative POU4F2 promoter, suggesting that POU4F2 is a direct transcriptional downstream target gene of Wt1.
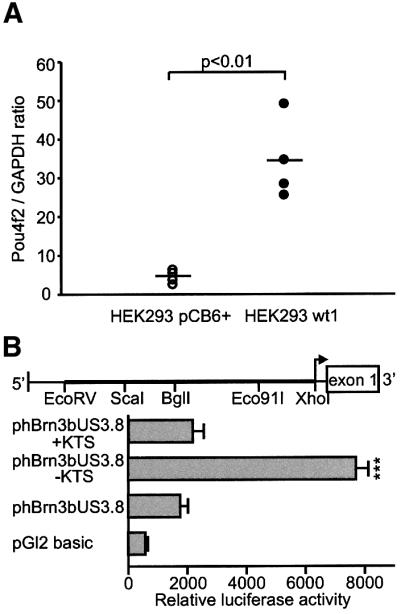
Fig. 5. (A) Expression of Pou4f2 in HEK293 cells stably transfected either with a Wt1(–KTS) expression construct or with the empty pCB6+ vector. Pou4f2 mRNA levels were quantified by real-time RT–PCR using the light cycler system (Roche Molecular Biochemicals) and normalized for GAPDH transcripts. Shown are the data obtained from four independent clones each of pCB6+ and Wt1(–KTS) transfected cells. Note that stable expression of Wt1(–KTS) increased Pou4f2 mRNA levels in HEK293 cells ∼8-fold. The horizontal bars indicate the mean values in each group. (B) Relative luciferase activities measured in the lysates of U2OS human osteosarcoma cells. U2OS cells were transiently co-transfected with phBrn3bUS3.8, Wt1 expression constructs encoding two different splice variants (+KTS/–KTS isoforms), and a cytomegalovirus-promoter driven β-galactosidase expression vector that was used for normalization of transfection efficiencies. Plasmid phBrn3bUS3.8 contained an ∼3.8 kb EcoRV–XhoI genomic sequence from the 5′-regulatory region of the human Pou4f2 gene (schematic drawing) in the pGL2 basic reporter vector. Values shown are means ± SEM of n = 7 experiments each performed in duplicate. P <0.05 was considered statistically significant (ANOVA).
Retinal defects of the Wt1–/– embryos are rescued by transgenic YAC complementation
Finally, we investigated whether transgenic delivery of a yeast artificial chromosome (YAC) carrying the human WT1 gene was able to rescue the ocular phenotype of the Wt1 null embryos. We have recently established YAC transgenic mouse lines expressing a LacZ reporter gene under the control of the human WT1 locus (Moore et al., 1998). LacZ expression in these lines recapitulated the endogenous pattern of Wt1 (Moore et al., 1998), including expression in the embryonic retina (A.W.Moore, personal communication). Transgenic lines were generated by pronuclear injection of amplified YAC DNA spanning 280 kb of the human WT1 locus (Moore et al., 1998). Notably, no retinal defects were visible in the WT280 transgenic embryos at E18 (Figure 6). In particular, Pou4f2 expression and optic nerve fiber growth were restored in the eyes of WT280–YAC transgenic embryos (Figure 6). Normal morphology of the WT280–YAC retinas is also reflected in a low number of apoptotic cells, which was not significantly different from that of Wt1+/+ retinas at stage E18 (Figure 3I).

Fig. 6. Rescue of retinal phenotype by YAC complementation of the human WT1 gene in Wt1–/– E18 embryos. Note that normal histology of the developing retina (A), as well as Pou4f2 immunoreactivity in the ganglion cell layer (B) and optic nerve fiber growth (C), are re-established by transgenic expression of a 280 kb YAC construct carrying the human WT1 gene in Wt1–/– embryos. The eyes of WT280– YAC transgenic embryos are indistinguishable by morphological means from their wild-type counterparts at stage E18. gcl, ganglion cell layer; onf, optic nerve fibers. Scale bars, 50 µm (A), 100 µm (B) and 200 µm (C), respectively.
Discussion
In this study, we demonstrate for the first time that the Wilms’ tumor gene Wt1 encodes a regulator of neuron development. The abnormalities in the Wt1–/– mutant retinas indicate that Wt1 is required for at least two critical functions during retinogensis: (i) proliferation of the progenitor cells; and (ii) development of the retinal ganglion cells.
