

Abstract
Hepatitis C virus proteins are synthesized as a polyprotein cleaved by a signal peptidase and viral proteases. The behaviour of internal signal sequences at the C-terminus of the transmembrane domains of hepatitis C virus envelope proteins E1 and E2 is essential for the topology of downstream polypeptides. We determined the topology of these transmembrane domains before and after signal sequence cleavage by tagging E1 and E2 with epitopes and by analysing their accessibility in selectively permeabilized cells. We showed that, after cleavage by signal peptidase in the endoplasmic reticulum, the C-terminal orientation of these transmembrane domains changed from luminal to cytosolic. The dynamic behaviour of these transmembrane domains is unique and it is linked to their multifunctionality. By reorienting their C-terminus toward the cytosol and being part of a transmembrane domain, the signal sequences at the C-terminus of E1 and E2 contribute to new functions: (i) membrane anchoring; (ii) E1E2 heterodimerization; and (iii) endoplasmic reticulum retention.
Keywords: hepatitis C virus/membrane topology/signal sequence/transmembrane domain/viral envelope protein
Introduction
The biogenesis of most membrane proteins involves targeting the nascent protein to the endoplasmic reticulum (ER), integration into the ER membrane and maturation into a final product. Most eukaryotic membrane proteins are cotranslationally integrated into the ER membrane at sites termed translocons. Translocons are composed of several ER membrane proteins that associate to form an aqueous pore through which secretory proteins and luminal domains of membrane proteins pass from the cytosol to the ER (for a review, see Johnson and van Waes, 1999). The translocon also plays a major role in integration of membrane proteins (Martoglio et al., 1995; Do et al., 1996; Mothes et al., 1997; Heinrich et al., 2000), and hence in the topology of these proteins. The mechanism by which the topology of a protein is directed by the cellular machinery of translocation is rather complex and not fully understood. Indeed, a simple single membrane-spanning protein needs to translocate certain domains into the ER lumen, leave others in the cytosol, properly orient the transmembrane (TM) segment, and move it from the aqueous translocation channel into the lipid bilayer. This is even more complex for polytopic proteins, which have to deal with more than one TM segment. Features such as the length and hydrophobicity of membrane-spanning segments, charge distribution of flanking residues, and size and folding state of surrounding domains have each been documented to affect topology (von Heijne, 1989; Beltzer et al., 1991; Parks and Lamb, 1991; Sakaguchi et al., 1992; Spiess, 1995; Gafvelin et al., 1997; Wahlberg and Spiess, 1997). Signal sequences, which direct the complete translocation of polypeptides into the lumen of the ER, are usually located at the N-terminus of precursor proteins and have relatively short hydrophobic segments (usually 7–12 amino acids; von Heijne, 1990). TM domains, which direct the integration of membrane proteins, generally consist of 20–25 non-polar amino acids, a length sufficient to span the hydrophobic lipid bilayer (Lemmon et al., 1997). Because they are often involved in other functions, e.g. heterodimerization, subcellular localization, signal sequence or fusion (Cleverley and Lenard, 1998; Cocquerel et al., 2000), the TM domains of viral envelope proteins have additional interesting features.
Hepatitis C virus (HCV) is the causal agent of hepatitis C, which is a major health problem worldwide. HCV is a positive-stranded RNA virus that belongs to the Flaviviridae family. Its genome encodes a single polyprotein of ∼3000 amino acid residues that is co- and post-translationally cleaved to generate at least 10 polypeptides (Reed and Rice, 2000). The two envelope glycoproteins, E1 and E2, are released from HCV polyprotein precursor after cleavage by host signal peptidase(s) (Figure 1A) and they assemble to form a non-covalent E1E2 heterodimer (Dubuisson, 2000). These membrane proteins are composed of a large N-terminal ectodomain and a C-terminal hydrophobic anchor. This latter domain is <30 amino acid residues long in both proteins and is composed of two stretches of hydrophobic residues separated by a short segment containing at least one fully conserved positively charged residue (Cocquerel et al., 2000) (Figure 1B). Because HCV synthesizes its polypeptides as a polyprotein that is cleaved co- or post-translationally by viral and host proteases, internal signal peptides must be used to target viral membrane proteins, e.g. the envelope glycoproteins, to the ER. As a consequence, after signal sequence cleavage, the signal peptide of these proteins remains bound to the C-terminus of the protein located upstream on the polyprotein. Such a sequence can then play other functions in the protein to which it is bound. In the case of HCV envelope glycoproteins, such signal sequences become part of their TM domain and contribute to the anchor function of these domains as well as to other functions (Cocquerel et al., 2000).

Fig. 1. (A) Schematic representation of HCV polyprotein. Processing of the N-terminal region of HCV polyprotein generates the envelope proteins E1 and E2. The arrows indicate the sites that are cleaved by a signal peptidase. Sp1, Sp2 and Sp7 are the signal sequences of E1, E2 and the small polypeptide p7, respectively. (B) Sequences and organization of the TM domains of E1 and E2. The TM domains are underlined and charged residues are indicated in bold. The positions of amino acid residues indicated above the sequences correspond to their position in the polyprotein (strain H of HCV).
Owing to the small size of their genome, viruses have evolved by packing a maximum of information in a minimum of polypeptide sequence. As a consequence, many of the proteins or protein domains encoded by viruses are multifunctional. The TM domains of HCV envelope glycoproteins are extreme examples of such multifunctionality. Indeed, besides their role as membrane anchors, these TM domains bear ER retention signals, contain a signal sequence function and are involved in E1E2 heterodimerization (Cocquerel et al., 1998, 1999, 2000; Op de Beeck et al., 2000). The presence of so many functions in sequences of <30 amino acids makes these domains very challenging tools to study the early steps of membrane protein biogenesis. A major question in the study of these domains concerns the topology that they can adopt. The presence of a putative anchor sequence and a signal sequence function separated by charged residues in the TM domains of E1 and E2 envelope glycoproteins had suggested to us that these domains were composed of two membrane-spanning segments with the charged residues facing the cytosol. However, amino acid sequence analysis (Cocquerel et al., 2000) and recent data from our laboratory suggest that, after E1E2 heterodimerization, these TM domains probably exhibit a single membrane-spanning topology (Op de Beeck et al., 2000). In the present study, we tested whether the topology of the TM domains of E1 and E2 changes after cleavage from the polyprotein. We showed that, in the absence of signal sequence cleavage, the TM domains of HCV envelope proteins form a hairpin structure. In contrast, when E1 or E2 were tagged at their C-terminus with an epitope, their TM domain showed a single membrane-spanning topology. Together, these data imply that a change in the orientation of the second stretch of hydrophobic residues occurs after C-terminal signal sequence cleavage, leading ultimately to a single TM segment.
