

Abstract
Changes in motor activity are a basic response to filling of smooth muscle organs. Responses to gastric filling, for example, are thought to be regulated by neural reflexes. Here, we demonstrate a previously uncharacterized aspect of stretch-dependent responses in visceral smooth muscles that is mediated by mechanosensitive interstitial cells of Cajal. Length ramps were applied to the murine antral muscles while recording intracellular electrical activity and isometric force. Stretching muscles by an average of 27 ± 1% of resting length resulted in 5 mN of force. Increasing length caused membrane depolarization and increased slow-wave frequency. The responses were dependent on the rate of stretch. Stretch-dependent responses were not inhibited by neuronal antagonists or nifedipine. Increases in slow-wave frequency, but not membrane depolarization, were inhibited by reducing external Ca2+ (100 μM) and by Ni2+ (250 μM). Responses to stretch were inhibited by indomethacin (1 μM) and were absent in cyclooxygenase II-deficient mice, suggesting that cyclooxygenase II-derived eicosanoids may mediate these responses. Dual microelectrode impalements of muscle cells within the corpus and antrum showed that stretch-induced changes in slow-wave frequency uncoupled proximal-to-distal propagation of slow waves. This uncoupling could interfere with gastric peristalsis and impede gastric emptying. Stretch of antral muscles of W/WV mice, which lack intramuscular interstitial cells of Cajal, did not affect membrane depolarization or slow-wave frequency. These data demonstrate a previously uncharacterized nonneural stretch reflex in gastric muscles and provide physiological evidence demonstrating a mechanosensitive role for interstitial cells of Cajal in smooth muscle tissues.
Keywords: gastric compliance, pacemaker, stretch, slow waves, propagation
In the stomach, electrical slow waves are generated by a specialized network of cells, known as interstitial cells of Cajal (ICC), located in the myenteric region between the circular and longitudinal muscle layers (ICC-MY) (1-3). The stomach has a continuous network of ICC-MY from the orad corpus to the pyloric sphincter (1), and samples of muscle removed from each region demonstrate spontaneous activity that is characteristic of pacemaker cells in that region (4). Corpus ICC-MY typically serve as the dominant pacemaker because slow waves occur at the greatest frequency in this region (4, 5). Slow waves, initiated in the corpus, actively propagate around and along the length of the stomach and entrain the activity of distal pacemakers. ICC-MY are electrically coupled to smooth muscle cells of the gastric wall (6), and slow waves conduct into the musculature, causing depolarization of smooth muscle cells, an increase in the open probability of voltage-dependent Ca2+ channels, Ca2+ entry, and phasic contractions (7). Contractions initiated by slow waves are the basis for the gastric peristalsis that is essential for proper processing of solid foods and gastric emptying. The force of phasic contractions is regulated, in part, by enteric excitatory and inhibitory motor neurons. Neural control is superimposed on the intrinsic activity of the ICC/smooth muscle syncytium and regulates the responses of the musculature to slow-wave depolarizations.
Ingestion of food activates a variety of neural responses in the stomach, including a relaxation reflex to actively expand the volume of the proximal stomach (8-10) and an excitatory reflex to increase the force of peristaltic contractions in the distal stomach (11). These reflexes involve both inhibitory and excitatory intrinsic and central neural pathways. The end-organ regulatory inputs from the enteric nervous system are mediated by ICC that are interspersed within the circular and longitudinal muscle layers (ICC-IM) (12-14). Varicose terminals of enteric neurons make synaptic contacts with ICC-IM, and ICC-IM form gap junctions with neighboring smooth muscle cells (12, 15, 16). It has recently been demonstrated that vagal nerve stimulation, which activates enteric motor neurons, increases the frequency of slow-wave activity in the gastric antrum (17). The increase in frequency and the ability to pace slow waves through neural stimulation (18) are mediated through ICC-IM. Excitatory neural inputs to ICC-IM influences the slow-wave firing frequency of the ICC-MY network (17, 18).
Although neural regulation of gastric function has been studied in some detail, considerably less is known about the mechanisms that regulate the ICC/muscular responses. For example, the effect(s) of wall distension (stretch), a normal occurrence during gastric filling and emptying, on the function of ICC has never been reported. Here, we have used a specialized apparatus to precisely increase the length of gastric muscles and recorded the responses to stretch with transmembrane potential and isometric force measurements. By using mutant animals lacking ICC-IM, we have determined that responses to muscle stretch are mediated through this class of ICC. The consequences of stretch-dependent changes in antral slow-wave activity on the normal proximal-to-distal propagation of slow waves also were investigated.
