

Abstract
Analysis of multisubunit RNA polymerase (RNAP) structures revealed several elements that may constitute the enzyme’s functional sites. One such element, the ‘rudder’, is formed by an evolutionarily conserved segment of the largest subunit of RNAP and contacts the nascent RNA at the upstream edge of the RNA–DNA hybrid, where the DNA template strand separates from the RNA transcript and re-anneals with the non-template strand. Thus, the rudder could (i) maintain the correct length of the RNA–DNA hybrid; (ii) stabilize the nascent RNA in the complex; and (iii) promote or maintain localized DNA melting at the upstream edge of the bubble. We generated a recombinant RNAP mutant that lacked the rudder and studied its properties in vitro. Our results demonstrate that the rudder is not required for establishment of the upstream boundary of the transcription bubble during promoter complex formation, nor is it required for separation of the nascent RNA from the DNA template strand or transcription termination. Our results suggest that the rudder makes critical contributions to elongation complex stability through direct interactions with the nascent RNA.
Keywords: localized DNA melting/RNA polymerase/transcription complex/transcription complex stability/transcription termination
Introduction
The family of multisubunit RNA polymerases (RNAPs) includes eubacterial and chloroplast-encoded RNAPs, archeal RNAPs, eukaryal nuclear RNAPs, as well as RNAPs encoded by some eukaryal viruses such as vaccinia virus. All members of the family are related through common ancestry, share a common core subunit composition (α2ββ′ω), have similar structures and probably share many mechanistic features (reviewed by Ebright, 2000).
During transcription, DNA strands are separated in a short area around the RNAP catalytic center, forming the so-called transcription bubble. Since Watson–Crick interactions are required for template-dependent RNA synthesis, there should exist an RNA–DNA hybrid inside the transcription bubble during transcript elongation. The hybrid is transient, and the nascent RNA is displaced from the DNA template and the DNA strands re-anneal to form double-stranded upstream DNA. DNA–RNA cross-links in stalled elongation complexes suggest that the hybrid is 8–9 bp long (Nudler et al., 1997; Korzheva et al., 1998). The structural elements of RNAP that are responsible for local strand separation and for the maintenance of the proper length of the hybrid are not known.
Recent years have seen a tremendous increase in the amount of structural information about multisubunit RNAPs. An atomic structural model of catalytically active RNAP core enzyme from the thermophilic bacterium Thermus aquaticus revealed a molecule with a ‘crab-claw’ shape, with a central mass formed by residues of β′, β and the α dimer, and two prominent jaw-like projections formed by residues of β′ and β (Zhang et al., 1999; Figure 1B). The channel formed by the jaws has dimensions appropriate to accommodate double-stranded template DNA. Superposition of structural data from T.aquaticus RNAP and RNA– and DNA–RNAP cross-links obtained using Escherichia coli RNAP transcription complexes has made it possible to generate a structural model of the transcription elongation complex (Korzheva et al., 2000), and of the open promoter complex (Naryshkin et al., 2000). These models orient RNAP with respect to both the DNA and the RNA, and suggest specific functional roles for several elements of the structure. Atomic structures of yeast RNAP II (Cramer et al., 2001) and of an artificial elongation complex formed by yeast RNAP II (Gnatt et al., 2001) revealed an astonishing degree of structural similarity between prokaryotic and eukaryotic RNAPs (Ebright, 2000) and validated the modeling results of Korzheva et al. (2000).

Fig. 1. Genetic and structural context of the RNAP rudder. (A) The bar at the top represents the largest RNAP subunit. The lettered boxes indicate evolutionarily conserved segments (Allison et al., 1985), the white box represents a long insertion in T.aquaticus β′ that is absent from the E.coli β′. The T.aquaticus segment C sequence (T. a.) is expanded underneath and is aligned with the corresponding segments from E.coli (E. c.), and yeast RNAP II (Yp2). The deletion studied in this work and the two amino acid linker (EL) inserted instead of the deleted material are shown above the T.aquaticus sequence. (B) The structure of T.aquaticus RNAP core enzyme (Zhang et al., 1999). The view on the left panel shows the ‘downstream face’ of the enzyme and is roughly parallel to the axis of the DNA-binding channel of the enzyme. Right panel: the view on the left was rotated 90° clockwise about the vertical axis. β′ is in green, β in cyan, α2 in white, ω in gray. The active center Mg2+ is in magenta. The β′ rudder and the coiled-coil element from which the rudder emanates are indicated. (C) The position of the β′ rudder according to the structural model of the bacterial RNAP elongation complex (Korzheva et al., 2000). A stereo view of the rudder area (roughly corresponding to the orientation shown in B, right panel) is shown. RNA is in red, template DNA strand in yellow, non-template DNA in orange (only the upstream portion of non-template DNA is shown, for clarity) and the active-center Mg2+ in magenta. The coiled-coil element is shown in gray and the rudder in green. Two evolutionarily conserved arginine residues of the rudder are indicated and are shown in spacefill representation. Conserved arginine residues located C-terminal to the rudder (A) are indicated in cyan. (D) The position of the rudder in the structure of the artificial elongation complex formed by yeast RNAP II (Gnatt et al., 2001). A stereo view of the relevant section of the yeast RNAP II elongation complex is shown. The orientation and labeling correspond to those in (C). A portion of the rudder is not resolved on this structure.
One noteworthy structural element of T.aquaticus RNAP and yeast RNAP II, a loop-like structure that protrudes upwards into the main channel, is comprised of the conserved segment C of the largest subunit and is called the ‘rudder’ (Zhang et al., 1999; Gnatt et al., 2001; Figure 1). In E.coli RNAP elongation complexes, cross-links from the –8/–9 positions of the nascent RNA to the largest subunit, β′, were localized to amino acids of conserved segment C (Korzheva et al., 2000). Structural analysis of the yeast RNAP II artificial elongation complex also shows that the rudder makes direct contacts with the nascent RNA nine bases upstream of the 3′ end (Gnatt et al., 2001; Figure 1D).
