

Abstract
The human immunodeficiency virus (HIV) Tat protein plays an essential role in promoting efficient transcriptional elongation of viral transcripts. We report that the transcriptional co-activator PCAF and Tat interact and synergize to activate the HIV promoter. The binding of Tat and PCAF in vitro and in vivo is dependent on the acetylated state of Lys50 of Tat and on the PCAF bromodomain. Structural analysis of the acetylated Tat peptide bound to the PCAF bromodomain defined amino acids Y47 and R53 in Tat and V763, Y802, and Y809 in PCAF as critical interaction points between the two proteins. Mutation of each of these residues in either Tat or PCAF inhibited in a cumulative manner the Tat–PCAF interaction in vitro and in vivo, and abrogated the synergistic activation of the HIV promoter by both proteins. These observations demonstrate that acetylation of Tat establishes a novel protein–protein interaction domain at the surface of Tat that is necessary for the transcriptional activation of the HIV promoter.
Keywords: acetylation/bromodomain/HIV/PCAF/Tat
Introduction
The Tat protein of human immunodeficiency virus (HIV) is a unique viral transactivator that binds to the Tat-responsive element (TAR), an RNA stem–loop structure that forms at the 5′ extremity of all viral transcripts (Cullen, 1998; Karn, 1999). In the absence of Tat, HIV transcription is highly inefficient because the assembled RNA polymerase II complex (RNAPII) cannot elongate efficiently on the viral DNA template (Garber and Jones, 1999). The binding of Tat to TAR stimulates the production of full-length HIV transcripts, and the integrity of the Tat/TAR axis critically determines the dynamics of viral replication in infected cells.
Recent experiments have defined the role of Tat in HIV transcription as an adaptor that co-ordinates the recruitment of critical co-factors near the transcription start site in the HIV promoter. Distinct functional domains of Tat are involved in the recruitment of different co-factors. The N-terminal cysteine-rich region interacts directly with the cyclin T1/cyclin-dependent kinase 9 (CDK-9) complex (Wei et al., 1998). Recruitment of CDK-9 is thought to lead to hyperphosphorylation of the C-terminal domain of RNAPII and increased elongation efficiency. The C-terminal arginine-rich motif (ARM) in Tat is essential for RNA binding and nuclear localization. A single lysine residue in the ARM, K50, becomes acetylated by the transcriptional co-activator p300 in vitro and in vivo (Kiernan et al., 1999; Ott et al., 1999). Mutation of K50 to arginine, a mutation that conserves the local charge but prevents acetylation by p300, markedly decreases the synergistic activation of the HIV promoter by Tat and p300 (Ott et al., 1999).
Reversible acetylation of lysine residues was first identified in histone proteins and plays an essential role in transcriptional regulation. Non-histone proteins including the transcriptional regulators TFIIE, TFIIF, p53, EKLF, GATA-1, HMGI(Y), HMG17, ACTR, MyoD and E2F1 are also reversibly acetylated (for a review see Kouzarides, 2000). In the case of chromatin, the level of acetylation of distinct lysine residues in each histone protein is under the control of competing histone acetylases (HATs) and histone deacetylases. Histone hypoacetylation is generally associated with transcriptional repression, while histone hyperacetylation has been correlated with transcriptional activation. Early models proposed that histone acetylation leads to a global neutralization of positive charges on histones and loosening of the histone–DNA interaction at transcriptionally active sites. However, recent data suggest that acetylated lysine residues on histone tails serve as a recognition code for the co-ordinated recruitment of specific factors (the ‘histone code’ hypothesis; Strahl and Allis, 2000). According to this model, acetylated lysine residues in the histone tails interact with a specialized protein module, the bromodomain (Dhalluin et al., 1999; Jacobson et al., 2000). Bromodomains are present in a variety of proteins, including nuclear HAT proteins, kinases and chromatin remodeling factors (Jeanmougin et al., 1997).
To test the possibility that acetylated Tat also interacts with a bromodomain, we tested a variety of bromodomain-containing proteins for potential interaction with the acetylated ARM peptide of Tat. We observed that PCAF, a transcriptional co-activator containing a bromodomain, interacted specifically with acetylated Tat (Mujtaba et al., 2002).
The goal of this study was to investigate the Tat–PCAF interaction and to examine its relevance to the transcriptional activation process mediated by Tat. We demonstrate that Tat and PCAF interact specifically via the ARM domain of Tat and the bromodomain of PCAF in vitro and in vivo. This interaction is critically dependent on the acetylation of K50 in Tat. Tat and PCAF functionally synergize to activate the HIV promoter and amino acids that are critical for the interaction in vitro and in vivo are also important for the transcriptional activity of the Tat protein.
Results
Tat and the transcriptional co-activator PCAF interact functionally to activate the HIV promoter
The identification of the bromodomain of PCAF as a protein module that specifically recognizes the acetylated ARM peptide (Mujtaba et al., 2002) suggested that PCAF might play a role in the transcriptional activation of the HIV promoter. To test this possibility, we co-transfected expression vectors for PCAF and Tat and measured their combined effect on the activity of the HIV promoter. The HIV promoter was activated synergistically in response to Tat and PCAF (Figure 1A). This synergy was particularly striking at low Tat concentrations and decreased at higher Tat plasmid concentrations (not shown). This synergy was specific for the HIV promoter and was not observed when another retroviral promoter, from Rous sarcoma virus, was co-transfected (data not shown).

Fig. 1. Functional synergy and physical interaction between HIV Tat and PCAF. (A) Synergistic activation of the HIV promoter by Tat and PCAF. A PCAF expression vector (HA-PCAF) (Yang et al., 1996b) or a control empty vector was co-transfected with an HIV Tat expression plasmid (Tat-FLAG) (Ott et al., 1999) and the LTR-luciferase reporter construct into HeLa cells. Cells were harvested after 24 h, and the luciferase activity was measured. Luciferase values represent the mean ± SEM of three independent experiments. (B) Tat and PCAF co-immunoprecipitate from cellular lysates. HEK 293 cells were co-transfected with HA-PCAF and Tat-FLAG expression vectors (+) or the corresponding empty vector controls (–). In the left panel, cellular lysates analyzed by western blot (WB) show equal protein expression. Equal amounts of protein (500 µg/lane) were subjected to immunoprecipitation (IP) with the indicated antibodies and protein G–Sepharose. The immunoprecipitated material was analyzed by western blotting and antisera specific for the FLAG and HA epitopes (middle and right panels).