Methods
Animals. BALB/c, W/WV mutants, and their wild-type siblings (WBB6F1) were obtained from The Jackson Laboratory, and cyclooxygenase II (COX II)-deficient mice (B6;129P2-Ptgs2tm1Smi) were obtained from Taconic Farms. Animals between 30 and 60 days postpartum (age-matched) were anesthetized by using isoflurane (Baxter, Deerfield, IL) inhalation and were exsanguinated after cervical dislocation. For electrophysiological and force measurements, stomachs were opened along the lesser curvature, gastric contents were washed with Krebs-Ringer buffer, and the mucosa were removed, revealing the underlying circular muscle layer. The use and treatment of animals was approved by the Institutional Animal Use and Care Committee at the University of Nevada.
Physiological Studies. For electrophysiological recordings, gastric corpus and antrum were prepared as described in ref. 18. Pins were placed at the greater curvature of the antrum, and the other end of the segment of tissue was attached to an isometric force transducer (UC3, Gould, Cleveland). The transducer was placed on an electronically controlled platform (860 SC, Newport, Irvine, CA) so that tension changes of the circular layer could be applied in a controlled manner. In some experiments, dual microelectrode impalements were performed on corpus and antral regions that were mechanically isolated from each other by using fine dissecting pins, while a small strip of antral tissue was stretched. Electrical recordings were performed as described in ref. 18.
Data are expressed as means ± SEM. Differences in the data were evaluated by Student's t test. P < 0.05 was considered to be statistically significant. n is the number of muscles obtained from separate animals used for each protocol. A total of 102 BALB/c mice, 21 W/WV mice, 6 COX II-deficient mice, and 9 WBB6F1 strain-matched controls were used in this study.
Solutions and Drugs. Muscles were maintained in Krebs-Ringer buffer (37.5 ± 0.5°C; pH 7.3-7.4) containing 137.4 mM Na+, 5.9 mM K+, 2.5 mM Ca2+, 1.2 mM Mg2+, 134 mM Cl-, 15.5 mM 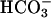 , 1.2 mM
, 1.2 mM 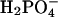 , and 11.5 mM dextrose, and were bubbled with 97% O2/3% CO2. Atropine sulfate, neurokinin-1 receptor antagonist (GR82334), tetrodotoxin (TTX), Nω-nitro-l-arginine, apamin, ω-conotoxin GVIA, and NiCl2 (Sigma) were dissolved in distilled H2O at 0.1 M-0.01 mM. Nifedipine indomethacin (Sigma) and the NK2 receptor antagonist SR48968 (Sanofi Recherche, France) were dissolved in ethanol at 0.01 M. All drugs were diluted in Krebs-Ringer buffer to the stated final concentrations.
, and 11.5 mM dextrose, and were bubbled with 97% O2/3% CO2. Atropine sulfate, neurokinin-1 receptor antagonist (GR82334), tetrodotoxin (TTX), Nω-nitro-l-arginine, apamin, ω-conotoxin GVIA, and NiCl2 (Sigma) were dissolved in distilled H2O at 0.1 M-0.01 mM. Nifedipine indomethacin (Sigma) and the NK2 receptor antagonist SR48968 (Sanofi Recherche, France) were dissolved in ethanol at 0.01 M. All drugs were diluted in Krebs-Ringer buffer to the stated final concentrations.
Results
We examined the effects of stretch on electrical parameters by increasing muscle length with length ramps (6.0 μm·s-1) until an isometric force of 5 mN was reached (length ramps reached this level of force in 297 ± 21 s). The controlled stretches increased muscle length by an average of 27 ± 1.0% of resting length and depolarized circular muscle cells from -63 ± 1.0 to -59 ± 1 mV. During stretch, slow-wave frequency increased from 2.5 ± 0.3 to 3.6 ± 0.3 cycles·min-1, and slow waves were reduced in amplitude from 24 ± 1 to 14 ± 2 mV. Depolarization and increased slow-wave frequency in response to stretch were transient (n = 15), and during maintained stretch (e.g., at 320 ± 6 s), resting membrane potential (RMP) partially recovered to prestretch levels and the muscles relaxed by an average of 2.1 ± 0.2 mN. Although RMP recovered, the waveforms of slow waves differed from those of control and had elevated plateau potentials (20 ± 1 mV) and shorter durations (12.1 ± 0.5 s; see Fig. 1). Phasic contractions increased in association with the increased level of depolarization during the plateau phase (i.e., from 0.2 ± 0.03 mN to 1.4 ± 0.20 mN; P < 0.01, compared with control).
Fig. 1.

Effects of antral stretch on MP and contractile activity. (A and B) Changes in electrical activity and isometric force in response to stretch of the gastric antrum. Stretching the antrum induced membrane depolarization increased slow-wave frequency and increased the amplitude and frequency of contractions. (C) The length ramp applied (0.0164 mN·s-1). Time bar in C applies to A-C. Break in records represents 5 min for recovery. (D) Examples of slow waves and contractions at an increased sweep speed at different stages of the stretch protocol (times denoted in A = *no.). Note the increase in frequency and contractions during the period of stretch (dotted line under MP trace). Increasing the rate of stretch increased the magnitude of the responses (see text for details). During periods of increased length, slow-wave frequency and MP recovered partially. Summaries of the effects of stretch on RMP and slow-wave frequency are shown in E and F, respectively.
