

Abstract
MLL (mixed-lineage leukemia) is a histone H3 Lys-4 specific methyltransferase that is a positive regulator of Hox expression. MLL rearrangements and amplification are common in acute lymphoid and myeloid leukemias and myelodysplastic disorders and are associated with abnormal up-regulation of Hox gene expression. Although MLL is expressed throughout hematopoiesis, Hox gene expression is sharply down-regulated during differentiation, suggesting that either the activity of MLL or its association with target promoters must be regulated. Here we show that MLL associates with actively transcribed genes but does not remain bound after transcriptional down-regulation. Surprisingly, MLL is associated not only with promoter regions but also is distributed across the entire coding regions of genes. MLL interacts with RNA polymerase II (pol II) and colocalizes with RNA pol II at a subset of actively transcribed target in vivo. Loss of function Mll results in defects in RNA pol II distribution. Together the results suggest that an intimate association between MLL and RNA pol II occurs at MLL target genes in vivo that is required for normal initiation and/or transcriptional elongation.
Keywords: histone methyltransferase, Hox genes, transcription
Proper expression of the clustered HOX genes is essential for normal embryonic development. In addition, over expression of select HOX genes such as HOXA9 and the HOX cofactor MEIS1 has been implicated in human myelodysplastic disorders as well as acute lymphoid and myeloid leukemias. HOXA9 and MEIS1 are normally expressed only in early hematopoietic lineages, but during later stages of differentiation, expression is down-regulated to undetectable levels (1). The mixed-lineage leukemia protein MLL, which is homologous to Drosophila trithorax, is one important regulator of HOXA9 expression. MLL-knockout mice show severe hematopoietic defects associated with defects in Hox gene (including Hoxa9) expression (2-5). Conversely, MLL rearrangements are commonly associated with lymphoid and myeloid leukemias (6-8). Translocations involving MLL delete the sequences most conserved with D. trithorax and replace them with an in frame fusion to 1 of >40 different translocation partners (9, 10). MLL fusion proteins enforce persistent expression of HOXA9 and MEIS1, which appears to be critical for leukemogenesis (11). By itself, over-expression of HOXA9 induces stem cell expansion and is associated with poor-prognosis acute myeloid leukemia (12, 13). However, when coexpressed with MEIS1, HOXA9 is acutely transforming (14).
Recently, we and others found that the C-terminal SET domain of MLL protein is a histone methyltransferase that is specific for histone H3 Lys-4 (15, 16). MLL binds directly to Hox gene promoters and promotes transcriptional activation by methylating histone H3 on Lys-4 (15, 16) and also by recruiting MOF, a histone H4 Lys-16 specific acetyltransferase (17). Although an H3 Lys-4 demethylase has recently been identified (18), in general Lys-4 methylation is a long-lasting mark for sustained transcription (19). One unresolved question is how the activity of MLL is regulated. Although MLL is expressed throughout hematopoiesis, MLL target genes including HOXA9 and MEIS1 are down-regulated. One possibility is that MLL remains continually associated with targets but that the methyltransferase activity or recruitment of coactivators such as MOF (17), CBP (20), or the SWI/SNF complex (21) is inhibited. Alternatively, MLL could be regulated through modulation of its binding. To distinguish between these possibilities, we analyzed expression patterns of MLL target genes in different cell types and then determined how MLL association with these targets correlated with transcriptional activity. These studies show that MLL is associated with actively transcribed target genes but does not remain bound after transcriptional down-regulation. Surprisingly, MLL is associated not only with promoter regions but is also distributed across the entire coding regions of genes, overlapping the distribution of RNA polymerase (pol) II. MLL interacts with RNA pol II, and loss-of-function MLL results in defects in RNA pol II distribution. Together the results suggest that an intimate association between MLL and RNA pol II occurs at a subset of actively transcribed targets in vivo that is required for normal transcriptional initiation and/or elongation. The finding that MLL does not associate with many actively transcribed genes indicates that MLL is not a general transcription factor and that other mechanisms, possibly interactions with specific transcriptional coactivators and histone modifications, contribute to target gene specificity.
Materials and Methods
Cells and Cell Lines. MLL-ENL and MLL-AF9 cell lines and Mll+/+,-/- and Mll-/- lines expressing FLAG-tagged exogenous MLL (lines no. 6 and 16) are described in refs. 11, 15, and 22. Neutrophils were enriched from C57/Bl6 mouse whole bone marrow cells by using Ficoll-Hypaque (Becton Dickinson) density gradient centrifugation resulting in 80-90% purity.
Chromatin Immunoprecipitation (ChIP). ChIPs were performed as described in ref. 22 except that mouse primary antibodies (Abs) were incubated overnight, then incubated with 2 μg of anti-mouse IgG for 7 h, then incubated with agarose A for 4 h, all at 4°C. ChIP was quantified by using Real-time PCR (Applied Biosystems) as described in ref. 22. Taqman primer and probe sequences are available from the authors on request. The source of Abs for Western blot and immunoprecipitation are provided in Supporting Text, which is published as supporting information on the PNAS web site.
