

Abstract
Human Tsg101 plays key roles in HIV budding and in cellular vacuolar protein sorting (VPS). In performing these functions, Tsg101 binds both ubiquitin (Ub) and the PTAP tetrapeptide ‘late domain’ motif located within the viral Gag protein. These interactions are mediated by the N-terminal domain of Tsg101, which belongs to the catalytically inactive ubiquitin E2 variant (UEV) family. We now report the struc ture of Tsg101 UEV and chemical shift mapping of the Ub and PTAP binding sites. Tsg101 UEV resembles canonical E2 ubiquitin conjugating enzymes, but has an additional N-terminal helix, an extended β-hairpin that links strands 1 and 2, and lacks the two C-terminal helices normally found in E2 enzymes. PTAP-containing peptides bind in a hydrophobic cleft exposed by the absence of the C-terminal helices, whereas ubiquitin binds in a novel site surrounding the β-hairpin. These studies provide a structural framework for understanding how Tsg101 mediates the protein–protein interactions required for HIV budding and VPS.
Keywords: late domain/Tsg101/ubiquitin E2 variant/vacuolar protein sorting/virus budding
Introduction
Human Tsg101 has recently been identified as the functional receptor required for budding of the enveloped human immunodeficiency (HIV) and Ebola viruses (Garrus et al., 2001; Martin-Serrano et al., 2001; VerPlank et al., 2001; Demirov et al., 2002). Tsg101 is recruited to the sites of virus budding by binding to a P(S/T)AP tetrapeptide motif (the ‘late domain’) located within the p6 region of HIV Gag and the Ebola Vp40 matrix structural proteins. Once there, Tsg101 appears to recruit other cellular factors that help to complete the budding process (Garrus et al., 2001; Martin-Serrano et al., 2001). In the absence of Tsg101, the final membrane fission step(s) fails and HIV release is arrested at a very late stage, in which the assembled viral particles remain attached to the plasma membrane (and to one another) via thin membrane stalks (Garrus et al., 2001).
The direct interaction between Tsg101 and the PTAP late domain motifs on the viral structural proteins is mediated by the N-terminal ubiquitin E2 variant (UEV) domain of Tsg101 (Garrus et al., 2001; Martin-Serrano et al., 2001; VerPlank et al., 2001), whereas the remaining C-terminal two-thirds of Tsg101 interacts with other proteins (Figure 1A). Like other UEV domains, Tsg101 UEV shows significant sequence similarity to E2 ubiquitin ligases but is unable to catalyze ubiquitin transfer as it lacks the active site cysteine that forms the transient thioester bond with the C-terminus of ubiquitin (Ub) (Koonin and Abagyan, 1997; Ponting et al., 1997) (Figure 1B). Nevertheless, at least some UEVs have retained the ability to bind Ub, and appear to act either as cofactors in ubiquitylation reactions, or as ubiquitin sensors (Hofmann and Pickart, 1999; Garrus et al., 2001; Katzmann et al., 2001; VanDemark et al., 2001; VerPlank et al., 2001). As discussed below, UEV domains also frequently contain other protein recognition motifs, and may generally serve to couple protein and Ub binding functions to facilitate the formation of multiprotein complexes.

Fig. 1. Sequence and structure of Tsg101. (A) Domain organization of Tsg101, showing the approximate domain boundaries of the UEV domain, proline-rich domain (PRD), putative coiled coil domain (COIL) and ‘steadiness box’ (SBOX) (Feng et al., 2000). The internal PTAP sequence between the coiled-coil domain and the steadiness box is shown explicitly. (B) Structure-based sequence alignment of UEV and E2 proteins. The secondary structures of Tsg101 UEV, uncomplexed human Mms2 (Moraes et al., 2001) and uncomplexed yeast Ubc13 (VanDemark et al., 2001) are shown at the top, middle and bottom, respectively. Conserved residues are shaded gray, identical residues black. The active site cysteine of catalytic E2 enzymes and the equivalent residues in UEV proteins are green. Tsg101 UEV residues that are important for binding PTAP and Ub are colored red and blue, respectively. (C) Stereo view superposition of the final 20 NMR structures of the Tsg101 UEV domain. Secondary structures are colored as in (B).
Tsg101 normally functions in the cellular vacuolar protein sorting (VPS) pathway, which coordinates the sorting of membrane-associated proteins through a series of endosomal compartments for eventual delivery to the lysosome (vacuole in yeast) (Dupre et al., 2001; Hicke, 2001; Piper and Luzio, 2001). A key decision-making step in this pathway occurs when membrane patches containing proteins destined for destruction bud as small vesicles into the lumen of the late endosome, creating an organelle called the ‘multivesicular body’ (MVB). Subsequent fusion of the MVB with the lysosome delivers these vesicles and all associated cargo to the lumen of the lysosome, where they are degraded by proteolysis. In contrast, proteins that remain in the limiting membrane of MVB are delivered to the limiting membrane of lysosomes and therefore escape degradation.
Recent work, particularly in the yeast system, has revealed that Tgs101 (yeast Vps23p) performs an important role in selecting which proteins enter the MVB lumen and which remain on the limiting membrane. In yeast, proteins destined for the lumen of the vacuole are covalently modified with monoubiquitin and bound by a 350-kDa protein complex called ESCRT-I, which contains the proteins Vps23p, Vps28p and Vps37p (Katzmann et al., 2001). ESCRT-I appears to function as the receptor and/or sorting complex that selects ubiquitylated proteins for incorporation into MVB vesicles. Consistent with this model, ESCRT-I can bind ubiquitin in vitro, and proteins that would otherwise remain on the limiting membrane of the late endosome are targeted into the lumen by addition of a ubiquitylation signal (Katzmann et al., 2001). Although the human system is less well characterized, Tsg101 also binds Ub in vitro (Garrus et al., 2001), forms a soluble 350-kDa complex that includes Vps28 (Babst et al., 2000; Bishop and Woodman, 2001), and is required for the delivery of cathepsin D and endocytosed receptors to the lysosome (Babst et al., 2000). Thus, it appears that ESCRT-I complex function is conserved from yeast to humans, and that recognition of ubiquitylated proteins is an important element in this function. Unlike Tsg101, however, Vps23p is not known to bind PTAP, and this may be an important difference between the yeast and mammalian systems.
Intriguingly, Ub also plays an important role in the budding of retroviruses and other enveloped viruses (Harty et al., 2000; Patnaik et al., 2000; Schubert et al., 2000; Strack et al., 2000; Vogt, 2000; Harty et al., 2001; Kikonyogo et al., 2001). Although the precise role of Ub in virus budding is not yet established, retroviruses contain high Ub levels and 2–5% of Gag proteins in the virion are monoubiquitylated (Putterman et al., 1990; Ott et al., 1998, 2000). Moreover, treatment of infected cells with proteasome inhibitors, which decreases the intracellular concentration of free Ub, also inhibits virus release at a late stage (Schubert et al., 2000; Harty et al., 2001). In some cases, virus release can be partially rescued either by overexpressing free Ub or by fusing Ub to the C-terminal end of the viral Gag protein (Patnaik et al., 2000).
Taken together, these observations have led us to hypothesize that Tsg101 may perform similar roles in facilitating virus particle budding from the plasma membrane and vesicle budding from the surface of MVB (Patnaik et al., 2000; Garrus et al., 2001). In this model, the essential difference between viral budding and MVB formation is that the PTAP signals on the viral proteins cause Tsg101 to relocalize to the sites of virus budding on the plasma membrane (Martin-Serrano et al., 2001). Once there, Tsg101 could recognize the viral proteins as ‘cargo’ to be incorporated into a budding vesicle and recruit the additional MVB machinery necessary to complete the budding process. This model further suggests that HIV Gag, like cellular proteins destined for the lumen of the MVB, may undergo a cycle of ubiquitylation and deubiquitylation during viral budding. Although this model remains speculative, it emphasizes the importance of understanding how Tsg101 interacts with the PTAP motif and with Ub. We have therefore used nuclear magnetic resonance (NMR) spectroscopy to determine the structure of the N-terminal UEV domain of Tsg101 and to map the sites of Ub and PTAP binding.