In the muscles of wild-type WBB6F1 mice, length ramps averaging 328 ± 16 s caused a 5-mN change in force in WBB6F1 muscles. Stretch caused depolarization from -65 ± 1 to -60 ± 1 mV, and slow-wave frequency increased from 2.8 ± 0.1 to 3.8 ± 0.2 cycles·min-1 (n = 9). Slow-wave amplitude and duration changed from 27 ± 2 to 17 ± 2 mV and from 12.2 ± 0.7 to 9.8 ± 0.4 s, respectively. These effects were not statistically different from the effects of stretch on BALB/c muscles (Fig. 1).
The rate of stretch was varied (e.g., 1.9, 6.0, 7.1, and 31 μm·s-1) in another series of experiments. Muscles were stretched until isometric force was increased by 5 mN (n = 6). Increasing ramp velocity enhanced depolarization and chronotropic responses to stretch. At 1.9 μm·s-1, 5 mN in force was reached in 771 ± 47 s (0.007 mN·s-1) and caused depolarization from -66 ± 1 to -63 ± 2 mV. This rate of stretch did not increase in slow-wave frequency significantly (e.g., 3.1 ± 0.3 to 3.6 ± 0.3 cycles·min-1; P > 0.05). At 7.1 μm·s-1, 5 mN of force was reached in 211 ± 6 s (0.024 mN·s-1), membrane depolarized from -68 ± 1 to -61 ± 1 mV (P < 0.05), and slow-wave frequency increased to 3.8 ± 0.3 cycles·min-1 (P < 0.05). The fastest ramps tested, 31.0 μm·s-1, depolarized membrane potential (MP) from -65 ± 1.0 to -57 ± 1 mV (P < 0.05) and increased slow-wave frequency to 3.9 ± 0.2 cycles·min-1 (P < 0.05; Fig. 1 E and F; n = 6).
Responses to stretch accommodated with time, so repetitive stretch/relaxation cycles were tested to determine whether this protocol caused more sustained effects on the slow-wave cycle. Repetitive changes in muscle length (1 min-1 at 31 μm·s-1) caused reproducible depolarization in MP from -66 ± 1 to -58 ± 2 mV (P < 0.05) and increased slow-wave frequency from 2.9 ± 0.2 to 4.1 ± 0.3 cycles·min-1 (n = 6; P < 0.05; Fig. 2). Membrane depolarization and the decrease in inter-slow-wave interval (i.e., increase in slow-wave frequency) occurred within one slow-wave cycle, suggesting that these responses could occur in vivo if antral stretch occurs repetitively during gastric peristalsis. Stretching at more rapid rates also decreased the rate-of-rise of the upstroke of slow waves and revealed a discharge of unitary potentials. Appearance of unitary potentials suggests reduced coordination between pacemakers in gastrointestinal (GI) muscles (20). After relaxation of the length ramps, MP returned to control and unitary potentials were no longer evident (Fig. 2D).
Fig. 2.

Effect of repetitive antral stretch on MP, slow-wave frequency, and contractions. (A and B) Electrical and mechanical activities in response to three stretch episodes, delivered at 1 ramp min-1 (C). Repetitive stretching of the antrum caused reproducible depolarization and increased slow-wave frequency. Selected slow waves (*no. in A) are shown at an increased sweep speed in D. Note the occurrence of unitary potentials superimposed on the slow waves.
The involvement of neural reflexes in responses to stretch was investigated by testing the effects of neural blockers. TTX (0.3 μM; n = 7), atropine (1 μM; n = 9), neurokinin-1 and neurokinin-2 receptor antagonists (GR82334 and SR48968; n = 6), Nω-nitro-l-arginine (100 μM; n = 9), apamin (0.1 μM; n = 7), and ω-conotoxin GVIA (0.1 μM; n = 4) did not affect stretch-dependent changes in MP and slow-wave frequency (data not shown).
Length ramps were applied in the presence of Ca2+ channel-blocking drugs to determine the role of Ca2+ entry mechanisms on stretch-mediated responses. Nifedipine (1.0 μM) had no effect on slow-wave activity, but it blocked the phasic contractions associated with slow waves. Nifedipine did not significantly affect the depolarization and increase in slow-wave frequency caused by stretch. During the length ramps, RMP depolarized to -58 ± 1 mV, and the frequency of slow waves increased from 2.6 ± 0.6 to 4.8 ± 0.3 cycles·min-1 (P < 0.05). Nifedipine increased compliance and increased the time required to reach 5 mN of force (297 ± 21 s before and 408 ± 70 s in the presence of nifedipine) (P < 0.05). Contractions were not observed during length ramps in the presence of nifedipine (data not shown).