Results
The C-terminal half of the TM domains of HCV envelope proteins possesses a signal sequence function
It has been shown that hydrophobic sequences at the C-terminus of E1 and E2 contain a signal sequence function that is involved in translocation of the protein located immediately downstream on the polyprotein (Figure 1; for a review, see Reed and Rice, 2000). The TM domain of E1 is involved in translocation of the ectodomain of E2, and the TM domain of E2 is supposed to be involved in translocation and integration of a small hydrophobic polypeptide of unknown function called p7. By analysing the sequences of the TM domains of E1 and E2, a sequence having the features of a signal peptide (von Heijne, 1990) can clearly be identified in the C-terminal half of these domains (Figure 1B). We then tried to determine whether, by themselves, these sequences behave as signal sequences, by analysing the glycosylation of reporter proteins fused to them as an indication that translocation has occurred.
To analyse the role of the putative signal sequence present in the TM domain of E1, i.e. Sp2, E2 protein was expressed with or without Sp2 (Figure 2A). The proteins of interest were expressed by using vaccinia virus recombinants and analysed by SDS–PAGE after immunoprecipitation. E2 expressed with Sp2 migrated as a 66 kDa band when analysed by SDS–PAGE, and migrated faster after PNGase F treatment (Figure 2B, compare Sp2E2 with Sp2E2+PNGaseF), indicating that, when expressed with Sp2, E2 was N-glycosylated. These results were very similar to those observed when E2 was expressed from HCV polyprotein (Dubuisson and Rice, 1996). However, in the absence of Sp2 (E2*), a fast migrating band that comigrated with the deglycosylated form of E2 was observed (Figure 2B, compare E2* with Sp2E2+PNGaseF), indicating that, in the absence of Sp2, E2 was not glycosylated (Figure 2B, E2*). Together, these data confirm that Sp2 is a signal sequence, leading to the translocation of E2 into the ER lumen.
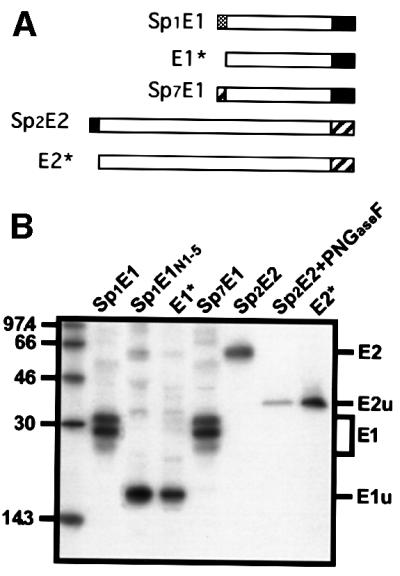
Fig. 2. Signal sequence function of the TM domains of E1 and E2. (A) Wild-type E1 and E2 proteins (Sp1E1 and Sp2E2) are schematically represented. E1* and E2* correspond to E1 and E2 proteins without their signal sequence, respectively. Sp7E1 corresponds to the C-terminal half of the TM domain of E2 followed by the ectodomain and TM domain of E1. (B) HepG2 cells were coinfected with vTF7-3 and the appropriate vaccinia virus recombinant at a multiplicity of infection of 5 p.f.u./cell. At 4.5 h post-infection, infected cells were labelled for 1 h with 35S-Protein Labelling Mix (NEN Life Science Products). Cell lysates were immunoprecipitated with A4 (anti-E1) or H47 (anti-E2) mAbs and treated or not with PNGaseF. Sp1E1N1–5 is an E1 protein that has been mutated at its potential sites of N-glycosylation. Samples were analysed by SDS–PAGE (10% polyacrylamide) and autoradiography. E1u, unglycosylated form of E1; E2u, unglycosylated form of E2.
Because p7 is not glycosylated and no antibody against this hydrophobic polypeptide is available, we used E1 as a reporter protein to analyse the role of the putative signal sequence present in the TM domain of E2 (Sp7). E1 was expressed with or without Sp7. As shown in Figure 2B (Sp7E1), E1 expressed with Sp7 migrated as three bands of 28–32 kDa when analysed by SDS–PAGE. This pattern is similar to the migration profile observed for E1 expressed with its own Sp1 signal sequence (Figure 2B, compare Sp7E1 with Sp1E1) and it corresponds to different glycoforms of E1, as observed previously (Dubuisson et al., 2000). However, in the absence of Sp7, a band comigrating with a mutated form of E1 lacking its N-glycosylation sites was detected, indicating that E1 was not glycosylated (Figure 2B, compare E1* with Sp1E1N1–5). Together, these data confirm that Sp7 is a signal sequence, leading to the translocation of a reporter protein (E1) into the ER lumen.
The C-terminus of the TM domains of E1 and E2 is oriented toward the cytosol when HCV envelope glycoproteins are expressed alone
A signal sequence that directs the complete translocation of a polypeptide adopts an Ncyt/Clum (cyt, cytosol; lum, luminal) orientation in the translocon (Mothes et al., 1998). Since the C-terminal halves of the TM domains of E1 and E2 act as signal sequences, the C-terminus of these domains should be oriented toward the luminal side of the ER. In addition, since the ectodomain of E1 is translocated into the lumen of the ER, the N-terminus of its TM domain should also be oriented toward the luminal side of the ER. The same is true for E2. For these reasons, it has been thought that the TM domains of E1 and E2 should adopt a hairpin structure, with their N- and C-termini oriented toward the luminal side of the ER membrane. However, recent data from alanine scanning insertion mutagenesis suggest that, after E1E2 heterodimerization, these domains might form single TM segments (Op de Beeck et al., 2000).
To determine the topology of the TM domains of E1 and E2, these proteins were tagged at their C-terminus with an HA (Figure 3A, Sp1E1†HA and Sp2E2†HA) or a Myc epitope, and antibody accessibility to these epitopes was analysed by immunofluorescence in selectively permeabilized cells. To avoid signal sequence cleavage between the C-terminus of HCV envelope glycoproteins and the tag, the last residue (Ala) of these proteins was replaced by an Arg. However, we first verified that neither the presence of the epitope tags nor the Ala to Arg mutations imposed a change in the topology of the TM domains of HCV envelope glycoproteins. Assuming that a change in topology imposed by the tag would alter the functions of the TM domains of HCV envelope glycoproteins, we used a heterodimerization assay to test the impact of the tag on their topology. Indeed, we have recently shown that the functions of these domains are very sensitive to mutagenesis (Cocquerel et al., 2000), with heterodimerization being the most sensitive (Op de Beeck et al., 2000). HCV proteins were coexpressed by vaccinia virus recombinants and analysed in pulse–chase experiments followed by immunoprecipitation with a conformation-sensitive, E2-specific monoclonal antibody (mAb; H53) that precipitates E1E2 heterodimers, as shown previously (Deleersnyder et al., 1997; Op de Beeck et al., 2000). Since mAb H53 is E2 specific, and because E2 can fold independently of E1 (Dubuisson, 2000), the amount of E1 coprecipitated with E2, when using mAb H53, is a good indicator of the assembly of the non-covalent heterodimer. As shown in Figure 3B, the presence of an HA epitope at the C-terminus of E1 or E2 did not modify the amount of E1 coprecipitated with E2, indicating that, in the presence of the epitope, no detectable effect on the assembly of the heterodimer was observed. Similar results were obtained with E1 and E2 proteins tagged with a Myc epitope (data not shown). It is worth noting that, due to the presence of the epitope at the C-terminus of E1, the E1†HA protein migrated slightly more slowly than E1. Together, these results indicate that the epitope tag does not alter the functionality of the TM domains of E1 and E2, suggesting that the topology of these domains is not affected by the presence of the epitope.