In the structural model of the prokaryotic RNAP elongation complex, the rudder has been placed at the upstream edge of the RNA–DNA hybrid, where the DNA template strand separates from the RNA transcript and re-anneals with the non-template strand (Figure 1C; Zhang et al., 1999; Korzheva et al., 2000; see also Naryshkin et al., 2000). Although the models of both Korzheva et al. and Naryshkin et al. propose contact between the rudder and nascent RNA, the two models differ in proposed contacts between the rudder and DNA (due to a difference in modeled positions of the upstream DNA duplex). The model of Korzheva et al. predicts that the rudder contacts the upstream separation point of the DNA non-template and template strands. The model of Naryshkin et al. predicts that the rudder does not contact the upstream separation point of the DNA non-template and template strands.
Based on its location in the transcription complex, the rudder could (i) initiate and/or maintain localized DNA melting at the upstream boundary of the transcription bubble, and prevent transcription bubble collapse (Korzheva et al., 2000), (ii) determine the length of the RNA–DNA hybrid by peeling the RNA away from the hybrid (Korzheva et al., 2000) or (iii) stabilize the nascent RNA in the elongation complex by direct protein–RNA interactions (Korzheva et al., 2000; Naryshkin et al., 2000). Various combinations of these mechanisms are also possible. The following phenotypes are therefore expected for an RNAP mutant with lesions in the rudder area. (i) There may be defects in the initiation or maintenance of the transcription bubble during both transcription initiation and transcription elongation. (ii) During transcription elongation, a persistent RNA–DNA hybrid may form. (iii) The stability of the nascent RNA in the elongation complex could decrease due to the lack of favorable protein–RNA contacts.
Here, we tested these predictions experimentally by removing the rudder from recombinant T.aquaticus RNAP β′ and analyzing the behavior of the mutant enzyme in discriminative in vitro transcription assays. Our results indicate that the most significant role of the rudder is to stabilize the nascent RNA in the RNAP elongation complex through protein–RNA interactions.
Results
Construction of the T.aquaticus RNAP mutant
The β′ rudder is part of the evolutionarily conserved segment C of the largest RNAP subunit (Figure 1A; Allison et al., 1985). At the left-hand side, the rudder is preceded by the segment C sequence that forms the so-called coiled-coil element that in bacterial RNAPs constitutes the primary binding site for promoter specificity subunit σ (Figure 1; Burgess et al., 1998). On the left-hand side, the rudder is followed by a stretch of segment C amino acids which contain several conserved positively charged amino acids (Figure 1A, indicated by cyan in Figure 1C and D). These residues may be involved in protein–nucleic acid interactions with the template DNA strand downstream of the catalytic center (Figure 1D; Gnatt et al., 2001).
Recently, we described a system for production of recombinant T.aquaticus RNAP by co-expressing T.aquaticus rpo genes from an E.coli plasmid (Minakhin et al., 2001). We used this system to remove site-specifically rpoC codons 585–601, that constitute the β′ rudder (Figure 1A; Zhang et al., 1999). To bridge the ∼6 Å gap between the points of the rudder attachment to the coiled-coil element, we introduced a two amino acid linker, LeuGlu, instead of 17 rudder amino acids removed by the deletion. The mutant enzyme was purified from overexpressing E.coli cells, and its properties were compared with control recombinant wild-type T.aquaticus RNAP in a panel of in vitro transcription assays designed to test the predictions of the structural model of the transcription elongation complex (Zhang et al., 1999; Korzheva et al., 2000).
Promoter complex formation by mutant RNAP
The mutant and the wild-type T.aquaticus core enzymes were combined with the recombinant T.aquaticus σ subunit (Minakhin et al., 2001), and the resultant holoenzymes were tested in the abortive transcription initiation assay on the A1 and A2 promoters of bacteriophage T7, which are strong promoters recognized by E.coli σ70 holoenzyme. The mutant enzyme was completely inactive in the abortive synthesis of CpApU from the CpA primer and UTP substrate on the T7 A1 promoter-containing DNA fragment (Figure 2A, lane 2) and was also inactive in abortive synthesis of GpCpU from GpC and UTP on T7 A2 (data not shown). In contrast, the wild-type enzyme was highly active on both promoters (Figure 2A, lane 1, and data not shown), as expected (Minakhin et al., 2001).

Fig. 2. Thermus aquaticus RNAP mutants harboring lesions in the β′ rudder form inactive promoter complexes. (A) Abortive transcription by the wild-type and mutant T.aquaticus RNAPs. The indicated enzymes were combined with the T7 A1 promoter-containing DNA fragment in the presence of CpA primer and [α-32P]UTP substrate. The reaction proceeded for 15 min at 65°C, and reaction products were resolved by denaturing PAGE and revealed by autoradiography. (B) Promoter complexes formed by the T.aquaticus rudder mutant are partially melted and are similar to complexes formed by E.coli RNAP with lesions in β dispensable region 1. A 170 bp DNA fragment containing the T7 A2 promoter (–83 to +87) 32P end-labeled on the bottom strand was combined with the indicated enzymes. Reactions were pre-incubated for 15 min at 37°C (E.coli RNAP), or for 15 min at 65°C (T.aquaticus RNAP), and probed with KMnO4. Reaction products were resolved on a 6% sequencing gel and visualized by autoradiography. On the right, phosphoimager traces of lanes 1, 2, 3 and 4 are presented. (C) Deletion of the rudder does not stabilize promoter complexes at suboptimal temperatures. The T7 A2 promoter complexes were formed at temperatures ranging from 5 to 75°C with 10°C increments and probed with KMnO4. Reaction products were analyzed as above.