Ni2+ (100 μM) had no effect on stretch-dependent responses (n = 5; data not shown). Increasing Ni2+ to 250 μM caused waveform changes in slow waves but did not alter basal frequency. In these experiments, control stretch ramps depolarized cells from -67 ± 1 to -60 ± 2 mV and increased slow-wave frequency from 2.7 ± 0.2 to 4.2 ± 0.2 cycles·min-1 (n = 5; P < 0.005). With Ni2+ (250 μM), stretch depolarized cells from -71.0 ± 1.6 mV to -64.1 ± 1.9 mV (P < 0.05) but did not increase the frequency of slow waves (i.e., from 2.7 ± 0.2 to 2.9 ± 0.1 cycles·min-1; P > 0.05; Fig. 3 C and D). Ni2+ (250 μM) did not affect the compliance of the gastric muscles (269 ± 10 s to reach 5 mN before Ni2+ and 269 ± 15 s in the presence of Ni2+).
Fig. 3.

Effect of Ca2+ channel blockade and low extracellular Ca2+ concentration ([Ca2+]o) on responses to induced responses. (A) Control responses before reduction in [Ca2+]o from 250 to 100 μM, which abolished slow-wave activity (B) but did not inhibit the depolarization in response to stretch (B). (C) Control responses before addition of Ni2+ (250 μM), which blocked the increase in slow-wave frequency in response to stretch but did not alter the stretch-induced depolarization (D). Time bars under length ramps refer to all traces in that panel.
Stretch-induced changes in MP and slow waves were tested before and after reduction in [Ca2+]o from 2.5 μM to 250 and 100 μM. Reduction in [Ca2+]o reduced the frequency of slow waves from 3.4 ± 0.2 to 0.4 ± 0.3 cycles·min-1 (n = 7; P < 0.05; Fig. 3 A and B). Stretch in low [Ca2+]o depolarized cells from -66 ± 1 to -59.0 ± 2 mV (P > 0.5, compared with control) but had no effect on slow-wave frequency (0.4 ± 0.3 cycles·min-1 during the length ramp; P > 0.5; Fig. 3C). Low [Ca2+]o did not affect the compliance of muscles (312 ± 16 s to reach 5 mN in 250 μM[Ca2+]o and 315 ± 14 s in 100 μM [Ca2+]o).
We wondered whether the antral response to stretch could lead to breakdown in the proximal-to-distal frequency gradient in the stomach (4, 5) and disrupt corpus-to-antrum slow-wave propagation. Experiments to test this question were performed on intact gastric sheets, and recordings were made from cells in the corpus and antrum simultaneously to monitor coupling between these regions. The corpus was mechanically isolated from the antrum by miniature pins to avoid direct effects of stretch on the corpus. Under control conditions, there was 1:1 coordination between slow waves in the corpus and antrum (i.e., 3.9 ± 0.1 cycles·min-1 in both regions; Fig. 4 A, B, and E). Slow waves were generated in the corpus and propagated to the antrum at 0.9 ± 0.1 mm·s-1. Stretching the antrum (6.0 μm·s-1) caused depolarization from -68 ± 1 to -60 ± 1 mV and increased slow-wave frequency in antral cells from 3.9 ± 0.1 to 5.0 ± 0.2 cycles·min-1. The records showed obvious uncoupling of slow-wave activity in the two regions (n = 6; Fig. 4 A, E2, and E3).
Fig. 4.

Antral stretch disrupts corpus-to-antral propagation. (A and B) Dual microelectrode recordings from a smooth muscle cell in the corpus (A) and one in the antrum (B). (C) Contractile responses of the antral muscle. (D) Stretching the antral wall produced depolarization in MP and increased slow-wave frequency. (E) Representative segments of the record in A and B (denoted by *no.). Under control conditions, corpus slow-wave occurred first and propagated to the antrum at an average conduction velocity of 0.9 ± 0.1 mm·s-1 (n = 6; E*1, arrows). During stretch, corpus and antral slow-waves occurred at the same frequency and were occasionally in phase (E*2); however, there were periods in which the events in the two regions went out of phase (E*3), demonstrating uncoupling of the pacemakers in the two regions. After stretch was removed, normal slow-wave frequency and propagation velocity were restored (E*4, arrows). MPs for corpus and antrum are denoted by dashed and solid lines, respectively.