To determine whether Tat and PCAF can interact within cells, we co-transfected an expression vector for an epitope (FLAG)-tagged Tat protein with a hemagglutinin (HA)-tagged PCAF expression vector. Western blot analysis of lysates with antisera for FLAG and HA demonstrated comparable expression levels of both proteins after transfection (Figure 1B). Cellular extracts of transfected cells were subjected to immunoprecipitation with antisera specific for the FLAG or HA epitope. The immunoprecipitated material was analyzed by western blotting analysis for both Tat and PCAF. PCAF co-immunoprecipitated with Tat in both directions. These experiments suggested that both proteins stably interact within cells and that this interaction might be important for their synergistic activation of the HIV promoter.
Tat K50 and K51 are necessary for functional synergy and interaction between Tat and PCAF
To test our original hypothesis that Tat binding to PCAF is mediated, at least in part, by the specific recognition of acetylated Tat by the bromodomain of PCAF, we replaced Tat residues K50 and K51 with arginines. This amino acid substitution blocks acetylation and conserves the local charge environment in the Tat protein. Comparison of the transcriptional activities of wild-type Tat (Tat-wt) and the Tat mutant (Tat-RR) showed that inhibition of Tat acetylation decreased the synergy between Tat and PCAF by 70% (Figure 2A). In agreement with the functional data, we observed that mutation of K50 and K51 partially suppressed the interaction between Tat and PCAF in vivo (Figure 2B).
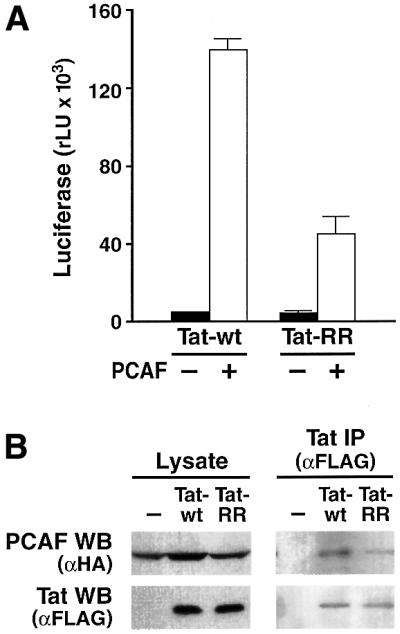
Fig. 2. Tat K50 is important for the synergy with PCAF and for Tat binding to PCAF. (A) Tat expression vector (Tat-wt or Tat-RR, in which K50 and K51 were replaced with arginine) was co-transfected with a PCAF expression vector and an LTR-luciferase reporter in HeLa cells. Luciferase values are the mean ± SEM of three independent transfection experiments. (B) Expression vectors for Tat-wt and Tat-RR (both FLAG tagged) were co-transfected with PCAF-HA. In the left panel, cellular lysates analyzed by western blot (WB) show equal protein expression. Equal amounts of protein (500 µg/lane) were subjected to immunoprecipitation (IP) with the anti-FLAG antiserum and protein G–Sepharose. The immunoprecipitated material was analyzed by western blotting and antisera specific for the FLAG and HA epitopes (right panel).
Inhibition of Tat transactivation of the HIV promoter by an antiserum specific for the bromodomain of PCAF
To further assess the role of the PCAF bromodomain in Tat activity, we raised a polyclonal antiserum using a recombinant PCAF bromodomain expressed in Escherichia coli. This antiserum specifically recognized PCAF by western blot in cellular extracts, and did not cross-react with cellular proteins (Figure 3A). It also exhibited high specificity, since the closely related bromodomain from the transcriptional co-activator CBP was not recognized, even when the antiserum was used at high concentration in an ELISA (Figure 3B).

Fig. 3. Inhibition of Tat transactivation by an antiserum specific for the PCAF bromodomain. (A) Western blot analysis of cellular lysates (50 µg/lane) of HEK 293 cells transfected with HA-PCAF with an antiserum specific for the PCAF bromodomain (αPCAF-BD) (1:100). The same membrane was probed with an anti-HA antiserum as control to visualize PCAF-HA (αHA). (B) Purified anti-PCAF bromodomain IgGs recognize recombinant PCAF bromodomain (PCAF-BD) and not the closely related CBP bromodomain (CBP-BD) proteins. Bromodomain proteins (500 ng of protein) were coated onto polystyrene plates and developed by ELISA with different concentrations of the anti-PCAF bromodomain IgGs (10–0.3 mg/ml). (C) HeLa cells stably expressing Tat protein (HeLa-Tat) and standard HeLa cells were microinjected with the HIV LTR-luciferase reporter. Another construct containing GFP under the control of the CMV promoter was co-injected as a control. Four hours post-injection, GFP-positive cells were counted, and cells were harvested for luciferase activity measurements. Average luciferase values (relative light units rLU ± SEM; left panel) and average number of GFP-positive cells (± SEM; middle panel) from five independent experiments are shown. In the right panel, HeLa-Tat cells microinjected with the HIV LTR-luciferase construct were treated with 0, 10 or 50 µM 5,6-dichlorobenzimidazole riboside (DRB), an inhibitor of the Tat-associated CDK-9, and luciferase activity was measured. (D) Anti-PCAF BD or pre-immune IgGs (5 µg/ml) were microinjected into HeLa-Tat cells with the HIV LTR-luciferase reporter construct, and luciferase values were measured as described above. Luciferase values are the mean ± SEM of three independent experiments. (E) Anti-PCAF BD or pre-immune IgGs (5 µg/ml) were pre-incubated with recombinant PCAF BD in vitro. The resulting mixture was microinjected into HeLa-Tat cells with the HIV LTR-luciferase reporter construct, and luciferase values were measured as described above. Luciferase values are the mean ± SEM of three independent experiments. (F) Constructs corresponding to 5× Gal4 binding site upstream of the TK promoter (Puigserver et al., 1999) or the HIV LTR both driving the luciferase reporter gene were microinjected in HeLa cells with either a plasmid encoding Gal4 or a Gal4–VP16 fusion protein. A representative experiment is shown. (G) A plasmid corresponding to 5× Gal4 binding site upstream of the TK promoter (Puigserver et al., 1999) was microinjected in HeLa cells with a vector encoding the Gal4–VP16 fusion protein in the presence of anti-PCAF BD or pre-immune IgGs (5 µg/ml). Luciferase values were measured as described above. Luciferase values are the mean ± SEM of three independent experiments.
