

Abstract
Activation of the caspase cascade is a pivotal step in apoptosis and can occur via death adaptor-mediated homo-oligomerization of initiator procaspases. Here we show that c-FLIPL, a protease-deficient caspase homolog widely regarded as an apoptosis inhibitor, is enriched in the CD95 death-inducing signaling complex (DISC) and potently promotes procaspase-8 activation through hetero-dimerization. c-FLIPL exerts its effect through its protease-like domain, which associates efficiently with the procaspase-8 protease domain and induces the enzymatic activity of the zymogen. Ectopic expression of c-FLIPL at physiologically relevant levels enhances procaspase-8 processing in the CD95 DISC and promotes apoptosis, while a decrease of c-FLIPL expression results in inhibition of apoptosis. c-FLIPL acts as an apoptosis inhibitor only at high ectopic expression levels. Thus, c-FLIPL defines a novel type of caspase regulator, distinct from the death adaptors, that can either promote or inhibit apoptosis.
Keywords: apoptosis/caspase activation/caspase-8/CD95 (Fas/APO-1)/c-FLIP
Introduction
The key mediators of apoptosis are a group of cysteinyl, aspartate-specific proteases known as caspases (Chang and Yang, 2000). Produced as latent precursors or procaspases, these proteases become activated during apoptosis through proteolytic processing at critical aspartate residues. The activation often occurs sequentially in a cascade, with an initiator procaspase being activated first, which then cleaves and activates executioner procaspases. Active executioner caspases cleave a critical set of cellular proteins to dismantle a cell. Activation of the initiator procaspases is a key regulatory step in apoptosis, and to date all proteins known to promote this activation directly are death adaptors, including the mammalian proteins FADD and Apaf-1 and the Caenorhabditis elegans protein CED-4. During apoptosis, these adaptors bind to the N-terminal prodomain region of procaspases, and facilitate the oligomerization of the C-terminal protease domain.
CD95 (APO-1/Fas) is a cell surface death receptor belonging to the TNFR (tumor necrosis factor receptor) superfamily (Peter et al., 1998). Upon binding to the trimeric CD95 ligand or agonistic antibodies, CD95 recruits FADD/MORT1 and procaspase-8 (FLICE/MACH/Mch5) to form the death-inducing signaling complex (DISC), in which procaspase-8 becomes activated (Kischkel et al., 1995; Medema et al., 1997). The precise mechanism of procaspase-8 activation in the DISC is not well understood, although homo-oligomerization of procaspase-8 alone can induce its activation (Martin et al., 1998; Muzio et al., 1998; Yang et al., 1998a). Of particular interest is the role of a procaspase-8-like, protease-deficient protein c-FLIP (Goltsev et al., 1997; Han et al., 1997; Hu et al., 1997; Inohara et al., 1997; Irmler et al., 1997; Shu et al., 1997; Srinivasula et al., 1997; Rasper et al., 1998). c-FLIP is expressed mainly in a long (c-FLIPL) and a short (c-FLIPS) splice form. The latter contains only two tandem repeats of DEDs and inhibits procaspase-8 activation in the DISC (Krueger et al., 2001a). In contrast, c-FLIPL shares extensive homology with procaspase-8, with a C-terminal domain that is highly homologous to the procaspase-8 protease domain yet enzymatically inactive due to the lack of key active site residues.
To date, the role of c-FLIPL in apoptosis remains controversial. In most reports, c-FLIPL has been described as anti-apoptotic, largely due to its ability to inhibit apoptosis at high levels of ectopic expression (reviewed in Krueger et al., 2001b). However, in cell lines that have been examined quantitatively, the level of endogenous c-FLIPL is merely 1% of that of endogenous procaspase-8 (Scaffidi et al., 1999). It is unclear what role c-FLIPL plays in these cells. Mice deficient in c-FLIP (which lack both c-FLIPL and c-FLIPS) were generated recently. Embryonic fibroblasts (MEFs) derived from the mice (through an in vitro selection process for cell growth) were shown to be more sensitive to CD95-induced apoptosis than the wild-type MEFs. However, inconsistent with these results, c-FLIP–/– mice showed developmental defects that strikingly resembled those of caspase-8–/– or FADD–/– mice (Yeh et al., 2000). These mice all died between E10.5 and E11.5 with a failure in heart formation and extreme hemorrhage, suggesting a function for c-FLIPL that is similar to that of caspase-8 and FADD. Furthermore, transient overexpression of c-FLIPL could induce as well as inhibit apoptosis, and this pro-apoptotic function required the c-FLIPL protease-like domain (Goltsev et al., 1997; Han et al., 1997; Inohara et al., 1997; Shu et al., 1997). However, it remains undetermined whether c-FLIPL can promote apoptosis at endogenous expression levels and how this might be possible without a protease activity. In the current study, we show that expression of c-FLIPL at levels comparable with the endogenous protein enhances procaspase-8 activation in the DISC and CD95-mediated apoptosis through hetero-dimerization with the protease domain of caspase-8 and enrichment at the DISC, whereas it inhibits apoptosis at high levels of expression. c-FLIPL is therefore a dual-function regulator for caspase-8 activation and apoptosis dependent on its level of expression.
Results
c-FLIPL potently enhances procaspase-8 activation upon their hetero-dimerization
During CD95-mediated apoptosis, both c-FLIPL and procaspase-8 are recruited to the DISC (Scaffidi et al., 1999). To assess the effect of c-FLIPL on procaspase-8 activation, we first mimicked their interaction in the DISC using an inducible dimerization system based on FK506-binding protein (FKBP) and its divalent ligand. We fused the protease domain of procaspase-8 to a derivative of FKBP called Fv (Clackson et al., 1998) (Fv-CASP-8, Figure 6A). After 4 h of treatment with a divalent Fv ligand AP20187, in vitro-translated, [35S]methionine-labeled Fv-CASP-8 underwent autoproteolytic processing (Figure 1A, lane 2). The activation of procaspase-8 in this system closely resembled that which occurred in the DISC (Medema et al., 1997), because it not only yielded the same final products but also proceeded with the same order of events (D.W.Chang, Z.Xing, V.L.Capacio, M.E.Peter and X.Yang, submitted for publication). Interestingly, when Fv-FLIP was included in the reaction, the processing of Fv-CASP-8 was markedly enhanced; >50% of Fv-CASP-8 was cleaved after only 2 h of AP20187 treatment (Figure 1A, lanes 7–9), whereas in the absence of Fv-FLIP virtually no zymogen cleavage was observed at this time point (Figure 1A, lanes 1 and 4). AP20187 could induce not only the Fv-FLIP:Fv-CASP-8 hetero-dimerization but also the homo-dimerization of each of these two Fv fusion proteins. To confirm that the increase in procaspase-8 processing was indeed due to the procaspase-8:c-FLIPL hetero-interaction, we took advantage of rapamycin-induced hetero-dimerization of FKBP and FRB (the rapamycin-binding region of the FKBP– rapamycin-associated protein; Chen et al., 1995). As shown in Figure 1B, forced hetero-dimerization of Fv-CASP-8 with FRB–FLIP (Figure 6A) resulted in the processing of procaspase-8. Interestingly, AP20187- induced procaspase-8:c-FLIPL dimerization also led to the cleavage of c-FLIPL, resulting in the release of its small subunit (Figure 1C, lane 2), and this processing was similar to that which occurred in the DISC (see below).