Intramuscular ICC (ICC-IM) generate unitary potentials (20, 21), and unitary potentials were enhanced during stretch (see Fig. 2). Thus, we next examined the role of ICC-IM in stretch responses. Responses to stretch were compared by using muscles of W/WV mice, which lack ICC-IM (12, 13, 18, 22) and strain-/age-matched controls. Slow-wave amplitudes and durations in W/WV muscles averaged 27 ± 2 mV amplitude, 11.3 ± 0.4 s duration, and 3.4 ± 0.3 cycles·min-1 frequency (P = 0.17, compared with wild-type muscles). W/WV muscles' slow waves generated phasic contractions averaging 0.19 ± 0.03 mN (P > 0.2, compared with wild-type muscles). Application of the 5-mN-length ramp at 6.0 μm·s-1 produced a 29 ± 1% change in resting length in 306 ± 19 s (n = 10). These data were not statistically different from wild-type animals. Depolarization and the increase in slow-wave frequency in response to stretch did not occur in W/WV muscles. After stretch, RMP averaged -66 ± 1.0 mV, and slow waves 26 ± 2 mV in amplitude and 10 ± 1.0 s in duration occurred at 3.5 ± 0.5 cycles·min-1 (Fig. 5; these values were not different from data before stretch; P > 0.5 for all parameters). W/WV muscles also were pretreated with TTX and Nω-nitro-l-arginine. These drugs had no significant effects on tissue compliance, as observed in BALB/c and WBB6F1 control muscles. Wild-type controls required 336 ± 10 s to attain 5 mN of force before and 278 ± 20 s after the addition of TTX (0.3 μM; P < 0.05; n = 4). W/WV muscles required 304 ± 10 s before and 297 ± 8 s after TTX (0.3 μM) to attain 5 mN of force (n = 6) and 299 ± 18 s before and 292 ± 3.8 after the addition of Nω-nitro-l-arginine (100 μM) to reach 5 mN of force (n = 5).
Fig. 5.

Responses to stretch are mediated by ICC-IM. Shown are antral slow waves (A) and isometric force (B) before, during, and after the application of a length ramp (C) in an antral muscle of a W/WV animal. These animals lack ICC-IM. Typical responses to stretch, membrane depolarization and increased slow-wave frequency, were not observed in W/WV muscles. (D) Individual slow waves at an increased sweep speed at various points during the protocol as indicated by the *no. in A. Note the reduction in unitary potentials in these records, compared with Fig. 2.
ICC-IM of the gastric antrum express COX isoforms, and inhibitors of COX reduce slow-wave frequency and potentiate phasic contractions in antral muscles (23). EP3-receptor agonists mimic the depolarization and chronotropic responses to stretch (24). Thus, we examined whether stretch-dependent responses were mediated by a COX-dependent mechanism. In these experiments, MP averaged -66 ± 1 mV and slow waves averaged 28 ± 2 mV in amplitude, 13.2 ± 0.5 s in duration, and occurred at a frequency of 3.6 ± 0.2 cycles·min-1. Stretch caused depolarization to -60 ± 1 mV and reduced slow-wave amplitude to 19 ± 1 mV. During stretch, slow-wave frequency increased to 4.5 ± 0.2 cycles·min-1 (n = 5). After indomethacin (1 μM for 30 min), MP averaged -65 ± 1 mV, and slow waves were 32 ± 2 mV in amplitude, 12.6 ± 1.4 s in duration (P > 0.05 for each parameter), and frequency decreased to 2.8 ± 0.3 cycles·min-1 (P < 0.05). Indomethacin also enhanced phasic contractions from 0.20 ± 0.03 to 0.33 ± 0.05 mN (n = 5), as reported in ref. 23. In the presence of indomethacin, stretching muscles did not affect RMP (-65 ± 1 mV) or slow-wave frequency (2.9 ± 0.3 cycles·min-1). Stretch caused a marked increase in the phasic contractions from 0.97 ± 0.23 (control response) to 8.29 ± 0.91 mN, and the increase in contractile amplitude was sustained for the duration of the length change (Fig. 6; P < 0.05).
Fig. 6.

COX mediates stretch-dependent responses in the antrum. (A-C) Stretch-dependent membrane depolarization, increased slow-wave frequency (A), and contractions (B) in antral tissues in response to stretch (C) under control conditions. (D-F) Loss of stretch-dependent responses in the same muscle as in A-C after pretreatment with indomethacin (1 μM for 30 min). Indomethacin reduced slow-wave frequency before and during length ramps and increased the force of contractions. (G-I) Loss of stretch-dependent responses in antral muscles of COX II-deficient mice. Slow waves and MP (G), force measurement (H), and the application of the 5-mN-length ramp (I). Depolarization and chronotropic response were not observed in response to stretch in muscles of COX II-deficient mice.
The role of COX II in stretch-dependent responses was tested in experiments using muscles from COX II-deficient mice. In these studies, RMP of COX II-deficient gastric muscles averaged -68 ± 1 mV, and slow waves, 31 ± 2 mV in amplitude and 11.5 ± 0.5 s in duration, occurred at 3.1 ± 0.1 cycles·min-1. The 5-mN-length ramp at 6.0 μm·s-1 had no significant effect on RMP or slow-wave amplitude, duration, and frequency (e.g., at the peak of stretch RMP = -67 ± 1 mV; amplitude = 30 ± 2 mV; duration = 12 ± 1 s and frequency = 3.2 ± 0.4 cycles·min-1; n = 6, P > 0.05 for all parameters). These data demonstrate the importance of COX II in mediating stretch-dependent responses in the antrum.