Results
Structure of the Tsg101 N-terminal UEV domain
The Tsg101 UEV domain was defined as residues 1–145 on the basis of preliminary sequence and NMR analyses (see Materials and methods). This construct (Tsg101 UEV) was expressed and purified, and its NMR solution structure calculated from a total of 1861 NOE interproton restraints, 76 hydrogen bonding restraints derived from amide proton exchange protection experiments, and 146 φ and ψ dihedral angle restraints. A stereo view of the superposition of the 20 lowest-penalty structures is shown in Figure 1C. The structures superimpose over the mean coordinate positions of all ordered residues of the domain (4–142) with root mean square deviations (r.m.s.ds) of 0.69 and 0.99 Å for backbone and non-hydrogen atoms, respectively (see Table I for structural statistics).
Table I. Structure statistics for Tsg101 UEV.
| <TAD>a | <CNS>a | |
|---|---|---|
| NOE distance restraintsb (Å) | 1861 | 1861 |
| Intraresidue | 426 | 426 |
| Sequential (|i–j| = 1) | 603 | 603 |
| Medium range (2<|i–j|<5) | 407 | 407 |
| Long range (|i–j|>5) | 425 | 425 |
| Hydrogen bond distance restraintsc (Å) | 76 | 76 |
| Hydrogen bonds | 38 | 38 |
| Dihedral angle restraints (°) | 146 | 146 |
| Phi | 86 | 86 |
| Psi | 60 | 60 |
| Stereospecific assignments | 98 | 98 |
| DYANA target function (Å4) | 0.57 ± 0.08 | N/A |
| CNS energy | ∼10000 ± 2000d | 131 ± 7 |
| Residual distance restraint violations | ||
| Number of violations ≥0.1 Å | 1 ± 1 | 0 |
| Sum of violations (Å or kcal/mol)e | 2.2 ± 0.3 | 19 ± 3 |
| Maximum violation (Å) | 0.16 | |
| Residual dihedral angle restraint violations | ||
| Number of violations ≥1° | 0 ± 0 | 0 |
| Sum of violations (° or kcal/mol)e | 0.1 ± 0.1 | 0.64 ± 0.09 |
| Maximal violation (°) | 0.07 | |
| Van der Waals violations | ||
| Number ≥ 0.1 Å | 0 ± 1 | 0 |
| Sum of violations (Å or kcal/mol)e | 3.5 ± 0.3 | 38 ± 3 |
| Maximal violation (Å) | 0.24 | |
| Ramachandran statisticsf | ||
| Favored (%) | 73.3 | 75.2 |
| Allowed (%) | 24.3 | 22.2 |
| Generously allowed (%) | 2.2 | 2.6 |
| Disallowed (%) | 0.1 | 0.0 |
| R.m.s.ds to the average coordinatesg (Å) | ||
| Residues 4–142 | ||
| Backbone | 0.69 ± 0.28 | 0.69 ± 0.27 |
| Heavy atoms | 1.01 ± 0.46 | 0.99 ± 0.44 |
| Residues 4–90 and 124–142h | ||
| Backbone | 0.61 ± 0.25 | 0.61 ± 0.24 |
| Heavy atoms | 0.93 ± 0.46 | 0.92 ± 0.44 |
| Secondary structural elements | ||
| Backbone | 0.60 ± 0.27 | 0.60 ± 0.25 |
| Heavy atoms | 0.88 ± 0.39 | 0.86 ± 0.37 |
a<TAD> is the ensemble of 20 lowest-penalty structures calculated using the program DYANA (Güntert et al., 1997). <CNS> is the same ensemble after 1000 steps (15 ps each) of simulated annealing at 2000 K, 1000 slow-cooling steps to 0 K and 2000 steps of restrained Powell minimization in cartesian space (anneal.inp protocol) (Brünger et al., 1998).
bOnly meaningful and non-redundant restraints as determined by the DYANA CALIBA function.
cTwo upper-limit distance restraints were used to define each hydrogen bond.
dEnergies for structures input into CNS (from DYANA) were estimated within the generate_easy.inp program after the first regularization without restraints.
eViolation energies from DYANA have units of Å or °, while energies from CNS are in kcal/mol.
fDetermined using PROCHECK-NMR (Laskowski et al., 1996).
gSuperposition and overall r.m.s.ds were calculated using the program MOLMOL (Koradi et al., 1996).
hAll structured residues minus the vestigial active site loop (residues 91–123).
The Tsg101 UEV fold is generally similar to E2 ligases and to Mms2, the only other structurally characterized UEV (Cook et al., 1992, 1993, 1997; Tong et al., 1997; Worthylake et al., 1998; Huang et al., 1999; Jiang and Basavappa, 1999; Zheng et al., 2000; Hamilton et al., 2001; Moraes et al., 2001; VanDemark et al., 2001). This ‘E2 fold’ consists of four helices packed against one side of a four-stranded antiparallel β-sheet (Figure 2). The structural similarity is greatest around the central active site regions of the proteins (residues 53–138), where Tsg101 UEV superimposes with hMms2 (Figure 2B) and yUbc13 (a canonical E2; Figure 2C) with r.m.s.ds over equivalent Cα positions of 2.22 and 1.97 Å, respectively (Figure 2D). However, there are important differences between Tsg101 UEV and other E2 and UEV proteins outside of this region.

Fig. 2. Structures of E2-fold proteins. (A–C) Ribbon representations of Tsg101 UEV (A), Mms2 (B) and Ubc13 (C). Secondary structure elements are colored as in Figure 1. Black arrows indicate residues that correspond to the active site cysteine of Ubc13. (D) Superposition of the three structures. Tsg101 is colored yellow, Mms2 green and Ubc13 purple. The three major structural differences between Tsg101 and the other two proteins are highlighted by arrows: the extra N-terminal helix (red arrow), the β-hairpin ‘tongue’ (blue arrow) and the missing C-terminal helices (green arrow).
Most significantly, Tsg101 UEV lacks the two C-terminal helices found in all structurally characterized E2 proteins (Figure 2C and D, gray helices and green arrow, respectively). Mms2 also lacks these helices, and sequence analyses suggest that they are missing from other UEV domains (Koonin and Abagyan, 1997), indicating that this is a general difference between UEV and canonical E2 proteins (Figure 1B). Although both Mms2 and Tsg101 UEV lack C-terminal helices, their structures differ in this region because the C-terminal residues of human and yeast Mms2 adopt extended, but ordered conformations that occupy the same site as the final E2 helix, whereas the equivalent residues in Tsg101 UEV project off into solution and are disordered. We considered the possibility that these residues might be ordered in longer Tsg101 constructs, and therefore analyzed proteins that contained an additional 5 or 12 residues (Tsg1011–150 and Tsg1011–157, respectively), but could find no evidence for additional order in these longer constructs (see Materials and methods). Thus, the C-terminal end of Tsg101 UEV differs from other known proteins with E2 folds and, as discussed below, this difference allows Tsg101 UEV to bind PTAP-containing peptides.