Normal cell proliferation is one major determinant of retinal development. The reduced cell numbers and the weaker PCNA and BrdU stainings of the mutant retinas suggest that Wt1 is important for normal neuroblast division during early retinogenesis. Stimulation of cell proliferation by WT1 was demonstrated previously in hematopoietic cells (Yamagami et al., 1996). Thus, inhibition of endogenous WT1 by treatment with antisense oligonucleotides arrested K562 leukemia cells in the G2/M phase, suggesting that WT1 is critical for normal progression through the cell cycle (Yamagami et al., 1998). On the other hand, inducible expression of Wt1 in osteosarcoma cell lines triggered an initial G1 phase arrest that was followed by apoptosis (Englert et al., 1995a). These findings suggest that the effect of WT1 on cell proliferation and growth may be cell type specific and depend on additional co-factors that are differentially expressed between tissues. It is interesting to note that hypocellularity of the Wt1–/– retinas at E12 was less severe in embryos on a mixed MF1×B6 genetic background than in C57BL/6 (B6) mice (our unpublished observation). This observation is in agreement with our previous study on the role of Wt1 in spleen development, indicating that the penetrance of the Wt1–/– phenotype depends on the existence of one or more modifier gene(s) (Herzer et al., 1999).
Severe abnormalities were also found in the Wt1–/– retinas at later stages of development. Consistent with the expression of Wt1 in the inner portion of the Wt1+/+ retina, a significant fraction of cells were lost by apoptosis in the developing retinal ganglion cell layer of mutant E18 embryos. Defects in the remaining ganglion cells in the Wt1–/– retinas are indicated by the failure of these cells to give rise to normally growing optic nerve fiber bundles. This phenotype of the Wt1–/– retinas is reminiscent of the abnormalities seen in mice, which lack the POU-domain transcription factor Pou4f2 (also known as Brn-3b) (Erkman et al., 1996; Gan et al., 1996; Wang et al., 2000). In fact, several important findings of this study suggest that Pou4f2 is among the candidate genes that mediate the effects of Wt1 in retinogenesis. First, Pou4f2 (mRNA and protein) is lost in the Wt1–/– retinas. Secondly, Pou4f2 expression and the retinal defects are rescued by transgenic delivery of the human WT1 gene into Wt1–/– embryos. Thirdly, endogenous Pou4f2 transcripts in human embryonic kidney cells are up-regulated by forced expression of Wt1. And finally, Wt1 can transactivate the promoter of the human POU4F2 gene. Notably, the activity of the POU4F2 promoter was stimulated only by the Wt1(–KTS) isoform, which functions as a transcriptional regulator (reviewed in Lee and Haber, 2001). In contrast, the Wt1(+KTS) isoform, which is thought to be involved in post-transcriptional mRNA processing (Englert et al., 1995b; Larsson et al., 1995; Davies et al., 1998; Landomery et al., 1999), had no significant effect. These latter results identify POU4F2 as a novel candidate downstream target of the Wt1 transcription factor. Notably, we found that Pou4f2 and Wt1 proteins were co-expressed in the developing retina and also in glomerular podocytes of adult mouse kidney (our unpublished observation). The highly differentiated podocytes, which have been implicated in the glomerular filtration process, constitute the only cell type that expresses Wt1 in mature kidneys (Mundlos et al., 1993). Given this overlapping cellular distribution of the two proteins, it will be challenging to investigate whether interaction between Wt1 and Pou4f2 is important for the differentiation of non-neuronal tissues.
Even though the phenotype of the Wt1–/– and Pou4f2–/– retinas at stage E18 is remarkably similar, our findings raise several controversial issues that need to be addressed in more detail. For instance, the selective loss of ∼70% of retinal ganglion cells in the Pou4f2–/– retina is more severe than the 40% reduction in cell numbers that we observed in the ganglion cell layer of the Wt1–/– embryos at E18. This difference appears even more dramatic if one considers that not all cells in the ganglion cell layer of the developing retina are bona fide ganglion cells (reviewed in Cepko et al., 1996). In view of this seeming discrepancy, it has to be recalled that apoptotic cell death in the developing Pou4f2–/– retina was found to be variable with regard to its time of onset and duration. Thus, in one study using a lacZ reporter gene knocked into the mouse Pou4f2 locus to follow the fate of Pou4f2-mutant retinal cells, a significant increase in apoptosis was detected in mutant retinas from E15.5 to E18.5 (Gan et al., 1999). Postnatal changes in apoptotic cell death were not examined in this investigation. In another study, however, enhanced apoptosis in Pou4f2–/– retinas was found at E18.5, P0 and P1, indicating that premature cell death continued during the early postnatal period (Xiang, 1998). Since the Wt1–/– embryos normally die before end-gestation, their failure to express Pou4f2 in the retina may therefore not become fully effective in terms of reduced retinal ganglion cell survival.