To determine whether the mutant enzyme forms inactive open promoter complexes, we used KMnO4 probing, which is specific for thymines in single-stranded (i.e. melted) DNA. In open complexes formed by the wild-type T.aquaticus RNAP on the T7 A2 promoter, thymines at –12, –11, –9, –7, –5, –4 and –3 were modified by KMnO4 (Figure 2B, lane 3). The wild-type T.aquaticus complexes probed at 65°C were indistinguishable from E.coli RNAP complexes probed at 37°C (Figure 2B, lanes 3 and 1), indicating that the architecture of promoter compexes is conserved in evolution (Minakhin et al., 2001). In contrast, in promoter complexes formed by the mutant, rudderless enzyme, downstream thymines were relatively resistant, and only positions –12, –11, –9 and –7 were modified by KMnO4 (Figure 2B, lane 4). Previously, we characterized E.coli RNAP mutants with lesions in the N-terminal dispensable region 1 of the second largest β subunit (Severinov and Darst, 1997; Nechaev et al., 2000). The mutant enzymes formed promoter complexes that were shortened in the downstream direction, but were able to maintain DNA strand separation at temperatures as low as 0°C, when the wild-type complexes were fully closed. Comparison of T.aquaticus mutant complexes with complexes formed by one of the E.coli mutants, Δ(186–433), revealed that patterns of KMnO4 modification were indeed similar: there was little or no modification of downstream thymines –5, –4 and –3, and the extent of modification at –11 was increased, compared with the wild-type complexes (Figure 2B, lanes 2 and 4). However, the extent of modification of thymine at position –7 was significantly lower in mutant E.coli RNAP complexes. Moreover, the mutant T.aquaticus RNAP complexes were as sensitive to a decrease in the reaction temperature as the wild-type complexes (Figure 2C). Be that as it may, we conclude that during promoter complex formation, initial promoter melting or distortion in the upstream portion of the –10 box can occur even in the absence of the β′ rudder.
Mutant RNAP is active on mismatched bubble promoter templates
In order to study the consequences of rudder removal on RNAP function, we needed to find conditions where the mutant enzyme is catalytically active. The results of KMnO4 probing suggested that the catalytic deficiency of the T.aquaticus RNAP mutant could be explained by the inability to propagate localized DNA melting all the way towards the transcription initiation start point (deHaseth and Helmann, 1995). If this were the case, the mutant enzyme could be made functional on artificial templates containing pre-formed transcription bubbles. To test this idea, promoter complexes were formed on the T7 A1 promoter-based templates containing a mismatched segment spanning positions –12 to +1, i.e. the entire transcription bubble length, as well as on two templates containing shorter mismatched bubbles, from –12 to –6 (‘upstream bubble’) and from –7 to +1 (‘downstream bubble’). Steady-state transcription reactions were performed with mismatched bubble templates and the fully double-stranded control template, and the results are presented in Figure 3. As can be seen, the rudderless mutant displayed substantial levels of RNA synthesis on templates containing the complete bubble and the downstream bubble. We conclude that the mutant, rudderless, RNAP is catalytically proficient and that in the absence of the β′ rudder the propagation of localized DNA melting downstream of the –10 promoter box does not occur.
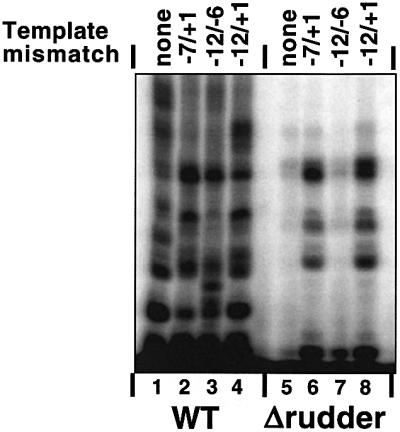
Fig. 3. Transcription by T.aquaticus RNAP mutants from mismatched bubble templates. The indicated RNAP holoenzymes were combined with the T7 A1 promoter (–46 to +20) synthetic DNA fragment (lanes 1 and 5) or the T7 A1 promoter with the synthetic mismatched bubble templates indicated at the top of the figure. Reactions were supplemented with CpApUpC primer, 50 µM NTPs and [α-32P]CTP. The reaction proceeded for 10 min at 55°C, and reaction products were resolved by denaturing PAGE and revealed by autoradiography.
Affinity labeling of mutant RNAP on minimal scaffold templates
Current structural models of bacterial RNAP transcrip tion complexes were built by superimposing structural information from T.aquaticus RNAP and cross-linking information obtained from E.coli RNAP transcription complexes. Though E.coli and T.aquaticus RNAPs are clearly homologous, significant functional and sequence differences do exist (Zhang et al., 1999; Minakhin et al, 2001). To demonstrate that the β′ rudder position in T.aquaticus RNAP transcription complexes is consistent with the structural model of the elongation complex (Korzheva et al., 2000), we performed affinity labeling experiments using artificial elongation complexes assembled on minimal nucleic acid scaffolds (Korzheva et al., 1998). Such complexes retain many essential features of E.coli RNAP elongation complexes, and protein–nucleic acid cross-linking information coming from studies of such complexes forms the basis of the structural model of the transcription elongation complex (Korzheva et al., 2000). Preliminary experiments demonstrated that the rudderless RNAP was as active as the wild-type enzyme in transcription from minimal scaffold templates (data not shown), which is expected, since the DNA melting step is bypassed.
Wild-type E.coli and T.aquaticus core RNAPs, as well as the rudderless mutant and ΩEc, a hybrid enzyme that contained E.coli rudder sequence incorporated instead of the T.aquaticus sequence, were incubated with a 28 base DNA oligonucleotide which served as a template strand. Complementary 18 base DNA oligonucleotide was used to create a stretch of double-stranded downstream DNA, which is thought to be necessary for complex stability (Figure 4A; Nudler et al., 1996). An eight base RNA complementary to the single-stranded portion of the template DNA oligonucleotide was next added to the reaction. The 5′ end of RNA was derivatized with a cross-linkable, lysine-specific aldehyde group. The enzymes were allowed to extend the RNA with radioactive nucleotides specified by the template strand, and cross-linking between the protein and radioactive nine base RNA was induced. In E.coli RNAP complexes, β′ Lys321 and Lys325 are cross-linked in this situation with high efficiency (Korzheva et al., 2000; Figure 4A, lane 1). Little or no cross-linking is observed in the wild-type T.aquaticus complexes (Figure 4A, lane 2). This is expected, since T.aquaticus β′ residues corresponding to E.coli β′ Lys321 and Lys325 are arginines (Figure 1A; Zhang et al., 1999) and the cross-linking reagent used is specific for lysines only. In contrast, a high level of cross-linking was observed with the hybrid enzyme (Figure 4A, lane 4), consistent with the idea that the E.coli rudder sequence assumed the expected position in the T.aquaticus transcription complex. Cross-link mapping performed using limited proteolysis with Met-specific CNBr (Grachev et al., 1987) revealed that the β′ cross-link in the hybrid enzyme was contained between Met573 and Met648 (Figure 4B), and therefore included the rudder area (amino acids 585–601). Thus, it appears that E.coli residues substituted for the T.aquaticus rudder assume a position identical to that found in E.coli RNAP complexes, contacting, or being close to the nascent RNA nine bases upstream of the catalytic center. By extension, we assume that the T.aquaticus rudder also occupies the same position in T.aquaticus RNAP elongation complexes.