Discussion
Electrical slow waves increase the open probability of voltage-dependent Ca2+ channels in GI muscles and time the phasic contractions of gastric peristalsis (see refs. 19 and 25). Superimposed on slow-wave activity are neurally mediated reflexes that modulate the force of peristaltic contractions. Stretching the walls of GI organs can activate mechanosensitive enteric neurons, including intrinsic primary type II afferent neurons (26) and mechanosensory S type-I neurons (27). Integration of the afferent inputs and discharge to efferent motorneurons can alter motility. For example, during ingestion of a meal, gastric accommodation occurs as the compliance of the proximal stomach increases by nitrergic inhibition of the fundus and orad corpus (e.g., refs. 28 and 29). Another feature of the gastric phase of digestion and antral distention is increased cholinergic input that increases the force of contractions that helps reduce the size of solid food particles in the distal stomach (e.g., refs. 30-32). Stimulation of cholinergic neurons also can increase the frequency of slow waves in the gastric antrum (17). In the present study, we have described a form of stretch-dependent regulation that did not involve neural pathways. Blockers of nerve action potentials (TTX), neurotransmitter release (conotoxin-GIVA), and postjunctional receptors did not affect the stretch-dependent depolarization and the positive chronotropic effects that we observed, suggesting that these responses were nonneural, possibly “myogenic” in nature.
Increasing muscle length initiated significant changes in resting potentials and the intrinsic frequency of antral pacemakers. Both of these responses could have significant effects on the performance of antral muscles during gastric emptying. Stretch-dependent responses in the antrum are mediated by ICC-IM because neither response was observed in antral muscles of W/WV mice, which lack ICC-IM (12). Previous authors have suggested a “sensory” role for ICC, and possibly sensitivity to stretch, from morphological considerations (33, 34); however, this study demonstrates a physiological role for ICC in stretch-dependent responses in the GI tract.
Inhibition of COX blocked responses to stretch, suggesting that products of arachidonic acid metabolism may mediate stretch-dependent responses. Support for this idea also was provided by experiments on muscles of COX II-deficient mice. These experiments showed that COX II, which is constitutively expressed in ICC-IM in the gastric antrum (23), provides a critical step in the stretch-dependent responses. We have shown previously that the arachidonic acid metabolite prostaglandin E2 (PGE2) has positive chronotropic effects in gastric muscles (24, 38), and synthesis of prostaglandins contributes to gastric arrhythmias (35-37). Thus, enhanced production of PGE2 might mediate the chronotropic effects of stretch. The chronotropic effects of stretch are mimicked by sulprostone and ONO-AE-248 (EP3 agonists) in cultured ICC and intact antral muscles. Thus, it is possible that EP3 receptors might mediate stretch-dependent responses in ICC-IM (24). Many studies have reported that a variety of ionic conductances are regulated by prostaglandins, and either suppression of outward currents or activation of inward currents could produce depolarization, as we observed in response to stretch. Nonselective cation conductances have been reported to be activated by PGE2 in neurons (e.g., refs. 39-41), and delayed rectifier-like K+ currents have been shown to be inhibited by PGE2 in neurons (42) and vascular smooth muscle cells (43). The nature of the conductance responsible for the depolarization responses in ICC-IM has not been identified.
At present, we have not fully isolated the stretch sensor that regulates stretch-dependent responses of ICC-IM. We identified some features of the stretch response that may be properties of the mechanosensor. The stretch sensor is rate-dependent because length ramps of increasing velocity produced progressively larger responses. The sensor, or downstream mechanisms, accommodated, because responses to sustained stretch relaxed within 104 ± 3 s. The effectiveness of the stretch sensor recovered within 18 ± 1 s because repetitive stretch responses at 1 per min were of equal magnitude. One possibility for the stretch transducer could be activation of mechanosensitive nonselective cation channels in ICC. Channels of this type have been observed in a variety of cells (44, 45). Inward current via such a conductance could depolarize cells, and Ca2+ entry could enhance slow-wave frequency. Persistence of the responses, however, in low external Ca2+ argues against Ca2+ entry as the initiating factor in the stretch-dependent responses of ICC-IM. Phospholipase A2 (PLA2), which liberates arachidonic acid, is mechanosensitive in some cells (46, 47). Thus, it is possible that mechanotransduction coupling, linked through activation of PLA2, could be a previously unrecognized property of ICC-IM. Studies to determine the expression of PLA2 isoforms and the mechanosensitivity of these enzymes will be needed to determine whether PLA2 provides the primary stretch-sensor in ICC-IM.