Another question arising from our observations relates to the expression of Pou4f1 and Pou4f3 (formerly Brn-3a and Brn-3c), which seemed to be normal in the Wt1–/– retinas. For comparison, down-regulation of Pou4f1 and, in particular, Pou4f3 was reported in the Pou4f2–/– mutant retina (Erkman et al., 1996; Gan et al., 1996; Xiang et al., 1998). Furthermore, Pou4f1- and Pou4f2-expressing cells were largely coincident in wild-type retina, and most of the Pou4f3-positive cells also contained Pou4f1 and Pou4f2 (Xiang et al., 1995). Cross-regulation between the POU-domain factors is suggested from a recent study demonstrating enhanced Pou4f3 expression in the developing chick retina upon retroviral transfection of each of the three Pou4f genes (Liu et al., 2001). Comparing these seeming discrepancies to our results, one has to note that most, but by far not all, Pou4f1- and Pou4f3-expressing cells were absent from the Pou4f2–/– retinas at E18.5 (Erkman et al., 1996; Gan et al., 1996; Xiang, 1998). The latter finding suggests that a population of retinal cells may exist in which the expression of Pou4f1 and Pou4f3 is regulated independently of Pou4f2 (Erkman et al., 1996). Thus, it is conceivable that Pou4f1 and Pou4f3 expression was maintained in the Wt1–/– retinas by activation of Pou4f2-independent signaling pathways. One also has to consider that the Wt1–/– and Pou4f2–/– mutations were performed on mouse strains with different genetic backgrounds. Therefore, the variable influence of modifier gene(s) could account for the differences in Pou4f1 and Pou4f3 expression between the Wt1–/– and Pou4f2–/– retinas.
Our findings may also have implications for human genetic disease. The risk of Wilms’ tumor patients developing ocular disorders is increased (Nelson et al., 1984; Bickmore and Hastie, 1989). Aniridia and, in rare cases, optic nerve hypoplasia in these patients can result from inactivation of the aniridia gene PAX6, which lies telomeric of WT1 on chromosome 11p13 (Ton et al., 1991). As the transcriptional mechanisms underlying retinogenesis are conserved, at least in part, among species (for a review see Jean et al., 1998), our observations raise the possibility that some of the eye abnormalities in patients with WAGR mutations (Wilms’ tumor, aniridia, genitourinary malformations, mental retardation) may arise through effects on WT1 itself. Notably, the morphology of the retina was only disturbed in mice that had both Wt1 alleles inactivated, but not in heterozygous animals. Moreover, the total number of Wt1-expressing cells in the developing retina is relatively small compared with other organs (e.g. the kidneys), and the chance of inactivating WT1 mutations occurring is therefore relatively low.
In summary, this is the first study to demonstrate a requirement for the Wilms’ tumor gene Wt1 in neuronal development. Our findings indicate that Wt1 has multiple functions during formation of the vertebrate retina. During early retinogenesis, Wt1 is necessary for continuous proliferation of the progenitor cells; at the later stages, Wt1 rescues a subset of retinal ganglion cells from apoptosis and is critical for normal growth of the optic nerve. Finally, our results suggest that Wt1 can activate transcription of the POU-domain factor, Pou4f2, which mediates, at least in part, the effects of Wt1 on retinal ganglion cell survival and differentiation.
Materials and methods
Animals
A heterozygous (Wt1+/–) breeding pair [C57BL/6 (B6) strain] was obtained from The Jackson Laboratory (Bar Harbor, ME) and genotyped according to the protocol provided. The Wt1 mutation was crossed into the MF1×B6 genetic background as described previously (Herzer et al., 1999).
Histology and immunohistochemistry
Staged embryos (the morning of vaginal plug was considered E0) were fixed overnight at 4°C in paraformaldehyde [3% in phosphate-buffered saline (PBS)] and either embedded in paraffin for conventional histology or snap-frozen in pre-chilled isopentane and then embedded in Tissue-Tek® optimal cutting temperature (OCT) compound (Sakura Finetek) for immunohistochemical analysis. Notably, examination of tissue from Wt1–/– embryos revealed no gross pathological changes, indicating that the retinal cell defects were not simply due to an overall poor health of these animals. Four-micrometer tissue sections were cut and transferred onto gelatin-coated glass slides. Tissue sections were permeabilized with 0.1% Triton X-100 in PBS and blocked by incubation for 1 h in 10% normal goat serum [in PBS, 0.1% Triton X-100, 3% bovine serum albumin (BSA)]. Following treatment (16 h, 4°C) with primary antibody and 3 × 15 min washes in PBS, the slides were incubated for 1.5 h with biotinylated secondary antibodies (1:150 dilutions in PBS, 1% BSA; Vector Laboratories, Inc.) and streptavidin–Cy3 complex (Sigma). The slides were viewed under an epifluorescence microscope (Axioplan 2; Zeiss) connected to a digital camera (Spot RT™Slider, Diagnostic Instruments) using the Metamorph V4.1.2 software (Universal Imaging Corp.). The following primary antibodies were used: WT1 polyclonal antibody diluted 1:150 (C-19; cat.# sc-192; Santa Cruz Biotechnology), Pou4f1 monoclonal antibody diluted 1:150 (14A6; cat.# sc-8429; Santa Cruz Biotechnology), Pou4f2 polyclonal antibody diluted 1:150 (C-13; cat.# sc-6026; Santa Cruz Biotechnology), Pou4f3 polyclonal antibody diluted 1:150 (BabCO, Richmond, CA), NF200 polyclonal antibody diluted 1:150 (cat.# N 4142; Sigma), PCNA monoclonal antibody diluted 1:150 (PC10; cat.# sc-56; Santa Cruz Biotechnology), and BrdU polyclonal antibody diluted 1:150 (BMC9318; Roche Molecular Biochemicals).