RNA Expression Analysis. Real-time PCR quantification of gene expression was performed as described in ref. 22. Briefly, standard curves were used to compare expression levels of individual genes with either Gapdh or β-actin. Unit values represent ratios so that comparisons of absolute levels of gene expression are not possible. Taqman probe and primer sequences will be provided by the authors on request.
GST Pull-Down Assay. For GST pull-down assays with HeLa nuclear extract, binding reactions contained 2 μg of GST-tagged protein and 500 μl of HeLa nuclear extract. Reaction mixtures included 150 mM KCl, 0.05% Nonidet P-40, 20 mM Tris, 20% glycerol, and protease inhibitor mixture (Roche Diagnostics). Binding was carried out at 4°C for 4 h, and beads were washed three times with BC150.
C-Terminal MLL (MLLC) Immunoprecipitations. Eight hundred micrograms of MLL-AF9 nuclear extracts prepared from large-scale cultures were incubated either with 5 μg of the MLLC Ab or with 5 μg of rabbit anti-mouse IgG (Upstate Biotechnology, Lake Placid, NY, catalog no. 06-371) overnight at 4°C. Ab/protein complexes were collected with agarose A beads at 4°C for 1 h, and beads were washed three times with 150 mM KCl, 0.05% Nonidet P-40, 20 mM Tris, 20% glycerol, and protease inhibitor mixture (Roche Diagnostics). Proteins were eluted by boiling beads for 10 min in a 1× SDS loading buffer (pH 6.8).
Results
Hox and Meis1 Transcription Varies with Cell Type and Stage of Hematopoietic Differentiation. Early hematopoietic progenitors express a variety of Hox genes including Hoxa9, Hoxa7, and Meis1 (1), whereas in more differentiated cell types these genes are down-regulated. Different Hoxa9 transcripts are expressed depending on the cell type including a long transcript a9a with an upstream start site and two smaller alternatively spliced transcripts a9b and a9T (Fig. 1A) (23-25). All three Hoxa9 transcripts in addition to Hoxa7 and Meis1 are highly expressed in MLL-AF9 and MLL-ENL transformed cells and down-regulated in neutrophils (Fig. 1B). Although MLL is expressed in all cell types (Figs. 1B and 2 D and E), Hoxc8, which is expressed and regulated by MLL in mouse embryonic fibroblasts (MEFs), is not expressed in any hematopoietic cells (Fig. 1B).
Fig. 1.

Expression of MLL target genes in hematopoietic cells. (A) Schematic of the Hoxa9 locus showing three major Hoxa9 transcripts, one upstream transcript (a9a) and two downstream transcripts (a9b and a9T). The a9b and a9T transcripts originate from different start sites (data not shown). The positions of all Taqman primer/probe sets (1-11) used for Hoxa9 ChIP experiments are indicated by arrowheads above the schematic. The homeodomain (HD) is noted as a hatched box and is contained in an exon that is common to all three transcripts. Dashed lines indicate multiple transcription start sites. Open boxes on the line represent exons, black bars beneath the line are CpG-rich regions, light gray boxes beneath the line are AT-rich regions, and medium gray bars indicate regions of highest homology between mouse and human. Distances (in kb) are indicated below the schematic. (B) Meis1, Hoxa7, and all three Hoxa9 isoforms are expressed in MLL-ENL (4) and MLL-AF9 (5) transformed lines and are minimally expressed in neutrophils. Mll is expressed, but Hoxc8 is silent, in all three cell types.
Fig. 2.

Assessment of MLL binding to target loci in hematopoietic cells. (A) Schematic of the Meis1, Hoxa1, and Hoxa7 loci. Gray bars on the line represent exons; arrows and arrowheads indicate location of probe/primer sets used for quantitative PCR, and black bars represent CpG-rich regions. (B) Legend for C-E. Blue and pink represent the myeloblast cell lines MLL-ENL and MLL-AF9, respectively, and yellow indicates neutrophils. (C) ChIP using Abs specific for MLLC shows that MLL binds directly to the coding region of Meis1 and the promoter region of Hoxa7 in myeloblast cell lines but not in neutrophils. The Hoxa1 and Gapdh loci are not MLL binding targets. Error bars are shown for all, but in some cases they are too small to be visible. (D) Western blot showing MLLC expression (top band, arrowhead) in neutrophils. The lower band is a nonspecific band reported by Hsieh et al. (26). (E) Western blot showing MLLC expression (top band, arrowhead) in MLL-AF9 cells. (F) ChIP showing distribution of MLLC across the Hoxa9 and Hoxc8 loci in myeloblastic cell lines and neutrophils. MLLC binds to Hoxa9 upstream and coding regions, but not to Hoxc8, which is not expressed in the myeloblastic lines. No binding of MLLC is seen in neutrophils to either the silenced Hoxa9 or Hoxc8 loci. The white line marks the ATG start site. (G) ChIP using Abs specific for the RNA pol II CTD shows that the distribution of RNA pol II closely corresponds to MLL localization.