Next, we established a nuclear microinjection assay to measure Tat transactivation quantitatively. HeLa cells stably expressing a full-length Tat protein (HeLa-Tat) or control HeLa cells were microinjected with a construct in which the luciferase reporter is under the control of the HIV promoter. A construct expressing enhanced green fluorescent protein (GFP) under the control of the cytomegalovirus promoter (CMV-GFP) was co-injected to assess the number and viability of injected cells before harvest. In a representative experiment, luciferase values were 500-fold higher in HeLa-Tat cells than in control HeLa cells (Figure 3C). On average, 100 cells were injected per condition, and the survival rate was ∼75% in both cell lines (Figure 3C, middle panel). In agreement with previous observations, Tat transactivation in this system was inhibited in a dose-dependent manner by 5,6-dichlorobenzimidazole riboside (DRB), a CDK-9 inhibitor that blocks Tat function (Mancebo et al., 1997; Figure 3C, right panel). Microinjection of an antiserum specific for the bromodomain of PCAF inhibited Tat-mediated transactivation of the HIV promoter in this system, whereas microinjection of the pre-immune serum or no antibody had no effect (Figure 3D). Additional control experiments were performed to ensure the specificity of this inhibition. First, we observed that pre-incubation of PCAF BD antiserum with an excess of recombinant PCAF BD protein prior to microinjection blocked the suppressive effect of this antiserum on HIV transcription (Figure 3E). Secondly, we tested the effect of the PCAF BD antiserum on another Tat-independent promoter. We used a plasmid containing five Gal4 binding sites upstream of the thymidine kinase (TK) promoter (Puigserver et al., 1999). This promoter can be induced transcriptionally by a fusion protein between the DNA binding domain of Gal4 and the transactivating domain of VP16, but not by the DNA binding domain of Gal4 alone (Figure 3F). Microinjection of the anti-PCAF BD antiserum had no effect on the transactivation mediated by the Gal4–VP16 fusion protein (Figure 3G). These experiments demonstrate that the bromodomain of PCAF and the ARM region of Tat are important for the transactivation of the HIV promoter and are consistent with a direct interaction between Tat and PCAF mediated by the bromodomain of PCAF and acetylated K50 of Tat.
Direct interaction between acetylated Tat and the bromodomain of PCAF in vitro
As discussed above, preliminary experiments using a GST–PCAF bromodomain fusion protein indicated that a Tat peptide corresponding to the ARM region of Tat (GGLGISYGRKKRRQRRRP) could bind directly to the recombinant PCAF bromodomain.
We demonstrated independently the ability of Tat to interact directly with the PCAF bromodomain with two distinct biochemical approaches. First, we used the recombinant PCAF bromodomain to coat microwell plates and tested its ability to bind a biotinylated peptide corresponding to amino acids 41–58 of Tat. Reactions were developed colorimetrically after incubation with streptavidin–horseradish peroxidase (HRP). We detected a dose-dependent binding of K50-acetylated Tat peptide. In contrast, the same Tat peptide containing unacetylated K50 showed lower, dose-independent binding, consistent with a non-specific interaction (Figure 4A). To determine whether full-length Tat also interacts with the PCAF bromodomain, we used a GST–PCAF bromodomain fusion protein and two full-length synthetic Tat proteins, one unacetylated and the other acetylated at K50. Both proteins activated the HIV promoter transcriptional activity after microinjection into HeLa cells (M.Ott and E.Verdin, unpublished observations), and the chemical synthesis ensured the biochemical homogeneity of the protein preparations. Both proteins were incubated either with purified GST or GST–PCAF bromodomain proteins in vitro. K50-acetylated Tat bound to the GST–PCAF bromodomain but not to GST alone. This interaction was strictly dependent on the presence of an acetyl group on K50, since the unacetylated Tat protein did not bind to the PCAF bromodomain (Figure 4B).
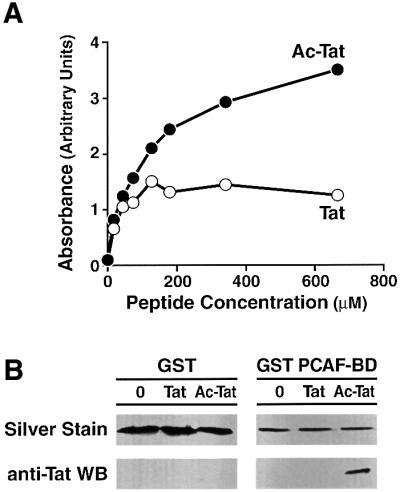
Fig. 4. PCAF bromodomain directly interacts with acetylated Tat in vitro. (A) An ELISA was used to demonstrate direct binding of the ARM peptide of Tat with recombinant bromodomain protein. Plates were coated with the bromodomain and incubated with biotinylated ARM peptide corresponding to Tat amino acids 43–58 containing either acetylated or unacetylated Tat. The binding of the peptide to the plate was measured with streptavidin–HRP and a colorimetric assay. A representative experiment is shown (duplicates). (B) Recombinant GST–PCAF bromodomain or GST protein was incubated with fully synthetic Tat proteins (non-acetylated or acetylated on K50). GST–PCAF and GST proteins were bound to glutathione beads, washed and eluted in Laemmli buffer. Eluted proteins were analyzed by silver staining to visualize the eluted GST and GST PCAF BD proteins, and by western blotting with an antiserum specific for Tat.