Fig. 4. Affinity labeling of T.aquaticus RNAP mutants on minimal scaffold templates. (A) Affinity labeling of RNAP β′ using a lysine-specific aldehyde-based reagent on the 5′ end of the nascent RNA. Artificial elongation complexes were assembled using the indicated RNAP core enzymes and the minimal nucleic acid scaffold templates schematically shown at the top of the figure. The 8mer RNA was elongated with radioactive CTP specified by the template. The position of the radioactive residue is indicated by an asterisk. The 5′ end of RNA was derivatized with a lysine-specific cross-linkable aldehyde group. Protein–RNA cross-links were induced by treatment with 10 mM NaBH4, and reaction products were resolved by SDS–PAGE and revealed by autoradiography. (B) The results of single-hit CNBr treatment of the major radioactive band from (A), lane 4, are shown. The distribution of N-terminal radioactively labeled CNBr fragments based on the known T.aquaticus β′ sequence is illustrated schematically on the right. M is a marker lane prepared using T.aquaticus β′ cross-linked to radioactively labeled RNA at an N-terminal site (labeled by an open circle on the schematics on the right).
Deletion of the rudder does not affect the length of the RNA–DNA hybrid
We next tested the effect of β′ rudder removal on the length of the RNA–DNA hybrid in transcription elongation complexes. Two types of experiments were performed. First, we repeated the experiment of Korzheva et al. (1998), to monitor the efficiency of RNA–DNA cross-linking with a cross-linking group positioned at the 5′ end of the nascent RNA. Transcription was initiated using wild-type T.aquaticus RNAP, T.aquaticus mutant RNAP and wild-type E.coli RNAP from the T7 A1 promoter-based mismatched bubble template using RNA primer UpCpApUpC. The primer was derivatized at its 5′ end with an azido group that can cross-link to both protein and DNA. The enzymes were allowed to extend the primer in the presence of ATP and GTP. Since CTP was missing from the reaction, transcription was stalled in front of the first C, when the RNA length reached 13 bases. Stalled elongation complexes were washed from unincorporated nucleotides and were subjected to limited pyrophosphorolysis. As a result, a population of transcription complexes containing the nascent RNAs with identical 5′ ends, but with different 3′ ends, was obtained. The complexes were washed and cross-linking was induced. RNA–protein and RNA–DNA cross-linking products were separated by SDS–PAGE and purified. Next, cross-links were reversed, and radioactively labeled RNAs recovered from RNA– protein and RNA–DNA adducts were compared with each other and with the initial population of RNAs in complexes that underwent pyrophosphorolysis by denaturing PAGE (Figure 5A). It is expected that efficient RNA–DNA cross-linking can only occur when RNA is close to DNA, i.e. is in the hybrid. The distribution of cross-linked RNA was asymmetrical and similar for all three enzymes: RNAs of eight, nine and 10 nucleotides in length preferentially cross-linked to DNA; longer RNAs preferentially cross-linked to RNAP (Figure 5B). We note that the efficiency of RNA–DNA cross-linking in the rudder mutant complexes was lower than in the wild-type T.aquaticus or E.coli RNAP complexes. We do not know the reason for this difference. However, based on the qualitative similarity of RNA–DNA cross-linking distributions in the wild-type and mutant T.aquaticus enzymes, we conclude that the length of the RNA–DNA hybrid is unchanged in the absence of the β′ rudder.

Fig. 5. Determining the length of the RNA–DNA hybrid in the RNAP rudder mutant. (A) The experiment was performed as described in the text (see also Korzheva et al., 1998). Lanes 1, 5 and 9, RNA in initial complexes; lanes 2, 6 and 10, RNA in washed complexes that underwent limited pyrophosphorolysis. Lanes labeled β, β and DNA show RNAs cross-linked to the largest RNAP subunits and template DNA, respectively. (B) Quantification of the results in (A). The calculated efficiency of RNA–DNA cross-linking as a function of RNA length is presented. The height of the bars reflects the fraction of the RNA product cross-linked to DNA related to the total amount of the corresponding RNA in the complex (100% – RNA cross-linked to protein and DNA). (C) Elongation complexes stalled at position +11 were prepared using the indicated core RNAP enzymes and mismatched bubble templates fused to the λ tR2 terminator. Transcription was resumed by the addition of 250 µM NTPs. Reactions proceeded for 10 min at 55°C and were terminated by the addition of formamide-containing buffer (lanes 1 and 3). In lanes 2 and 4, 2.5 U of RNase H was added to completed transcription reactions for 10 min at 37°C prior to the addition of loading buffer. Lanes 5 and 6 are controls establishing that RNase H efficiently recognized products of in vitro transcription reactions (lane 5) annealed to a complementary DNA fragment, resulting in shorter RNA products (indicated by arrows in lane 6). The reaction products were resolved by denaturing PAGE and visualized by autoradiography.