MLL Selectively Binds to Transcriptionally Active Target Genes. ChIP was used to map MLLC binding sites at target loci in the myeloblastic cell lines and in MEFs. The MLLC Ab recognizes only wild-type MLL and does not cross-react with MLL fusion proteins. Minimal Mll binding was detected at silenced genes including Hoxa9 in neutrophils (Fig. 2F) and the Hoxc8 locus in all cell types (Fig. 2F). Conversely, in MLL-AF9 and MLL-ENL transformed cells, which express Hoxa9, MLL binding is detected across the Hoxa9 locus (Fig. 2F). Similarly, MLL bound to the Hoxa7 and Meis1 target genes (Fig. 2C) but not in neutrophils where these genes are not expressed (Fig. 2C). MLL is proteolytically processed into two fragments, N-terminal MLL (MLLN) (320 kDa) and MLLC (180 kDa), that noncovalently associate in the nucleus (16, 26-28). The loss of binding in neutrophils is unlikely to represent lack of proteolytic processing because MLLC protein levels are similar in neutrophils (Fig. 2D) compared with MLL-AF9 cells (Fig. 2E). In addition, in all ChIP experiments binding of MLLC and MLLN was highly correlated (Fig. 3; see also Figs. 6 and 7, which are published as supporting information on the PNAS web site), suggesting that MLL is most likely regulated by changes in MLLN binding rather than by modulating the association between MLLC and MLLN.
Fig. 3.

Mll association with the Hoxa9 locus in fibroblasts. (A) Quantitative PCR shows that Hoxa9b and Hoxa9T are expressed at higher levels in Mll+/+ fibroblasts relative to Mll-/- fibroblasts. Reexpression of MLL in Mll-/- cells (F-MLL#6 and F-MLL#16) up-regulates transcription of the Hoxa9a and a9T transcripts. The upstream a9a transcript, which is expressed in hematopoietic cells, is not expressed in any of these fibroblast cells. (B) Legend for C-E. Blue and pink represent Mll+/+ and Mll-/- fibroblast cells, respectively, and yellow represents the FMLL#16 line. The white line in C-E marks the ATG start of the Hoxa9. (C) MLLC binding. ChIP using Abs specific for the C-terminal (MLLC) proteolytic fragment of MLL shows highest levels of binding in the Hoxa9 coding and promoter region in Mll expressing cells and no binding at the transcriptionally inactive upstream region. Similar results were obtained in ChIP experiments by using Abs against MLLN. (D) MLLN binding. ChIP for the RNA pol II CTD shows that RNA pol II is distributed across the coding region of Hoxa9 in Mll+/+ cells and F-MLL#16 cells corresponding to peaks of Mll binding. A small but reproducible peak of RNA pol II is seen centered on the downstream Hoxa9 promoter (at ≈5 Kb) in Mll-/- cells. Stable reexpression of MLL results in redistribution of pol II in the downstream Hoxa9 coding region. Schematic indicates position of two alternative start sites. (E) RNA pol II binding.
In contrast to myeloblastic cells, in Mll+/+ MEFs, only the downstream Hoxa9b and a9T transcripts are expressed (Fig. 3A). Expression of these transcripts is decreased in Mll-/- cells and is rescued by MLL reexpression (F-MLL#6 and F-MLL#16) indicating that both are MLL targets (Fig. 3A). In contrast, the a9a transcript expressed in hematopoietic cells is not expressed in Mll+/+ cells and is not reactivated by MLL (Fig. 3A). MLL is associated with the a9b and a9T, but not a9a, start sites, indicating that MLL associates only with loci when they are actively transcribed. Furthermore, MLL association with transcriptionally active genes is specific, because no binding was detected at either the Gapdh or Hoxa1 loci (Fig. 2C), which are not regulated by MLL (15).
MLL Binding Interacts with and Colocalizes with RNA pol II. Collectively our results indicate that MLL binding is dynamic. MLL binds to actively transcribed regions, but not inactive loci, even when those loci are regulated by MLL in other cell types. Therefore, MLL recruitment to target must involve more than DNA sequence recognition, such as protein-protein interactions. One feature recognized by MLL complexes could be covalent histone modifications such as acetylation or histone methylation. The MLL bromodomain and SET domain could bind acetylated histones. In addition, WDR5, a core component of the MLL methyltransferase complex, preferentially interacts with dimethylated histone H3 Lys-4 (29).