Mutations in the PCAF bromodomain and the ARM domain of Tat cumulatively inhibit their interaction in vivo
The PCAF bromodomain structure consists of a left-handed, four-helix bundle (helices aZ, aA, aB and aC; Dhalluin et al., 1999). Analysis of the Tat/PCAF bromodomain structure revealed that while the overall three-dimensional structure of the bromodomain was preserved, the ZA and BC loops, which compose the acetyllysine binding site, underwent significant conformational changes when bound to Tat. The Tat peptide adopted an extended conformation and lay across a pocket formed between the ZA and BC loops. The side-chain of the acetyllysine residue was located in the protein hydrophobic cavity and interacted extensively with several PCAF bromodomain residues, including V752, Y760, I764, Y802 and Y809. Peptide residues flanking Tat K50 also contacted the protein. Tat residues G48, R49 and R53 showed intermolecular nuclear Overhauser effects (NOEs) on the protein, while Y47 and Q54 formed extensive pair-wise interactions with V763 and E756 of PCAF, respectively (for full description and discussion of these observations, see Mujtaba et al., 2002). In vitro mutagenesis analysis of PCAF based on this structural information confirmed that residues Y809, Y802, V752 and F748 of PCAF were essential for acetyllysine binding, whereas V763 and E756 were important for recognition of Tat residues Y47, R53 and Q54. Together, these specific interactions result in a highly selective association between Tat and the bromodomain of PCAF.
To examine the in vivo relevance of these amino acids to the binding of Tat to PCAF in vivo, we introduced point mutations in PCAF at positions 809 (Y809A), and 763 and 802 (V763+Y802A) with the aim of destabilizing the Tat–PCAF interaction. We also introduced corresponding mutations in the Tat protein at amino acids that participate in the interaction with the bromodomain of PCAF (Y47A, R53A, R53E and Y47A+R53A). These constructs were co-transfected into HeLa cells, and the Tat–PCAF interaction was tested after immunoprecipitation of Tat. All recombinant proteins, Tat and PCAF, wild-type and mutants, were expressed at the same level after transfection (Figure 5A). Tat-wt immunoprecipitated PCAF and PCAF-bromodomain mutants (V763+Y802A and Y809) to nearly the same extent (Figure 5). Introduction of mutations in Tat in amino acids involved in bromodomain recognition (Y47A, R53A, R53E and Y47A+R53A) significantly reduced the ability of Tat to immunoprecipitate wild-type PCAF (Figure 5). Combining Tat mutations with PCAF mutations completely abolished the Tat–PCAF interaction (Figure 5). This experiment confirmed the crucial role of the PCAF bromodomain and the Tat ARM domain in the PCAF/Tat interaction. Amino acids defined as critical by structural analysis and by in vitro binding assays are also critical for Tat–PCAF interaction in vivo.

Fig. 5. Differential binding of Tat-wt and ARM mutants to the PCAF BD and to TAR RNA. (A) HA-PCAF and Tat-FLAG expression vectors (wild-type or mutant proteins) were transfected in HEK 293 cells. In the two upper panels, cellular lysates were directly assayed by western blotting (WB) with the indicated monoclonal antibodies (αHA for PCAF and αFLAG for Tat) to assess the level of protein expression after transfection (Pre-IP Lysate). In the lower two panels, cellular lysates were subjected to Tat immunoprecipitation with anti-FLAG antiserum, and the immunoprecipitated material was analyzed by western blot (WB) with the indicated monoclonal antibodies. (B) Binding of Tat-wt and ARM mutants to TAR. Tat and mutant Tat proteins were translated and biolabeled in vitro, incubated with a synthetic RNA corresponding to nucleotides 1–57 of TAR-biotin. After incubation, TAR and bound proteins was bound to streptavidin–agarose beads, centrifuged and washed. The bound proteins were eluted and analyzed by SDS–PAGE and autoradiography.
Since the region targeted by our mutations is also involved in RNA binding of Tat to TAR, we assessed the effect of each mutation on TAR binding. In vitro translated Tat-wt was incubated with a synthetic TAR RNA incorporating a biotin label at its 5′ extremity. The TAR RNA was bound to agarose–streptavidin beads, centrifuged and the bound protein eluted by boiling in Laemmli buffer. The eluted material was analyzed by SDS–PAGE analysis followed by autoradiography. This analysis showed that three mutants, K50/51RR, Y47A and R53A, showed relative increased binding to TAR. In contrast, the two remaining mutants, R53E and Y47A/R53A, showed moderately decreased binding (40%) in comparison with Tat-wt (Figure 5B).
Mutation that suppress the Tat–PCAF interaction in vitro and in vivo also inhibit Tat-mediated transactivation
To further define the role of the Tat–PCAF interaction in relation to the Tat-mediated transactivation process, we examined the effect of the same Tat and PCAF mutations on Tat transactivation. First, we examined the effect of these mutations without adding exogenous PCAF. Since the ARM region studied here is also the nuclear/nucleolar localization signal, mutations in this domain might interfere with the subcellular localization of Tat. To exclude this possibility, all transfections were performed both with Tat-FLAG constructs and with fusion constructs in which the nuclear localization signal of SV40 large T antigen was added to the N-terminus of Tat-FLAG (NLS-Tat). NLS-Tat protein was fully competent to activate the HIV promoter (Figure 6A), and all mutants in the ARM domain of Tat expressed as NLS fusion proteins were transported to the nucleus with the same efficiency, as determined by immunofluorescence microscopy (data not shown). Both individual and combined mutations in the ARM domain of Tat inhibited the transcriptional activity of NLS-Tat on the HIV LTR with the following efficiency: R53E > Y47A+R53A > Y47A > R53A (Figure 6A). Since a partial defect in RNA binding was noted for both the R53E and Y47A/R53A mutants, the defect in transactivation is difficult to interpret. However, the R53A and Y47A single mutants bound TAR with normal or increased affinity; therefore, the defect in transactivation is likely to occur as a result of defective PCAF binding. Similar results were observed when the same mutations were examined in the context of Tat protein not fused to the SV40 large T antigen NLS (data not shown).