Fig. 6. The entire protease-like domain is required for c-FLIPL function. (A) Schematic structures of caspase-8, c-FLIPL, c-FLIPS and various constructs used in this study. DED, death effector domain. Amino acids present in each construct are labeled. Regions of c-FLIPL and caspase-8 in the chimeric constructs are indicated by shaded and open boxes, respectively. The domains that form the mature caspases as well as the corresponding domain on c-FLIPL (p10, p18, etc.) are marked. Fv, a derivative of FK506-binding protein (Clackson et al., 1998); FRB, the minimal rapamycin- binding domain of the FKBP–rapamycin-associated protein (FRAP) (Chen et al., 1995); asterisk, caspase-8 active site Cys360 to serine mutation; arrowhead, c-FLIPL processing site Asp376 to alanine mutation. (B) Capase-8 processing induced by c-FLIPL mutants and c-FLIPL:caspase-8 chimeras. Top panel: 1 µl of [35S]Fv-CASP-8 was treated with or without AP20187, in the presence of 1 µl of the indicated unlabeled Fv fusion proteins. Deduced domain structures are shown on the left. Bottom panel: the Fv fusion proteins used in the top panel were labeled with [35S]methionine to confirm equal expression of protein. (C) Effects of c-FLIPL mutants and c-FLIPL:caspase-8 chimeras on caspase-8-induced apoptosis. HeLa cells were transfected with Fv-CASP-8 (25 ng) and the indicated Fv-FLIP mutants (500 ng), and apoptosis was determined as in Figure 1E.

Fig. 1. c-FLIPL enhances caspase-8 activation upon induced proximity. (A) The c-FLIPL protease-like domain enhances caspase-8 processing. A 1 µl aliquot of in vitro translated, 35S-labeled Fv-CASP-8 was treated with 100 nM AP20187 for the indicated time (lanes 1 and 2), or treated with vehicle (–) or 100 nM AP20187 (+) for 2 h in the presence of the indicated amounts of in vitro translated, non-radioisotope-labeled Fv-FLIP (lanes 3–10). The reaction mixtures were then resolved by SDS–PAGE, and 35S-labeled products were visualized by autoradiography. The deduced domain structures of the marked bands are shown on the left and the order of processing is marked above. Molecular weight standards (in kDa) are shown on the right. (B) Enhancement of caspase-8 processing by c-FLIPL is due to their hetero-dimerization. A 1 µl aliquot of [35S]Fv-CASP-8 was treated with 1 mM rapamycin (Rap.) for 8 h in the presence of 1 µl of unlabeled FRB-FLIP. The products were analyzed as in (A). No processing was observed in the absence of FRB-FLIP (not shown). Domain structures are shown on the left and molecular weight standards on the right. (C) Dimerization induces processing of Fv-FLIP by caspase-8. A mixture of 1 µl of [35S]Fv-FLIP and 1 µl of unlabeled Fv-CASP-8 or Fv-CASP-8(C360S) was treated with or without AP20187 for 2 h and analyzed as in (A). The deduced domain structures are shown on the left. (D) Caspase-8 processing in transfected cells. 293 cells were transfected with the indicated combinations of Fv-CASP-8 (1 µg), Fv-FLIP (1 µg) and pRK5-CrmA (2 µg). At 13 h after transfection, cells were treated with vehicle (–) or 50 nM AP20187 (+) for 5 h. Cell extracts were analyzed by immunoblotting analysis using an anti-FLAG antibody. (E) Fv-FLIP enhances the cell death activity of caspase-8 in mammalian cells. HeLa cells were transfected with the indicated combinations of Fv constructs (in ng) and pRK5-crmA (1.5 µg, lane 6), together with pCMV-lacZ. At 6 h after transfection, cells were incubated with AP20187 (final concentration 125 nM), FK506 (200 nM) and z-DEVD (5 µM) for 10 h and scored for apoptosis.
Activation of procaspase-8 by c-FLIPL and processing of c-FLIPL by caspase-8 were not in vitro phenomena since co-expression of Fv-CASP-8 and Fv-FLIP in 293 cells also led to the processing of both proteins, generating the signature 10 kDa fragment p10 (Figure 1D, lane 2 versus lanes 1 and 3). The low level of processing seen here in the absence of AP20187 might represent the basal tendency of these fusion proteins to engage each other upon overexpression (see below). Addition of AP20187 further decreased the levels of transfected proteins (Figure 1D, lane 5). Because cell death among transfected cells was increased by AP20187 treatment (see below), which might cause the decrease in protein levels, we co-transfected these two proteins together with crmA, a poxviral caspase inhibitor that blocks caspase-8-induced apoptosis but not the generation of p10 (Medema et al., 1997; Yang et al., 1998a). When apoptosis was prevented by crmA, the amount of p10 was increased upon AP20187 treatment (Figure 1D, lane 11 versus lane 8). Taken together, these results show that c-FLIPL catalyzes autoprocessing of procaspase-8 upon their hetero-dimerization.
To examine the effect of c-FLIPL on caspase-8 cyto toxic activity, we transfected Fv-CASP-8 and Fv-FLIP individually or in combination into HeLa cells and quantified apoptosis in these cells. In this experiment, small amounts of Fv-CASP-8 caused minimal cell death even in the presence of AP20187 (Figure 1E, column 3). However, co-transfection with increasing amounts of Fv-FLIP caused a dose-dependent enhancement of apoptosis in >30% of transfected cells. (Figure 1E, columns 4 and 5, open bars). Cell death was increased further by addition of AP20187 (Figure 1E, columns 4 and 5, closed bars) and inhibited by the monomeric FKBP ligand, FK506 (data not shown). In addition, apoptosis was diminished by the caspase inhibitors crmA (Figure 1E, column 6) and zDEVD-fmk (data not shown), and could not be induced by the active site mutant of Fv-CASP-8 [Fv-CASP-8 (C/S), Figure 1E, column 7]. Similar results were observed when 293 cells were used for transfection (data not shown). This experiment also showed that transfection of ∼8-fold more procaspase-8 DNA was required to induce cell death equivalent to that observed with co-transfection of Fv-CASP-8 and Fv-FLIP (Figure 1E, column 1 versus columns 4 and 5). Therefore, c-FLIPL is a potent activator of caspase-8-mediated killing.
Ectopic expression of c-FLIPL at physiologically relevant levels enhances CD95-mediated apoptosis
To test the effect of c-FLIPL on CD95-dependent and -independent apoptosis, we introduced increasing amounts of c-FLIPL DNA into HeLa cells. The effect of c-FLIPL was dependent on its expression levels, which can be functionally grouped into three distinct concentration ranges. Very small amounts of c-FLIPL sensitized these cells to CD95-mediated apoptosis (region I in Figure 2A). The pro-apoptotic function of c-FLIPL required the caspase-like domain, as a c-FLIPL mutant lacking the large subunit (c-FLIPLΔ, see Figure 6A) did not show this activity (data not shown). These results raised the possibility that the function of endogenous c-FLIPL in HeLa cells is pro-apoptotic. Consistent with published reports, high levels of ectopic c-FLIPL expression led to decreased CD95 sensitivity (region II in Figure 2A), while at very high expression levels c-FLIPL induced apoptosis on its own (region III in Figure 2A). Similar dosage-dependent effects of c-FLIPL were also observed in another CD95-sensitive cell line MCF7-CD95 (data not shown). At very high expression levels achievable by transient expression, c-FLIPL may associate with and activate procaspase-8 outside of the DISC through both their DED domains and their protease/protease-like domain, which explains c-FLIPL-enhanced apoptosis independent of the DISC.