Retinal ganglion cell counts
Ten-micrometer serial sections were made close to the optic disk level and stained with HE to estimate the number of cells in the developing ganglion cell layer of wild-type and Wt1 null mutant E18 retinas. Twenty retinal sections each obtained from five different Wt1+/+, Wt1–/– and WT280–YAC transgenic embryos were counted under the microscope (Axiovert S100; Zeiss) at 200-fold magnification.
In situ mRNA hybridization
Non-radioactive in situ mRNA hybridization was carried out on 10-µm paraformaldehyde (3% in PBS) fixed cryostat sections of staged mouse eyes as described by Braissant and Wahli (1998). Briefly, digoxigenin-labeled RNA probes were synthesized in vitro by transcription with T3 RNA polymerase (Roche Molecular Biochemicals) from BamHI-linearized plasmid pWt1-ISH. Plasmid pWt1-ISH contained the complete mouse Wt1 cDNA coding sequence (Buckler et al., 1991) on a 1.5 kb Sau3AI piece ligated into the blunt XbaI site of pBluescript II KS+. Sense RNA transcripts were generated using T7 RNA polymerase after linearization of pWt1-ISH with NotI. Hybridization was performed for 48 h at 58°C in a 50% formamide, 5× SSC solution supplemented with 60 µg/ml salmon sperm DNA (Promega). The tissue sections were washed at high stringency (1 h in 0.1× SSC at 65°C), and mRNA hybrids were detected by incubation with alkaline phosphatase-coupled anti-digoxigenin antibody and staining with NBT/BCIP (Roche Molecular Biochemicals). Stained tissue sections were viewed under an Axiovert S100 microscope (Zeiss).
Detection of apoptotic cells
Apoptotic cells were detected by TUNEL assay in the neural retinas of paraformaldehyde-fixed mouse embryos using the In Situ Cell Death Detection Kit (Roche Molecular Biochemicals). Five 10 µm retinal sections near the optic disk level were obtained from each animal to determine the number of apoptotic cells in Wt1+/+, Wt1 null mutant and WT280–YAC transgenic embryos. Five animals were studied in each group at the indicated time points (E12, E14, E18).
BrdU labeling and analysis
Timed pregnant mice were injected intraperitoneally with a single dose of BrdU (5 mg/100 g body weight; Roche Molecular Biochemicals). The E12 embryos were removed after 3 h from ether-anesthetized animals and fixed overnight at 4°C in a 3% paraformaldehyde–PBS solution. BrdU incorporation was detected by immunofluorescent staining of cryostat sections using anti-BrdU antibody at 1:150 dilution (BMC9318; Roche Molecular Biochemicals) according to the protocol described above.
RT–PCR and real-time RT–PCR
First strand cDNA synthesis was performed with 2 µg of total RNA using oligo(dT) primers and superscript II reverse transcriptase (Life Technologies). One-tenth of the reaction product was used for PCR amplification carried out in a thermal cycler (Biometra, UNO-II, Göttingen, Germany): DNA denaturing at 94°C, primer annealing at 55°C, primer extension at 72°C. The following gene-specific oligonucleotide primers were used for PCR amplification: Pou4f1, 5′-CATGATCGCGCTCAAGCCCAT-3′ (forward primer), 5′-CCGCTTCTGCTTCTGTCTCTG-3′ (reverse primer); Pou4f2, 5′-GCAATATATTCGGCGGGCTGG-3′ (forward primer), 5′-TTAGGTGCTCAAGCAGCTCGC-3′ (reverse primer); Pou4f3, 5′-GAAGCTTTGATGAGAGCCTGC-3′ (forward primer), 5′-GATGTGCTCAAGTAAGTCGCC-3′ (reverse primer); β-actin, 5′-ACCAAGGTGTGATGGTGGGAATG-3′ (forward primer), 5′-CGCTCGTTGCCAATAGTGATG-3′ (reverse primer). The expected PCR products had lengths of 274 bp (Pou4f1), 300 bp (Pou4f2), 229 bp (Pou4f3) and 642 bp (β-actin), respectively. The number of PCR cycles was adjusted to 28 to be within the exponential phase of the PCR. Real-time RT–PCR was performed with the use of the light cycler system (Roche Molecular Biochemicals) as described in detail elsewhere (Hammes et al., 2001). POU4F2 mRNA obtained from the HEK293 cell line was amplified after reverse transcription using the following oligonucleotide primers: 5′-GGAGAAGAAGCGCAAGC-3′ (forward primer); 5′-TCTGGAGAGGCCAAGAGTC-3′ (reverse primer). For normalization of POU4F2 transcripts, the housekeeping gene GAPDH was co-amplified in each sample: 5′-AACAGCGACACCCACTCCTC-3′ (forward primer); 5′-GGAGGGGAGATTCAGTGTGGT-3′ (reverse primer).