Fig. 6. Functional synergy between Tat and PCAF is disrupted by mutations that inhibit interactions between the PCAF bromodomain and acetylated Tat. (A) Tat-FLAG-expression vectors and mutants were fused to the NLS of the SV40 large T antigen and transfected into Jurkat cells with the LTR-luciferase reporter construct. Similar results were obtained with expression vectors for Tat proteins not fused to the SV40 large T antigen (data not shown). (B) Expression vectors for Tat and mutants and PCAF were co-transfected into HeLa cells with the LTR-luciferase reporter construct. Cells were harvested after 24 h, and luciferase activity was measured. Luciferase values are the mean ± SEM of three independent transfections. (C) Expression vectors for Tat and PCAF were co-transfected into HeLa cells with the LTR-luciferase reporter construct. Cells were harvested after 24 h, and luciferase activity was measured. Luciferase values are the mean ± SEM of three independent transfections.
Next, we examined the ability of each Tat mutant to synergize with PCAF. As shown above, co-transfection of Tat with wild-type PCAF led to synergistic activation of the HIV promoter (Figure 6B). Mutations in Tat decreased the synergy with wild-type PCAF in the same manner as in the absence of exogenous PCAF: R53E > Y47A+R53A > Y47A > R53A (Figure 6B). Finally, the effect of wild-type PCAF and mutant PCAF Y809A were compared both in the presence of Tat-wt and mutant Tat proteins (Figure 6C). This experiment showed that the PCAF Y809 mutation aggravated the defect caused by the Tat mutations alone. In fact, transfection of the PCAF mutant suppressed Tat-mediated transactivation in comparison to the control transactivation (–PCAF; Figure 6C). This observation suggest that PCAF Y809A functions as a dominant-negative mutant in Tat transactivation and suppresses partially the activity of endogenous wild-type PCAF.
Discussion
The discovery that the HIV Tat protein is modified by reversible acetylation raised new questions regarding the mechanism of Tat transactivation. Here, we present evidence that acetylated Tat specifically interacts with the transcriptional co-activator PCAF. This interaction is direct and mediated by the bromodomain of PCAF, a conserved protein module previously described to contain an acetyllysine binding pocket for histone peptides (Dhalluin et al., 1999). Point mutations of amino acid residues Y47 and R53 in Tat or residues V763 and Y802 in PCAF severely impaired both the binding of Tat to PCAF in vivo and the transcriptional synergy observed between the two proteins on the HIV promoter. These findings support the model that the recruitment of PCAF by acetylated Tat plays an important role in the regulation of HIV transcription.
PCAF was originally identified as a factor that competes with E1A for binding to p300 (Yang et al., 1996b). PCAF exhibits HAT activity (Ogryzko et al., 1996) and resides in cells in multiprotein complexes containing other transcriptional regulatory proteins, including counterparts of the yeast ADA2, ADA3 and SPT3 proteins (Ogryzko et al., 1998), histone-like TAFs (Ogryzko et al., 1998) and the transcriptional co-activators p300 and ACTR/Src1 (Yang et al., 1996b; Chen et al., 1997). PCAF is also associated with the elongation-competent form of RNA polymerase II (Cho et al., 1998) and plays an important role in several biological functions, including differentiation, cell cycle progression and gene-specific transcriptional regulation.
Although the acetylated ARM domain and the PCAF bromodomain were sufficient to mediate a Tat–PCAF interaction, other domains in each protein might further contribute to binding of the two proteins in vivo. Tat reportedly binds to p300 (Benkirane et al., 1998; Hottiger and Nabel, 1998; Marzio et al., 1998), which interacts with PCAF (Yang et al., 1996b). However, we have been unable to detect p300 in PCAF and Tat co-immunoprecipitation experiments and conclude that p300 does not participate in the Tat–PCAF interaction. Benkirane et al. (1998) reported that PCAF interacts with Tat in GST pull-down experiments and synergizes with Tat to activate the HIV promoter. The HAT domain of PCAF was essential for PCAF-mediated activation of the HIV promoter, and this activation was only observed on chromatinized templates. Our observations confirm the functional synergy between Tat and PCAF and further demonstrate that the Tat–PCAF interaction in vitro and in vivo and their transcriptional synergy are mediated by the specific recognition of acetylated Tat by the PCAF bromodomain. In particular, the inhibition of Tat activity by microinjection of a specific anti-PCAF bromodomain antiserum supports the in vivo relevance of the observed Tat–PCAF bromodomain interaction.
The role of the PCAF HAT domain in the acetylation of histones or Tat remains to be investigated further. PCAF has been reported to acetylate K28 of Tat (Kiernan et al., 1999). However, a Tat peptide containing acetylated K28 does not specifically interact with the PCAF bromodomain (Mujtaba et al., 2002). We have also observed that recombinant PCAF acetylates Tat on K50 in the ARM domain with weak efficiency (W.Dormeyer, A.Dorr, M.Schnolzer and M.Ott, manuscript in preparation). However, given the marked discrepancy in the efficiency of Tat acetylation by p300 and PCAF, we do favor the hypothesis that Tat acetylation is mediated by p300.
The specific interaction of acetylated Tat with the bromodomain of PCAF could modulate the enzymatic activity of PCAF on histones or other substrates. Recently, another viral transactivator, the protein E1A of adenovirus, was shown to bind to several HAT proteins and to modulate their enzymatic and biological activities (Chakravarti et al., 1999; Hamamori et al., 1999). Such an effect of Tat on PCAF and other bromodomain-containing proteins could potentially explain the pleiotropic effects exerted by Tat on the expression of many cellular genes (reviewed in Rosenblatt et al., 1995).
While the data presented here establish a potential mechanism for the Tat–PCAF synergy, it will be critical to determine at what step in the transactivating process this interaction takes place. Our working model is that Tat becomes acetylated after binding to TAR and coming into close contact with p300 bound to the HIV promoter (Marzio et al., 1998; Ott et al., 1999). According to this model, the interaction between Tat and PCAF is restricted to Tat associated with the HIV promoter. It is not entirely clear whether PCAF can interact with Tat bound to TAR or whether the Tat–PCAF interaction causes Tat to dissociate from TAR. In vitro titration experiments indicate that PCAF competes efficiently against TAR RNA for binding to the K50-acetylated Tat peptide (Mujtaba et al., 2002). In vitro RNA gel shift experiments have also failed to show the binding of PCAF to TAR in the presence or the absence of Tat, whereas Tat bound to TAR efficiently (data not shown). These results imply that the PCAF bromodomain interaction with acetylated K50 on Tat may lead to the release of acetylated Tat from TAR RNA. Such dissociation could help in transferring Tat from TAR onto the elongating RNAPII, as reported previously (Wu-Baer et al., 1995; Yang et al., 1996a; Cujec et al., 1997). PCAF specifically associates with the elongating form of RNAPII and is thought to play a role in the hyperacetylation of histones in transcribed domains of chromatin (Cho et al., 1998). In contrast, p300 is associated with the hypophosphorylated, initiation-competent form of RNAPII (Cho et al., 1998). It is conceivable that acetylated Tat helps in the loading of PCAF to the elongating polymerase (Wu-Baer et al., 1995; Yang et al., 1996a; Cujec et al., 1997). According to this scenario, acetylated Tat could serve as a specific adaptor between PCAF and the elongation-competent RNAPII, thereby facilitating transcriptional elongation. Future experiments will test this hypothesis and should further increase our understanding of HIV transcriptional regulation by the transactivator protein Tat.