Fig. 3. Tagging of HCV envelope glycoproteins with an HA epitope. (A) Schematic representation of tagged proteins, Sp1E1†HA and Sp2E2†HA. The HA epitope is shown as a triangle at the C-terminal end. (B) Heterodimerization of E1 and E2 expressed with or without the HA epitope. HepG2 cells coinfected with vTF7-3 and the appropriate vaccinia virus recombinant at a multiplicity of infection of 5 p.f.u./cell were pulse-labelled for 10 min and chased for 4 h. Cell lysates were immunoprecipitated with H53 mAb (conformation-sensitive anti-E2 mAb) and the samples were analysed by SDS–PAGE (10% polyacrylamide) under non-reducing conditions.
The orientation of the epitope tags was determined by immunofluorescence using digitonin-permeabilized cells. Low concentrations of digitonin selectively permeabilize the plasma membrane because of its higher concentration of cholesterol compared with intracellular membranes (Lange, 1991). Triton X-100-permeabilized cells were analysed in parallel as a control. After Triton X-100 treatment, cells expressing Sp1E1†HA or Sp2E2†HA were fluorescent when revealed with mAbs A4 and H53, which are directed against the ectodomain of E1 and E2, respectively (Figure 4, A4 and H53, Triton). In contrast, when treated with digitonin, no specific fluorescence was detected above background in cells expressing Sp1E1†HA or Sp2E2†HA (Figure 4, A4 and H53, digitonin). Therefore, A4 and H53 epitopes were accessible to their respective antibody in Triton X-100-permeabilized cells, but not in digitonin-permeabilized cells. These data show that following digitonin treatment, the antibodies had no access to the ectodomains of E1 and E2 in the ER lumen, indicating that the integrity of the ER membrane was preserved. To analyse the accessibility of the HA epitope of the tagged proteins, a rabbit anti-HA polyclonal antibody was used in the same conditions. Cells expressing Sp1E1†HA or Sp2E2†HA were fluorescent when revealed with the anti-HA antibody in both digitonin- and Triton X-100-treated cells (Figure 4, α-HA, digitonin and Triton X-100). Therefore, the HA epitope was accessible to its antibody in both digitonin-permeabilized and Triton X-100-permeabilized cells expressing Sp1E1†HA or Sp2E2†HA, indicating that the HA epitope is oriented toward the cytosolic side of the ER membrane. Similar results were obtained with HCV envelope glycoproteins tagged with a Myc epitope (data not shown). It is worth noting that similar results were also observed when E1†HA was coexpressed with E2 and when E2†HA was coexpressed with E1 (data not shown).
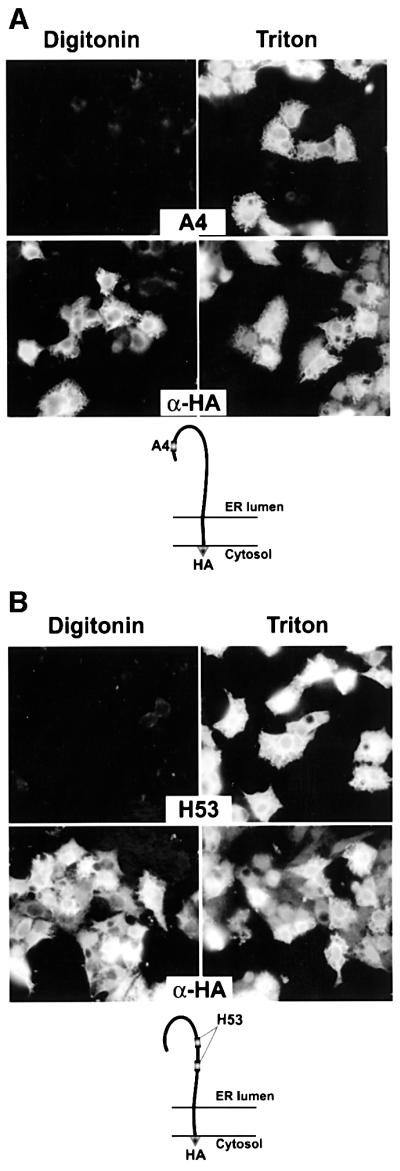
Fig. 4. Topology of the TM domains of E1 and E2 envelope proteins expressed alone. HepG2 cells were coinfected with vTF7-3 and vaccinia virus recombinant expressing Sp1E1†HA (A) or Sp2E2†HA (B) at a multiplicity of infection of 5 p.f.u./cell. At 6 h post-infection, cells were fixed with paraformaldehyde, permeabilized with digitonin or Triton X-100 and immunostained with A4 (anti-E1), H53 (anti-E2) or anti-HA mAbs. HepG2 cells infected with vTF7-3 alone were used as a control to determine the background levels of fluorescence (data not shown). The deduced topology of E1†HA and E2†HA proteins is shown at the bottom.
Altogether, these results indicate that the TM domains of E1 and E2 proteins span the membrane once with their N-terminus oriented toward the luminal side of the ER and their C-terminus oriented toward the cytosolic side of the ER membrane.
The TM domains of HCV envelope glycoproteins form a hairpin structure before signal sequence cleavage
The presence of a signal sequence in the second half of the C-terminus of the TM domains of E1 and E2 does not fit with a single membrane-spanning topology. However, as HCV proteins are synthesized as a polyprotein, it is conceivable that, before cleavage of the polyprotein, the topology of the TM domains of E1 and E2 is different. The topology of these domains was therefore analysed in the absence of signal sequence cleavage.
To analyse the topology of the TM domain of E1 before signal sequence cleavage, we worked with an E1E2 polyprotein truncated at the C-terminus of E2 and having the last C-terminal residue of E1 mutated (Ala→Arg) to abolish signal sequence cleavage (Sp1E1†E2661Myc; Figure 5A). In addition, the ectodomain of E2 was tagged at its C-terminus with a Myc epitope. A glycosylated polyprotein of the expected size was detected by SDS–PAGE after immunoprecipitation with an anti-E1 or anti-E2 mAb (data not shown). The orientation of the uncleaved polyprotein was determined by immunofluorescence using digitonin-permeabilized cells as above. Triton X-100-permeabilized cells were also analysed in parallel experiments as a control. mAb A4 directed against the ectodomain of E1 was used as a control of the integrity of the ER membrane. Indeed, E1 was accessible to A4 antibody in Triton X-100-permeabilized cells, but not in digitonin-permeabilized cells (Figure 5A, A4). The orientation of the C-terminus of the TM domain of E1 was determined in digitonin-permeabilized cells by using anti-Myc and anti-E2 (H47) mAbs. mAb H47 is a conformation-insensitive antibody that is able to detect misfolded E2. It is thus useful for the potential detection of E2 in the cytosol, a location that does not allow folding of this protein. As shown in Figure 5A (H47 and α-Myc), the H47 and Myc epitopes were accessible to their respective antibody in Triton X-100-permeabilized cells, but not in digitonin-permeabilized cells, indicating that the ectodomain of E2 and the Myc tag are located in the lumen of the ER. It should be remembered that the Ala→Arg mutation at the C-terminus of E1 had no effect on the topology of the TM domain of E1, because the same mutation was introduced in E1†HA and it did not affect the function of the TM domain of E1 (see above). The association of E1†E2661Myc with the ER membrane was confirmed by alkaline extraction. Approximately 50% of the polyprotein was resistant to alkaline extraction (data not shown). Together, these data indicate that, in the absence of cleavage between E1 and E2, the TM domain of E1 spans the membrane twice, leading to the translocation of the ectodomain of E2 into the ER lumen.