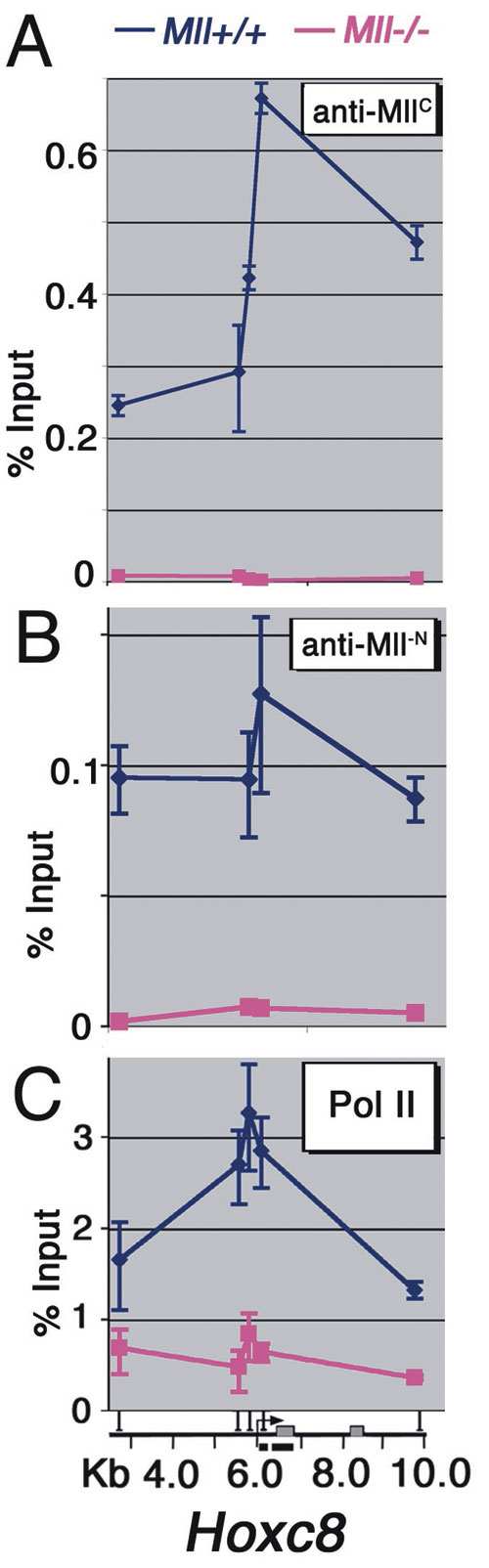
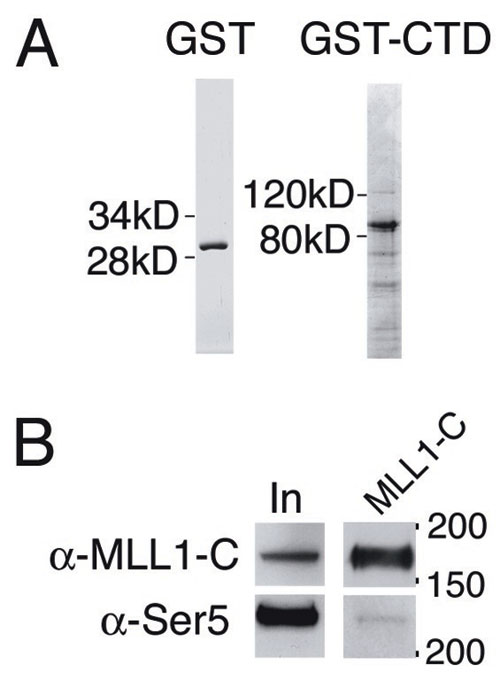
MLL binding also may involve interactions with promoter-bound transcription factors or with RNA pol II. We tested whether MLL interacted with the RNA pol II C-terminal domain (CTD) by GST pull-downs of nuclear extracts. In this assay, both MLLN and MLLC interact with the RNA pol II CTD (Fig. 4A, GST-CTD lane). As reported in ref. 30, we also detected an interaction between GST-CTD and menin and MLL2 (Fig. 4A, GST-CTD lane). Immunoprecipitations with an MLLC-specific Ab effectively precipitated MLL1 (Fig. 4B, MLL1-C lane) along with the phospho-Ser-5 form of RNA pol II (Fig. 4B, Ser-5) and menin (Fig. 4B, menin). Similar results were seen in HeLa cells (Fig. 7B). ChIP with Abs specific for the CTD of RNA pol II at MLL target genes ChIP also showed striking colocalization of RNA pol II and MLL in vivo (Fig. 2 F vs. G). In contrast, MLL and pol II show decreased or absent binding at the Hoxa9 locus in neutrophils (Fig. 2 F vs. G) and at the Hoxc8 locus in all cell types (Fig. 2 F vs. G). Similarly, MLL and RNA pol II colocalize at Hoxa9 and Hoxc8 in fibroblasts, with peaks of binding in the downstream coding region of Hoxa9 (Fig. 3E) and the promoter region of Hoxc8 (Fig. 6C). In the F-MLL#16 line, F-MLL binding peaks near the putative TATA box at Hoxa9 (Fig. 3 C and D, yellow line) and is reduced at the more 3′ end of the gene. This binding pattern is paralleled by reduced pol II binding at the 3′ end of Hoxa9 in F-MLL#16 cells (Fig. 3E, yellow line).
Fig. 4.