Cloning of the 5′-regulatory region of the human POU4F2 gene
A bacterial artificial chromosome (BAC) spanning the human POU4F2 gene (NCBI accession No. AC027193) was obtained from the German Resource Center for Genome Research (RZPD clone ID RPCIB753D12667Q2). The Escherichia coli DH10B host strain was grown overnight at 37°C in LB liquid medium supplemented with 20 µg/ml chloramphenicol. The BAC DNA was isolated by alkaline lysis and subsequent purification according to the manufacturer’s protocol. Twenty micrograms of plasmid DNA were double digested with XhoI and EcoRV, separated on a 0.8% agarose gel, and blotted onto positively charged nylon membrane (Roche Molecular Biochemicals) with 10× SSC as a transfer buffer. Southern hybridization was performed overnight at 65°C with a 32P-labeled DNA probe (PrimeIt II labeling kit; Stratagene) as described previously (Scholz et al., 1997). The probe consisted of a 387 bp sequence located ∼2.4 kb upstream of the predicted transcription start site in the human POU4F2 gene. The following PCR primers were used for generation of the probe: 5′-ACACAG TGAAGCGCGGTGTCT-3′ (forward primer); 5′-GTAGCCGAAGGG AACTCCTAG-3′ (reverse primer). The 32P-labeled probe hybridized specifically with an ∼3.8 kb EcoRV–XhoI genomic DNA fragment, which was identified by dideoxy sequencing of both strands, and blunt-end ligated into the XhoI restriction site of the pGL2basic reporter plasmid (Promega). This construct was designated phBrn3bUS3.8.
Cell transfections and reporter gene assays
Three micrograms of the phBrn3bUS3.8 reporter construct together with Wt1 expression vectors (16 µg) and 1 µg of a cytomegalovirus-driven β-galactosidase plasmid were transiently co-transfected into U2OS osteosarcoma cells using the calcium phosphate precipitation technique (Gorman et al., 1982). The Wt1 expression constructs contained cDNA encoding two alternatively spliced variants (+KTS/ –KTS isoforms) in the pCB6+ plasmid (Haber et al., 1991). U2OS cells were grown to 60% confluence on 100 mm dishes in Dulbecco’s modified Eagle’s medium (Life Technologies) supplemented with 10% fetal calf serum (Biochrom KG), 100 IU/ml penicillin (Life Technologies) and 100 µg/ml streptomycin (Life Technologies). The cells were lysed 48 h after the transfections, and luciferase reporter gene activities were measured in a luminometer (Microlite TLX1; Dynatech Laboratories Inc.) with beetle luciferin as substrate (Promega). β-galactosidase activities were determined spectrophotometrically (Beckman DU 540 spectrophotometer) using a commercial kit according to the manufacturer’s instructions (Promega). Values are given as relative light units normalized to β-galactosidase activities as an internal control for transfection efficiencies. Data shown are means ± SEM of n = 7 experiments each performed in duplicate. P <0.05 was considered statistically significant (ANOVA).
Generation and molecular characterization of YAC transgenic mice
The YAC construct WT280, which was used for complementation of the ocular defects in Wt1–/– mice, is described in detail elsewhere (Menke et al., 1998). WT280 contained the entire human WT1 locus (∼50 kb) flanked by ∼220 kb upstream and ∼10 kb downstream sequence on either side. This construct was generated by truncation of a larger 470 kb clone (WT470) at a position ∼10 kb downstream of the WT1 termination site (Menke et al., 1998). Transgenic lines were obtained by injection of amplified WT280–YAC DNA into pronuclei of fertilized mouse oocytes as described (Schedl et al., 1996). Positive lines were identified by PCR and Southern blot analysis (Moore et al., 1999).