Fig. 5. Topology of the TM domain of E1 and E2 before signal sequence cleavage. The polyproteins constructed to study the topology of the TM domains of E1 and E2 before signal sequence cleavage are shown at the top of (A) and (B), respectively. Sp1E1†E2661Myc is a polyprotein containing E1 with its signal peptide, followed by the ectodomain of E2 in fusion with a Myc epitope (A). The last residue (Ala) at the C-terminus of E1 was replaced by an Arg to avoid signal sequence cleavage. Sp2E2†E1346Myc is a polyprotein containing E2 with its signal peptide, followed by the ectodomain of E1 in fusion with a Myc epitope (B). The last residue (Ala) at the C-terminus of E2 was replaced by an Arg to avoid signal sequence cleavage. HepG2 cells were coinfected with vTF7-3 and the vaccinia virus recombinant expressing Sp1E1†E2661Myc (A) or Sp2E2†E1346Myc (B) at a multiplicity of infection of 5 p.f.u./cell. At 6 h post-infection, cells were fixed with paraformaldehyde, permeabilized with digitonin or Triton X-100 and immunostained with A4 (anti-E1), H47 (anti-E2) or anti-Myc mAbs (A), or with H53 (anti-E2), AP21.010 (anti-E1) or anti-Myc mAbs (B). HepG2 cells infected with vTF7-3 alone were used as a control to determine the background levels of fluorescence (data not shown). The deduced topology of E1†E2661Myc and E2†E1346Myc polyproteins is shown at the bottom of (A) and (B), respectively.
A similar strategy was used to study the topology of the TM domain of E2 before signal sequence cleavage. In this case, we used a polyprotein having the position of E1 and E2 inverted (Cocquerel et al., 2000), with E1 being used as a reporter protein. The E2E1 polyprotein was truncated at the C-terminus of E1 and had the last C-terminal residue of E2 mutated (Ala→Arg) to abolish signal sequence cleavage (Sp2E2†E1346Myc; Figure 5B). In addition, the ectodomain of E1 was tagged at its C-terminus with a Myc epitope. A glycosylated polyprotein of the expected size was detected by SDS–PAGE after immunoprecipitation with an anti-E1 or anti-E2 mAb (data not shown). mAb H53 directed against the properly folded ectodomain of E2 was used as a control of the integrity of the ER membrane. Indeed, the H53 epitope was accessible to its antibody in Triton X-100-permeabilized cells, but not in digitonin-permeabilized cells (Figure 5B, H53). The orientation of the C-terminus of the TM domain of E2 was determined in digitonin-permeabilized cells by using anti-Myc and anti-E1 (AP21.010) mAbs. Note that the AP21.010 mAb was used instead of A4 because the epitope recognized by the latter is located at the N-terminus of E1 and is poorly accessible in the absence of cleavage. As shown in Figure 5B (AP21.010 and α-Myc), the AP21.010 and Myc epitopes were accessible to their respective antibody in Triton X-100-permeabilized cells, but not in digitonin-permeabilized cells, indicating that the ectodomain of E1 and the Myc tag are located in the lumen of the ER. The association of E2†E1346Myc with the ER membrane was confirmed by alkaline extraction. Approximately 50% of the polyprotein was resistant to alkaline extraction (data not shown). Together, these results indicate that, in the absence of cleavage between E2 and E1, the TM domain of E2 adopts a double membrane-spanning topology, leading to the translocation of the ectodomain of E1 into the ER lumen.
Altogether, these results indicate that, before processing of the viral polyprotein by signal peptidases, the TM domains of E1 and E2 adopt a double membrane-spanning topology. This topology is probably necessary for signal sequence interaction with the translocon.
Dynamic change in the orientation of the internal signal peptides
The single membrane-spanning topology observed for E1 and E2 tagged with the HA epitope at their C-terminus (Figure 4), together with the hairpin structure observed in the absence of cleavage, suggest that a reorientation of the signal sequence occurs after cleavage at the C-terminus of these domains. Indeed, assuming that termination of translation after this short epitope essentially mimicks signal cleavage, its ultimate position in the cytosol is supportive of a reorientation. To further support the hypothesis of dynamic changes in the orientation of these internal signal peptides, we analysed whether the HA epitope at the C-terminus of E1 and E2 would be transiently translocated into the lumen of the ER. As an indication of such a transient translocation, we analysed whether the HA epitope at the C-terminus of E1 and E2 would be cleaved off by the signal peptidase when the cleavage site between the viral glycoproteins and the tag was not mutated (E1-HA and E2-HA). As shown in Figure 6, immunoprecipitation with the anti-HA antibody did not bring down any protein when the signal sequence cleavage site was not altered, indicating that the HA epitope has been cleaved off by the signal peptidase and hence the HA epitope has been translocated into the lumen of the ER. Similar results were observed for E2 (data not shown). The difference in the intensity of the proteins precipitated by A4 and the anti-HA antibodies is probably due to a difference in the affinities of the antibodies. The slight difference in the migration of E1†HA immunoprecipitated by the anti-HA antibody is probably due to the presence of the light chain of this antibody, which slows down the migration of E1†HA. It should be remembered that the Ala→Arg mutation at the C-terminus of E1 in E1†HA had no effect on the topology of the TM domain of E1, because a different topology was observed for this domain in E1†E2661Myc despite the presence of the same mutation (see above). Together with the observation that the TM domains of E1†HA and E2†HA adopt a single membrane-spanning topology, these data indicate that a reorientation of the signal sequence occurs after cleavage at the C-terminus of these domains.

Fig. 6. Cleavage of the HA epitope at the C-terminus of E1 by the signal peptidase. Plasmids expressing E1 tagged with the HA epitope and containing (E1†HA) or not (E1-HA) a mutation in the signal sequence cleavage site between E1 and the HA epitope tag were used for in vitro transcription–translation in the presence of microsomal membranes. Reaction mixtures were analysed by immunoprecipitation with anti-HA or anti-E1 (A4) antibodies and the samples were analysed by SDS–PAGE (10% polyacrylamide).