MLL binds to RNA pol II and affects histone modifications and transcription elongation. (A) Interaction of MLL1-N and -C (indicated on the right) with either GST alone or RNA pol II C-terminal tail GST fusion (GST-CTD, indicated at the top). Consistent with previous experiments (30), an interaction of MLL2 and menin with the GST-CTD also was seen. In, input. (B) Immunoprecipitations performed with the Mll1-C Ab coprecipitates menin as well as the phospho-Ser-5 form of RNA pol II. Immunoprecipitations were performed in MLL-AF9 nuclear extracts. Control IPs with rabbit IgG using the same extracts also are shown. (C and D) ChIP using Abs specific for either Ser-2 (C) or Ser-5 (D) phosphorylated RNA pol II at the Hoxa9 TATA box, first exon and homeodomain. Taqman primer/probe sets used are shown in H. ChIP shows that Ser-2 is increased in the coding region (exon, HD) relative to the promoter (TATA), whereas Ser-5 is instead increased at the promoter (TATA) in Mll+/+ (blue) and F-MLL#16 (yellow) cells. Conversely, in Mll-/- cells (pink), Ser-2 and -5 are concentrated in small peaks at the promoter, suggesting a defect in transcription elongation. (E-H) ChIP for various histone modifications shows high levels of each mark at the promoter and in the coding regions of the Hoxa9 locus in Mll+/+ cells (blue). Each mark is drastically reduced in Mll-/- cells (pink) and is partially restored by MLL reexpression (yellow). Distances across the Hoxa9 locus (in kb) are shown across the bottom of each image. Positions of Taqman primer/probe sets are shown at the bottom of G and H. (E) Histone H3 dimethyl Lys-4 ChIP. (F) Histone H3 trimethyl Lys-4 ChIP. (G) Histone H3 acetyl Lys-9 ChIP. (H) Histone H3 dimethyl Lys-79 ChIP.
Mll-Null Cells Show Defects in Transcriptional Elongation and Loss of Multiple Histone Modifications. The colocalization of MLL and RNA pol II at target loci in vivo raises the possibility that MLL promotes transcriptional elongation. If so, the distribution of RNA pol II would be expected to be abnormal in Mll-null cells. In yeast, one of the earliest events in transcriptional elongation is phosphorylation of RNA pol II CTD at Ser-5, which is followed by dephosphorylation at Ser-5 and phosphorylation at Ser-2 (31). Similarly, in Mll-/- cells, both Ser-2 and -5 phosphorylated forms of RNA pol II are abnormally enriched at the TATA box (Fig. 4 C and D) (32, 33). Importantly, reexpression of MLL restored a normal pol II phosphorylation pattern (Mll+/+ and F-MLL cells) with Ser-2 phosphorylation increased in the coding region relative to the TATA box (Fig. 4C) and Ser-5 phosphorylation increased near the TATA box relative to the Hoxa9 coding region (Fig. 4D).
In yeast, successful transcriptional elongation entails association of Set1, a histone H3 Lys-4 methyltransferase and Dot1 a histone H3 Lys-79 methyltransferase with the elongating pol II complex (34, 35). Whether a similar interaction occurs in mammalian cells is not known. In MEF cells, histone H3 Lys-4 di- and trimethylation is increased along with Lys-79 methylation at the Hoxa9 locus (Fig. 4 E and F) in a pattern that colocalizes with both MLL (Fig. 3 C and D) and RNA pol II (Fig. 3E). This result suggests that in mammalian cells, H3 Lys-4 and -79 methylation results from successful transcriptional elongation across coding regions. Smaller increases in Lys-4 and -79 methylation in the F-MLL #16 line correlate with lower levels in the Hoxa9 coding region (Fig. 3 C and D).
Histone acetylation also changes across the Hox loci in an MLL-dependent way. Recently, we showed that histone H4 Lys-16-specific acetylation by MOF contributes to Hox gene expression (17). ChIP with Abs to other specific acetyl residues shows that both histone H3 Lys-9 (Fig. 4G) and Lys-27 (data not shown) acetylation are dramatically increased across the Hoxa9 locus and colocalize with MLL binding. Conversely, histone H3 Lys-14 acetylation appears to be Mll-independent. We did not identify any in vitro histone H3 acetyltransferase activity in purified MLL complexes (17), which suggests that increases in histone H3 acetylation are downstream of MLL-mediated Lys-4 methylation and MOF-mediated Lys-16 acetylation.
MLL Is Not a Member of the General pol II Transcription Machinery. A recent report (36) suggests that MLL is a global transcriptional regulator that is recruited to virtually all sites of RNA pol II activity. To explore this theory, we compared MLL binding patterns with RNA pol II binding patterns at a number of MLL target genes, all of which are expressed in MEF cells. We confirmed that Hoxa7 and Meis1 are direct target genes of MLL. In addition, here we show FoxC1 and FoxC2 are two additional MLL target genes, which we originally identified through microarray expression analysis of Mll+/+ and Mll-/- cells (data not shown). We also identified genes that are not regulated by MLL, including the Hox cofactors Pbx1 and Pbx3, as well as Hoxa1 and Gapdh, both previously identified to be MLL independent (15). MLLC, MLLN, and menin all bind directly to Hoxa7, Meis1, FoxC1, and FoxC2 (Fig. 5 A and B; see also Fig. 8, which is published as supporting information on the PNAS web site). In Mll-/- cells expression of these four genes is reduced but not completely abolished (data not shown). This reduction is accompanied by a parallel reduction in RNA pol II binding (Fig. 5C). Conversely, MLL and menin do not bind to Hoxa1, Gapdh, Pbx1, Pbx3, or the endogenous Mll locus (Fig. 5 A and B). All five of these genes are expressed at approximately equal levels in Mll+/+ and Mll-/- cells (data not shown). Although ample RNA pol II is localized to these genes, no MLL binding was detected (Fig. 5C).