Materials and methods
Cells and plasmids
HeLa and HEK 293 cells were from the American Type Culture Collection, Jurkat 1G5 cells were obtained from Aguilar-Cordova and colleagues through the AIDS Research and Reference Reagent Program (Division of AIDS, NIAID, NIH). HeLa-Tat cells were a gift from P.Krammer, Heidelberg, Germany (Westendorp et al., 1995). The N-terminally HA-tagged PCAF construct was amplified from the PCAF open reading frame (ORF) (Yang et al., 1996b) with a 5′ primer containing the coding sequence for influenza HA and cloned into pCI (Promega). The full-length (101 amino acid) C-terminally FLAG-tagged Tat-wt and Tat-RR have been described previously (Ott et al., 1999). HA-PCAF and Tat-FLAG constructs were used as templates for QuickChange site-directed mutagenesis with primers carrying the indicated mutations (Stratagene). After mutagenesis, the Tat ORF was fully sequenced. For PCAF, a PvuII–KpnI fragment containing the HAT region and the bromodomain was subcloned, sequenced and cloned back into the wild-type construct. The LTR-luciferase construct has been described previously (Emiliani et al., 1996). The CMV-GFP construct was obtained from Clontech.
Transfections and luciferase assays
Initial co-transfections with Tat-, Tat-RR- and PCAF-expressing plasmids were performed in HeLa cells as described previously (Ott et al., 1999). The co-transfections with Tat mutants and PCAF mutants were performed in HeLa cells with the calcium phosphate precipitation method. Cells were harvested 24 h after transfection of 1.5 µg of plasmid DNA per well (6-well plates) and analyzed for luciferase activity. Transient transfections into Jurkat 1G5 cells were performed with DEAE–dextran (225 ng of DNA/3 × 105 cells). Cells were harvested after 24 h, and luciferase activity was measured (Ott et al., 1999).
Co-immunoprecipitations
HEK 293 cells were transfected in duplicate with expression vectors for Tat-FLAG, HA-PCAF, or empty vector constructs (total 1 µg of DNA) using 8 µl LipofectAMINE reagent (Invitrogen Life Sciences) for 6 h. After 24 h, cells were lysed in 250 mM NaCl, 0.1% NP-40, 20 mM NaH2PO4 pH 7, 5 mM EDTA, 30 mM sodium pyrophosphate, 10 mM NaF and protease inhibitor cocktail tablets (Roche). Duplicates were pooled and equal amounts of total protein were immunoprecipitated with anti-FLAG (M2; Sigma) or anti-HA (Roche) monoclonal antibodies (10 µg/ml each) together with protein G–Sepharose (Amersham Pharmacia) for 6 h at 4°C. Pellets were washed five times in lysis buffer, resuspended in Laemmli buffer, and analyzed by western blot with anti-FLAG (M2; Sigma) or anti-HA (Roche) monoclonal antibodies (10 µg/ml each).
Tat synthesis and purification
Solid-phase peptide synthesis of full-length 72 amino acid one-exon Tat was performed with a sequence derived from the isolate HIV-1BRU on an Applied Biosystems 433A peptide synthesizer by standard Fmoc-Strategy. For synthesis of the acetylated Tat protein (Nε-Lys50), the Nε group of K50 was protected by the ivDde group [Nε-1(4,4 dimethyl-2,6-dioxocyclohex-1-ylidene)-3-methylbutyl]. To deprotect the ivDde group in K50, the peptide was treated with 5% hydrazine in dimethylformamide for 5 min and one part of the product was acetylated with acetic anhydride. Both acetylated and non-acetylated Tat peptides were fully deprotected with trifluoracetic acid, containing 3% triisopropylsilane and 5% water. The peptides were purified to homogeneity by reverse-phase high-pressure liquid chromatography. The correct molecular weights of 8340 Da for the Tat and 8382 Da for the acetylated Tat were established by positive-ion ESI mass spectra recorded on an ion trap Finnigan LCQ mass spectrometer.
Antibody generation and use
Recombinant CBP and PCAF bromodomain proteins were generated as previously reported (Dhalluin et al., 1999). PCAF bromodomain protein was conjugated to activated keyhole limpet hemocyanin (Pierce). CHB rabbits were immunized subcutaneously with 150 µg of conjugated protein in complete Freund’s adjuvant (Sigma) followed by four boosts in incomplete Freund’s adjuvant. Immunoglobulin G (IgG) fractions were purified with Gammabind plus Sepharose (Amersham Pharmacia Biotech). For ELISA, recombinant PCAF BD or CBP BD proteins were coated overnight at 4°C on Maxisorp polystyrene plates (Nalge Nunc International) and blocked with milk (5% skimmed dry milk in T-TBS). After incubation with anti-PCAF BD lgGs at the indicated concentrations, reactions were developed with 10 µg/ml anti-rabbit IgG-HRP (Jackson ImmunoResearch Laboratories) and 5 mg of orthophenylenediamine dihydrochloride (OPD)/ml H2O (Sigma) as a colorimetric reagent. Reactions were stopped after 30 min, and absorbances read at 492 and 620 nm on a Multiskan MS ELISA plate reader (Labsystems). For western blot analysis, 50 µg of total cell lysate of HEK 293 cells transfected with HA-PCAF or the vector control were analyzed by SDS–PAGE and developed with anti-PCAF BD antiserum (1:100) or anti-HA monoclonal antibody (Roche).