Charged residues located between the two stretches of hydrophobic residues play a major role in the pre-cleavage topology of the TM domains of E1 and E2
The TM domains of E1 and E2 glycoproteins are composed of two stretches of hydrophobic residues separated by a short segment containing at least one fully conserved positively charged residue (Figure 1B). Replacement of these charged residues by alanine residues has been shown to alter the various functions played by the TM domains of E1 and E2, including the signal sequence function (Cocquerel et al., 2000). Indeed, the mutations of the Lys residue and the polar Asn residue of the TM domain of E1 introduced in the E1E2 polyprotein (Sp1E1NKE2) partially abolished the cleavage between E1 and E2. Similarly, the mutations of the Asp and Arg residues of the TM domain of E2 in the context of the E2E1 polyprotein (Sp2E2DRE1) totally abolished the cleavage between E2 and E1. Since these charged residues are crucial for the multifunctionality of the TM domains of E1 and E2 (Cocquerel et al., 2000; Op de Beeck et al., 2000), we wanted to know whether these charged residues play a crucial role in the topology of the TM domains of HCV envelope glycoproteins. For this purpose, we analysed the topology of the TM domains of E1 and E2 in the context of the mutated polyproteins, Sp1E1NKE2 and Sp2E2DRE1. Selectively permeabilized cells expressing Sp2E2DRE1 were analysed by immunofluorescence with H53 and AP21.010 mAbs. As above, mAb H53 was used as a control of the integrity of the ER membrane (Figure 7, H53). As shown in Figure 7, the AP21.010 epitope was accessible to its antibody in both digitonin-permeabilized and Triton X-100-permeabilized cells, indicating that when the charged residues of the TM domain of E2 are mutated, E1 is not translocated into the ER lumen and the C-terminus of the TM domain of E2 is oriented toward the cytosol. Similar results were obtained with the TM domain of E1 analysed in the context of the Sp2E1NKE2 polyprotein (data not shown), suggesting that the mutation of the Asn and Lys residues in the TM domain of E1 led to a defect in translocation of E2. However, these mutations had only a partial effect on the signal sequence function (Cocquerel et al., 2000), and the intensity of the fluorescence of E2 was lower in digitonin-permeabilized cells than in Triton X-100-permeabilized cells (data not shown).
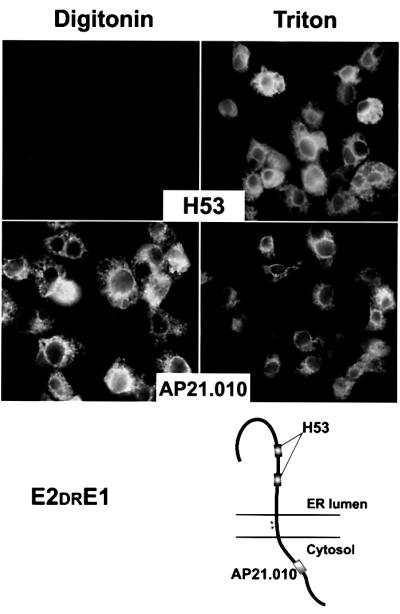
Fig. 7. Topology of the TM domain of E2 in the context of the mutated polyprotein Sp1E2DRE1. HepG2 cells were coinfected with vTF7-3 and the vaccinia virus recombinant expressing Sp1E2DRE1 at a multiplicity of infection of 5 p.f.u./cell. At 6 h post-infection, cells were fixed with paraformaldehyde, permeabilized with digitonin or Triton X-100 and immunostained with H53 (anti-E2) or AP21.010 (anti-E1) mAbs. HepG2 cells infected with vTF7-3 alone were used as a control to determine the background levels of fluorescence (data not shown). The deduced topology of E2DRE1 polyprotein is shown at the bottom. Stars indicate the positions of the residues that have been mutated.
In agreement with previous data (Cocquerel et al., 2000), these observations indicate that these charged residues play an major role in the signal sequence function of the TM domains of HCV envelope glycoproteins.
Discussion
Owing to their multifunctionality, the TM domains of HCV envelope glycoproteins are very attractive tools to challenge the limits of our understanding of the early steps in membrane protein biogenesis. The presence of an anchor sequence and a signal sequence functions in the TM domains of E1 and E2 indicates that these domains form a hairpin structure before signal sequence cleavage. However, our work shows that a reorientation of the signal sequence occurs after cleavage at the C-terminus of these domains. This dynamic event is essential for the sequential appearance of other functions played by the transmembrane domains of HCV envelope glycoproteins.
When E1 and E2 are expressed alone, their TM domains form a single TM segment with the charged residues located in the middle of the membrane-spanning sequence. This is in agreement with our previous work on alanine scanning insertion mutagenesis in these domains (Op de Beeck et al., 2000). This also fits very well with the prediction of a single TM helical segment from residue 353 to 379 for E1 and from residue 718 to 742 for E2 by amino acid sequence analysis (Cocquerel et al., 2000). Indeed, a minimum of 16 leucines are required to form a transmembrane α-helix (Monne et al., 1999), but stretches of 20–25 hydrophobic amino acids are generally observed in integral membrane proteins. The two hydrophobic stretches in the TM domains of E1 and E2 are too short to form, by themselves, TM α-helices. Hence, each of the two small hydrophobic stretches would have to adopt an extended structure to cross the membrane. Such an extended structure is very unfavourable in a membrane environment because of the absence of stabilization by hydrogen bonds, while the hydrogen-bond network of an α-helix offers the most thermodynamically favoured conformation. This is supported by our recent NMR structure analysis, which demonstrated that the N-terminal hydrophobic stretch of E1 forms an α-helix in a membrane environment (Op de Beeck et al., 2000). Moreover, preliminary structural analyses of chemically synthesized peptides corresponding to the TM domains of E1 and E2 clearly indicate that both domains adopt an α-helical fold in any membrane mimetic environment tested (data not shown). Although the presence of charged residues within the centre of these TM domains seems to be less favourable in a membrane environment, it is possible that they could form ion pairs after heterodimerization, as already described for several membrane proteins (Kaback et al., 1997). Alternatively, as the pK of charged residues is generally largely shifted in the hydrophobic environment of the membrane, the carboxyl and amine groups could be simply neutralized by protonation and deprotonation, respectively. In addition, these charged residues have been shown to play a major role in the ER retention function of the TM domains of HCV envelope glycoproteins (Cocquerel et al., 2000). This is in agreement with the usual features of ER retention by TM sequences, which bear one or several hydrophilic residues located in the middle of the TM domain (Bonifacino et al., 1991).