We also determined whether in the absence of the β′ rudder a persistent RNA–DNA hybrid is formed behind transcribing RNAP. Stalled elongation complexes containing radioactive 13mer RNA were prepared on mismatched bubble templates fused to the well-characterized tR2 terminator (see Materials and methods) and were restarted synchronously by the addition of unlabeled NTPs. The mutant enzyme was able to reach the end of the template efficiently (i.e. was processive), but terminated transcription on tR2 more efficiently than the wild-type enzyme (the termination efficiency for the wild-type and the mutant enzymes was 18 and 43%, respectively, Figure 5C, lanes 1 and 3). The significance of increased termination efficiency is not clear, however, since transcription termination by the mutant enzyme at the λ tR′ terminator was less affected (data not shown). The addition of RNase H to completed transcription reactions had no effect on the length of RNA products synthesized by either the wild-type or the mutant RNAP, suggesting that no extensive RNA–DNA hybrid was formed in either case (Figure 5C, lanes 2 and 4). The following control experiment demonstrated that RNase H recognized and destroyed RNA–DNA hybrids, as expected. RNA from the completed transcription reaction was annealed to synthetic template DNA corresponding to positions –46 to +20 of the promoter. Treatment of such a reaction with RNase H resulted in shortening of the terminated and run-off transcripts by ∼20 bases, as expected (Figure 5C, compare lanes 5 and 6). We conclude that in agreement with RNA–DNA cross-linking results, no extensive RNA– DNA hybrid is formed during transcription by the rudderless RNAP.
Deletion of the β′ rudder does not prevent transcription termination but severely destabilizes elongation complexes
Stalled elongation complexes obtained on semi-synthetic mismatched bubble template fused to the tR2 terminator were allowed to elongate RNA chains in the presence of increasing concentrations of KCl in the buffer. Wild-type RNAP efficiently extended the initial 12mer RNA at salt concentrations as high as 0.5 M (Figure 6A, compare lanes 2 and 7). Likewise, the hybrid enzyme containing the rudder from E.coli (above) elongated the nascent RNA chains in the presence of high concentrations of salt in the transcription buffer (data not shown). In contrast, the rudderless mutant was unable to extend the nascent 12mer RNA efficiently when the KCl concentration was increased above 200 mM (Figure 6A, compare lanes 9 and 11). As a result, the amount of transcripts produced by the mutant enzyme decreased at high salt, while the amount of the unchased initial 12 nucleotide RNA increased. All of the unchased 12mer was found to be released from the complex (data not shown), indicating that the mutant complex is unstable at high salt concentrations in the buffer.

Fig. 6. Transcription complexes formed by RNAP without the β′ rudder are unstable at elevated salt concentrations. (A) Transcription elongation in the presence of increasing concentrations of KCl. Stalled elongation complexes were formed as described in the legend of Figure 5C, and transcription was resumed by the addition of 250 µM NTPs and in the presence of the indicated concentration of KCl in the buffer. The reaction products were resolved by denaturing PAGE, visualized by autoradiography and quantified by phosphoimagery. (B) The rudder contributes to the ability of the transcription complex to withstand high concentrations of salt in the absence of the non-template DNA strand. Immobilized transcription complexes containing radioactively labeled nine nucleotide long RNA were assembled using scaffold template shown schematically at the top of the figure. The complexes were then transferred into a buffer containing 0.5 NaCl, incubated for the times indicated, and the amount of RNA remaining in the complex was revealed by denaturing PAGE and autoradiography. The gels were also quantified by phosphoimagery and the results are presented at the right of the figure. At the bottom of the figure, the results of a similar experiment conducted at low (40 mM) NaCl concentration in the buffer are presented.
In principle, increased sensitivity of the rudderless enzyme to salt can be due to (i) loss of RNA–protein contacts that stabilize the complex, or (ii) an increased tendency of the non-template strand to invade the heteroduplex in the absence of the β′ rudder. Differences in the stability of the transcription complex in the absence of the non-template strand should reflect the contribution of RNA–protein contacts to complex stability. The results presented in Figure 6B, top, demonstrate that artificial elongation complexes formed by the rudderless enzyme on a nucleic acid scaffold lacking the non-template strand upstream of the catalytic center position remained highly sensitive to salt (half-life <1 min in the presence of 0.5 M NaCl), while the wild-type complexes were resistant (half-life of ∼60 min). In contrast, complexes formed by the rudderless enzyme were as stable as the wild-type enzyme complexes at low concentration of salt in the buffer (Figure 6B, bottom). Thus, the β′ rudder is required for RNAP elongation complex stability even in the absence of the non-template strand, consistent with the idea that the rudder’s interactions with the nascent RNA 8–9 bases upstream of the 3′ end critically contribute to the elongation complex stability.
Discussion
In this work, we set out to investigate the role of the largest RNAP subunit’s rudder element in transcription. Our results clearly identify several processes for which the rudder is or is not required. First, the rudder is not necessary for initial melting or distortion of –10 box DNA during open promoter complex formation. Secondly, the rudder is not required for the maintenance of the correct length of the RNA–DNA hybrid. Thirdly, the rudder is not required for processive transcript elongation and transcription termination at intrinsic terminators. We note that available structure–function models of the transcription elongation complex imply that the rudder is required for at least one of these processes (Korzheva et al., 2000; Gnatt et al., 2001).
Our results reveal that deletion of the β′ rudder results in dramatic defects in transcription initiation and severely destabilizes RNAP elongation complexes. KMnO4 probing revealed that the transcription bubble in promoter complexes formed by RNAP without the rudder is shortened in the downstream direction. Two scenarios can account for the observed melting defects of the mutant RNAP in initiation. First, deletion of the rudder can change the specificity of the interaction of the σ subunit with promoter DNA and thus affect melting. The primary binding site of the σ subunit has been mapped to the coiled-coil element of β′, from which the rudder emanates (Burgess et al., 1998). Thus, it is not inconceivable that changes in the rudder may alter RNAP core–σ interactions. However, limited proteolysis and native PAGE failed to detect any differences between the wild-type and mutant holoenzymes (data not shown), suggesting that large-scale changes between the mutant and the wild-type enzymes are unlikely. Further, the mutant holoenzymes are fully proficient in promoter recognition, which is governed by σ, and we failed to detect differences between the wild-type and mutant RNAP complexes by DNase I footprinting (data not shown). Alternatively, the rudder may play an active role in the downstream propagation of melting by directing the template DNA towards the catalytic center and/or positioning evolutionarily conserved positively charged segment C amino acids (indicated by cyan in Figure 1C and D) to allow favorable electrostatic interactions with the template strand downstream of the catalytic center. Interestingly, E.coli RNAP harboring mutations in the β subunit also form promoter complexes that are shortened in the downstream direction and appear to be similar to mutant T.aquaticus RNAP complexes. The fact that similar complexes can be obtained by mutating different subunits in RNAPs from different organisms lends further support to the idea that these unusual promoter complexes represent real structural intermediates of the opening pathway (Nechaev et al., 2000).