Tsg101 UEV differs from Mms2 and canonical E2 proteins in two additional ways: (i) Tsg101 contains two N-terminal helices (colored red and orange in Figure 2A), whereas Ubc13 and Mms2 contain only one (Figure 2D, red arrow). The general disposition of Tsg101 UEV helix 2 (orange) is similar to the N-terminal helices of Ubc13 and Mms2, but is shifted slightly towards the center of the sheet (Figure 2D). (ii) The disposition of the first two β-strands in Tsg101 UEV is quite different from that of Mms2 and Ubc13. In Tsg101, strand 1 is twisted and displaced toward the N-terminal end of strand 2, and these two strands form an extended β-hairpin ‘tongue’ that projects 11 residues beyond the main body of the domain and terminates in a type I turn (Figure 2D, blue arrow). This β-tongue is well defined by strong interstrand Hα-to-Hα and Hα-to-HN NOEs, as well as interstrand backbone hydrogen bonds that protect the backbone amide protons from rapid deuterium exchange (not shown). As discussed below, the extended β-tongue appears to be a functionally important feature because it forms part of the Ub binding site. The disposition of the first two β-strands also allows extensive packing interactions between the Tsg101 UEV N-terminal helices and the β-sheet, which are not possible in Ubc13 or Mms2. For example, the loop connecting Tsg101 helices 1 and 2 inserts a tyrosine side-chain hydroxyl (Tyr15) between strands 2 and 3, bridging the hydrogen bonding interaction between the amide proton of Met53 (strand 2) and the carboxyl oxygen of Lys76 (strand 3), thereby terminating the hydrogen bonding network of the β-sheet.
In summary, Tsg101 UEV is similar to canonical E2 enzymes in the hydrophobic core and ‘active site’ regions, but differs significantly at both its N- and C-termini. Although some of these structural differences are shared by the Tsg101 and Mms2 UEV domains (e.g. the lack of C-terminal helices), it is apparent that Tsg101 has diverged even further from the canonical E2 fold than has Mms2.
p6 binding
NMR chemical shift perturbation experiments were used to map the binding site of the HIV-1 p6 protein on the Tsg101 UEV domain (Figure 3). Our previous binding studies showed that the Tsg101 UEV domain binds p6 with moderate affinity under physiological conditions (Kd = 27 ± 5 µM) (Garrus et al., 2001), and the interaction is even tighter under the low salt and slightly acidic conditions used in our NMR titration experiments (Kd = 4.3 ± 1.6 µM at 50 mM NaCl, 20 mM sodium phosphate pH 6; Table II), suggesting an ionic component to the interaction. Heteronuclear single quantum coherence (HSQC) spectra of 15N-labeled Tsg101 UEV were collected during stepwise addition of unlabeled p6, and sites of 1H–15N chemical shift changes were monitored. The majority of Tsg101 UEV amide protons (71/130) changed chemical shift significantly, suggesting the possibility that local conformational changes may accompany binding (normalized chemical shift, δ ≥ 0.5; Cheever et al., 2001). A nine-residue peptide spanning the p6 PTAP motif (5PEPTAPPEE13) produced very similar chemical shift changes as full-length p6 (Figure 3A), and bound with the same affinity as full-length p6 (Figure 3B). Thus, we conclude that this PTAP peptide contains all of the relevant residues for Tsg101 binding. Upon addition of 0.5 molar equivalents of either p6 or PTAP peptide, two distinct peaks were observed for each shifted amide, indicating that both complexes were in slow exchange on the NMR time scale (Figure 3A, lower panels). As expected, complex formation proceeded to completion upon addition of a second 0.5 molar equivalent of peptide, and additional shifts were not observed when supersaturating levels of p6 (>4-fold) were added (not shown).

Fig. 3. Chemical shift mapping of the PTAP binding site on Tsg101 UEV. (A) Overlay of the 1H/15N-HSQC spectra of Tsg101 UEV in the absence (gray) or presence of the 1.0 molar equivalent of PTAP peptide (red). The lower panels show an expansion of the boxed region with an additional titration point indicating that the Tsg101 UEV/PTAP complex is in slow exchange. (B) Biosensor binding of Tsg101 UEV to immobilized full-length p6 (upper left panel and closed circles) and PTAP peptide to immobilized Tsg101 UEV (lower left panel and open circles). The near superimposition of the two curves demonstrates that Tsg101 UEV binds p6 and the PTAP peptide with the same affinity. (C) The 27 residues with the greatest change in chemical shift (δ ≥ 1.8) mapped onto a surface representation of the Tsg101 UEV structure. Residues surrounding the PTAP binding groove include: Val61, Tyr63, Arg64, Asn66, Tyr68, Ile70, Val89, Pro91, Met95, Thr96, Pro139, Pro140, Val141, Phe142 and Arg144. (D) Ribbon representation of Tsg101 UEV showing residues important for binding. The Mms2 (green) and Ubc13 (purple) structures have been superimposed in this figure, but only the C-terminal residues are shown, to illustrate how the binding groove of Tsg101 UEV is filled in cis by C-terminal residues in E2 enzymes and in Mms2 UEV. The C-terminus of Tsg101, which points away from the PTAP binding site, is indicated by the arrowhead.
Table II. Binding of wild-type and mutant Tsg101 UEV domains to HIV-1 p6, Ub and a p6–Ub fusion construct.
| Relative affinitya |
|||
|---|---|---|---|
| p6 | p6–Ub | Ubb | |
| Wild type | 27 ± 5 µMc | 2.1 ± 0.7 µMc | 635 ± 82 µMc |
| 16 ± 2 µMd | 1.9 ± 0.3 µMd | 354 ± 24 µMd | |
| 2.8 ± 0.8 µMe | |||
| 4.3 ± 1.6 µMf | |||
| p6 binding | |||
| Y63A | 14 | 32 | 0.9 |
| V89A | 0.8 | 1.2 | 1.0 |
| M95A | 52 | 290 | 1.6 |
| V141A | 2.5 | 3.5 | 1.4 |
| Ub binding | |||
| V43A | 1.0 | 3.7 | 3.1 |
| F44A | 1.1 | 1.4 | 1.4 |
| N45A | 1.3 | 7.1 | 8.0 |
| D46A | 0.8 | 4.4 | 5.1 |
| W75A | 1.1 | 1.3 | 1.6 |
| F88A | 1.6 | 4.2 | 3.7 |
aAffinities of wild-type Tsg101 UEV domain for p6, p6–Ub and Ub are reported as dissociation constants (Kd), averaged from 16, 8 and 3 independent measurements, respectively. Affinities of mutant constructs are reported as fold decreases relative to wild-type binding.
bExtrapolated to 50% binding.
cMeasured at 20 mM sodium phosphate pH 7.2, 150 mM NaCl. All mutant binding affinities were measured under these conditions.
dMeasured at 20 mM sodium phosphate pH 7.2, 50 mM NaCl.
eMeasured at 20 mM sodium phosphate pH 7.2, 0 mM NaCl.
fMeasured at 20 mM sodium phosphate pH 6, 50 mM NaCl, the conditions used for NMR chemical shift perturbation.
Tsg101 UEV residues exhibiting the greatest chemical shift changes upon PTAP binding (δ ≥ 1.8) were clustered about a groove defined by residues from three different structural elements: (i) the loop connecting strands 2 and 3; (ii) the N-terminal third of the vestigial active site loop; and (iii) the C-terminal residues of the domain (Figure 3C and D). Several intermolecular NOEs between the peptide and the protein have been assigned in preliminary studies, and are consistent with PTAP peptide binding in this groove (data not shown). The groove is opened up by a five-residue insertion in the active site loop (Figure 1B), displacement of the loop connecting strands 2 and 3 (Figure 2D), and irregularities in C-terminal helix geometry created by two glycine residues (Gly126 and Gly136). Hydrophobic residues from strands 3 and 4 (Ile70, Val89, Pro91) line the bottom of the groove, while one end is flanked by charged residues (Arg64, Arg144). This asymmetric charge distribution may help orient the bound peptide, possibly by interacting with complementary acidic residues that flank the PTAP motif. We have shown previously that single alanine point mutations of both p6 Glu6 and Glu13 (but not Glu12) residues reduce the overall binding energy (Garrus et al., 2001).