We demonstrated here that in the absence of signal sequence cleavage at their C-terminus, the TM domains of HCV envelope glycoproteins form a hairpin structure. As presented in our model (Figure 8), this topology probably occurs in the environment of the translocon and before integration into the lipid bilayer. The translocon appears to be an active participant in the integration process of TM segments into the lipid bilayer. Indeed, because it serves as the entry point for the integration of TM sequences into the lipid bilayer, the translocon is directly involved in the recognition, orientation, lateral movement and insertion of TM sequences. The translocon must recognize the TM sequence to position it properly for its lateral move into the bilayer. Once oriented properly, the non-polar TM sequence will minimize its exposure to water by associating with non-polar components of the translocon and/or lipids (Martoglio et al., 1995; Do et al., 1996; Mothes et al., 1997). Recently, it has been shown that TM domains containing at least one charge can be crosslinked with TRAM, a protein associated with the translocon (Heinrich et al., 2000). TRAM can also interact with short hydrophobic sequences because it can be crosslinked to all signal sequences examined so far (Görlich et al., 1992). It is therefore likely that the TM domains of HCV envelope glycoproteins interact with this component of the translocon before signal sequence cleavage at their C-terminus. In addition, because the TM domains of HCV envelope glycoproteins have at least one charged residue that is located in the centre of the TM sequence after cleavage and reorientation, these domains might remain in the environment of the translocon, where assembly of E1E2 might preferentially occur. This would fit with the observation that membrane integration of E2 is improved when this protein is coexpressed with E1 (Cocquerel et al., 2001). This is also supported by the observation that the folding of E1 is assisted by E2 (Dubuisson, 2000).

Fig. 8. Model of the behaviour of the TM domains of HCV envelope glycoproteins E1 and E2 during the early steps of their biogenesis. (1) The N-terminus of E1 is translocated into the lumen of the ER and also orients the N-terminus of the TM domain of E1 toward the ER lumen. The C-terminal half of the TM domain of E1 is the signal sequence of E2 and has its C-terminus oriented toward the lumen of the ER to allow the translocation of E2. At this step and before signal sequence cleavage, the TM domain of E1 forms a hairpin structure that is likely to be located in the environment of the translocon. (2) After signal sequence cleavage between E1 and E2, the signal sequence present in the C-terminal half of the TM domain of E1 is reoriented toward the cytosol and E1 probably remains very close to the translocon. (3) As shown for the TM domain of E1, the TM domain of E2 transiently adopts a hairpin structure to allow the translocation of p7. (4) After signal sequence cleavage between E2 and p7, the signal sequence present in the C-terminal half of the TM domain of E2 is reoriented toward the cytosol, the TM domains of E1 and E2 interact and move in the lipid bilayer. It is worth noting that, in the context of HCV polyprotein, the cleavage between E2 and p7 is delayed (T½ ≈15 min) (Dubuisson, 2000). Owing to the absence of experimental data on its topology, p7 is not included in step 4.
The reorientation of the second stretch of hydrophobic residues leading to a single TM segment probably occurs immediately after signal sequence cleavage. How can such a change in orientation occur? The precise molecular understanding of this reorientation mechanism will require knowledge of the atomic structures and the intermolecular interactions of the various partners. However, taking into account that the most stable thermodynamic folding of the TM domains of E1 and E2 is the α-helix (see above), a putative scenario can be drawn. The signal sequence is probably a dominant topogenic signal before cleavage. However, from a geometric point of view, the hairpin conformation of the TM domains of E1 and E2 before signal peptidase cleavage imposes that some backbone CO and NH groups are not involved in intramolecular α-helix hydrogen bonds. This probably yields a local unstable hot spot that can be tentatively localized in the middle of the TM domains of E1 and E2 at the level of the charged/polar residues. These conformational constraints should be released by signal peptidase cleavage at the C-terminus, allowing the reorganization of the hydrogen bond network toward the stabilization of the helical structure in the hydrophobic context of the lipid bilayer–translocon interface. It is tempting to speculate that the propagation of the α-helical folding from the N-terminal end of the TM domains could stabilize this region by setting up the complete hydrogen bond network of transmembrane α-helices. As a result, the C-terminus of the TM domains would be translocated from the luminal side to the cytosolic side of the membrane, possibly through the aqueous channel of the translocon. From a thermodynamic standpoint, it is reasonable to think that the reorganization of the hydrogen bond network toward α-helix formation imposed by the hydrophobic environment is the principal driving force of the reorientation process.
The dynamic event observed in the topology of the TM domains of HCV envelope glycoproteins is novel and it is linked to the multifunctionality of these TM domains. A general feature of positive strand RNA viruses is that they synthesize their polypeptides as one or more polyprotein(s). This implies that internal signal peptides are used to target viral membrane proteins to the ER. As a consequence, after signal sequence cleavage, the signal peptide of these proteins remains bound to the C-terminus of the protein located upstream on the polyprotein. By reorienting their C-terminus toward the cytosol and being part of a TM domain, the signal sequences at the C-terminus of E1 and E2 acquire new functions. As for other envelope proteins that play a major role in virus assembly and entry (Hernandez et al., 1996; Garoff et al., 1998), the biogenesis of HCV envelope glycoproteins is very complex. Besides the fact that they have to heterodimerize, these proteins are retained in the ER, where the viral particle is supposed to acquire its envelope (Duvet et al., 1998). These pivotal functions make the TM domains of E1 and E2 major actors in the biogenesis of HCV envelope glycoproteins. In addition, as shown for other viruses, these domains probably play a major role in viral fusion (Cleverley and Lenard, 1998). Retaining all these functions is probably essential for HCV. A detailed molecular knowledge of the TM domains of HCV envelope glycoproteins is therefore not only interesting to understand the complexity of the biogenesis of membrane proteins, but is also essential for finding new approaches to fight this major viral pathogen.
Materials and methods
Cell culture
The HepG2 cell line was obtained from the American Type Culture Collection (ATCC). Cell monolayers were grown in Dulbecco’s modified essential medium supplemented with 10% fetal bovine serum.
Antibodies
mAbs A4 (anti-E1), H53 (anti-E2), H47 (anti-E2) and anti-Myc (ATCC CRL-1725) were produced in vitro using a MiniPerm apparatus (Heraeus) as recommended by the manufacturer. Polyclonal anti-HA (HA11) antibody was purchased from Babco. mAb AP21.010 (anti-E1) was kindly provided by A.H.Patel (Institute of Virology, Glasgow, UK).
Plasmid constructs
Plasmids were constructed using standard methods. Briefly, DNA sequences of protein domains were introduced into plasmid pTM1 (Moss et al., 1990) by PCR with appropriate oligonucleotides and templates. Plasmids pTM1/E1* and pTM1/E2* contain the entire sequence of the E1 and E2 proteins, respectively, without their signal sequence. Plasmid pTM1/Sp7E1 contains the C-terminal half of the TM domain of E2 followed by the sequence of the E1 protein. Plasmids pTM1/Sp1E1-HA and pTM1/Sp2E2-HA contain the entire sequence (including the signal sequence) of the E1 and E2 proteins, respectively, in fusion with the HA9 epitope (YPYDVPDYA) preceded by three Gly residues. In plasmids pTM1/Sp1E1†HA and pTM1/Sp2E2†HA, the last residue (Ala) at the C-terminus of E1 and E2 was replaced by an Arg to avoid signal sequence cleavage. Plasmids pTM1/Sp1E1†Myc and pTM1/Sp2E2†Myc contain the entire sequence (including the signal sequence) of the E1 and E2 proteins, respectively, in fusion with a Myc epitope (EQKLISEEDL) preceded by three Gly residues. The last residue (Ala) at the C-terminus of E1 and E2 was replaced by an Arg to avoid signal sequence cleavage. Plasmid pTM1/Sp1E1†E2661Myc contains the entire sequence of E1 with its signal sequence, followed by the sequence of the ectodomain of E2 in fusion with a Myc epitope. The last residue (Ala) at the C-terminus of E1 was replaced by an Arg to prevent cleavage between E1 and E2. Plasmid pTM1/Sp2E2†E1346Myc contains the entire sequence of E2 with its signal sequence, followed by the sequence of the ectodomain of E1 in fusion with a Myc epitope. The last residue (Ala) at the C-terminus of E2 was replaced by an Arg to prevent cleavage between E2 and E1.