Fig. 5.

Mll and pol II association with other target genes in fibroblasts. (A) MLLC binding. ChIP using Abs specific for MLLC shows MLL binding at the Hoxa7, Meis1, FoxC1, and FoxC2 loci but not at Hoxa1, Pbx1, Pbx3, Mll or Gapdh. Positions of Taqman primer/probe sets used for quantification are shown in Figs. 2 and 8. (B) Menin binding. Menin binds to the same target genes as Mll. (C) RNA pol II binding. RNA pol II shows an Mll and menin-dependent increase in binding for the Hoxa7, Meis1, FoxC1, and FoxC2 target loci, but pol II binding is independent of Mll and menin at the Hoxa1, Pbx1, Pbx3, Mll, and Gapdh genes.
Discussion
Despite intensive study, the factors that govern the recruitment of MLL to target genes promoters have remained elusive. The data presented here indicate that MLL associates with transcriptionally active genes and with RNA pol II. At the same time, our data show recruitment is highly specific and cannot simply be the result of RNA pol II recruitment of MLL to all genes. MLL recognition of targets almost certainly involves DNA interactions. MLL contains AT hook motifs, which bind to AT-rich DNA (37), and a DNA methyltransferase homology region that binds to CpG-rich DNA (38). However to date the AT hook or DNA methyltransferase homology regions are known to have only general affinity for AT- and GC-rich sequences. Although many Mll targets contain CpG rich regions within their first exons (Figs. 1 and 2), non-Mll targets such as Hoxa1 and Gapdh also contain CpG-rich sequences. This finding suggests that the CpG binding activity of MLL by itself is insufficient for recruitment to target genes and that other, more specific interactions are involved. In addition, our data show that MLL associates only with transcriptionally active promoters and therefore is cell-type and differentiation-stage specific.
We suggest that the specificity of MLL for a subset of transcriptionally active promoters involves combinatorial interactions with specific transcription factors as well as specific histone modifications that ultimately result in localization of MLL to specific targets. Much of the support for this model comes from our finding that MLL is associated with p53 and, most importantly, that p53-dependent MLL recruitment is required for transcriptional activation in vitro (17). Although the putative transcriptional cofactors involved in Hox regulation remain to be identified, it is noteworthy that the MLL complex contains sequence-specific transcription factors Max (17) and E2F6, which reportedly associates with Bmi-1, a known regulator of Hox gene expression (39). We propose that Hoxa9 is down-regulated and MLL is dissociated from the locus in neutrophils as the concentration specific transcription factors required decrease with differentiation.
Preexisting patterns of histone modifications may provide additional affinity for MLL localization to targets. SET domains have been shown to show selective binding and activity with specific histone modifications. We found that MLL preferentially methylates histone tails that are already acetylated (15). In addition, the trx SET domain binds more avidly to acetylated nucleosomes (40). Another possibility is that the WDR5 protein, a component of the MLL complex, targets MLL to loci dimethylated at histone H3 Lys-4 (29). It is noteworthy in this regard that the Hox loci show large blocks of dimethylation encompassing coding regions in contrast to many other genes where modification is localized to the promoter (41)
The mechanism by which MLL and trx promote transcription is largely unknown Our previous experiments showing deletion of the MLL SET domain abolished the ability of MLL to activate Hox gene expression provide strong evidence that histone H3 Lys-4 methylation is important. However, recently we found that both histone H4 acetylated mediated by MOF and histone H3 Lys-4 methylation contribute to transcriptional activation in an in vitro transcription assay (17). In addition, MLL recruitment of SWI/SNF complexes to target genes (21) also could be involved in overcoming the nucleosomal barrier to transcriptional elongation. The patterns of RNA pol Ser-2 and -5 phosphorylation in Mll-/- cells furthermore suggest that the transcriptional activating effects of Mll may be mediated at the level of promoting transcriptional elongation as has been proposed for trx (42). Additional experiments will be needed to determine whether this mechanism involves physical association of MLL and progressive RNA pol or whether it represents an indirect effect on elongation and how leukemogenic fusion proteins alter or replace the activity of wild-type MLL on transcription initiation and elongation. The Hox and Meis1 loci and the experimental approaches outlined here provide an ideal approach for exploring this mechanism in detail.