Fig. 2. c-FLIPL functions as an activator for CD95-mediated apoptosis. (A) Dosage-dependent effect of c-FLIPL on CD95-mediated apoptosis. HeLa cells seeded in 6-well plates were transfected with the indicated amounts of c-FLIPL/pcDNA3 construct plus the vector DNA to make the total amount of DNA constant. After 16 h, cells were treated with CH11 (75 ng/ml) plus cycloheximide (1 µg/ml) for 6 h and scored for apoptosis. (B) CD95-mediated apoptosis in MCF7-CD95 cells stably expressing c-FLIPL. Left: expression of c-FLIPL, caspase-8, FADD and CD95 in the MCF7-CD95 cells expressing no exogenous c-FLIPL (MCF7-C) or stably expressing low (MCF7-FLIP-Lo) or high levels (MCF7-FLIP-Hi) of c-FLIPL. The exogenous c-FLIPL (HA-c-FLIPL) migrated more slowly than the endogenous c-FLIPL on SDS–PAGE due to the tags. Right: MCF7-CD95 cells were treated with the indicated concentrations of anti-APO-1 plus protein A (5 ng/ml) for 3 h and scored for apoptosis. These cells were similarly sensitive to staurosporine-induced apoptosis (data not shown). (C) Activation of overall caspase-8 in the MCF7-CD95 cells expressing different levels of c-FLIPL. Different MCF7 cells were treated with anti-APO-1 as in (B) for the indicated times, and whole-cell lysates were analyzed by immunoblotting with the anti-caspase-8 mAb C15, which recognized a region in the large subunit. No p18 band was detected in MCF7-FLIP-Hi cells even after a much longer exposure. (D) Endogenous c-FLIPL is required for CD95-mediated apoptosis. HeLa cells cultured in 12-well plates were transfected with the indicated amount of the c-FLIPL antisense plasmid plus 0.15 µg of pEGFP-N3. The total amount of DNA used for transfection was made constant using vector DNA. Top panel: 24 h after transfection, cells were treated with anti-APO-1 (0.4 ng/ml) plus protein A (5 ng/ml) for 4 h, and green fluorescent protein (GFP)-positive cells were scored for apoptosis. Without CD95 stimulation, the background cell death in each condition was approximately the same and <5%. Bottom panels: expression levels of endogenous c-FLIPL, caspase-8 and actin detected at 24 h post-transfection. The ratio of c-FLIP proteins in different lanes was determined using a Bio-Rad Densitometer GS-700, which was then adjusted according to the transfection efficiency (∼70% as judged by EGFP expression) to obtain the ratio of c-FLIPL in transfected (trans.) cells.
To further analyze the effect of c-FLIPL on CD95-mediated apoptosis, we generated MCF7-CD95 cells that stably expressed exogenous c-FLIPL. In the clones that expressed exogenous c-FLIPL at levels comparable with the endogenous protein, sensitivity to CD95-mediated apoptosis increased >2-fold, whereas, in clones that had a much higher expression level of c-FLIPL, apoptosis was almost completely blocked (Figure 2B). The effect of c-FLIPL on apoptosis correlated with overall procaspase-8 activation in these cells (Figure 2C). In the cells expressing low levels of exogenous c-FLIPL, overall processing of procaspase-8 was enhanced compared with control cells. In contrast, in cells expressing high levels of c-FLIPL, no mature caspase-8 subunit p18 was detected even though generation of the processing intermediates p43/p41 was evident (Figure 2C). This latter result confirmed a recent report that c-FLIPL at high expression levels blocks the full activation of procaspase-8 (Krueger et al., 2001a). To eliminate effects that could be caused by clonal variation, we also constructed MCF7-CD95 cells that express exogenous c-FLIPL under the control of an inducible metallothionein promotor. Again, maximal apoptosis sensitivity in these inducible cells was observed when the exogenous c-FLIPL level was near that of the endogenous protein, whereas c-FLIPL inhibited apoptosis at higher concentrations (data not shown). Taken together, our results suggest that in both HeLa and MCF7 cells, c-FLIPL at endogenous levels promotes CD95-mediated apoptosis.
To test directly for a pro-apoptotic role for endogenous c-FLIPL, we downregulated endogenous c-FLIPL in HeLa cells using an antisense c-FLIPL construct. A 77% reduction in the expression level of endogenous c-FLIPL with no change in expression of caspase-8 resulted in a decrease of CD95 apoptosis sensitivity by ∼50% (Figure 2D). This inhibition occurred in spite of a concurrent reduction of the anti-apoptotic splice variant c-FLIPS (data not shown). This result indicates that in HeLa cells, c-FLIPL is required for maximal apoptosis induced by CD95.
Processing of c-FLIPL depends on caspase-8 and occurs very early during CD95-mediated apoptosis
In the DISC of most cells, only the processed p43 fragment of c-FLIPL can be detected, suggesting that most of the processed c-FLIPL is cleaved at the DISC. A comparison of the cleavage kinetics between c-FLIPL and procaspase-8 revealed that in MCF7-CD95 cells, processing of c-FLIPL preceded that of procaspase-8 during CD95-mediated apoptosis (Figure 3A). In contrast, c-FLIPS was not cleaved (Figure 3A). The cleavage of c-FLIPL was dependent mainly on caspase-8 because it was severely delayed in Jurkat cells lacking functional caspase-8 (Juo et al., 1998; Figure 3B). Taken together, these results are consistent with the notion that c-FLIPL is involved in procaspase-8 activation in the DISC at the initiation step of CD95-mediated apoptosis.

Fig. 3. Processing of procaspase-8 and c-FLIPL during CD95-mediated apoptosis. (A) Processing of caspase-8 and c-FLIPL in MCF7-CD95 cells. MCF7-CD95 cells were treated with anti-APO-1 (0.5 µg/ml) plus protein A (10 ng/ml) for the indicated times. Whole-cell lysates were analyzed by immunoblotting with anti-caspase-8 (top and middle) or anti-c-FLIP (bottom) antibody. Molecular weight standards are shown on the right. (B) Processing of c-FLIPL requires caspase-8 activity. Wild-type or caspase-8 deficient Jurkat cells were treated with anti-APO-1 and the cell lysates were analyzed as in (A). The small amount of c-FLIPL processing after prolonged treatment was probably caused by caspase-10, which is also recruited to the DISC (Figure 5C).
c-FLIPL enhances procaspase-8 activation in the DISC
To study directly c-FLIPL’s activity in the DISC in living cells, we analyzed the activation of procaspase-8 at the DISC in cells expressing exogenous c-FLIPL either stably or inducibly under the control of the metallothionein promotor. Similar to the Fv-mediated procaspase-8 activation in vitro (Figure 1A), the activation of procaspase-8 in the DISC occurs in two proteolytic steps, with the first cleavage separating the large and small subunits and the second step separating the large subunit and the DISC-binding prodomain. This second step leads to the release of the mature caspase into the cytosol. When higher levels of exogenous c-FLIPL were expressed in the inducible cells, more c-FLIPL was recruited subsequently to the DISC, and concurrently more procaspase-8 was partially processed (Figure 4A, lanes 3 and 4 versus lane 2). Similar results were observed in the c-FLIPL stable transfectants; in cells stably expressing high levels of c-FLIPL, the DISC complex predominantly contained partially processed caspase-8 (Figure 4B).