Acknowledgments
Acknowledgements
The expert technical assistance of I.Grätsch and A.Richter, as well as the help of J.Ward and N.Howells from the animal facility in Karlsruhe is gratefully acknowledged. We thank Dr D.Haber for the Wt1 expression constructs. We are grateful to Drs R.Grantyn, A.Rodriguez-Tébar, D.Engelkamp and R.E.Anderson for their critical reading of the manuscript. Retinal tissue specimens obtained at autopsy of a 19-week-gestation human embryo were kindly provided by Dr P.Bovolenta. This study was supported by grants from the Deutsche Forschungs gemeinschaft (grant no. Scho 634/3-1 to H.S., EN 280/2-4 to C.E. and Sche 5641/1 to A.S.).
References
- Armstrong J.F., Pritchard-Jones,K., Bickmore,W.A., Hastie,N.D. and Bard,J.B. (1993) The expression of the Wilms’ tumor gene, WT1, in the developing mammalian embryo. Mech. Dev., 40, 85–97. [DOI] [PubMed] [Google Scholar]
- Bennington J. and Beckwith,J. (1975) Tumors in the kidney, renal pelvis and ureter. In Atlas of Tumor Pathology. Series 2, fascile 12. Armed Forces Institute of Pathology, Washington, DC.
- Bickmore W.A. and Hastie,N.D. (1989) Aniridia, Wilms’ tumor and human chromosome 11. Ophthalmic Paediatr. Genet., 10, 229–248. [DOI] [PubMed] [Google Scholar]
- Braissant O. and Wahli,W. (1998) A simplified in situ hybridization protocol using non-radioactively labeled probes to detect abundant and rare mRNAs on tissue sections. Biochemica, 1, 10–16. [Google Scholar]
- Buckler A.J., Pelletier,J., Haber,D.A., Glaser,T. and Housman,D.E. (1991) Isolation, characterization and expression of the murine Wilms’ tumor gene (WT1) during kidney development. Mol. Cell. Biol., 11, 1707–1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cepko C.L., Austin,C.P., Yang,X., Alexiades,M. and Ezzeddine,D. (1996) Cell fate determination in the vertebrate retina. Proc. Natl Acad. Sci. USA, 93, 589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies R.C., Calvio,C., Bratt,E., Larsson,S.H., Lamond,A.I. and Hastie,N.D. (1998) WT1 interacts with the splicing factor U2AF65 in an isoform-dependent manner and can be incorporated into spliceosomes. Genes Dev., 12, 3217–3225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Englert C., Hou,X., Maheswaran,S., Bennett,P., Ngwu,C., Re,G.G., Garvin,A.J., Rosner,M.R. and Haber,D.A. (1995a) WT1 suppresses synthesis of the epidermal growth factor receptor and induces apoptosis. EMBO J., 14, 4662–4675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Englert C., Vidal,M., Maheswaran,S., Ge,Y., Ezzel,R., Isselbacher,K.J. and Haber,D.A. (1995b) Truncated WT1 mutants alter the subnuclear localization of the wild-type protein. Proc. Natl Acad. Sci. USA, 92, 11960–11964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Englert C., Maheswaran,S., Garvin,A.J., Kreidberg,J. and Haber,D.A. (1997) Induction of p21 by the Wilms’ tumor suppressor gene WT1. Cancer Res., 57, 1429–1434. [PubMed] [Google Scholar]
- Erkman K. et al. (1996) Role of transcription factors Brn-3.1 and Brn-3.2 in auditory and visual system development. Nature, 381, 603–606. [DOI] [PubMed] [Google Scholar]
- Gan K., Wang,S.W., Huang,Z. and Klein,W.H. (1999) POU domain factor Brn-3b is essential for retinal ganglion cell differentiation and survival but not for initial cell fate specification. Dev. Biol., 210, 469–480. [DOI] [PubMed] [Google Scholar]