Mutational analyses were used to confirm that residues in the binding groove make energetically significant interactions in the Tsg101 UEV/p6 complex (Figures 3D and 4). Four Tsg101 UEV residues located around the putative PTAP binding site were mutated to alanine and tested for their effect on Tsg101 binding affinity in BIAcore biosensor experiments (Garrus et al., 2001). As expected, alanine substitutions in three residues that surround the rim of the groove (Tyr63, Met95 and Val141) all reduced p6 binding significantly (Figure 4 and Table II). Surprisingly, mutation of Val89, which is located at the bottom of the pocket, had no effect on p6 binding. Therefore, although the Val89 amide showed the greatest chemical shift change upon addition of p6 (Figure 3A), the Val89 methyl groups do not contribute appreciable binding energy, perhaps because the PTAP peptide does not reach down to the very bottom of the pocket. Importantly, the Tyr63→Ala and Met95→Ala mutations, which decreased p6 binding affinity by 14- and 52-fold, respectively, also significantly impaired the ability of Tsg101 to support HIV-1 budding, whereas the Val89→ Ala mutation had no effect on budding (J.Garrus, personal communication). The excellent correspondence between the viral budding phenotypes and in vitro binding activities of both PTAP and Tsg101 UEV mutants strongly supports the idea that PTAP-mediated recruitment of Tsg101 to HIV-1 budding sites at the plasma membrane is a prerequisite for the efficient release of virus particles from infected cells.

Fig. 4. Mutagenic analysis of Tsg101 UEV binding to p6, p6–Ub and Ub. Biosensor binding isotherms are shown for putative PTAP-binding mutants (left) and Ub-binding mutants (right). Binding isotherms for p6 (top panels), p6–Ub (middle panels) and Ub (bottom panels) are shown. Mutants that significantly reduced p6 binding are shown in red, while those that significantly reduced Ub binding are shown in blue (see Table II). Mutations that had no significant effect are in green. Wild-type binding isotherms are in black.
Ubiquitin binding
The Tsg101 UEV domain also binds ubiquitin, although the 1 : 1 interaction is quite weak (Kd ≈ 500 µM) (Garrus et al., 2001). The Ub and PTAP binding sites appear to be distinct, and indeed can be simultaneously occupied as evidenced by the fact that direct fusion of Ub to p6 enhances the binding affinity over that of either ligand alone (i.e. the ligands bind cooperatively; Figure 4). As compared with p6, titration with Ub produced chemical shift changes in a distinct, but slightly overlapping subset of Tsg101 UEV amides, consistent with the idea that PTAP and Ub bind to different sites (compare Figures 3A and 5A). Significant amide chemical shift changes (δ ≥ 1.8) were observed for only 13 residues, suggesting that Ub does not contact a large surface area or cause large conformational changes upon binding (Figure 5A). Consistent with the weak affinity of Tsg101 UEV for (unconjugated) ubiquitin, the complex was in fast exchange. As shown in Figure 5A, single resonances were always observed for the perturbed amides and their chemical shift changes increased as the Ub protein ratio increased. Residues undergoing the greatest chemical shift changes were located throughout the β-tongue and at the vestigial active site loop, which flank a large hydrophobic patch on the protein surface formed by side-chains from strands 3 and 4 (Figure 5B and C).

Fig. 5. Mapping the Tsg101 UEV and Ub interaction sites. (A) Overlay of the 1H/15N-HSQC spectra of Tsg101 UEV in the absence (gray) or presence of 2.0 molar equivalents of Ub (blue). The lower panels show an expansion of the boxed region, with an additional titration point, indicating that the Tsg101 UEV/Ub complex is in fast exchange. (B) The 13 residues with the greatest changes in chemical shift (δ ≥ 1.8) are mapped onto a surface representation of the Tsg101 UEV structure. (C) Ribbon representation of Tsg101 UEV showing residues important for binding. (D) Overlay of the 1H/15N-HSQC spectra of Ub in the absence (gray) or presence of Tsg101 UEV (magenta). The lower panels show an expansion of the boxed region showing that the Lys48 amide is shifted. (E and F) The 15 residues with the greatest change in chemical shift (δ ≥ 1.0) are mapped onto a surface (E) and ribbon (F) representation of Ub. Lys48 and Lys63 are shown explicitly in yellow (F).
The importance of this region for Ub binding was tested by introducing a series of alanine point mutations and quantitating their effects on Ub, p6–Ub and p6 binding (Figure 4 and Table II). Alanine substitutions of β-tongue residues (Val43, Asn45, Asp46) and a hydrophobic sheet residue (Phe88) significantly reduced Tsg101 UEV binding to both p6–Ub and Ub alone (>3-fold) without affecting p6 binding. Substitution of Trp75, which is exposed on the hydrophobic surface, did not significantly affect the ubiquitin-binding activity of Tsg101 UEV. This was unexpected, as the indole N-H of Trp75 is significantly shifted during Ub titration. This difference notwithstanding, our mutagenesis and NMR chemical shift mapping experiments are generally in good agreement and define the location of the Tsg101 UEV Ub-binding site.
The complementary Tsg101 UEV-interacting surface on ubiquitin was also mapped by chemical shift perturbation experiments. As expected, titration of unlabeled Tsg101 UEV into 15N-labeled Ub shifted a small subset of Ub amides, and the complex was again in fast exchange (Figure 5D). The greatest chemical shift changes (δ ≥ 1.0) mapped to the two shortest strands (3 and 4) of the antiparallel β-sheet of Ub, and the N-terminal end of strand 5 (Figure 5E and F). This region contains a number of polar and charged residues, which may form ionic and/or hydrogen bonding interactions with Tsg101 UEV β-tongue residues. The shifted Ub residues include Lys48, which is used for the formation of polyubiquitin chains that target proteins for proteasomal degradation (Hershko and Ciechanover, 1998).
Discussion
PTAP binding
The structure presented here demonstrates that the N-terminal UEV domain of human Tsg101 conforms to the general E2 fold, but with several distinctive features that allow it to bind PTAP and ubiquitin. Tsg101 UEV is the first example of an E2/UEV domain with a peptide binding motif, and our work shows that residues from five different secondary structural elements come together to create a PTAP binding ‘groove’. Analogous sites in other E2/UEV proteins are buried, either by a C-terminal helix (in canonical E2 enzymes) or by the ordered C-terminal residues of Mms2 UEV (Figure 3B), explaining why these domains do not have peptide binding activities.
The importance of the Tsg101/PTAP interaction in the release of HIV particles seems clear because mutations in either the p6 PTAP motif (Göttlinger et al., 1991; Huang et al., 1995) or in Tsg101 UEV (J.Garrus, personal communication) that inhibit the interaction also block virus release. In principle, the Tsg101 UEV domain represents an attractive target for therapeutic intervention against HIV and Ebola as these viruses require Tsg101 for budding (Garrus et al., 2001; Martin-Serrano et al., 2001) and because the cellular Tsg101 protein presumably would not develop drug resistance as rapidly as a viral target. Our structure therefore provides the first glimpse of potential binding sites for small molecule inhibitors designed to block viral egress by inhibiting Tsg101 binding.
The role(s) of PTAP binding in normal cellular Tsg101 functions remains an open question. A number of candidate cellular Tsg101-binding proteins also carry P(S/T)AP motifs (Strack et al., 2000; Garrus et al., 2001), including plasma membrane proteins known to be downregulated via the vacuolar protein sorting pathway (Berthoud et al., 2000; Strack et al., 2000). A PSAP motif is also found in hepatocyte growth factor-regulated tyrosine kinase substrate (Hrs/Hgs, the ortholog of yeast Vps27) (Garrus et al., 2001), a protein required for tyrosine kinase receptor downregulation and MVB formation in Drosophila (Komada and Kitamura, 2001; Lloyd et al., 2002). This suggests the intriguing possibility that viral late domains may act by mimicking the normal protein recruiting functions of Hrs during vacuolar protein sorting and multivesicular body formation. Consistent with this idea, human Hrs has very recently been shown to be ubiquitylated (Polo et al., 2002). Hrs also contains a PPEY sequence, which is another motif that can function as a late domain in other enveloped viruses (Vogt, 2000).