Fig. 4. c-FLIPL enhances caspase-8 activation in the DISC. (A) Effects of c-FLIPL on caspase-8 processing in the DISC in MCF7-CD95 cells inducibly expressing c-FLIPL. MCF7-FLIP-In cells were treated with the indicated concentration of ZnCl2 for 5 h. Cells were then stimulated with anti-APO-1 (+) or left untreated (–), and DISC complexes were isolated. Cell lysates and DISC were subjected to immunoblotting analysis using anti-c-FLIP (top two panels) or anti-caspase-8 antibody (bottom two panels). **IgG heavy chain. (B) Caspase-8 processing in MCF7 transfectants stably expressing c-FLIPL. Different MCF7 cells were stimulated with anti-APO-1 or left untreated. Cell lysates and DISC were analyzed by immunoblotting with anti-c-FLIP (top panels) or anti-caspase-8 (bottom panels) antibody. (C) c-FLIPL increases caspase-8 activity in the DISC. A total of 2 × 107 MCF7-c-FLIP-In cells were treated with ZnCl2 for 2 h, washed and kept without ZnCl2 for another 4 h at 37°C. The DISC was isolated and the caspase-8 activity in the DISC was assayed using IETD-AFC. The data shown are representative of three independent experiments.
To confirm that the processed caspase-8 was enzymatically active, we directly assayed the activity of caspase-8 in the DISC using a fluorogenic caspase-8 substrate IETD-AFC. Caspase-8 activity in the DISC was enhanced in cells expressing exogenous c-FLIPL, with the highest activity observed when exogenous c-FLIPL was expressed at a level similar to the endogenous c-FLIPL (Figure 4C, lane 3). However, even high expression levels of c-FLIPL did not completely block the caspase activity of the DISC, and the activity of the c-FLIPL-containing DISC was still higher when compared with the DISC of cells expressing only endogenous c-FLIPL (Figure 4C, lane 4 versus lane 1). This discrepancy was probably due to c-FLIPL’s ability to enhance the protease activity of the zymogen form of caspase-8 (see below). Furthermore, in cells stably expressing high levels of c-FLIPL, the amount of caspase-8 in the DISC was only reduced by ∼50% (Figure 4B, lane 5 versus lane 1). This modest decrease in the steady-state level of caspase-8 in the DISC did not appear to account for the nearly complete inhibition of apoptosis in these cells (Figure 2B), but rather the lack of generation of mature caspase-8 subunits in these cells upon CD95 treatment did (Figure 2C), consistent with previous studies by us and others (Scaffidi et al., 1999; Krueger et al., 2001a). Therefore, high levels of c-FLIPL, although promoting the first cleavage of procaspase-8 in the DISC, prevent recruitment of additional cytosolic procaspase-8 to the DISC, thereby inhibiting apoptosis. In contrast, low levels of c-FLIPL expression enhance procaspase-8 activation in the DISC and allow processed caspase-8 to be released from the DISC, resulting in enhanced activation of cytosolic procaspase-8 and apoptosis (Figures 2 and 4).
c-FLIPL is a specific activator for procaspases engaged by death receptors
To determine whether c-FLIPL affects the activation of other initiator procaspases, we tested its effect on the processing of procaspase-9 and -10. Procaspase-9 is linked to mitochondria-mediated apoptosis, while procaspase-10 is another tandem DED-containing caspase implicated in death receptor signaling. Similar to procaspase-8, dimerization of either procaspase-9 or -10 led to their self-processing (Figure 5A and B; D.W.Chang, Z.Xing, V.L.Capacio, M.E.Peter and X.Yang, submitted for publication). However, c-FLIPL enhanced procaspase-10 but not procaspase-9 activation. These data suggested that caspase-10 might be involved in CD95-mediated apoptosis as a DISC component. We indeed found procaspase-10, together with c-FLIPL, to be part of the endogenous DISC of type I cells that we have characterized previously (Scaffidi et al., 1998; Figure 5C), which is consistent with recent observations (Kischkel et al., 2001; Wang et al., 2001). Interestingly, even in these cell lines which are highly sensitive to CD95-mediated apoptosis, the DISC also contained only the p43 fragment of c-FLIPL but not the full-length c-FLIPL, consistent with the notion that c-FLIPL is an important activator for CD95-mediated apoptosis. Together, these results show that c-FLIPL could be a specific regulator of the two procaspases directly engaged by death receptors.
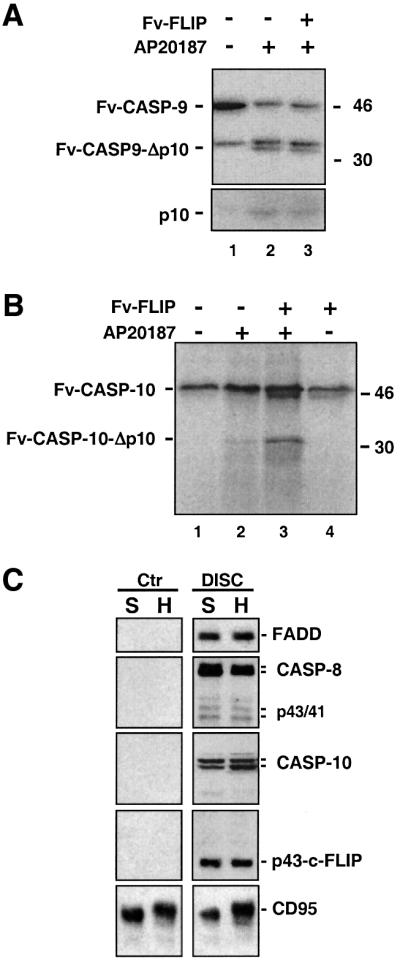
Fig. 5. Specific effect of c-FLIPL on caspase-8 and -10 and identification of caspase-10 as a DISC component. (A and B) Fv-FLIP enhances caspase-10 but not caspase-9 processing. A 1 µl aliquot of [35S]Fv-CASP-9 (A) or [35S]Fv-CASP-10 (B) was treated with 100 nM AP20187, in the presence or absence of 1 µl of unlabeled Fv-FLIP. The reaction mix was analyzed as in Figure 1A. Domain structures are shown on the left, and molecular weight standards on the right. (C) Identification of caspase-10 as a DISC component. CD95 was immunoprecipitated from stimulated (DISC) or unstimulated (Ctr) SKW6.4 (S) and H9 (H) cells. CD95 and the associated proteins were analyzed by immunoblotting using appropriate monoclonal antibodies.