Our most important conclusions come from the analysis of transcription elongation and termination by the T.aquaticus rudder mutant. These experiments were conducted using RNAP core enzymes to ensure that no σ-specific defects are observed. Our results demonstrate that at low and physiological salt concentrations, transcription elongation and transcription termination can occur normally in the absence of the β′ rudder. Moreover, no persistent RNA–DNA hybrid elongation complexes are formed by the rudderless enzyme. Though we did not test specifically localized DNA melting at the upstream edge of the transcription bubble during elongation, the relatively mild effects of rudder removal on transcription elongation and termination under physiological conditions suggest that the rudder is also dispensable for this function.
The β′ rudder critically contributes to the elongation complex stability when the ionic strength is increased. Transcription complexes formed by the rudderless enzyme fall apart when the concentration of salt in the transcription buffer is increased above 200 mM. Thus, the rudder appears to be equivalent to the tight-binding RNA site, whose existence was postulated from theoretical considerations (Chamberlin, 1992), biochemical studies (Korzheva et al., 1998) and a structural model of the elongation complex (Korzheva et al., 2000; Korzheva and Mustaev, 2001). It appears that the rudder contributes to elongation complex stability through favorable protein–RNA interactions at the upstream edge of the RNA–DNA hybrid. The stabilizing effect of protein–RNA contacts between the rudder and nascent RNA 8–9 nucleotides upstream of the catalytic center is fully consistent with previous biochemical results, which indicated that RNA has to achieve a critical length of 7–8 nucleotides for the elongation complex to acquire stability (Korzheva et al., 1998). Interestingly, the most conserved amino acids of the rudder are two positively charged amino acids, lysines or arginines. In the structure of the elongation complex with yeast RNAP II, one of these residues, Lys323, is engaged in interactions with RNA in the hybrid, while the second one, Arg320, is not seen (Figure 1C). We postulate that the ability of either of the two conserved positively charged rudder residues to interact with the nascent RNA in an alternative and ordered manner and to stabilize RNA in the elongation complex, coupled with lateral mobility of the rudder, provides a mechanism that allows processive elongation to occur. Future experiments involving point mutations of these residues should help verify this hypothesis.
Materials and methods
Construction of T.aquaticus RNAP mutants
The following multistep procedure was used to create plasmid pET28TaqABCΔR, co-overproducing T.aquaticus α, β and His-tagged β′ lacking the rudder. At the first step, two PCRs were performed to amplify T.aquaticus rpoC regions at the left- and right-hand sides of the deletion site. The left-hand side non-mutagenic PCR primer corresponded to T.aquaticus rpoC positions 1289–1313 and contained a BglII site corresponding to the natural rpoC site at position 1289. The right-hand side non-mutagenic primer was complementary to rpoC positions 2868–2887 and contained a KpnI site corresponding to the natural site at position 2882. The mutagenic primers corresponded to rpoC positions 1729–1752 and 1804–1827 for the left-hand side flanking PCR and the right-hand side PCR, respectively. Each mutagenic primer also contained an in-frame XbaI site. The products of each PCR were purified, treated with XbaI and ligated. The ∼1.6 kbp ligation product was cloned into the pT7blue blunt vector (Novagen) and sequenced to confirm the presence of the desired mutation and the absence of undesirable mutations generated by PCR. The entire mutant rpoC gene was assembled next by sequentially ligating the 5′ end-terminal NdeI–BglII rpoC fragment, and the 3′ end-terminal KpnI–EcoRI fragment from the pET28TaqC plasmid (Minakhin et al., 2001) to an appropriately treated pT7blue-based plasmid harboring the BglII–KpnI rpoC fragment with mutation. The resultant pT7blue-based plasmid, pT7blueTaqCΔR, contained the entire rpoC gene bounded by NdeI and EcoRI sites and had a unique XbaI site at the site of the rudder deletion. The plasmid pET28TaqABCΔR co-expressing Taq rpoA, rpoB and rpoC was assembled next using a procedure similar to that described previously (Minakhin et al., 2001); details on plasmid construction are available from the authors upon request. pT7blueTaqCΔR was also used for construction of a second-generation insertion mutant by treating it with XbaI and ligating it to an appropriately selected double-stranded synthetic linker corresponding to the E.coli rpoC sequence coding for the rudder. The resulting plasmid pT7blueTaqCΩEcR was used to create co-expression plasmid pET28TaqABCΩEcR as described above.
Protein purification
A single colony containing pET28TaqABC or its derivatives was inoculated in 2 ml of LB containing 25 mg/ml kanamycin and grown for 2–3 h at 37°C. The starter culture was then used to inoculate 1 l of LB containing 25 mg/ml kanamycin. Cells were grown at 37°C with vigorous agitation for 18–20 h and harvested by centrifugation. The cell pellet was resuspended in 15 ml of buffer containing 40 mM Tris–HCl pH 7.7, 10 mM EDTA, 0.1 M NaCl, 5% glycerol, 2 mM β-mercaptoethanol and 0.2 mM phenylmethylsulfonyl fluoride (PMSF). Cells were lysed by sonication as described (Borukhov and Goldfarb, 1993). The lysate was cleared by low-speed centrifugation, and the volume was adjusted to 80 ml with the buffer, following by a 0.5 h incubation at 65°C with occasional mixing. The precipitate was removed by repeated low-speed centrifugation, and discarded. The supernatant was loaded onto a 1 ml HiTrap heparin column (Pharmacia) equilibrated in TGE buffer (40 mM Tris–HCl pH 7.7, 1 mM EDTA, 5% glycerol, 2 mM β-mercaptoethanol, 0.2 mM PMSF) and attached to a Waters 650 FPLC. The column was washed with TGE containing 0.3 M NaCl, and RNAP was eluted with TGE containing 0.6 M NaCl and precipitated with ammonium sulfate (40 g/100 ml). The ammonium sulfate pellet was dissolved in 0.35 ml of TGE and loaded onto a Superose-6 column (Pharmacia) equilibrated in TGE containing 0.1 M NaCl. The column was developed with the same buffer, and RNAP-containing fractions were pooled and loaded onto a 1 ml Resource-Q column (Pharmacia) equilibrated in TGE containing 0.1 M NaCl. The column was developed with a 20 ml linear gradient of NaCl concentration (from 0.2 to 1 M) in TGE. RNAP-containing fractions were pooled and dialyzed against storage buffer containing 40 mM Tris–HCl pH 7.7, 0.2 M KCl, 50% glycerol, 1 mM dithiothreitol (DTT), 1 mM EDTA.