We have also noticed that Tsg101 contains its own PTAP motif, which is located in a predicted loop region connecting the apparent binding sites for the Vps37 and Vps28 proteins (Figure 1A). We hypothesize that the Tsg101 UEV domain could fold back and bind to this PTAP sequence, perhaps creating an ‘auto-inhibited’ conformation. Thus, we speculate that regulated PTAP binding may be important for normal Tsg101 function, and viruses like HIV and Ebola are simply mimicking this natural binding interaction.
Ubiquitin binding
Previous biochemical and modeling studies have suggested that ubiquitin can bind to E2/UEV domains in two distinct modes. Remarkably, our data indicate that ubiquitin binds in yet another, third way to the Tsg101 UEV domain, although the biological relevance of this interaction remains to be tested. As illustrated in Figure 6, both previously known modes of ubiquitin binding are utilized by the heterodimeric Ubc13/Mms2 complex. This complex can assemble polymeric chains of Ub molecules by binding and catalyzing the linkage of ‘acceptor’ and ‘donor’ ubiquitin molecules. The most general mode of Ub binding is illustrated by the interaction between the donor ubiquitin and the catalytically active Ubc13 E2 domain. As in other E2 enzymes, the active site cysteine of Ubc13 forms a transient thioester bond with the C-terminus of Ub in the process of transferring Ub from an E1 enzyme onto the lysine side chain of a protein substrate (Lys63 of another Ub molecule in this case). This covalent chemistry implies that the C-terminal ‘tail’ of Ub must bind near the active site cysteine residue, and there is direct evidence that tail residues form the major contacts when Ub binds to the Ubc1 and Ubc2b E2 enzymes (Miura et al., 1999; Hamilton et al., 2001).
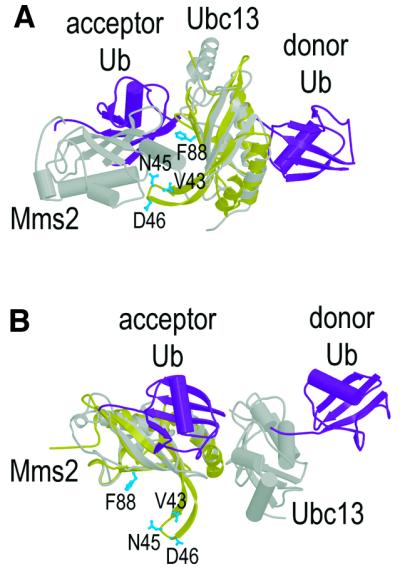
Fig. 6. Comparison of the ubiquitin binding site (blue residues) on Tsg101 UEV (yellow) with models for the two distinct ubiquitin binding sites (purple) on the Ubc13/Mms2 heterodimer (gray) (VanDemark et al., 2001). To create the figure, Tsg101 UEV was superimposed on either the Ubc13 E2 (A) or the Mms2 UEV (B) subunits of the Ubc13/Mms2 heterodimer. The figure illustrates the fact that the Ub binding surface on Tsg101 is distinct from previously characterized Ub binding sites on either E2 or UEV domains, but does roughly correspond to the Mms2 binding site on Ubc13.
Ubiquitin binds in quite a different mode to the catalytically inactive Mms2 UEV subunit of the Ubc13/Mms2 complex. The role of Mms2 is to position the acceptor Ub Lys63 side-chain to attack the C-terminus of the donor ubiquitin. Although two different models for the Mms2/Ub have been proposed (Moraes et al., 2001; VanDemark et al., 2001), mutagenesis studies are most consistent with Ub binding in a channel that straddles the Mms2/Ubc13 interface (Chan and Hill, 2001; VanDemark et al., 2001), with the Mms2 binding surface defined by the C-terminal half of helix 1, the outer edge of strand 1, and the loop that connects strands 1 and 2.
In contrast, our chemical shift mapping and mutagenesis experiments indicate that ubiquitin binds to the concave ‘lower’ half of the four-stranded sheet of Tsg101 UEV. The most prominent feature of this binding site is the β-tongue projection, and mutations of residues at the tip of the tongue reduce Ub binding. Although this site is distinct from both previously characterized Ub binding surfaces in E2/UEV proteins, it does roughly match the Mms2 binding site on the Ubc13 protein (Figure 6A). Ub binds Tsg101 UEV using a convex surface that encompasses strands 3 and 4, and we therefore propose that shape complementarity may play an important role in recognition. The Lys48 residue at the N-terminal end of Ub strand 4 is likely to be sequestered at the Tsg101 UEV/Ub interface, which probably precludes polyubiquitylation via Lys48 linkages (the targeting signal for proteasomal degradation).
In summary, the Ub-binding surface of Tsg101 differs from that used by other E2/UEV proteins, leading to the surprising conclusion that although E2/UEV domains utilize a conserved three-dimensional fold to bind ubiquitin, they have evolved at least three distinct surfaces to mediate these interactions.
Materials and methods
Tsg101 UEV domain
Tsg101 UEV domain constructs were designed based on an alignment of Tsg101 homologs obtained from the NCBI protein database (Arabidopsis thaliana, BAB03147; Caenorhabditis elegans, AAC25822; Chelonia mydas, AAF87776; Drosophila melanogaster, AAG29564; Homo sapiens, AAC52083; Mus musculus, AAH05424; Saccharomyces cerevisiae, AAB62820). Since canonical E2 enzymes are ∼150 residues in length, gaps in the alignment near Tsg101 residue 150 were interpreted as potential domain boundaries. Based on the alignments, constructs spanning residues 1–145, 1–150, 1–157 and 1–161 of Tsg101 were cloned and expressed from a pET11d vector (Novagen, Madison, WI). The three shortest constructs expressed soluble proteins at high levels in BL21(DE3) Escherichia coli cells, but 1–161 (and longer constructs) expressed poorly and were insoluble. Although we were unable to make these longer protein constructs, we conclude that Tsg101 residues 151–180 are unlikely to form the two helices seen in canonical E2 enzymes because the protein sequence in this region (151PPYQATGPPNTSYMPGMPGGISPYPSGYPP180) is 50% Pro/Gly (15/30) and has an extremely low helical propensity. Moreover, this sequence is poorly conserved in alignments of Tsg101 proteins from multiple species (Bishop and Woodman, 2001), and cannot be aligned meaningfully with the C-terminal helices of E2 enzymes (Koonin and Abagyan, 1997; Ponting et al., 1997).
To purify the soluble Tsg101 UEV proteins, cells were lysed using lysozyme and sonication, and insoluble material was removed from the lysate by centrifugation. Tsg101 UEV was precipitated from the soluble fraction with ammonium sulfate at 28–55% saturation. The precipitate was redissolved in buffer containing 25 mM MOPS pH 6.5 and fractionated on a methyl sulfonate (SP)-Sepharose column (Pharmacia, Uppsala, Sweden). Tsg101 UEV eluted at an ∼300 mM NaCl linear salt gradient (50–1000 mM NaCl over 400 ml). Fractions containing Tsg101 were pooled, adjusted to 1 M ammonium sulfate and refractionated on a phenyl-Sepharose column (Pharmacia). Pure Tsg101 UEV eluted near the beginning of a linear salt gradient (1–0 M ammonium sulfate over 200 ml), with typical yields of 15–20 mg pure protein per liter of E.coli culture. N-terminal amino acid sequencing and mass spectrometry analyses showed that the N-terminal methionine was quantitatively removed during expression, and the masses of all purified proteins were within 1–3 Da of those expected (not shown). The 1H/15N-HSQC spectra of Tsg1011–145, Tsg1011–150 and Tsg1011–157 were completely superimposable (except for C-terminal residues), and amide protons beyond Phe142 displayed limited proton chemical shift dispersion and sharp peaks with half-height linewidths of 14–19 Hz (versus 21–24 Hz for structured residues of the domain), indicating that they are disordered. In addition, all three UEV constructs bound with same affinities to p6, p6–Ub and Ub (data not shown). Tsg101 residues 1–145 therefore appear to encompass the entire ordered UEV domain. Hence, all reported structural and biochemical analyses were performed on this defined domain. Mutant Tsg101 UEV constructs were cloned using the megaprimer PCR method (Picard et al., 1994), and purified as described for the wild type.