An intact protease-like domain is required for c-FLIPL function
To study the mechanism by which c-FLIPL enhances caspase activation, we first examined which region(s) of the c-FLIPL caspase-like domain, which shares 60% similarity with the procaspase-8 protease domain, is responsible for its activity. We deleted either the large or the small subunit of Fv-FLIP (Figure 6A). Neither deletion mutant induced procaspase-8 processing in vitro or its cell death activity when transfected into HeLa cells (Figure 6B, lane 4 and 5, and C, columns 2 and 3), suggesting that both subunits of c-FLIPL are required to engage procaspase-8. In addition, despite their high homology, replacement of either the large or the small subunit of c-FLIPL with the corresponding subunit of procaspase-8 (Figure 6A) rendered c-FLIPL inactive (Figure 6B, lanes 7–9, and C, columns 5 and 6). These results indicate that c-FLIPL is not simply an active site mutant of tandem DED pro caspases. Its active subunit-like domains, instead, have a specific and relevant function.
To determine the importance of c-FLIPL cleavage for procaspase-8 activation, we mutated the cleavage site Asp376 to glutamate (D/E) (Figure 6A). Both the in vitro procaspase-8 processing assay and the in vivo apoptosis assay showed that the mutation also impaired the proapoptotic function of c-FLIPL (Figure 6B, lane 6, and C, column 4). Thus, many aspects of the c-FLIPL caspase-like domain appear to be designed specifically to mediate its catalysis of procaspase-8 activation.
c-FLIPL is recruited preferentially to the DISC
Previous studies showed that initiator procaspases are activated by induced dimerization of the zymogen molecules. We recently found that this activation requires the formation of a structural intermediate, comprised of two stably associated procaspase molecules, that possesses caspase activity (D.W.Chang, Z.Xing, V.L.Capacio, M.E.Peter and X.Yang, submitted for publication). This mechanism does not require both dimeric partners to possess enzymatic activity, and thus would allow a proteolytically inactive caspase homolog like c-FLIPL to promote procaspase activation allosterically. We thus compared the heterophilic interaction between the c-FLIPL protease domain and the procaspase-8 protease domain with the homophilic interaction of the procaspase-8 protease domain. The procaspase-8 protease domain or the c-FLIPL protease-like domain tagged with a hemagglutinin (HA) epitope was co-expressed in 293 cells with the procaspase-8 protease domain tagged with FLAG epitope (the caspase-8 active site mutant was used to prevent caspase activation). Cell lysates were immunoprecipitated with an anti-FLAG antibody and the precipitates were then subjected to immunoblotting analysis using an anti-HA antibody. Interestingly, the heterophilic c-FLIPL:caspase-8 interaction was significantly stronger than the homophilic caspase-8:caspase-8 interaction (Figure 7A). A similar result was observed when in vitro-translated proteins were used for the co-immunoprecipitation assay (data not shown).

Fig. 7. The c-FLIPL protease-like domain induces the enzymatic activity of the caspase-8 zymogen. (A) The c-FLIPL protease-like domain interacts with the caspase-8 protease domain more efficiently than the caspase-8 protease domain does with itself. 293 cells were transfected with HA-tagged caspase-8 protease domain (PD) or HA-tagged c-FLIPL protease-like domain together with FLAG-tagged caspase-8-PD. The cell lysates were immunoprecipitated with an anti-FLAG antibody. The immunoprecipitated proteins and the lysates were analyzed by immunoblotting as indicated. (B) Ratios of c-FLIPL versus caspase-8 in cell lysate and in the DISC. The DISC complexes and whole-cell lysates (lys.) were made from 3 × 106 MCF7-CD95 cells. Thirty percent of these DISC complexes and 0.3% of the lysate (lys.) were resolved by SDS–PAGE together with a c-FLIPL (top) or a caspase-8 (bottom) standard (Std), and analyzed by immunoblotting (IB) with the indicated antibodies. For making protein standards, HA-tagged c-FLIPL and caspase-8 proteins were generated by in vitro translation. The relative concentration of these two proteins was determined by anti-HA immunoblotting, and equal amounts of these two proteins were used. The ratio of c-FLIPL/caspase-8 in the DISC was higher when the expression level of c-FLIPL increased. In the MCF7-CD95 cells stably expressing low and high levels of c-FLIPL, the ratio is ∼1:2 and 10:1, respectively (Figure 4B, lanes 3 and 5). (C and D) Fv-FLIP enhances the caspase activity of non-cleavable Fv-CASP-8(DD/AA). (C) Recombinant Fv-CASP-8 fusion proteins (100 ng) were incubated with the indicated amount of Fv-FLIP protein (in ng). Caspase-8 activity was measured using IETD-AFC. The data shown were representative of three independent experiments done in duplicate or triplicate. The insert shows analysis of recombinant Fv-CASP-8 (1), Fv-CASP-8(DD/AA) (2), Fv-CASP(C/S) (3) and Fv-FLIP (4) purified from bacteria. About 100 ng of each protein was resolved by SDS–PAGE and visualized by Coomassie Blue staining. (D) Fv-CASP-8 proteins were labeled with biotin-DEVD in the presence or absence of Fv-FLIP. The mixes were then resolved by SDS–PAGE and immunoblotted with either avidin–HRP (top) or anti-caspase-8 mAb C15 (bottom).
The expression of endogenous c-FLIPL in many cell types is ∼1% of that of procaspase-8 (Scaffidi et al., 1999). A confounding question is how such a small amount of c-FLIPL could make a significant difference in death receptor-mediated apoptosis. Assuming that c-FLIPL binds to procaspase-8 with higher affinity than procaspase-8 does itself, the c-FLIPL concentration after recruitment to the DISC may be higher than that of procaspase-8 when compared with their cytoplasmic concentration. We therefore quantified the amounts of c-FLIPL and procaspase-8 in the cytosol and in the DISC of MCF7-CD95 cells. This analysis revealed that c-FLIPL indeed was recruited more efficiently to the DISC upon CD95 stimulation than procaspase-8; c-FLIPL was enriched 18-fold in the DISC when compared with procaspase-8. As a result, the ratio of c-FLIPL to procaspase-8 in the DISC reached ∼1:5 (Figure 7B).
The protease-like domain of c-FLIPL induces the activity of the caspase-8 zymogen
The correlation between the binding efficiency of c-FLIPL to the procaspase-8 protease domain and its ability to promote procaspase-8 activation suggested that c-FLIPL may enhance procaspase-8 activity by adopting a structure that is more efficient in inducing the activity of caspase-8 zymogen than the procaspase-8 protease domain itself. To test whether c-FLIPL could induce the activity of procaspase-8 without causing its cleavage, we expressed recombinant Fv-FLIP and a non-processable form of Fv-CASP-8 [Fv-CASP-8(DD/AA)], along with Fv-CASP-8 and Fv-CASP-8(C/S) from bacteria and purified them to homogeneity. As expected, the wild-type Fv-CASP-8 was partially processed, while Fv-CASP-8(DD/AA) and Fv-CASP-8(C/S) were in the precursor form (insert of Figure 7C). The protease activity of Fv-CASP-8(DD/AA) was significantly induced by Fv-FLIP in a dose-dependent manner (Figure 7C). This enhancement was found to be AP20187 independent, due to the very high binding efficiency of two proteins at the concentrations used (data not shown). No activity was detected when the catalytically inactive mutant of caspase-8 was used (Figure 7C, column 5), and c-FLIP could not enhance the activity of already processed Fv-CASP-8, suggesting that it only acts on the caspase-8 proform (Figure 7C, column 7).