KMnO4 probing
The T7 A2 promoter-containing DNA fragment (–97 to +87) was PCR amplified from the pT7blueT7A2 plasmid (K.Kuznedelov, unpublished) and treated with EcoRI. The fragment was 32P end-labeled by filling-in EcoRI sticky ends with Klenow enzyme in the presence of [α-32P]dATP. The bottom strand labeled fragment (–83 to +87) was purified by a Qiagen PCR kit. Promoter complexes were formed in 20 µl reactions containing 200 nM wild-type or mutant RNAP, 100 nM 32P end-labeled DNA fragment, 40 mM Tris–HCl pH 7.9, 40 mM KCl and 10 mM MgCl2. Reactions were pre-incubated for 15 min at 37°C (for E.coli RNAP complexes) or at 65°C (for T.aquaticus RNAP complexes). For KMnO4 probing, promoter complexes were treated with KMnO4 (1 mM) for 15 s at 37°C or at 65°C for the E.coli and T.aquaticus RNAP complexes, respectively. Reactions were terminated by addition of β-mercaptoethanol to 300 mM followed by phenol extraction, ethanol precipitation and 10% piperidine treatment.
To footprint promoter complexes formed at different temperatures, RNAP and 32P end-labeled T7 A2 promoter-containing fragments were combined on ice, transferred to the assay temperature, incubated for an additional 15 min and the footprinting reaction was performed. The same conditions were used for KMnO4 probing at different temperatures. Control experiments demonstrated that KMnO4 modification was complete after 15 s at all temperatures used.
Products of footprinting reactions were analyzed by urea–PAGE (8 M urea, 6% polyacrylamide), followed by autoradiography.
In vitro transcription reactions
The RNAP core enzyme (0.1–0.2 mg/ml) was pre-incubated for 15 min in a 10 µl reaction containing 40 mM Tris–HCl pH 8.4, 40 mM KCl, 5 mM MgSO4, in the presence or absence of 0.025 mg/ml recombinant T.aquaticus σ subunit (Minakhin et al., 2001) at 65°C. The appropriate DNA template was added to the final concentration of 100–500 nM, and the incubation was continued for an additional 10–15 min. Transcription was initiated by the addition of 5 µl containing 40 mM Tris–HCl pH 8.4, 40 mM KCl, 5 mM MgSO4, 100–500 nM CpA (for the T7 A1 promoter), or GpC (for the T7 A2 promoter) primer and 0.25 µl of [α-32P]UTP (for abortive initiation), or 10–50 nM CpApUpC, 0.25 µl of [α-32P]ATP (3000 Ci/mmol, 10 mCi/ml) and 50 µM GTP (to obtain elongation complexes stalled at the +11 position of the template). Reactions were allowed to proceed for 5–10 min at 55°C. To study transcription elongation, stalled elongation complexes were restarted by the addition of 250 µM unlabeled NTPs. Reactions were terminated by the addition of formamide-containing loading buffer and products were analyzed by denaturing PAGE and revealed by autoradiography and phosphoimager analysis.
RNA and DNA components of nucleic acid scaffolds (10–50 pmol each; Korzheva et al., 2000) were mixed in 20–50 µl of transcription buffer (TB) containing 20 mM Tris pH 7.9, 40 mM KCl and 10 mM MgCl2 at 50°C, and the mixture was allowed to cool down to 24°C over 10–30 min. A 5–10 pmol aliquot of annealed scaffold was combined with 2 pmol of His6-tagged core RNAP (either E.coli or T.aquaticus) immobilized on Ni2+-NTA–agarose in 20 µl of TB and reactions were incubated at 20°C for 10 min to allow complex formation. After washing (4 × 1 ml TB), immobilized complexes were incubated with [α-32P]UTP (0.6 µM) for 10 min at 37°C. The beads were washed, as above, and radioactive RNA products were analyzed on a 23% denaturating urea gel.
Cross-linking reactions
Two kinds of reactive groups (chemical structures shown in Korzheva et al., 2000) were used for RNA cross-linking: the fluorinated aromatic azide, which reacts non-selectively with any type of amino acid and DNA, and the aldehyde, which cross-links exclusively to lysine residues. All cross-linking was performed using minimal RNA–DNA scaffolds with the cross-linking probe attached to the 5′ terminus of the RNA oligonucleotide. The RNAP–scaffold complexes labeled at the 3′ end of the RNA were obtained as described above and the cross-links achieved by either irradiation at 254 nm for 2 min at 24°C (in the case of the photoreactive reagent) or treatment with 10 mM NaBH4 for 15 min at 24°C (in the case of the aldehyde reagent). Products were analyzed by 4% SDS–PAGE.
Mapping of the RNA–protein cross-links
To map RNA cross-links in β′, radiolabeled, cross-linked adduct was subjected to single-hit chemical degradation by BrCN (Grachev et al., 1987), and products were analyzed by 8–16% SDS–PAGE.