Other protein samples
GST–p6 constructs and the free p6 domain of the HIV-1 Gag protein were purified as described previously (Jenkins et al., 2001). The PTAP peptide (NH-PEPTAPPEE-COOH) was obtained by solid-phase synthesis and purified using reverse phase high performance liquid chromatography. Bovine ubiquitin, which is identical in sequence to human ubiquitin, was purchased from Sigma and purified using size exclusion chromatography. 15N-labeled ubiquitin was purchased from VLI Research, Inc., (Malvern, PA).
NMR spectroscopy
Samples for structure determination were ∼1.5 mM Tsg101 UEV in 20 mM sodium phosphate, 50 mM NaCl pH 5.5 in 90% H2O/10% D2O. Spectra were recorded at 25°C on a Varian Inova 600 MHz spectrometer equipped with a triple-resonance 1H/13C/15N probe and z-axis pulsed field gradient capability. Backbone and side-chain assignments were made using the following NMR experiments: 15N/1H HSQC (Mori et al., 1995), HNCACB (Wittekind, 1993), HNCO (Kay et al., 1994), 13C/1H CT–HSQC (Santoro and King, 1992; Vuister, 1992), 3D 15N-edited TOCSY–HSQC (Zhang et al., 1994), H(CCO)NH, (H)C(CO)NH (Grzesiek et al., 1993) and 4D 13C/13C-edited HMQC–NOESY–HMQC (Vuister et al., 1993). The following NOE data were used to generate distance restraints: 3D 15N-edited NOESY–HSQC (Zhang et al., 1994; Mori et al., 1995) and 3D 13C-edited NOESY–HSQC (Muhandiram et al., 1993; Pascal et al., 1994). NOESY mixing times were 80 ms. Three-bond coupling constants (3JHN-HA) were obtained from a 3D HNHA experiment (Kuboniwa et al., 1994). Hydrogen-bonded amides were identified using deuterium exchange as described previously (Bai et al., 1997). All spectra were processed using FELIX (MSI).
Structure determination
Backbone and side-chain correlations were assigned and NOE intensities were integrated using the tools in SPARKY (T.D.Goddard and D.G.Kneller, University of California, San Francisco). Structures were calculated using the torsion angle dynamics approach in DYANA (Güntert et al., 1997). Initial structures were calculated from ∼1500 NOEs. Iterative rounds of structure calculation and NOE assignments led to 10 models that superimposed over secondary structure backbone atoms with an average r.m.s.d. of ∼0.5 Å. Hydrogen bonding restraints (two per hydrogen bond) and phi and psi dihedral angle restraints were then added. These restraints did not alter the structure, but improved convergence. To generate the reported structures, 200 structures were calculated using DYANA and geometries of the 20 lowest penalty structures were regularized using the crystallography and NMR system (CNS) (Brünger et al., 1998). The structures were analyzed using PROCHECK-NMR (Laskowski et al., 1996), MOLMOL (Koradi et al., 1996) and INSIGHT II (Molecular Simulations Inc.; www.accelrys.com/life/) (Table I). Structure figures were created with MOLSCRIPT (Kraulis, 1991), BOBSCRIPT (Esnouf, 1997) and GRASP (Nicholls et al., 1991).
Chemical shift perturbation
Titration experiments were performed at 20°C in 20 mM sodium phosphate, 50 mM NaCl pH 6 in 90% H2O/10% D2O. To identify the PTAP and Ub binding sites on the UEV domain, unlabeled p6, PTAP peptide or Ub were titrated into 0.5 mM 15N-labeled UEV domain. To identify the UEV binding site on Ub, unlabeled UEV domain was titrated into 0.2 mM 15N-labeled Ub. Normalized chemical shift changes were calculated using the equation: δ = 25[(δHN)2 – (δN/5)2]0.5 (Cheever et al., 2001). Amides displaying the greatest normalized chemical shift changes were displayed using cutoffs of δ ≥1.8 (for Tsg101 UEV amides) or ≥1.0 (for Ub amides). Amide chemical shift assignments for human Ub were obtained from the VLI Research, Inc. website (www.vli-research.com).
Binding experiments
Binding affinities of purified wild-type and mutant Tsg101 UEV domains for immobilized GST–p61–27, GST–p61–27-Ub and GST–Ub were quantified using a BIAcore biosensor as described previously (Garrus et al., 2001). Measurements were performed at 20°C in 20 mM sodium phosphate, 150 mM NaCl, 0.05% bovine serum albumin, 0.01% P20 (pH 7.2).
Acknowledgments
Acknowledgements
We thank Brittany Chamberlin, Lianghuey Liu, Peter Stenlund and Dennis Edwards for technical support, Bob Schackmann for peptide synthesis, and Eric Ross for computer support. We thank Andrew VanDemark for providing the coordinates of the model for the Ubc13-Mms2/Ub2 ternary complex, and Chris Hill, Andrew VanDemark and Marty Rechsteiner for critical reading of the manuscript. This work was supported by National Institutes of Health (NIH) funding to W.I.S. The Utah Biomolecular NMR Facility is supported by grants from the NIH and the National Science Foundation. Structure coordinates and chemical shifts have been deposited at the Protein Data Bank (1KPP, CNS ensemble; 1KPQ, DYANA ensemble).