To confirm that the measured caspase-8 activity originated from the full-length caspase-8 protein and not from aberrant processing products, we used a biotinylated, irreversible inhibitor of caspases (biotin-VAD-fmk) that covalently modifies the active site, to allow direct visualization of the activation of unprocessed caspase-8 by c-FLIPL. Upon incubation with Fv-FLIPL, full-length Fv-CASP-8(DD/AA) protein was found to be affinity labeled with this inhibitor (Figure 7D, lane 2). As a negative control, Fv-FLIP had no effect on the catalytically inactive Fv-CASP-8(C/S) (Figure 7D, lane 4). Again Fv-FLIP did not enhance the activity of Fv-CASP-8, which represented mostly processed caspase-8 in the experiment (Figure 7D, lane 6). Taken together, these results suggest that c-FLIPL promotes caspase-8 activation through induction of the enzymatic activity of the zymogen but not of the caspase-8 active subunits.
Discussion
c-FLIP has been widely regarded as an inhibitor of death receptor-mediated apoptosis due to its ability to inhibit apoptosis at high levels of expression (Krueger et al., 2001b). By examining the effect of c-FLIPL on procaspase-8 activation in a controlled caspase activation system that mimics the DISC, in combination with direct analyses of cells expressing c-FLIPL at physiologically relevant levels and of cell lines with decreased c-FLIPL expression, we now show that c-FLIPL can also function as a novel type of caspase activator. Unlike death adaptors that associate with the N-terminal prodomain of procaspases to facilitate oligomerization of the C-terminal protease domain, c-FLIPL promotes procaspase-8 activation by acting directly on the protease domain.
We found that the effect of c-FLIPL on cells depends on its expression levels. Three different concentration-dependent effects could be distinguished, allowing us to propose the following model that could explain the contradictory findings in the literature on the function of c-FLIPL. At low expression levels (region I in Figure 2A), which are probably found in most cells, c-FLIPL enhances the DISC activity. At intermediate expression levels (region II in Figure 2A), possibly found in some cell types such as monocytes/macrophages and certain tumors (Irmler et al., 1997; Griffith et al., 1998; Perlman et al., 1999) and achieved in c-FLIPL transgenic mice (Medema et al., 1999; Van Parijs et al., 1999), c-FLIPL acts as an inhibitor of the DISC. Only at very high non-physiological concentrations achieved by transient overexpression (region III in Figure 2A) is c-FLIPL cytotoxic by itself without the need for stimulation of CD95, probably through a direct association with caspase-8 outside the DISC. In contrast to c-FLIPL, c-FLIPS has been shown to be a dedicated apoptosis inhibitor and it prevents the first cleavage of procaspase-8 activation in the DISC (Krueger et al., 2001a). Interestingly, stimulation of CD95-sensitive T cells through either the T-cell receptor or CD28 was shown to result in a specific upregulation of only c-FLIPS, demonstrating that c-FLIPL and c-FLIPS are not always co-regulated (Kirchhoff et al., 2000).
c-FLIP–/– MEFs were reported to show increased sensitivity to death receptor-mediated apoptosis. This result, which seems to contradict our study, might be reconciled by the possibility that the MEFs express levels of c-FLIPL high enough to inhibit death receptor-mediated apoptosis. An alternative but not mutually exclusive possibility is that c-FLIPS may play a major role in the inhibition of CD95-mediated apoptosis in these cells. Remarkably, mice deficient for either c-FLIP, FADD or caspase-8 all share strikingly similar developmental defects (Yeh et al., 2000). This observation on the level of whole animals clearly places these three DISC components on the same functional axis, and could be explained by c-FLIPL playing an essential role as a caspase-8 activator during development.
The two main apoptosis pathways are started by two large prodomain initiator caspases. Caspase-8 initiates the extrinsic and caspase-9 the intrinsic pathway. The intrinsic pathway is regulated by the ying:yang of pro-apoptotic proteins released from mitochondria, such as cytochrome c or Smac/Diablo-like proteins, and the expression levels of IAP proteins that regulate the activity of the caspases downstream of mitochondria. In comparison, without c-FLIP, the extrinsic pathway would appear to be regulated only by the intensity of the stimulus received through death receptors. Neither FADD nor procaspase-8 is significantly regulated on the transcriptional level and, at present, no inhibitor of the IAP class has been found that would act on caspase-8. The identification of c-FLIPL as a dual functional regulator for this pathway reveals a previously unexpected complexity of regulation that may be adapted to the specific functions of caspase-8. The modulation of apoptosis sensitivity by a protease-inactive caspase homolog like c-FLIPL, instead of a caspase itself, also represents a highly effective and safe mechanism for the regulation of apoptosis. It greatly decreases the risk of basal toxicity associated with high levels of caspases since c-FLIPL only acts on DISC-bound caspases. In addition, it allows a fine-tuned and versatile regulation of caspase activation in response to different stimuli. To our knowledge, c-FLIPL represents the first example of an inactive protease analog that acts as an activator of proteases.
Unlike caspase-8 or FADD, the expression of c-FLIPL appears to be highly regulated, for example by the mitogen-activated protein kinase pathway and the NF-κB pathway (Kreuz et al., 2001; Micheau et al., 2001). Upregulation of c-FLIPL was also reported in certain tumor cells, and these tumor cells may escape apoptosis induced through death receptors (Irmler et al., 1997; Griffith et al., 1998). The expression level of c-FLIPL is thus a focal point for apoptosis regulation, and it can not only determine whether the apoptosis pathway is turned on or off but also seems to allow the cell to switch between apoptosis and survival pathways. c-FLIPL may therefore represent an attractive target for therapeutic intervention with death receptor signaling.
Materials and methods
Cell lines and reagents
HeLa, 293, SKW6.4 (B lymphoblastoid) and H9 (T-cell leukemia) cells were obtained from ATCC. MCF7 cells stably expressing CD95 (MCF7-CD95) and the wild-type and caspase-8-deficient Jurkat cells were gifts from Drs E.Alnemri and J.Blenis, respectively. The following reagents were obtained from the indicated sources: AP20187 (ARIAD Pharmaceuticals); z-DEVD (z-Asp-Glu-Val-Asp) and biotin-z-VAD (Enzyme System Products); anti-CD95 CH11 (MBL Co.); anti-FLAG antibody and anti-CD95 (C20, for western blotting; Santa Cruz Biotech); protein A (Pierce); anti-caspase-10 4C1 (PanVer); anti-FADD A36620 (Transduction Lab); and anti-actin, cycloheximide, rapamycin and ZnCl2 (Sigma). Monoclonal antibodies specific for caspase-8 (C15) and c-FLIP (NF-6) (Scaffidi et al., 1999), and anti-APO-1 (Kischkel et al., 1995) have been described previously.
To generate c-FLIPL stable and inducible cell lines, c-FLIPL/pRK5 and c-FLIPL/pMT (see below) were transfected into MCF7-CD95 cells together with pBabe-Puro using SuperFect-mediated gene transfer (Qiagen). Cells were selected in Dulbecco’s modified Eagle’s medium/fetal bovine serum (DMEM/FBS) with 2 µg/ml of puromycin and screened for exogenous c-FLIPL expression by immunoblotting. Two independent MCF7-FLIP-Lo cell lines, several MCF7-FLIP-Hi cell lines and three MCF7-FLIP-In cell lines were obtained. Similar results were obtained with the different clones.