The normal pacemaker in isolated antral muscles is ICC within the myenteric region (i.e., ICC-MY between the circular and longitudinal muscle layers; see ref. 2). Cholinergic nerve stimulation, which can drive postprandial enhancement of gastric motility in vivo, elicits premature slow waves in antral muscles, and these effects are mediated by ICC-IM (18). Others have suggested that during vagal nerve stimulation, ICC-IM become the dominant pacemaker in antral muscles (17). Thus, ICC-IM play an important role in regulating postprandial motility responses in the distal stomach. Our data demonstrate another role for ICC-IM and suggest that antral distention may be an additional factor affecting antral motility. Sustained antral distension may not be an effective stimulus for stretch-dependent effects because the mechanism we characterized adapted rapidly to increases in length, but pulsatile distensions of the antrum, as might occur during peristaltic contractions, may influence the intrinsic frequency of antral pacemakers and the force of antral contractions. Stretch-dependent responses may negatively impact the normal proximal-to-distal progress of gastric peristalsis if the compliance of the antrum is abnormally increased.
In summary, our data suggest that stretch of gastric antral muscles activates arachidonic acid metabolism and formation of eicosanoids that have effects on MP and pacemaker mechanisms. Receptors and transduction pathways for the eicosanoid(s) mediating the response to stretch are expressed by ICC-IM, demonstrating a previously uncharacterized physiological role for ICC in the GI tract. When the antrum is distended, acceleration of intrinsic pacemaker frequency in antral muscles may disrupt the normal proximal-to-distal slow-wave frequency gradient and interfere with gastric peristalsis and gastric emptying. The stretch-dependent mechanism described is a previously uncharacterized “ICCgenic” form of regulation of gastric motility that may contribute to the tendency of some human patients to respond to gastric filling with antral arrhythmias (e.g., ref. 48).
Acknowledgments
This work was supported by National Institutes of Health Grant DK57236 from the National Institute of Diabetes and Digestive and Kidney Diseases.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: ICC, interstitial cells of Cajal; ICC-IM, intramuscular ICC; ICC-MY, myenteric ICC; COX II, cyclooxygenase II; TTX, tetrodotoxin; MP, membrane potential; RMP, resting MP; GI, gastrointestinal; [Ca2+]o, extracellular Ca2+ concentration.
References
- 1.Burns, A. J., Herbert, T., Ward, S. M. & Sanders, K. M. (1997) Cell Tissue Res. 290, 11-20. [DOI] [PubMed] [Google Scholar]
- 2.Dickens, E. J., Hirst, G. D. & Tomita, T. (1999) J. Physiol. 514, 515-531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ördog, T., Ward, S. M. & Sanders, K. M. (1999) J. Physiol. 518, 257-269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ördog, T., Baldo, M., Danko, R. & Sanders, K. M. (2002) Gastroenterology 123, 2028-2040. [DOI] [PubMed] [Google Scholar]
- 5.Kelly, K. A. & Code, C. F. (1971) Am. J. Physiol. 220, 112-118. [DOI] [PubMed] [Google Scholar]
- 6.Cousins, H. M., Edwards, F. R., Hickey, H., Hill, C. E. & Hirst, G. D. (2003) J. Physiol. 550, 829-844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Horowitz, B., Ward, S. M. & Sanders, K. M. (1999) Annu. Rev. Physiol. 61, 19-43. [DOI] [PubMed] [Google Scholar]
- 8.Cannon, W. B. & Lieb, C. W. (1911) Am. J. Physiol. 29, 267-273. [Google Scholar]
- 9.Paton, W. D. M. & Vane, J. R. (1963) J. Physiol. 165, 10-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hennig, G. W., Brookes, S. J. & Costa, M. (1997) J. Physiol. 501, 197-212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cannon, W. B. (1911) in The Mechanical Factors of Digestion, eds. Hill, L. & Bullock, W. (Longmans Green, New York), pp. 48-58.
- 12.Burns, A. J., Lomax, A. E., Torihashi, S., Sanders, K. M. & Ward, S. M. (1996) Proc. Natl. Acad. Sci. USA 93, 12008-12013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ward, S. M., Beckett, E. A., Wang, X., Baker, F., Khoyi, M. & Sanders, K. M. (2000) J. Neurosci. 20, 1393-1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Beckett, E. A., Horiguchi, K., Khoyi, M., Sanders, K. M. & Ward, S. M. (2002) J. Physiol. 543, 871-887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Seki, K. & Komuro, T. (2001) Cell Tissue Res. 306, 417-422. [DOI] [PubMed] [Google Scholar]
- 16.Horiguchi, K., Sanders, K. M. & Ward, S. M. (2003) Cell Tissue Res. 311, 299-313. [DOI] [PubMed] [Google Scholar]
- 17.Hirst, G. D., Dickens, E. J. & Edwards, F. R. (2002) J. Physiol. 541, 917-928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Beckett, E. A., McGeough, C. A., Sanders, K. M. & Ward, S. M. (2003) J. Physiol. 553, 545-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Szurszewski, J. H. (1987) in Physiology of the Gastrointestinal Tract, ed. Johnson, L. R. (Raven, New York), 2nd Ed., pp. 383-422.