References
- Babst M., Odorizzi,G., Estepa,E.J. and Emr,S.D. (2000) Mammalian tumor susceptibility gene 101 (TSG101) and the yeast homologue, Vps23p, both function in late endosomal trafficking. Traffic, 1, 248–258. [DOI] [PubMed] [Google Scholar]
- Bai Y., Karimi,A., Dyson,H.J. and Wright,P.E. (1997) Absence of a stable intermediate on the folding pathway of protein A. Protein Sci., 6, 1449–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berthoud V.M., Tadros,P.N. and Beyer,E.C. (2000) Connexin and gap junction degradation. Methods, 20, 180–187. [DOI] [PubMed] [Google Scholar]
- Bishop N. and Woodman,P. (2001) TSG101/mammalian VPS23 and mammalian VPS28 interact directly and are recruited to VPS4-induced endosomes. J. Biol. Chem., 276, 11735–11742. [DOI] [PubMed] [Google Scholar]
- Brünger A.T. et al. (1998) Crystallography and NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D, 54, 905–921. [DOI] [PubMed] [Google Scholar]
- Chan N.L. and Hill,C.P. (2001) Defining polyubiquitin chain topology. Nature Struct. Biol., 8, 650–652. [DOI] [PubMed] [Google Scholar]
- Cheever M.L., Sato,T.K., de Beer,T., Kutateladze,T.G., Emr,S.D. and Overduin,M. (2001) Phox domain interaction with PtdIns3P targets the Vam7 t-SNARE to vacuole membranes. Nature Cell Biol., 3, 613–618. [DOI] [PubMed] [Google Scholar]
- Cook W.J., Jeffrey,L.C., Sullivan,M.L. and Vierstra,R.D. (1992) Three-dimensional structure of a ubiquitin-conjugating enzyme (E2). J. Biol. Chem., 267, 15116–15121. [DOI] [PubMed] [Google Scholar]
- Cook W.J., Jeffrey,L.C., Xu,Y. and Chau,V. (1993) Tertiary structures of class I ubiquitin-conjugating enzymes are highly conserved: crystal structure of yeast Ubc4. Biochemistry, 32, 13809–13817. [DOI] [PubMed] [Google Scholar]
- Cook W.J., Martin,P.D., Edwards,B.F., Yamazaki,R.K. and Chau,V. (1997) Crystal structure of a class I ubiquitin conjugating enzyme (Ubc7) from Saccharomyces cerevisiae at 2.9 Å resolution. Biochemistry, 36, 1621–1627. [DOI] [PubMed] [Google Scholar]
- Demirov D.G., Ono,A., Orenstein,J.M. and Freed,E.O. (2002) Overexpression of the N-terminal domain of TSG101 inhibits HIV-1 budding by blocking late domain function. Proc. Natl Acad. Sci. USA, 99, 955–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupre S., Volland,C. and Haguenauer-Tsapis,R. (2001) Membrane transport: ubiquitylation in endosomal sorting. Curr. Biol., 11, R932–R934. [DOI] [PubMed] [Google Scholar]
- Esnouf R.M. (1997) An extensively modified version of MolScript that includes greatly enhanced coloring capabilities. J. Mol. Graph Model, 15, 132–134, 112–133. [DOI] [PubMed] [Google Scholar]
- Feng G.H., Lih,C.J. and Cohen,S.N. (2000) TSG101 protein steady-state level is regulated posttranslationally by an evolutionarily conserved COOH-terminal sequence. Cancer Res., 60, 1736–1741. [PubMed] [Google Scholar]
- Garrus J.E. et al. (2001) Tsg101 and the vacuolar protein sorting pathway are essential for HIV-1 budding. Cell, 107, 55–65. [DOI] [PubMed] [Google Scholar]
- Göttlinger H.G., Dorfman,T., Sodroski,J.G. and Haseltine,W.A. (1991) Effect of mutations affecting the p6 gag protein on human immunodeficiency virus particle release. Proc. Natl Acad. Sci. USA, 88, 3195–3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grzesiek S., Anglister,J. and Bax,A. (1993) Correlation of backbone amide and aliphatic side-chain resonances in 13C/15N-enriched proteins by isotropic mixing of 13C magnetization. J. Magn. Reson. B, 101, 114–119. [Google Scholar]
- Güntert P., Mumenthaler,C. and Wüthrich,K. (1997) Torsion angle dynamics for NMR structure calculation with the new program DYANA. J. Mol. Biol., 273, 283–298. [DOI] [PubMed] [Google Scholar]
- Hamilton K.S., Ellison,M.J., Barber,K.R., Williams,R.S., Huzil,J.T., McKenna,S., Ptak,C., Glover,M. and Shaw,G.S. (2001) Structure of a conjugating enzyme-ubiquitin thiolester intermediate reveals a novel role for the ubiquitin tail. Structure (Camb), 9, 897–904. [DOI] [PubMed] [Google Scholar]
- Harty R.N., Brown,M.E., Wang,G., Huibregtse,J. and Hayes,F.P. (2000) A PPxY motif within the VP40 protein of Ebola virus interacts physically and functionally with a ubiquitin ligase: implications for filovirus budding. Proc. Natl Acad. Sci. USA, 97, 13871–13876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harty R.N., Brown,M.E., McGettigan,J.P., Wang,G., Jayakar,H.R., Huibregtse,J.M., Whitt,M.A. and Schnell,M.J. (2001) Rhabdoviruses and the cellular ubiquitin-proteasome system: a budding interaction. J. Virol., 75, 10623–10629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershko A. and Ciechanover,A. (1998) The ubiquitin system. Annu. Rev. Biochem., 67, 425–479. [DOI] [PubMed] [Google Scholar]
- Hicke L. (2001) A new ticket for entry into budding vesicles-ubiquitin. Cell, 106, 527–530. [DOI] [PubMed] [Google Scholar]
- Hofmann R.M. and Pickart,C.M. (1999) Noncanonical MMS2-encoded ubiquitin-conjugating enzyme functions in assembly of novel polyubiquitin chains for DNA repair. Cell, 96, 645–653. [DOI] [PubMed] [Google Scholar]
- Huang L., Kinnucan,E., Wang,G., Beaudenon,S., Howley,P.M., Huibregtse,J.M. and Pavletich,N.P. (1999) Structure of an E6AP-UbcH7 complex: insights into ubiquitination by the E2-E3 enzyme cascade. Science, 286, 1321–1326. [DOI] [PubMed] [Google Scholar]
- Huang M., Orenstein,J.M., Martin,M.A. and Freed,E.O. (1995) p6Gag is required for particle production from full-length human immuno deficiency virus type 1 molecular clones expressing protease. J. Virol., 69, 6810–6818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins Y., Pornillos,O., Rich,R.L., Myszka,D.G., Sundquist,W.I. and Malim,M.H. (2001) Biochemical analyses of the interactions between human immunodeficiency virus type 1 Vpr and p6Gag. J. Virol., 75, 10537–10542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang F. and Basavappa,R. (1999) Crystal structure of the cyclin-specific ubiquitin-conjugating enzyme from clam, E2-C, at 2.0 Å resolution. Biochemistry, 38, 6471–6478. [DOI] [PubMed] [Google Scholar]
- Katzmann D.J., Babst,M. and Emr,S.D. (2001) Ubiquitin-dependent sorting into the multivesicular body pathway requires the function of a conserved endosomal protein sorting complex, ESCRT-I. Cell, 106, 145–155. [DOI] [PubMed] [Google Scholar]
- Kay L.E., Xu,G.Y. and Yamazaki,T. (1994) Enhanced-sensitivity triple-resonance spectroscopy with minimal H2O saturation. J. Magn. Reson., 109, 129–133. [Google Scholar]
- Kikonyogo A., Bouamr,F., Vana,M.L., Xiang,Y., Aiyar,A., Carter,C. and Leis,J. (2001) Proteins related to the Nedd4 family of ubiquitin protein ligases interact with the L domain of Rous sarcoma virus and are required for gag budding from cells. Proc. Natl Acad. Sci. USA, 98, 11199–11204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komada M. and Kitamura,N. (2001) Hrs and hbp: possible regulators of endocytosis and exocytosis. Biochem. Biophys. Res. Commun., 281, 1065–1069. [DOI] [PubMed] [Google Scholar]
- Koonin E.V. and Abagyan,R.A. (1997) TSG101 may be the prototype of a class of dominant negative ubiquitin regulators. Nature Genet., 16, 330–331. [DOI] [PubMed] [Google Scholar]