Expression constructs
pRK5-crmA (Dr D.Goeddel) and c-FLIPL/pcDNA3 (Dr S.Hu) were obtained from the indicated sources. c-FLIPL Δ/pcDNA3 expression plasmid was generated by deleting the c-FLIPL sequence between the BamHI and EcoRI sites. The full-length c-FLIPL was cloned into pRK5 and the plasmid pMT (containing the metallothionein promoter) with an HA tag at the N-terminus and a FLAG tag at the C-terminus. The antisense c-FLIPL construct, with the entire open reading frame expressed in the reversed orientation, was made in pRK7. The Fv and FRB fusion constructs were made in pRK5 similarly to the Fkp fusion constructs (Yang et al., 1998a). Fragments for various mutations and chimeras were generated by PCR. The expression constructs for the procaspase-8 protease domain (amino acids 206–480) and the c-FLIPL protease-like (amino acids 208–479) were made in pRK5 with a C-terminal HA or FLAG tag.
In vitro cleavage of caspases
In vitro transcription and translation were carried out with the TNT reticulocyte lysate system (Promega) in the presence of [35S]methionine or non-radioisotope-labeled methionine. A 1 µl aliquot of [35S]methio nine-labeled product was mixed with the indicated amounts of unlabeled product. The total volume of in vitro translated products was kept constant by adding in vitro translation reaction mix containing the parental plasmid. In vitro translated products were then mixed with an equal volume of 2× CED3 buffer (Yang et al., 1998a) containing AP20187, and incubated at 30°C for the indicated time.
Apoptosis assay
Cell death was measured as described, with modifications (Yang et al., 1998a). Anti-APO-1 (in combination with protein A) and CH11 (in combination with cycloheximide) were used to induce apoptosis through CD95. Transfection was performed using the calcium phosphate precipitation method. After transfection and/or treatment, cells were fixed and, for cells co-transfected with pCMV-lacZ, also stained with X-gal. Apoptosis was scored based on characteristic morphological changes including membrane blebbing, cell shrinkage and detachment from the plates. Data (mean ± SD) were obtained from three or more independent experiments, and in each experiment >300 cells/transfected cells were examined.
DISC analysis
The DISC analysis was performed essentially as described (Kischkel et al., 1995). A total of 3 × 106 MCF7-CD95 cells and 1 × 107 H9 and SKW6.4 cells were treated with 2 µg/ml anti-APO-1 for 20 min (MCF7-CD95 cells) or 5 min (H9 and SKW6.4 cells) at 37°C and then lysed in lysis buffer (Kischkel et al., 1995) (stimulated condition), or the cells were first lysed and then supplemented with 2 µg/ml of anti-CD95 (unstimulated condition). CD95 was then immunoprecipitated for 2 h at 4°C with protein A–Sepharose beads. To detect caspase-8 activity associated with the receptor, the DISC was incubated in cleavage buffer [20 mM HEPES pH 7.4, 100 mM NaCl, 10 mM dithiothreitol (DTT), 1 mM EDTA, 0.1% CHAPS, 10% sucrose] containing 40 µM caspase-8-specific fluorogenic substrate IETD-AFC for 1 h at 37°C. Caspase activity was determined using a fluorescence plate reader with a 400 nm excitation filter and 508 nm emission filter. Values of immunoprecipitated CD95 from unstimulated cells were taken as background and subtracted from those obtained with stimulated cells.
Assay of caspase-8 activity in vitro
Purification of bacterially expressed Fv-CASP-8 fusions was described elsewhere (D.W.Chang, Z.Xing, V.L.Capacio, M.E.Peter and X.Yang, submitted for publication), and Fv-FLIP was purified similarly by Ni-NTA and DEAE columns. Fv-caspase-8 protein was mixed with Fv-FLIP in 150 µl of CED3 buffer containing 7 µM IETD-AFC. After incubation at 37°C for 15 min, the reaction mix was measured for fluorescence in a Bio-Rad Versaflor fluorometer. Alternatively, Fv-CASP-8 and Fv-FLIP proteins were mixed and incubated in MDB buffer (10 mM HEPES pH 7.0, 50 mM NaCl, 2 mM MgCl2, 5 mM EDTA, 1 mM DTT) containing 100 µM biotinylated VAD-fmk at 37°C for 45 min. The proteins were then separated by SDS–PAGE, and biotinylated products were detected by immunoblotting with horseradish peroxidase (HRP)–avidin.
Co-immunoprecipitation assay
A total of 1 × 106 293 cells were transiently transfected with 3 µg of the indicated HA-tagged and FLAG-tagged protein expression constructs. After 16 h, cells were lysed with 1 ml of IP-lysis buffer (Yang et al., 1998b) containing a protease inhibitor cocktail. The lysates were immunoprecipitated with 20 µl of anti-FLAG M2 antibody conjugated to agarose beads (Sigma) for 3 h. The beads were then washed three times with lysis buffer and resuspended in loading buffer for SDS–PAGE and immunoblotting analyses.
Acknowledgments
Acknowledgements
We thank Drs D.Baltimore, C.B.Thompson, and H.Y.Chang for critically reading the manuscript, L.Gardner and V.Capacio for excellent technical assistance, J.Blenis for the caspase-8-deficient Jurkat cells, S.Hu for c-FLIPL cDNA, E.S.Alnemri for MCF7-CD95 cells, D.V.Goeddel for pRK7, pRK5 and pRK5-crmA plasmids, M.Lazar for pMT, and E.Brown for FRB cDNA. We also thank ARIAD Pharmaceuticals for providing the FKBP dimerization system. Supported by a start-up fund from University of Pennsylvania (to X.Y.) and NIH grants GM60911 (to X.Y.) and GM61712 (to M.P.). D.W.C. was supported by an NCI training grant.