Microinjection experiments
Subconfluent HeLa-Tat cells (70%) were grown on Cellocate coverslips (Eppendorf) and microinjected at room temperature with an automated injection system (Carl Zeiss). Samples were prepared as a 20 µl injection mix containing the LTR-luciferase (100 ng/ml) and CMV-GFP (50 ng/ml) constructs together with 5 mg/ml anti-PCAF BD IgGs or pre-immune IgGs. Live cells were examined on a Zeiss Axiovert microscope to determine the number of GFP-positive cells. Four hours after injection, cells were washed in cold phosphate buffer and processed for luciferase assays (Promega).
ELISA
Microwell plates were coated with the PCAF bromodomain (1 µg/ml in 10 mM Na or K phosphate, pH 7.0) for 2 h at room temperature. Non-specific binding sites were saturated with a 10% bovine serum albumin (BSA) solution in binding buffer (100 mM Na or K phosphate, 100 mM NaCl, 5 mM dithiothreitol) for 2 h at room temperature. Wells were washed twice with binding buffer containing 0.1% BSA and 0.05% Tween-20 (washing buffer). Increasing concentrations of the Tat peptide (biotin-GGLGISYGRK50KRRQRRRP, acetylated on K50 or not) was allowed to bind overnight at 4°C. Samples were washed four times with washing buffer. Bound peptide was revealed by incubating with a 0.1 µg/ml solution of streptavidin-conjugated HRP for 1 h at room temperature, followed by washes and incubation with tetramethyl benzidine (TMB) as an HRP substrate (Pierce). The reaction was stopped before saturation of the colorimetric reaction by adding 2 M H2SO4. The absorbance of the colored product was measured at 450 nm. Absorbance in each well was corrected for the blank obtained in a corresponding well subjected to the complete procedure but containing no PCAF BD. Results are the average of duplicate samples in one representative experiment.
Tat–PCAF interaction in vitro
Purified GST–PCAF bromodomain proteins (1 µg) were incubated with full-length synthetic acetylated or non-acetylated Tat proteins (5 µg) in binding buffer (97.6 mM NaH2PO4, 12.4 mM Na2HPO4, 250 mM NaCl, 30 mM Na pyrophosphate, 5 mM EDTA, 10 mM NaF, 0.1% NP-40) for 10 min at 30°C. Precleared and blocked (3% BSA) glutathione beads (Pharmacia) were added to the above mixture for another 30 min at 4°C, washed extensively and resuspended in Laemmli buffer. Binding of Tat to the GST–PCAF bromodomain was analyzed by silver staining or by western blot with a guinea pig anti-Tat antibody.
Tat-TAR binding in vitro
Streptavidin–Sepharose beads (Amersham Pharmacia Biotech, Uppsala, Sweden) were blocked for 1 h at 4°C in binding buffer containing 3% nuclease-free BSA (Amersham Pharmacia Biotech). After equilibration in binding buffer, 30 µl beads were incubated with 0.5 µg biotinylated TAR element (biotin-AATTCCAGATCTGAGCCTGGGAGCTCTCTGGA; Xeragon, Zurich, Switzerland) and incubated for 1 h at 4°C. TAR bound to beads was mixed with 2 µl lysates from Tat expression plasmids in vitro translated in the presence of 20 µCi [35S]methionine (Amersham Pharmacia Biotech) using the TNT T7 coupled reticulocyte Lysate System (Promega, Madison, WI). Reactions were incubated for 10 min at 30°C and twice washed with binding buffer prior the addition of loading buffer. Proteins were separated by SDS–PAGE, fixed and amplified (Amplify; Amersham Pharmacia Biotech) for 30 min, dried and exposed to a BioMax MR film (Kodak, Rochester, NY).
Acknowledgments
Acknowledgements
We thank Hajo Delius, Jan Eglinger, Katrin Kählcke, Prisca Kunert and Sebastian Luksch for technical help; Roger Fischer and Michael Trendelenburg for help with microinjections; Peter Krammer for the HeLa-Tat cell line; and Bruce Spiegelman for plasmids. We thank Harald zur Hausen and Warner Greene for support and discussions. We thank John Carroll for graphics, Heather Gravois for manuscript preparation, and Stephen Ordway and Gary Howard for editorial assistance. This work was supported by a strategy fund of the Herrmann von Helmholtz-Gemeinschaft deutscher Forschungszentren (HGF) (to M.O.) and a Public Health Service grant (NIAID AI40847 to E.V.).