- Koradi R., Billeter,M. and Wüthrich,K. (1996) MOLMOL: a program for display and analysis of macromolecular structures. J. Mol. Graph., 14, 51–55. [DOI] [PubMed] [Google Scholar]
- Kraulis P.J. (1991) MOLSCRIPT: A program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr., 24, 946–950. [Google Scholar]
- Kuboniwa H., Grzesiek,S., Delaglio,F. and Bax,A. (1994) Measurement of HN-Hα J couplings in calcium-free calmodulin using new 2D and 3D water-flip-back methods. J. Biomol. NMR, 4, 871–878. [DOI] [PubMed] [Google Scholar]
- Laskowski R.A., Rullmannn,J.A., MacArthur,M.W., Kaptein,R. and Thornton,J.M. (1996) AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR, 8, 477–486. [DOI] [PubMed] [Google Scholar]
- Lloyd T.E., Atkinson,R., Wu,M.N., Zhou,Y., Pennetta,G. and Bellen,H.J. (2002) Hrs regulates endosome membrane invagination and tyrosine kinase receptor signaling in Drosophila. Cell, 108, 261–269. [DOI] [PubMed] [Google Scholar]
- Martin-Serrano J., Zang,T. and Bieniasz,P.D. (2001) HIV-1 and Ebola virus encode small peptide motifs that recruit Tsg101 to sites of particle assembly to facilitate egress. Nature Med., 7, 1313–1319. [DOI] [PubMed] [Google Scholar]
- Miura T., Klaus,W., Gsell,B., Miyamoto,C. and Senn,H. (1999) Characterization of the binding interface between ubiquitin and class I human ubiquitin-conjugating enzyme 2b by multidimensional heteronuclear NMR spectroscopy in solution. J. Mol. Biol., 290, 213–228. [DOI] [PubMed] [Google Scholar]
- Moraes T.F., Edwards,R.A., McKenna,S., Pastushok,L., Xiao,W., Glover,J.N. and Ellison,M.J. (2001) Crystal structure of the human ubiquitin conjugating enzyme complex, hMms2-hUbc13. Nature Struct. Biol., 8, 669–673. [DOI] [PubMed] [Google Scholar]
- Mori S., Abeygunawardana,C., Johnson,M.O. and van Zijl,P.C. (1995) Improved sensitivity of HSQC spectra of exchanging protons at short interscan delays using a new fast HSQC (FHSQC) detection scheme that avoids water saturation. J. Magn. Reson. B, 108, 94–98. [DOI] [PubMed] [Google Scholar]
- Muhandiram D.R., Xy,G.Y. and Kay,L.E. (1993) An enhanced-sensitivity pure absorption gradient 4D 15N, 13C-edited NOESY experiment. J. Biomol. NMR, 3, 463–470. [Google Scholar]
- Nicholls A., Sharp,K.A. and Honig,B. (1991) Protein folding and association: insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins, 11, 281–296. [DOI] [PubMed] [Google Scholar]
- Ott D.E. et al. (1998) Ubiquitin is covalently attached to the p6Gag proteins of human immunodeficiency virus type 1 and simian immunodeficiency virus and to the p12Gag protein of Moloney murine leukemia virus. J. Virol., 72, 2962–2968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott D.E., Coren,L.V., Chertova,E.N., Gagliardi,T.D. and Schubert,U. (2000) Ubiquitination of HIV-1 and MuLV Gag. Virology, 278, 111–121. [DOI] [PubMed] [Google Scholar]
- Pascal S.M., Yamazaki,D.R., Forman-Kay,J.D. and Kay,L.E. (1994) Simultaneous acquisition of 15N- and 13C-edited NOE spectra of proteins dissolved in H2O. J. Magn. Reson., 191, 197–201. [Google Scholar]
- Patnaik A., Chau,V. and Wills,J.W. (2000) Ubiquitin is part of the retrovirus budding machinery. Proc. Natl Acad. Sci. USA, 97, 13069–13074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard V., Ersdal-Badju,E., Lu,A. and Bock,S.C. (1994) A rapid and efficient one-tube PCR-based mutagenesis technique using Pfu DNA polymerase. Nucleic Acids Res., 22, 2587–2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piper R.C. and Luzio,J.P. (2001) Late endosomes: sorting and partitioning in multivesicular bodies. Traffic, 2, 612–621. [DOI] [PubMed] [Google Scholar]
- Polo S., Sigismund,S., Faretta,M., Guidi,M., Capua,M.R., Bossi,G., Chen,H., De Camilli,P. and Di Fiore,P.P. (2002) A single motif responsible for ubiquitin recognition and monoubiquitination in endocytic proteins. Nature, 416, 451–455. [DOI] [PubMed] [Google Scholar]
- Ponting C.P., Cai,Y.D. and Bork,P. (1997) The breast cancer gene product TSG101: a regulator of ubiquitination? J. Mol. Med., 75, 467–469. [PubMed] [Google Scholar]
- Putterman D., Pepinsky,R.B. and Vogt,V.M. (1990) Ubiquitin in avian leukosis virus particles. Virology, 176, 633–637. [DOI] [PubMed] [Google Scholar]
- Santoro J. and King,G.C. (1992) A constant-time 2D Overbodenhausen experiment for inverse correlation of isotopically enriched species. J. Magn. Reson., 97, 202–207. [Google Scholar]
- Schubert U., Ott,D.E., Chertova,E.N., Welker,R., Tessmer,U., Princiotta,M.F., Bennink,J.R., Kräusslich,H.G. and Yewdell,J.W. (2000). Proteasome inhibition interferes with gag polyprotein processing, release and maturation of HIV-1 and HIV-2. Proc. Natl Acad. Sci. USA, 97, 13057–13062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strack B., Calistri,A., Accola,M.A., Palu,G. and Göttlinger,H.G. (2000). A role for ubiquitin ligase recruitment in retrovirus release. Proc. Natl Acad. Sci. USA, 97, 13063–13068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong H., Hateboer,G., Perrakis,A., Bernards,R. and Sixma,T.K. (1997) Crystal structure of murine/human Ubc9 provides insight into the variability of the ubiquitin-conjugating system. J. Biol. Chem., 272, 21381–21387. [DOI] [PubMed] [Google Scholar]
- VanDemark A.P., Hofmann,R.M., Tsui,C., Pickart,C.M. and Wolberger,C. (2001). Molecular insights into polyubiquitin chain assembly: crystal structure of the Mms2/Ubc13 heterodimer. Cell, 105, 711–720. [DOI] [PubMed] [Google Scholar]
- VerPlank L., Bouamr,F., LaGrassa,T.J., Agresta,B., Kikonyogo,A., Leis,J. and Carter,C.A. (2001) Tsg101, a homologue of ubiquitin-conjugating (E2) enzymes, binds the L domain in HIV type 1 Pr55Gag. Proc. Natl Acad. Sci. USA, 98, 7724–7729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt V.M. (2000) Ubiquitin in retrovirus assembly: actor or bystander? Proc. Natl Acad. Sci. USA, 97, 12945–12947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuister G.W. (1992) Resolution enhancement and spectral editing of uniformly 13C-enriched proteins by homonuclear broadband 13C decoupling. J. Magn. Reson., 98, 428–435. [Google Scholar]
- Vuister G.W.C., Gronenborn,A.M., Powers,R., Garrett,D.S., Tschudin,R. and Bax,A. (1993) Increased resolution and improved spectral quality of 4D 13C/13C-separated HMQC-NOESY-HMQC spectra using pulsed field gradients. J. Magn. Reson. B, 101, 210–213. [Google Scholar]
- Wittekind M. (1993) HNCACB, A high-sensitivity 3D NMR experiment to correlate amide-proton and nitrogen resonances with the alpha and beta carbon resonances in proteins. J. Magn. Reson. B, 101, 201–205. [Google Scholar]
- Worthylake D.K., Prakash,S., Prakash,L. and Hill,C.P. (1998) Crystal structure of the Saccharomyces cerevisiae ubiquitin-conjugating enzyme Rad6 at 2.6 Å resolution. J. Biol. Chem., 273, 6271–6276. [DOI] [PubMed] [Google Scholar]
- Zhang O., Kay,L.E., Olivier,J.P. and Forman-Kay,J.D. (1994) Backbone 1H and 15N resonance assignments of the N-terminal SH3 domain of drk in folded and unfolded states using enhanced-sensitivity pulsed field gradient NMR techniques. J. Biomol. NMR, 4, 845–858. [DOI] [PubMed] [Google Scholar]
- Zheng N., Wang,P., Jeffrey,P.D. and Pavletich,N.P. (2000) Structure of a c-Cbl-UbcH7 complex: RING domain function in ubiquitin-protein ligases. Cell, 102, 533–539. [DOI] [PubMed] [Google Scholar]