Generation and growth of viruses
Vaccinia virus recombinants were generated as described (Fournillier-Jacob et al., 1996). The following vaccinia virus recombinants have been described previously: vTF7-3 (expressing the T7 DNA-dependent RNA polymerase) (Fuerst et al., 1986), vE1E2 (expressing the signal sequence of E1, E1 and E2) (Fournillier-Jacob et al., 1996), vE1 (expressing the C-terminal 60 amino acids of the capsid protein and E1) (Fournillier-Jacob et al., 1996), vE1N1–5 (expressing a glycosylation mutant of E1 lacking its five sites of glycosylation) (Meunier et al., 1999), vE2 (expressing E2 with its signal sequence) (Cocquerel et al., 2000), vE1NKE2 (expressing E1E2 polyprotein in which Asn and Lys residues of the TMD of E1 were mutated into Ala) (Cocquerel et al., 2000) and vE2DRE1 (expressing inverted E2E1 polyprotein in which Asp and Arg residues of the TM domain of E2 were mutated into Ala) (Cocquerel et al., 2000). The genes of HCV proteins expressed in this work are under the control of a T7 promoter and expression of the proteins of interest is achieved by coinfection with vTF7-3.
Metabolic labelling and immunoprecipitation
Metabolic labelling and immunoprecipitation have been described previously (Dubuisson and Rice, 1996) and endoglycosidase were carried out as described by the manufacturer. Digested samples were mixed with an equal volume of 2× Laemmli sample buffer and analysed by SDS–PAGE.
Immunofluorescence
Subconfluent HepG2 cells grown on coverslips were infected by the appropriate vaccinia virus recombinants at a multiplicity of infection of 5 p.f.u./cell. At 6 h post-infection, cells were fixed for 10 min with paraformaldehyde [4% in phosphate-buffered saline (PBS)]. Cells were then permeabilized or not for 15 min at 4°C with buffer (20 mM HEPES pH 6.9, 0.3 M sucrose, 0.1 M KCl, 2.5 mM MgCl2, 1 mM EDTA) containing digitonin at a final concentration of 5 µg/ml. Cells were incubated for 30 min at room temperature with PBS containing 1% bovine serum albumin. Cells were then permeabilized or not for 30 min at room temperature with PBS containing 0.1% Triton X-100. The expression of the proteins of interest was revealed with mAbs A4 (anti-E1; dilution 1/1000), AP21.010 (anti-E1; dilution 1/200), H53 (anti-E2; dilution 1/1000), H47 (anti-E2; dilution 1/500), anti-HA (dilution 1/600) or anti-Myc (dilution 1/100) followed by anti-mouse (A4, AP21.010, H53, H47 and anti-Myc) or anti-rabbit (anti-HA) (rhodamine-conjugated) immunoglobulins (dilution 1/1000; Jackson Immunoresearch).
Acknowledgments
Acknowledgements
We thank M.Le Maire and F.Jacob-Dubuisson for critical reading of the manuscript, and Sophana Ung for excellent technical assistance. We are grateful to A.Patel for the gift of mAb AP21.010. This work was supported by the CNRS, the Institut Pasteur de Lille, a PRFMMIP grant from the French Ministry of Research, EU grant QLK2-1999-00356 and grant 5651 from the ARC. L.C. was successively supported by an MENRT and a FRM fellowship. A.O.d.B. was successively supported by an ARC and an ANRS fellowship.
References
- Beltzer J.P., Fiedler,K., Fuhrer,C., Geffen,I., Handschin,C., Wessels,H.P. and Spiess,M. (1991) Charged residues are major determinants of the transmembrane orientation of a signal-anchor sequence. J. Biol. Chem., 266, 973–978. [PubMed] [Google Scholar]
- Bonifacino J.S., Cosson,P., Shah,N. and Klausner,R.D. (1991) Role of potentially charged transmembrane residues in targeting proteins for retention and degradation within the endoplasmic reticulum. EMBO J., 10, 2783–2793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleverley D.Z. and Lenard,J. (1998) The transmembrane domain in viral fusion: essential role for a conserved glycine residue in vesicular stomatitis virus G protein. Proc. Natl Acad. Sci. USA, 95, 3425–3430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cocquerel L., Meunier,J.-C., Pillez,A., Wychowski,C. and Dubuisson,J. (1998) A retention signal necessary and sufficient for endoplasmic reticulum localization maps to the transmembrane domain of hepatitis C virus glycoprotein E2. J. Virol., 72, 2183–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cocquerel L., Duvet,S., Meunier,J.-C., Pillez,A., Cacan,R., Wychowski,C. and Dubuisson,J. (1999) The transmembrane domain of hepatitis C virus glycoprotein E1 is a signal for static retention in the endoplasmic reticulum. J. Virol., 73, 2641–2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cocquerel L., Wychowski,C., Minner,F., Penin,F. and Dubuisson,J. (2000) Charged residues in the transmembrane domains of hepatitis C virus glycoproteins play a key role in the processing, subcellular localization and assembly of these envelope proteins. J. Virol., 74, 3623–3633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cocquerel L., Meunier,J.C., Op de Beeck,A., Bonte,D., Wychowski,C. and Dubuisson,J. (2001) Coexpression of hepatitis C virus envelope proteins E1 and E2 in cis improves the stability of membrane insertion of E2. J. Gen. Virol., 82, 1629–1635. [DOI] [PubMed] [Google Scholar]
- Deleersnyder V., Pillez,A., Wychowski,C., Blight,K., Xu,J., Hahn,Y.S., Rice,C.M. and Dubuisson,J. (1997) Formation of native hepatitis C virus glycoprotein complexes. J. Virol., 71, 697–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do H., Falcone,D., Lin,J., Andrews,D.W. and Johnson,A.E. (1996) The cotranslational integration of membrane proteins into the phospholipid bilayer is a multistep process. Cell, 85, 369–378. [DOI] [PubMed] [Google Scholar]
- Dubuisson J. (2000) Folding, assembly and subcellular localization of HCV glycoproteins. Curr. Top. Microbiol. Immunol., 242, 135–148. [DOI] [PubMed] [Google Scholar]
- Dubuisson J. and Rice,C.M. (1996) Hepatitis C virus glycoprotein folding: disulfide bond formation and association with calnexin. J. Virol., 70, 778–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubuisson J., Duvet,S., Meunier,J.C., Op de Beeck,A., Cacan,R., Wychowski,C. and Cocquerel,L. (2000) Glycosylation of the hepatitis C virus envelope protein E1 is dependent on the presence of a downstream sequence on the viral polyprotein. J. Biol. Chem., 275, 30605–30609. [DOI] [PubMed] [Google Scholar]