- 20.Kito, Y., Ward, S. M. & Sanders, K. M. (2005) Am. J. Physiol. 288, C710-C720. [DOI] [PubMed] [Google Scholar]
- 21.Edwards, F. R., Hirst G. D. S. & Suzuki, H. (1999) J. Physiol. 519, 235-250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dickens, E. J., Edwards, F. R. & Hirst, G. D. (2001) J. Physiol. 531, 827-833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Porcher, C., Horowitz, B., Bayguinov, O., Ward, S. M. & Sanders, K. M. (2002) Gastroenterology 122, 1442-1454. [DOI] [PubMed] [Google Scholar]
- 24.Kim, T. W., Beckett, E. A, Hanna, R., Koh, S. D., Ördog, T., Ward, S. M. & Sanders, K. M. (2002) J. Physiol. 538, 145-157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Publicover, N. G. & Sanders, K. M. (1989) Am. J. Physiol. 256, G265-G274. [DOI] [PubMed] [Google Scholar]
- 26.Kunze, W. A., Furness, J. B., Bertrand, P. P. & Bornstein, J. C. (1998) J. Physiol. 506, 827-842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Spencer, N. J. & Smith, T. K. (2004) J. Physiol. 558, 577-596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Desai, K. M., Zembowicz, A., Sessa, W. C. & Vane, J. R. (1991) Proc. Natl. Acad. Sci. USA 88, 11490-11494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Takahashi, T. & Owyang, C. (1997) J. Physiol. 504, 479-488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mizumoto, A., Mochiki, E., Suzuki, H., Tanaka, T. & Itoh, Z. (1997) J. Smooth Muscle Res. 33, 211-222. [DOI] [PubMed] [Google Scholar]
- 31.Parkman, H. P., Trate, D. M., Knight, L. C. Brown, K. L., Maurer, A. H. & Fisher, R. S. (1999) Gut 45, 346-354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grundy, D., Hutson, D. & Scratcherd, T. (1986) J. Physiol. 381, 377-384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sternini, C., Su, D., Gamp, P. D. & Bunnett, N. W. (1995) J. Comp. Neurol. 358, 531-540. [DOI] [PubMed] [Google Scholar]
- 34.Thuneberg, L. & Peters, S. (2001) Anat. Rec. 262, 110-124. [DOI] [PubMed] [Google Scholar]
- 35.Sanders, K. M. (1984) Am. J. Physiol. 247, G117-G126. [DOI] [PubMed] [Google Scholar]
- 36.Kim, C. H., Azpiroz, F. & Malagelada, J. R. (1986) Gastroenterology 90, 421-427. [DOI] [PubMed] [Google Scholar]
- 37.Kohagen, K. R., Kim, M. S., McDonnell, W. M., Chey, W. D., Owyang, C. & Hasler, W. L. (1996) Gastroenterology 110, 3-11. [DOI] [PubMed] [Google Scholar]
- 38.Sanders, K. M. & Szurszewski, J. H. (1981) Am. J. Physiol. 241, G191-G195. [DOI] [PubMed] [Google Scholar]
- 39.Matsumura, K., Watanabe, Y., Onoe, H., Watanabe, Y., Tanaka, S., Shiraki, T. & Kobayashi, S. (1993) Brain Res. 626, 343-346. [DOI] [PubMed] [Google Scholar]
- 40.Sutarmo Setiadji, V., Shibuya, I., Kabashima, N., Ibrahim, N., Harayama, N., Ueta, Y. & Yamashita, H. (1998) J. Neuroendocrinol. 10, 927-936. [DOI] [PubMed] [Google Scholar]
- 41.Baba, H., Kohno, T., Moore, K. A. & Woolf, C. J. (2001) J. Neurosci. 21, 1750-1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Evans, A. R., Vasko, M. R. & Nicol, G. D. (1999) J. Physiol. 516, 163-178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ren, J., Karpinski, E. & Benishin, C. G. (1995) J. Pharmacol. Exp. Ther. 275, 710-719. [PubMed] [Google Scholar]
- 44.Sadoshima, J., Takahashi, T., Jahn, L. & Izumo, S. (1992) Proc. Natl. Acad. Sci. USA 89, 9905-9909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kizer, N., Guo, X. L. & Hruska, K. (1997) Proc. Natl. Acad. Sci. USA 94, 1013-1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vandenburgh, H. H., Shansky, J., Karlisch, P. & Solerssi, R. L. (1993) J. Cell. Physiol. 551, 63-71. [DOI] [PubMed] [Google Scholar]
- 47.Alexander, L. D., Alagarsamy, S. & Douglas, J. G. (2004) Kidney Int. 65, 551-563. [DOI] [PubMed] [Google Scholar]
- 48.Koch, K. L., Hong, S. P. & Xu, L. (2000) J. Clin. Gastroenterol. 31, 125-129. [DOI] [PubMed] [Google Scholar]