- Gan L., Xiang,M., Zhou,L., Wagner,D.S., Klein,W.H. and Nathans,J. (1996) POU domain factor Brn-3b is required for the development of a large set of retinal ganglion cells. Proc. Natl Acad. Sci. USA, 93, 3920–3925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorman C., Moffat,L.F. and Howard,B.H. (1982) Recombinant genomes which express chloramphenicol acetyltransferase in mammalian cells. Mol. Cell. Biol., 2, 1044–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haber D.A., Sohn,R.L., Buckler,A.J., Pelletier,J., Call,K.M. and Housman,D.E. (1991) Alternative splicing and genomic structure of the Wilms tumor gene WT1. Proc. Natl Acad. Sci. USA, 88, 9618–9622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammes A., Guo,J.K., Lutsch,G., Leheste,J.R., Landrock,D., Ziegler,U., Gubler,M.C. and Schedl,A. (2001) Two splice variants of the Wilms’ tumor 1 gene have distinct functions during sex determination and nephron formation. Cell, 106, 319–329. [DOI] [PubMed] [Google Scholar]
- Hastie N.D. (1994) The genetics of Wilms’ tumor—a case of disrupted development. Annu. Rev. Genet., 28, 523–558. [DOI] [PubMed] [Google Scholar]
- Herzer U., Crocoll,A., Barton,D., Howells,N. and Englert,C. (1999) The Wilms tumor suppressor gene Wt1 is required for development of the spleen. Curr. Biol., 9, 837–840. [DOI] [PubMed] [Google Scholar]
- Jean D., Ewan,K. and Gruss,P. (1998) Molecular regulators involved in vertebrate eye development. Mech. Dev., 76, 3–18. [DOI] [PubMed] [Google Scholar]
- Kreidberg J.A., Sariola,H., Loring,J.M., Maeda,M., Pelletier,J., Housman,D. and Jaenisch,R. (1993) WT-1 is required for early kidney development. Cell, 74, 679–691. [DOI] [PubMed] [Google Scholar]
- Landomery M.R., Slight,J., McGhee,S. and Hastie,N.D. (1999) Presence of WT1, the Wilms’ tumor suppressor gene product, in nuclear poly(A)+ ribonucleoprotein. J. Biol. Chem., 274, 36520–36526. [DOI] [PubMed] [Google Scholar]
- Larsson S.H., Charlieu,J.P., Miyagawa,K., Engelkamp,D., Rassoulzadegan, M., Ross,A., Cuzin,F., van Heyningen,V. and Hastie,N.D. (1995) Subnuclear localization of WT1 in splicing or transcription factor domains is regulated by alternative splicing. Cell, 81, 391–401. [DOI] [PubMed] [Google Scholar]
- Lee S.B. and Haber,D.A. (2001) Wilms tumor and the WT1 gene. Exp. Cell Res., 264, 74–99. [DOI] [PubMed] [Google Scholar]
- Lee S.B. et al. (1999) The Wilms’ tumor suppressor WT1 encodes a transcriptional activator of amphiregulin. Cell, 98, 663–673. [DOI] [PubMed] [Google Scholar]
- Liu W., Mo,Z. and Xiang,M. (2001) The Ath5 proneural genes function upstream of Brn3 POU domain transcription factor genes to promote retinal ganglion cell development. Proc. Natl Acad. Sci. USA, 98, 1649–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsunaga E. (1981) Genetics of Wilms’ tumor. Hum. Genet., 57, 231–246. [DOI] [PubMed] [Google Scholar]
- Maurer U., Jehan,F., Englert,C., Hubinger,G., Weidmann,E., DeLuca,H.F. and Bergmann,L. (2001) The Wilms’ tumor gene product (WT1) modulates the response to 1,25-dihydroxyvitamin D3 by induction of the vitamin D receptor (VDR). J. Biol. Chem., 276, 3727–3732. [DOI] [PubMed] [Google Scholar]
- Menke A., McInnes,L., Hastie,N.D. and Schedl,A. (1998) The Wilms’ tumor suppressor WT1: approaches to gene function. Kidney Int., 53, 1512–1518. [DOI] [PubMed] [Google Scholar]
- Moore A., Schedl,A., McInnes,L., Doyle,M., Hecksher-Sorensen,J. and Hastie,N.D. (1998) YAC analysis reveals Wilms’ tumour 1 gene activity in the proliferating coelomic epithelium, developing diaphragm and limb. Mech. Dev., 79, 169–184. [DOI] [PubMed] [Google Scholar]
- Moore A.W., McInnes,L., Kreidberg,J., Hastie,N.D. and Schedl,A. (1999) YAC complementation shows a requirement for Wt1 in the development of epicardium, adrenal gland and throughout nephrogenesis. Development, 126, 1845–1857. [DOI] [PubMed] [Google Scholar]
- Mundlos S., Pelletier,J., Darveau,A., Bachmann,M., Winterpacht,A. and Zabel,B. (1993) Nuclear localization of the protein encoded by the Wilms’ tumor gene WT1 in embryonic and adult tissues. Development, 119, 1329–1341. [DOI] [PubMed] [Google Scholar]
- Nelson L.B., Spaeth,G.L., Nowinski,T.S., Margo,C.E. and Jackson,L. (1984) Aniridia. A review. Surv. Ophthalmol., 28, 621–642. [DOI] [PubMed] [Google Scholar]