Supplementary Material
Acknowledgments
This work is dedicated to the memory of Dr. Stanley J. Korsmeyer. J.L.H. was supported by grants from the National Institutes of Health and by a Specialized Center of Research grant from the Leukemia and Lymphoma Society of America. T.A.M. is supported by a Fellowship from the Canadian Institute of Health Research.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: MLL, mixed-lineage leukemia; MEF, mouse embryonic fibroblast; pol, polymerase; CTD, C-terminal domain; ChIP, chromatin immunoprecipitation.
See Commentary on page 14481.
References
- 1.Pineault, N., Helgason, C. D., Lawrence, H. J. & Humphries, R. K. (2002) Exp. Hematol. 30, 49-57. [DOI] [PubMed] [Google Scholar]
- 2.Ernst, P., Fisher, J. K., Avery, W., Wade, S., Foy, D. & Korsmeyer, S. J. (2004) Dev. Cell 6, 437-443. [DOI] [PubMed] [Google Scholar]
- 3.Hess, J. L., Yu, B. D., Li, B., Hanson, R. & Korsmeyer, S. J. (1997) Blood 90, 1799-1806. [PubMed] [Google Scholar]
- 4.Yagi, H., Deguchi, K., Aono, A., Tani, Y., Kishimoto, T. & Komori, T. (1998) Blood 92, 108-117. [PubMed] [Google Scholar]
- 5.Yu, B. D., Hess, J. L., Horning, S. E., Brown, G. A. & Korsmeyer, S. J. (1995) Nature 378, 505-508. [DOI] [PubMed] [Google Scholar]
- 6.Armstrong, S. A., Staunton, J. E., Silverman, L. B., Pieters, R., den Boer, M. L., Minden, M. D., Sallan, S. E., Lander, E. S., Golub, T. R. & Korsmeyer, S. J. (2002) Nat. Genet 30, 41-47. [DOI] [PubMed] [Google Scholar]
- 7.Rozovskaia, T., Feinstein, E., Mor, O., Foa, R., Blechman, J., Nakamura, T., Croce, C. M., Cimino, G. & Canaani, E. (2001) Oncogene 20, 874-878. [DOI] [PubMed] [Google Scholar]
- 8.Yeoh, E. J., Ross, M. E., Shurtleff, S. A., Williams, W. K., Patel, D., Mahfouz, R., Behm, F. G., Raimondi, S. C., Relling, M. V., Patel, A., et al. (2002) Cancer Cell 1, 133-143. [DOI] [PubMed] [Google Scholar]
- 9.Ayton, P. M. & Cleary, M. L. (2001) Oncogene 20, 5695-5707. [DOI] [PubMed] [Google Scholar]
- 10.Hess, J. L. (2004) Trends Mol. Med. 10, 500-507. [DOI] [PubMed] [Google Scholar]
- 11.Zeisig, B. B., Milne, T., Garcia-Cuellar, M. P., Schreiner, S., Martin, M. E., Fuchs, U., Borkhardt, A., Chanda, S. K., Walker, J., Soden, R., et al. (2004) Mol. Cell. Biol. 24, 617-628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moskow, J. J., Bullrich, F., Huebner, K., Daar, I. O. & Buchberg, A. M. (1995) Mol. Cell. Biol. 15, 5434-5443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nakamura, T., Largaespada, D. A., Shaughnessy, J. D., Jr., Jenkins, N. A. & Copeland, N. G. (1996) Nat. Genet 12, 149-153. [DOI] [PubMed] [Google Scholar]
- 14.Kroon, E., Krosl, J., Thorsteinsdottir, U., Baban, S., Buchberg, A. M. & Sauvageau, G. (1998) EMBO J. 17, 3714-3725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Milne, T. A., Briggs, S. D., Brock, H. W., Martin, M. E., Gibbs, D., Allis, C. D. & Hess, J. L. (2002) Mol. Cell 10, 1107-1117. [DOI] [PubMed] [Google Scholar]
- 16.Nakamura, T., Mori, T., Tada, S., Krajewski, W., Rozovskaia, T., Wassell, R., Dubois, G., Mazo, A., Croce, C. M. & Canaani, E. (2002) Mol. Cell 10, 1119-1128. [DOI] [PubMed] [Google Scholar]
- 17.Dou, Y., Milne, T. A., Tackett, A. J., Smith, E. R., Fukuda, A., Wysocka, J., Allis, C. D., Chait, B. T., Hess, J. L. & Roeder, R. G. (2005) Cell, in press. [DOI] [PubMed]
- 18.Shi, Y., Lan, F., Matson, C., Mulligan, P., Whetstine, J. R., Cole, P. A. & Casero, R. A. (2004) Cell 119, 941-953. [DOI] [PubMed] [Google Scholar]
- 19.Lachner, M. & Jenuwein, T. (2002) Curr. Opin. Cell Biol. 14, 286-298. [DOI] [PubMed] [Google Scholar]