References
- Chang H.Y. and Yang,X. (2000) Proteases for cell suicide: functions and regulation of caspases. Microbiol. Mol. Biol. Rev., 64, 821–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J., Zheng,X.F., Brown,E.J. and Schreiber,S.L. (1995) Identification of an 11-kDa FKBP12–rapamycin-binding domain within the 289-kDa FKBP12–rapamycin-associated protein and characterization of a critical serine residue. Proc. Natl Acad. Sci. USA, 92, 4947–4951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clackson T. et al. (1998) Redesigning an FKBP–ligand interface to generate chemical dimerizers with novel specificity. Proc. Natl Acad. Sci. USA, 95, 10437–10442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goltsev Y.V., Kovalenko,A.V., Arnold,E., Varfolomeev,E.E., Brodianskii,V.M. and Wallach,D. (1997) CASH, a novel caspase homologue with death effector domains. J. Biol. Chem., 272, 19641–19644. [DOI] [PubMed] [Google Scholar]
- Griffith T.S., Chin,W.A., Jackson,G.C., Lynch,D.H. and Kubin,M.Z. (1998) Intracellular regulation of TRAIL-induced apoptosis in human melanoma cells. J. Immunol., 161, 2833–2840. [PubMed] [Google Scholar]
- Han D., Chaudhary,P.M., Wright,M.E., Friedman,C., Trask,B.J., Riedel,R.T., Baskin,D.G., Schwartz,S.M. and Hood,L. (1997) MRIT, a novel death-effector domain-containing protein, interacts with caspases and BclXL and initiates cell death. Proc. Natl Acad. Sci. USA, 94, 11333–11338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu S., Vincenz,C., Ni,J., Gentz,R. and Dixi,V.M. (1997) I-FLICE, a novel inhibitor of tumor necrosis factor receptor-1- and CD-95-induced apoptosis. J. Biol. Chem., 272, 17255–17257. [DOI] [PubMed] [Google Scholar]
- Inohara N., Koseki,T., Hu,Y., Chen,S. and Nunez,G. (1997) CLARP, a death effector domain-containing protein interacts with caspase-8 and regulates apoptosis. Proc. Natl Acad. Sci. USA, 94, 10717–10722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irmler M. et al. (1997) Inhibition of death receptor signals by cellular FLIP. Nature, 388, 190–195. [DOI] [PubMed] [Google Scholar]
- Juo P., Kuo,C.J., Yuan,J. and Blenis,J. (1998) Essential requirement for caspase-8/FLICE in the initiation of the Fas-induced apoptotic cascade. Curr. Biol., 8, 1001–1008. [DOI] [PubMed] [Google Scholar]
- Kirchhoff S., Muller,W.W., Krueger,A., Schmitz,I. and Krammer,P.H. (2000) TCR-mediated up-regulation of c-FLIPshort correlates with resistance toward CD95-mediated apoptosis by blocking death-inducing signaling complex activity. J. Immunol., 165, 6293–6300. [DOI] [PubMed] [Google Scholar]
- Kischkel F.C., Hellbardt,S.H., Behrmann,I., Germer,M., Pawlita,M., Krammer,P.H. and Peter,M.E. (1995) Cytotoxicity-dependent APO-1 (Fas/CD-95)-associated proteins form a death-inducing signaling complex (DISC) with the receptor. EMBO J., 14, 5579–5588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kischkel F.C. et al. (2001) Death receptor recruitment of endogenous caspase-10 and apoptosis initiation in the absence of caspase-8. J. Biol. Chem., 276, 46639–46646. [DOI] [PubMed] [Google Scholar]
- Kreuz S., Siegmund,D., Scheurich,P. and Wajant,H. (2001) NF-κB inducers upregulate cFLIP, a cycloheximide-sensitive inhibitor of death receptor signaling. Mol. Cell. Biol., 21, 3964–3973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger A., Schmitz,I., Baumann,S., Krammer,P.H. and Kirchhoff,S. (2001a) Cellular FLICE-inhibitory protein splice variants inhibit different steps of caspase-8 activation at the CD95 death-inducing signaling complex. J. Biol. Chem., 276, 20633–20640. [DOI] [PubMed] [Google Scholar]
- Krueger A., Baumann,S., Krammer,P.H. and Kirchhoff,S. (2001b) FLICE-inhibitory proteins: regulators of death receptor-mediated apoptosis. Mol. Cell. Biol., 21, 8247–8254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin D.A., Siegel,R.M., Zheng,L. and Lenardo,M.J. (1998) Membrane oligomerization and cleavage activates the caspase-8 (FLICE/MACHα1) death signal. J. Biol. Chem., 273, 4345–4349. [DOI] [PubMed] [Google Scholar]
- Medema J.P., Scaffidi,C., Kischkel,F.C., Shevchenko,A., Mann,M., Krammer,P.H. and Peter,M.E. (1997) FLICE is activated by association with the CD95 death-inducing signaling complex (DISC). EMBO J., 16, 2794–2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medema J.P., de Jong,J., van Hall,T., Melief,C.J. and Offringa,R. (1999) Immune escape of tumors in vivo by expression of cellular FLICE-inhibitory protein. J. Exp. Med., 190, 1033–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micheau O., Lens,S., Gaide,O., Alevizopoulos,K. and Tschopp,J. (2001) NF-κB signals induce the expression of c-FLIP. Mol. Cell. Biol., 21, 5299–5305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muzio M., Stockwell,B.R., Stennicke,H.R., Salvesen,G.S. and Dixit,V.M. (1998) An induced proximity model of caspase-8 activation. J. Biol. Chem., 273, 2926–2930. [DOI] [PubMed] [Google Scholar]
- Perlman H., Pagliari,L.J., Georganas,C., Mano,T., Walsh,K. and Pope,R.M. (1999) FLICE-inhibitory protein expression during macrophage differentiation confers resistance to fas-mediated apoptosis. J. Exp. Med., 190, 1679–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter M.E., Scaffidi,C., Medema,J.P., Kischkel,F. and Krammer,P.H. (1998) The death receptors. In Kumar,S. (ed.), Apoptosis: Biology and Mechanisms. Springer, Heidelberg, Germany, pp. 25–63.
- Rasper D.M. et al. (1998) Cell death attenuation by ‘Usurpin’, a mammalian DED-caspase homologue that precludes caspase-8 recruitment and activation by the CD-95 (Fas, APO-1) receptor complex. Cell Death Differ., 5, 271–288. [DOI] [PubMed] [Google Scholar]
- Scaffidi C., Fulda,S., Srinivasan,A., Friesen,C., Li,F., Tomaselli,K.J., Debatin,K.M., Krammer,P.H. and Peter,M.E. (1998) Two CD95 (APO-1/Fas) signaling pathways. EMBO J., 17, 1675–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaffidi C., Schmitz,I., Krammer,P.H. and Peter,M.E. (1999) The role of c-FLIP in modulation of CD95-induced apoptosis. J. Biol. Chem., 274, 1541–1548. [DOI] [PubMed] [Google Scholar]
- Shu H.B., Halpin,D.R. and Goeddel,D.V. (1997) Casper is a FADD- and caspase-related inducer of apoptosis. Immunity, 6, 751–763. [DOI] [PubMed] [Google Scholar]
- Srinivasula S.M. et al. (1997) FLAME-1, a novel FADD-like anti-apoptotic molecule that regulates Fas/TNFR1-induced apoptosis. J. Biol. Chem., 272, 18542–18545. [DOI] [PubMed] [Google Scholar]
- Van Parijs L., Refaeli,Y., Abbas,A.K. and Baltimore,D. (1999) Auto immunity as a consequence of retrovirus-mediated expression of C-FLIP in lymphocytes. Immunity, 11, 763–70. [DOI] [PubMed] [Google Scholar]
- Wang J., Chun,H.J., Wong,W., Spencer,D.M. and Lenardo,M.J. (2001) Caspase-10 is an initiator caspase in death receptor signaling. Proc. Natl Acad. Sci. USA, 98, 13884–13888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X., Chang,H.Y. and Baltimore,D. (1998a) Autoproteolytic activation of pro-caspases by oligomerization. Mol. Cell, 1, 319–325. [DOI] [PubMed] [Google Scholar]
- Yang X., Chang,H.Y. and Baltimore,D. (1998b) Essential role of CED-4 oligomerization in CED-3 activation and apoptosis. Science, 281, 1355–1357. [DOI] [PubMed] [Google Scholar]
- Yeh W.C. et al. (2000) Requirement for Casper (c-FLIP) in regulation of death receptor-induced apoptosis and embryonic development. Immunity, 12, 633–642. [DOI] [PubMed] [Google Scholar]