- Palmer R.E., Kotsianti,A., Cadman,B., Boyd,T., Gerald,W. and Haber,D.A. (2001) Wt1 regulates the expression of the major glomerular podocyte membrane protein Podocalyxin. Curr. Biol., 11, 1805–1809. [DOI] [PubMed] [Google Scholar]
- Pelletier J., Schalling,M., Buckler,A., Rogers,A., Haber,D.A. and Housman,D. (1991) Expression of the Wilms’ tumor gene Wt1 in the murine urogenital system. Genes Dev., 5, 1345–1356. [DOI] [PubMed] [Google Scholar]
- Pritchard-Jones K. et al. (1990) The candidate Wilms’ tumour gene is involved in genitourinary development. Nature, 346, 194–197. [DOI] [PubMed] [Google Scholar]
- Rackley R.R., Flenniken,A.M., Kuriyan,N.P., Kessler,P.M., Stoler,M.H. and Williams,B.R. (1993) Expression of the Wilms’ tumor suppressor gene Wt1 during mouse embryogenesis. Cell Growth Differ., 4, 1023–1031. [PubMed] [Google Scholar]
- Rauscher F.J. III (1993) The WT1 Wilms tumor gene product: a developmentally regulated transcription factor in the kidney that functions as a tumor suppressor. Adv. Exp. Med. Biol., 348, 23–29. [PubMed] [Google Scholar]
- Schedl A., Ross,A., Lee,M., Engelkamp,D., Rashbass,P., van Heyningen,V. and Hastie,N.D. (1996) Influence of PAX6 gene dosage on development: overexpression causes severe eye abnormalities. Cell, 86, 71–82. [DOI] [PubMed] [Google Scholar]
- Scholz H., Bossone,S.A., Cohen,H.T., Akella,U., Strauss,W.M. and Sukhatme,V.P. (1997) A far upstream cis-element is required for Wilms’ tumor-1 (WT1) gene expression in renal cell culture. J. Biol. Chem., 272, 32836–32846. [DOI] [PubMed] [Google Scholar]
- Sidman R.L. (1961) The development of the vertebrate retina: a comparative survey. In Smelser,G.K. (ed.), The Structure of the Eye. Academic Press, New York, NY, pp. 403–420.
- Ton C.C. et al. (1991) Positional cloning and characterization of a paired box- and homeobox-containing gene from the aniridia region. Cell, 67, 1059–1074. [DOI] [PubMed] [Google Scholar]
- Wagner K.D., Wagner,N., Sukhatme,V.P. and Scholz,H. (2001) Activation of vitamin D receptor by the Wilms’ tumor gene product mediates apoptosis of renal cells. J. Am. Soc. Nephrol., 12, 1188–1196. [DOI] [PubMed] [Google Scholar]
- Wang S.W., Gan,L., Martin,S.E. and Klein,W.H. (2000) Abnormal polarization and axon outgrowth in retinal ganglion cells lacking the POU-domain transcription factor Brn-3b. Mol. Cell. Neurosci., 16, 141–156. [DOI] [PubMed] [Google Scholar]
- Xiang M. (1998) Requirement for Brn-3b in early differentiation of postmitotic retinal ganglion cell precursors. Dev. Biol., 197, 155–169. [DOI] [PubMed] [Google Scholar]
- Xiang M., Zhou,L., Peng,Y.W., Eddy,R.L., Shows,T.B. and Nathans,J. (1993) Brn-3b: a POU domain gene expressed in a subset of retinal ganglion cells. Neuron, 11, 689–701. [DOI] [PubMed] [Google Scholar]
- Xiang M., Zhou,L., Macke,J.P., Yoshioka,T., Hendry,S.H.C., Eddy,R.L., Shows,T.B. and Nathans,J. (1995) The Brn-3 family of POU-domain factors: primary structure, binding specificity and expression in subsets of retinal ganglion cells and somatosensory neurons. J. Neurosci., 15, 4762–4785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang M., Gan,L., Li,D., Zhou,L., Chen,Z.-Y., Wagner,D., O’Malley,B.W., Klein,W. and Nathans,J. (1998) Role of the Brn-3 family of POU-domain genes in the development of the auditory/vestibular, somatosensory and visual systems. Cold Spring Harb. Symp. Quant. Biol., 62, 325–335. [PubMed] [Google Scholar]
- Yamagami T. et al. (1996) Growth inhibition of human leukemic cells by WT1 (Wilms’ tumor gene) antisense oligonucleotides: implications for the involvement of WT1 in leukemogenesis. Blood, 88, 2267–2278. [PubMed] [Google Scholar]
- Yamagami T. et al. (1998) Suppression of Wilms’ tumor gene (WT1) expression induces G2/M arrest in leukemic cells. Leukemia Res., 22, 383–384. [DOI] [PubMed] [Google Scholar]