References
- Benkirane M., Chun,R.F., Xiao,H., Ogryzko,V.V., Howard,B.H., Nakatani,Y. and Jeang,K.T. (1998) Activation of integrated provirus requires histone acetyltransferase. p300 and P/CAF are coactivators for HIV-1 Tat. J. Biol. Chem., 273, 24898–24905. [DOI] [PubMed] [Google Scholar]
- Chakravarti D., Ogryzko,V., Kao,H.Y., Nash,A., Chen,H., Nakatani,Y. and Evans,R.M. (1999) A viral mechanism for inhibition of p300 and PCAF acetyltransferase activity. Cell, 96, 393–403. [DOI] [PubMed] [Google Scholar]
- Chen H., Lin,R.J., Schiltz,R.L., Chakravarti,D., Nash,A., Nagy,L., Privalsky,M.L., Nakatani,Y. and Evans,R.M. (1997) Nuclear receptor coactivator ACTR is a novel histone acetyltransferase and forms a multimeric activation complex with P/CAF and CBP/p300. Cell, 90, 569–580. [DOI] [PubMed] [Google Scholar]
- Cho H., Orphanides,G., Sun,X., Yang,X.J., Ogryzko,V., Lees,E., Nakatani,Y. and Reinberg,D. (1998) A human RNA polymerase II complex containing factors that modify chromatin structure. Mol. Cell. Biol., 18, 5355–5363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cujec T.P., Cho,H., Maldonado,E., Meyer,J., Reinberg,D. and Peterlin,B.M. (1997) The human immunodeficiency virus trans activator Tat interacts with the RNA polymerase II holoenzyme. Mol. Cell. Biol., 17, 1817–1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullen B.R. (1998) HIV-1 auxiliary proteins: making connections in a dying cell. Cell, 93, 685–692. [DOI] [PubMed] [Google Scholar]
- Dhalluin C., Carlson,J.E., Zeng,L., He,C., Aggarwal,A.K. and Zhou,M.M. (1999) Structure and ligand of a histone acetyltransferase bromodomain. Nature, 399, 491–496. [DOI] [PubMed] [Google Scholar]
- Emiliani S., Van Lint,C., Fischle,W., Paras,P.,Jr, Ott,M., Brady,J. and Verdin,E. (1996) A point mutation in the HIV-1 Tat responsive element is associated with postintegration latency. Proc. Natl Acad. Sci. USA, 93, 6377–6381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garber M.E. and Jones,K.A. (1999) HIV-1 Tat: coping with negative elongation factors. Curr. Opin. Immunol., 11, 460–465. [DOI] [PubMed] [Google Scholar]
- Hamamori Y., Sartorelli,V., Ogryzko,V., Puri,P.L., Wu,H.Y., Wang,J.Y., Nakatani,Y. and Kedes,L. (1999) Regulation of histone acetyltransferases p300 and PCAF by the bHLH protein twist and adenoviral oncoprotein E1A. Cell, 96, 405–413. [DOI] [PubMed] [Google Scholar]
- Hottiger M.O. and Nabel,G.J. (1998) Interaction of human immuno deficiency virus type 1 Tat with the transcriptional coactivators p300 and CREB binding protein. J. Virol., 72, 8252–8256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson R.H., Ladurner,A.G., King,D.S. and Tjian,R. (2000) Structure and function of a human TAFII250 double bromodomain module. Science, 288, 1422–1425. [DOI] [PubMed] [Google Scholar]
- Jeanmougin F., Wurtz,J.M., Le Douarin,B., Chambon,P. and Losson,R. (1997) The bromodomain revisited. Trends Biochem. Sci., 22, 151–153. [DOI] [PubMed] [Google Scholar]
- Karn J. (1999) Tackling Tat. J. Mol. Biol., 293, 235–254. [DOI] [PubMed] [Google Scholar]
- Kiernan R.E. et al. (1999) HIV-1 Tat transcriptional activity is regulated by acetylation. EMBO J., 18, 6106–6118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouzarides T. (2000) Acetylation: a regulatory modification to rival phosphorylation? EMBO J., 19, 1176–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancebo H.S. et al. (1997) P-TEFb kinase is required for HIV Tat transcriptional activation in vivo and in vitro. Genes Dev., 11, 2633–2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzio G., Tyagi,M., Gutierrez,M.I. and Giacca,M. (1998) HIV-1 tat transactivator recruits p300 and CREB-binding protein histone acetyltransferases to the viral promoter. Proc. Natl Acad. Sci. USA, 95, 13519–13524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mujtaba S., He,Y., Zeng,L., Farooq,A., Carlson,J.E., Ott,M., Verdin,E. and Zhou,M.M. (2002) Structural basis of lysine-acetylated HIV-1 Tat recognition by PCAF bromodomain. Mol. Cell, 9, 575–586. [DOI] [PubMed] [Google Scholar]
- Ogryzko V.V., Schiltz,R.L., Russanova,V., Howard,B.H. and Nakatani,Y. (1996) The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell, 87, 953–959. [DOI] [PubMed] [Google Scholar]
- Ogryzko V.V., Kotani,T., Zhang,X., Schiltz,R.L., Howard,T., Yang,X.J., Howard,B.H., Qin,J. and Nakatani,Y. (1998) Histone-like TAFs within the PCAF histone acetylase complex. Cell, 94, 35–44. [DOI] [PubMed] [Google Scholar]
- Ott M., Schnolzer,M., Garnica,J., Fischle,W., Emiliani,S., Rackwitz,H.R. and Verdin,E. (1999) Acetylation of the HIV-1 Tat protein by p300 is important for its transcriptional activity. Curr. Biol., 9, 1489–1492. [DOI] [PubMed] [Google Scholar]
- Puigserver P., Adelmant,G., Wu,Z., Fan,M., Xu,J., O’Malley,B. and Spiegelman,B.M. (1999) Activation of PPARγ coactivator-1 through transcription factor docking. Science, 286, 1368–1371. [DOI] [PubMed] [Google Scholar]
- Rosenblatt J.D., Miles,S., Gasson,J.C. and Prager,D. (1995) Transactivation of cellular genes by human retroviruses. Curr. Top. Microbiol. Immunol., 193, 25–49. [DOI] [PubMed] [Google Scholar]
- Strahl B.D. and Allis,C.D. (2000) The language of covalent histone modifications. Nature, 403, 41–45. [DOI] [PubMed] [Google Scholar]
- Wei P., Garber,M.E., Fang,S.M., Fischer,W.H. and Jones,K.A. (1998) A novel CDK9-associated C-type cyclin interacts directly with HIV-1 Tat and mediates its high-affinity, loop-specific binding to TAR RNA. Cell, 92, 451–462. [DOI] [PubMed] [Google Scholar]
- Westendorp M.O., Frank,R., Ochsenbauer,C., Stricker,K., Dhein,J., Walczak,H., Debatin,K.M. and Krammer,P.H. (1995) Sensitization of T cells to CD95-mediated apoptosis by HIV-1 Tat and gp120. Nature, 375, 497–500. [DOI] [PubMed] [Google Scholar]
- Wu-Baer F., Sigman,D. and Gaynor,R.B. (1995) Specific binding of RNA polymerase II to the human immunodeficiency virus trans-activating region RNA is regulated by cellular cofactors and Tat. Proc. Natl Acad. Sci. USA, 92, 7153–7157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X., Herrmann,C.H. and Rice,A.P. (1996a) The human immunodeficiency virus Tat proteins specifically associate with TAK in vivo and require the carboxyl-terminal domain of RNA polymerase II for function. J. Virol., 70, 4576–4584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X.J., Ogryzko,V.V., Nishikawa,J., Howard,B.H. and Nakatani,Y. (1996b) A p300/CBP-associated factor that competes with the adenoviral oncoprotein E1A. Nature, 382, 319–324. [DOI] [PubMed] [Google Scholar]