- 20.Ernst, P., Wang, J., Huang, M., Goodman, R. H. & Korsmeyer, S. J. (2001) Mol. Cell. Biol. 21, 2249-2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rozenblatt-Rosen, O., Rozovskaia, T., Burakov, D., Sedkov, Y., Tillib, S., Blechman, J., Nakamura, T., Croce, C. M., Mazo, A. & Canaani, E. (1998) Proc. Natl. Acad. Sci. USA 95, 4152-4157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Martin, M. E., Milne, T. A., Bloyer, S., Galoian, K., Shen, W., Gibbs, D., Brock, H. W., Slany, R. & Hess, J. L. (2003) Cancer Cell 4, 197-207. [DOI] [PubMed] [Google Scholar]
- 23.Fujimoto, S., Araki, K., Chisaka, O., Araki, M., Takagi, K. & Yamamura, K. (1998) Gene 209, 77-85. [DOI] [PubMed] [Google Scholar]
- 24.Kim, M. H., Chang, H. H., Shin, C., Cho, M., Park, D. & Park, H. W. (1998) DNA Cell Biol. 17, 407-414. [DOI] [PubMed] [Google Scholar]
- 25.Dintilhac, A., Bihan, R., Guerrier, D., Deschamps, S., Pellerin I. (2004) Gene Expression Patterns 4, 215-222. [DOI] [PubMed] [Google Scholar]
- 26.Hsieh, J. J., Ernst, P., Erdjument-Bromage, H., Tempst, P. & Korsmeyer, S. J. (2003) Mol. Cell. Biol. 23, 186-194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yokoyama, A., Wang, Z., Wysocka, J., Sanyal, M., Aufiero, D. J., Kitabayashi, I., Herr, W. & Cleary, M. L. (2004) Mol. Cell. Biol. 24, 5639-5649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xia, Z. B., Anderson, M., Diaz, M. O. & Zeleznik-Le, N. J. (2003) Proc. Natl. Acad. Sci. USA 100, 8342-8347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wysocka, J., Swigut, T., Milne, T. A., Dou, Y., Zhang, X., Burlingame, A.L., Roeder, R. G., Brivanlou, A. H. & Allis, C. D. (2005) Cell 121, 859-872. [DOI] [PubMed] [Google Scholar]
- 30.Hughes, C. M., Rozenblatt-Rosen, O., Milne, T. A., Copeland, T. D., Levine, S. S., Lee, J. C., Hayes, D. N., Shanmugam, K. S., Bhattacharjee, A., Biondi, C. A., et al. (2004) Mol. Cell 13, 587-597. [DOI] [PubMed] [Google Scholar]
- 31.Gerber, M. & Shilatifard, A. (2003) J. Biol. Chem. 278, 26303-26306. [DOI] [PubMed] [Google Scholar]
- 32.Morillon, A., Karabetsou, N., O'Sullivan, J., Kent, N., Proudfoot, N. & Mellor, J. (2003) Cell 115, 425-435. [DOI] [PubMed] [Google Scholar]
- 33.Morillon, A., O'Sullivan, J., Azad, A., Proudfoot, N. & Mellor, J. (2003) Science 300, 492-495. [DOI] [PubMed] [Google Scholar]
- 34.Feng, Q., Wang, H., Ng, H. H., Erdjument-Bromage, H., Tempst, P., Struhl, K. & Zhang, Y. (2002) Curr. Biol. 12, 1052-1058. [DOI] [PubMed] [Google Scholar]
- 35.Krogan, N. J., Dover, J., Wood, A., Schneider, J., Heidt, J., Boateng, M. A., Dean, K., Ryan, O. W., Golshani, A., Johnston, M., et al. (2003) Mol. Cell 11, 721-729. [DOI] [PubMed] [Google Scholar]
- 36.Guenther, M. G., Jenner, R. G., Chevalier, B., Nakamura, T., Croce, C. M., Canaani, E. & Young, R. A. (2005) Proc. Natl. Acad. Sci. USA 102, 8603-8608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zeleznik-Le, N. J., Harden, A. M. & Rowley, J. D. (1994) Proc. Natl. Acad. Sci. USA 91, 10610-10614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Birke, M., Schreiner, S., Garcia-Cuellar, M. P., Mahr, K., Titgemeyer, F. & Slany, R. K. (2002) Nucleic Acids Res. 30, 958-965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Trimarchi, J. M., Fairchild, B., Wen, J. & Lees, J. A. (2001) Proc. Natl. Acad. Sci. USA 98, 1519-1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Katsani, K. R., Arredondo, J. J., Kal, A. J. & Verrijzer, C. P. (2001) Genes Dev. 15, 2197-2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bernstein, B. E., Kamal, M., Lindblad-Toh, K., Bekiranov, S., Bailey, D. K., Huebert, D. J., McMahon, S., Karlsson, E. K., Kulbokas, E. J., III, Gingeras, T. R., et al. (2005) Cell 120, 169-181. [DOI] [PubMed] [Google Scholar]
- 42.Smith, S. T., Petruk, S., Sedkov, Y., Cho, E., Tillib, S., Canaani, E. & Mazo, A. (2004) Nat. Cell Biol. 6, 162-167. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}