Determination of RNA–DNA hybrid size
Immobilized initiation complexes were formed on templates containing the T7 A1 promoter-based mismatched bubble at positions –12/+1 (above). Elongation complexes containing 13 nucleotide radioactive nascent RNA were next prepared by incubating immobilized promoter complexes at 55°C for 15 min in the presence of 10 µM UCAUC primer derivatized with fluorinated aromatic azide at the 5′ end, 0.6 µM [α-32P]ATP and 25 µM GTP. After ternary complex formation, the beads were washed, and RNA was allowed to undergo pyrophosphorolysis for 5 min at 24°C in the presence of 10 mM pyrophosphate. Pyrophos phorolysis was terminated by washing (as above). Next, cross-linking was induced by irradiation of the samples at 254 nm for 3 min. The cross-linked products (RNA–protein and RNA–DNA) were separated by 4–20% SDS–PAGE, and gel chips containing radioactive bands were excised from the gel, washed from the SDS, and incubated in 5% formic acid at 37°C for 2 h to reverse the cross-link. The supernatant, which contained RNA molecules detached from protein or DNA, was freeze-dried and samples were analyzed by 23% denaturing PAGE.
Acknowledgments
Acknowledgements
We are grateful to Dr Rachid Rahmouni for suggesting the alternating rudder–RNA contact mechanism. This work was supported by a Career Award in Biomedical Sciences from the Borroughs Welcome Fund for Biomedical Research. N.K. and A.M. were supported by NIH grants GM 30717 and GM49242 to Alex Goldfarb.
References
- Allison L.A., Moyle,M., Shales,M. and Ingles,C.J. (1985) Extensive homology among the largest subunits of eukaryotic and prokaryotic RNA polymerases. Cell, 42, 599–610. [DOI] [PubMed] [Google Scholar]
- Borukhov S. and Goldfarb,A. (1993) Recombinant Escherichia coli RNA polymerase: purification of individually overexpressed subunits and in vitro assembly. Protein Expr. Purif., 4, 503–511. [DOI] [PubMed] [Google Scholar]
- Burgess R.R., Arthur,T.M. and Pietz,B.C. (1998) Interaction of Escherichia coli σ70 with core RNA polymerase. Cold Spring Harb. Symp. Quant. Biol., 63, 277–287. [DOI] [PubMed] [Google Scholar]
- Chamberlin M.J. (1992) New models for the mechanism of transcription elongation and its regulation. Harvey Lect., 88, 1–21. [PubMed] [Google Scholar]
- Cramer P., Bushnell,D.A. and Kornberg,R.D. (2001) Structural basis of transcription: RNA polymerase II at 2.8 Å resolution. Science, 292, 1863–1876. [DOI] [PubMed] [Google Scholar]
- deHaseth P.L. and Helmann,J.D. (1995) Open complex formation by Escherichia coli RNA polymerase: the mechanism of polymerase-induced strand separation of double helical DNA. Mol. Microbiol., 16, 817–824. [DOI] [PubMed] [Google Scholar]
- Ebright R.H.E. (2000) RNA polymerase: structural similarities between bacterial RNA polymerase and eukaryotic RNA polymerase II. J. Mol. Biol., 304, 687–898. [DOI] [PubMed] [Google Scholar]
- Gnatt A.L., Cramer,P., Fu,J., Bushnell,D.A. and Kornberg,R.D. (2001) Structural basis of transcription: an RNA polymerase II elongation complex at 3.3 Å resolution. Science, 292, 1876–1882. [DOI] [PubMed] [Google Scholar]
- Grachev M.A., Kolocheva,T.I., Lukhtanov,E.A. and Mustaev,A.A. (1987) Studies on the functional topography of Escherichia coli RNA polymerase. Highly selective affinity labeling by analogues of initiating substrates. Eur. J. Biochem., 163, 113–121. [DOI] [PubMed] [Google Scholar]
- Korzheva N. and Mustaev,A. (2001) Transcription elongation complex: structure and function. Curr. Opin. Microbiol., 4, 119–125. [DOI] [PubMed] [Google Scholar]
- Korzheva N., Mustaev,A., Nudler,E., Nikiforov,V. and Goldfarb,A. (1998) Mechanistic model of the elongation complex of Escherichia coli RNA polymerase. Cold Spring Harb. Symp. Quant. Biol., 63, 337–345. [DOI] [PubMed] [Google Scholar]
- Korzheva N., Mustaev,A., Kozlov,M., Malhotra,A., Nikiforov,V., Goldfarb,A. and Darst,S.A. (2000) A structural model of transcription elongation. Science, 289, 619–625. [DOI] [PubMed] [Google Scholar]
- Minakhin L., Nechaev,S., Campbell,E.A. and Severinov,K. (2001) Recombinant Thermus aquaticus RNA polymerase—a new tool for structure-based analysis of transcription. J. Bacteriol., 183, 71–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naryshkin N., Revyakin,A., Kim,Y., Mekler,V. and Ebright,R.H. (2000) Structural organization of the RNA polymerase–promoter open complex. Cell, 101, 601–611. [DOI] [PubMed] [Google Scholar]
- Nechaev S., Chlenov,M. and Severinov,K. (2000) Dissection of two hallmarks of the open promoter complex by mutation in RNA polymerase core subunit. J. Biol. Chem., 275, 25516–25522. [DOI] [PubMed] [Google Scholar]
- Nudler E., Avetissova,E., Markovtsov,V. and Goldfarb,A. (1996) Tran scription processivity: protein–DNA interactions holding together the elongation complex. Science, 273, 211–217. [DOI] [PubMed] [Google Scholar]
- Nudler E., Mustaev,A., Lukhtanov,E. and Goldfarb,A. (1997) RNA– DNA hybrid maintains the register of transcription by preventing backtracking of RNA polymerase. Cell, 89, 33–41. [DOI] [PubMed] [Google Scholar]
- Severinov K. and Darst,S.A. (1997) A mutant RNA polymerase that forms unusual open promoter complexes. Proc. Natl Acad. Sci. USA, 94, 13481–13486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G., Campbell,E.A., Minakhin,L., Richter,C., Severinov,K. and Darst,S.A. (1999) Crystal structure of Thermus aquaticus core RNA polymerase at 3.3 Å resolution. Cell, 98, 811–824. [DOI] [PubMed] [Google Scholar]