- Duvet S., Cocquerel,L., Pillez,A., Cacan,R., Verbert,A., Moradpour,D., Wychowski,C. and Dubuisson,J. (1998) Hepatitis C virus glycoprotein complex localization in the endoplasmic reticulum involves a determinant for retention and not retrieval. J. Biol. Chem., 273, 32088–32095. [DOI] [PubMed] [Google Scholar]
- Fournillier-Jacob A., Cahour,A., Escriou,N., Girard,M. and Wychowski,C. (1996) Processing of the E1 glycoprotein of hepatitis C virus expressed in mammalian cells. J. Gen. Virol., 77, 1055–1064. [DOI] [PubMed] [Google Scholar]
- Fuerst T.R., Niles,E.G., Studier,F.W. and Moss,B. (1986) Eukaryotic transient-expression system based on recombinant vaccinia virus that synthesizes bacteriophage T7 RNA polymerase. Proc. Natl Acad. Sci. USA, 83, 8122–8126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gafvelin G., Sakaguchi,M., Andersson,H. and von Heijne,G. (1997) Topological rules for membrane protein assembly in eukaryotic cells. J. Biol. Chem., 272, 6119–6127. [DOI] [PubMed] [Google Scholar]
- Garoff H., Hewson,R. and Opstelten,D.J.E. (1998) Virus maturation by budding. Microbiol. Mol. Biol. Rev., 62, 1171–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Görlich D., Hartmann,E., Prehn,S. and Rapoport,T.A. (1992) A protein of the endoplasmic reticulum involved early in polypeptide translocation. Nature, 357, 47–52. [DOI] [PubMed] [Google Scholar]
- Heinrich S.U., Mothes,W., Brunner,J. and Rapoport,T.A. (2000) The Sec61p complex mediates the integration of a membrane protein by allowing lipid partitioning of the transmembrane domain. Cell, 102, 233–244. [DOI] [PubMed] [Google Scholar]
- Hernandez L.D., Hoffman,L.R., Wolfsberg,T.G. and White,J.M. (1996) Virus–cell and cell–cell fusion. Annu. Rev. Cell Dev. Biol., 12, 627–661. [DOI] [PubMed] [Google Scholar]
- Johnson A.E. and van Waes,M.A. (1999) The translocon: a dynamic gateway at the ER membrane. Annu. Rev. Cell Dev. Biol., 15, 799–842. [DOI] [PubMed] [Google Scholar]
- Kaback H.R., Voss,J. and Wu,J. (1997) Helix packing in polytopic membrane proteins: the lactose permease of Escherichia coli. Curr. Opin. Struct. Biol., 7, 537–542. [DOI] [PubMed] [Google Scholar]
- Lange Y. (1991) Disposition of intracellular cholesterol in human fibroblasts. J. Lipid Res., 32, 329–339. [PubMed] [Google Scholar]
- Lemmon M.A., MacKenzie,K.R., Arkin,I.T. and Engelman,D.M. (1997) Transmembrane α-helix interactions in folding and oligomerization of integral membrane proteins. In von Heijne,G. (ed.), Membrane Protein Assembly. Springer, Heidelberg, Germany, pp. 3–23.
- Martoglio B., Hofmann,M.W., Brunner,J. and Dobberstein,B. (1995) The protein-conducting channel in the membrane of the endoplasmic reticulum is open laterally toward the lipid bilayer. Cell, 81, 207–214. [DOI] [PubMed] [Google Scholar]
- Meunier J.-C., Fournillier,A., Choukhi,A., Cahour,A., Cocquerel,L., Dubuisson,J. and Wychowski,C. (1999) Analysis of the glycosylation sites of hepatitis C virus (HCV) glycoprotein E1 and the influence of E1 glycans on the formation of the HCV glycoprotein complex. J. Gen. Virol., 80, 887–896. [DOI] [PubMed] [Google Scholar]
- Monne M., Nilsson,I., Elofsson,A. and von Heijne,G. (1999) Turns in transmembrane helices: determination of the minimal length of a ‘helical hairpin’ and derivation of a fine-grained turn propensity scale. J. Mol. Biol., 293, 807–814. [DOI] [PubMed] [Google Scholar]
- Moss B., Elroy-Stein,O., Mizukami,T., Alexander,W.A. and Fuerst,T.R. (1990) New mammalian expression vectors. Nature, 348, 91–92. [DOI] [PubMed] [Google Scholar]
- Mothes W., Heinrich,S.U., Graf,R., Nilsson,I., von Heijne,G., Brunner,J. and Rapoport,T.A. (1997) Molecular mechanism of membrane protein integration into the endoplasmic reticulum. Cell, 89, 523–533. [DOI] [PubMed] [Google Scholar]
- Mothes W., Jungnickel,B., Brunner,J. and Rapoport,T.A. (1998) Signal sequence recognition in cotranslational translocation by protein components of the endoplasmic reticulum membrane. J. Cell Biol., 142, 355–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Op de Beeck A., Montserret,R., Duvet,S., Cocquerel,L., Cacan,R., Barberot,B., Le Maire,M., Penin,F. and Dubuisson,J. (2000) Role of the transmembrane domains of hepatitis C virus envelope proteins E1 and E2 in the assembly of the noncovalent E1E2 heterodimer. J. Biol. Chem., 275, 31428–31437. [DOI] [PubMed] [Google Scholar]
- Parks G.D. and Lamb,R.A. (1991) Topology of eukaryotic type II membrane proteins: importance of N-terminal positively charged residues flanking the hydrophobic domain. Cell, 64, 777–787. [DOI] [PubMed] [Google Scholar]
- Reed K.E. and Rice,C.M. (2000) Overview of hepatitis C virus genome structure, polyprotein processing and protein properties. Curr. Top. Microbiol. Immunol., 242, 55–84. [DOI] [PubMed] [Google Scholar]
- Sakaguchi M., Tomiyoshi,R., Kuroiwa,T., Mihara,K. and Omura,T. (1992) Functions of signal and signal–anchor sequences are determined by the balance between the hydrophobic segment and the N-terminal charge. Proc. Natl Acad. Sci. USA, 89, 16–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiess M. (1995) Heads or tails—what determines the orientation of proteins in the membrane. FEBS Lett., 369, 76–79. [DOI] [PubMed] [Google Scholar]
- von Heijne G. (1989) Control of topology and mode of assembly of a polytopic membrane protein by positively charged residues. Nature, 341, 456–458. [DOI] [PubMed] [Google Scholar]
- von Heijne G. (1990) The signal peptide. J. Membr. Biol., 115, 195–201. [DOI] [PubMed] [Google Scholar]
- Wahlberg J.M. and Spiess,M. (1997) Multiple determinants direct the orientation of signal-anchor proteins: the topogenic role of the hydrophobic signal domain. J. Cell Biol., 137, 555–562. [DOI] [PMC free article] [PubMed] [Google Scholar]