

Abstract
The p53 tumor suppressor is inhibited and destabilized by Mdm2. However, under stress conditions, this downregulation is relieved, allowing the accumulation of biologically active p53. Recently we showed that c-Abl is important for p53 activation under stress conditions. In response to DNA damage, c-Abl protects p53 by neutralizing the inhibitory effects of Mdm2. In this study we ask whether this neutralization involves a direct interplay between c-Abl and Mdm2, and what is the contribution of the c-Abl kinase activity? We demonstrate that the kinase activity of c-Abl is required for maintaining the basal levels of p53 expression and for achieving maximal accumulation of p53 in response to DNA damage. Importantly, c-Abl binds and phosphorylates Mdm2 in vivo and in vitro. We characterize Hdm2 (human Mdm2) phosphorylation at Tyr394. Substitution of Tyr394 by Phe394 enhances the ability of Mdm2 to promote p53 degradation and to inhibit its transcriptional and apoptotic activities. Our results suggest that phosphorylation of Mdm2 by c-Abl impairs the inhibition of p53 by Mdm2, hence defining a novel mechanism by which c-Abl activates p53.
Keywords: c-Abl/Mdm2/p53/tyrosine phosphorylation
Introduction
Exposure of cells to stress activates the p53 tumor suppressor protein. This activation is cardinal for the efficient response of cells to stress stimuli, such as DNA damage and hypoxia (Giaccia and Kastan, 1998; Oren, 1999; Vogt Sionov et al., 2001b). The response is either in the form of growth arrest, senescence or apoptosis (Vogt Sionov and Haupt, 1999). A loss of p53 function, either by mutations or by deregulation of regulatory proteins, is often detrimental, leading eventually to the development of cancer (reviewed in Vogt Sionov and Haupt, 1999). The p53 protein is subject to a tight regulation, keeping it labile and inactive under normal conditions. This regulation occurs at the levels of protein stability, post-translational modifications and subcellular localization (Ashcroft et al., 1999; Jimenez et al., 1999; Gottifredi and Prives, 2001).
The key inhibitor of p53 is the mdm2 proto-oncogene. Mdm2 (Hdm2 in humans) inhibits the transcriptional activity and growth suppression ability of p53 through an autoregulatory feedback loop (Momand et al., 2000; Michael and Oren, 2002). Importantly, Mdm2 promotes p53 for degradation through the ubiquitin system (Haupt et al., 1997; Kubbutat et al., 1997) by acting as an E3-ubiquitin ligase (Honda and Yasuda, 1999; Fang et al., 2000). The nuclear export of p53 is believed to be important for Mdm2-dependent degradation (Roth et al., 1998; Lain et al., 1999; Tao and Levine, 1999), although degradation within the nucleus has also been observed (Yu et al., 2000; Xirodimas et al., 2001). Nucleo-cytoplasmic shuttling depends on one or more nuclear export signals of p53 (Stommel et al., 1999; Zhang and Xiong, 2001) and requires the ubiquitylation of p53 by Mdm2 (Boyd et al., 2000; Geyer et al., 2000).
In order for p53 to be activated in response to stress, the inhibitory activities of Mdm2 on p53 have to be modulated. This modulation is achieved by multiple mechanisms, including specific modifications of both p53 (Ashcroft et al., 1999; Jimenez et al., 1999; Meek, 1999) and Mdm2. Mdm2 is phosphorylated on multiple sites by various kinases (Hay and Meek, 2000; see below). The inhibitory activities of Mdm2 are also neutralized by the action of partner proteins. In response to oncogenic activation, the major activator of p53 is the ARF tumor suppressor protein, which neutralizes Mdm2-mediated ubiquitylation and degradation of p53 (reviewed in Sherr and Weber, 2000). However, in response to ionizing radiation, the ataxia telangiectasia mutant (ATM) kinase plays a key role. ATM induces the phosphorylation of p53 directly and indirectly through the activation of the Chk2 kinase (reviewed by Kastan and Lim, 2000; Shiloh, 2001). Phosphorylation of Mdm2 by ATM also impairs its inhibitory effects on p53 (Khosravi et al., 1999; Maya et al., 2001).
Another pathway that is activated by ATM involves the c-Abl non-receptor tyrosine kinase. ATM phosphorylates and activates c-Abl in response to ionizing radiation (Baskaran et al., 1997; Shafman et al., 1997). In response to stress such as DNA damage, c-Abl promotes cell growth arrest and apoptosis, unlike its oncogenic forms such as Bcr-Abl, which counteracts these cellular responses (reviewed in Kharbanda et al., 1998; Van Etten, 1999; Shaul, 2000). The apoptotic activity of c-Abl is mediated partly via p73 (Agami et al., 1999; Gong et al., 1999; Yuan et al., 1999), and to a lesser extent through a p53-dependent pathway (Wen et al., 1996; Yuan et al., 1997). We have recently shown that c-Abl plays an important role in achieving maximal and rapid accumulation of p53 in response to DNA damage (Vogt Sionov et al., 2001a). c-Abl protects p53 by neutralizing the ability of Mdm2 to promote p53 for degradation (Vogt Sionov et al., 1999), most likely by impairing the ubiquitylation of p53 and its subsequent export to the cytoplasm (Vogt Sionov et al., 2001a). These actions of c-Abl explain how c-Abl enhances the transcriptional and biological activities of p53 (e.g. Goga et al., 1995; Wen et al., 1996; Vogt Sionov et al., 1999).
The regulatory role of c-Abl in the activation of p53 in response to stress, and its ability to neutralize Mdm2, prompted us to propose that these actions of c-Abl involve a direct interplay between c-Abl and Mdm2. We report here that c-Abl and Mdm2 physically interact in vitro and in vivo. We demonstrate that the kinase activity of c-Abl is important for maintaining the basal level of p53 expression and for its accumulation in response to DNA damage. Furthermore, c-Abl phosphorylates Mdm2 in vivo and in vitro, and the biochemical and functional characterization of the phosphorylation on Tyr394 is presented. Mutational analysis of Tyr394 suggests that phosphorylation of Tyr394 impairs the ability of Mdm2 to inhibit the transcriptional and apoptotic activities of p53, as well as its efficiency to promote p53 degradation. Thus, our results define a new mechanism by which c-Abl regulates p53.
Results
c-Abl and Mdm2 interact in vivo and in vitro
In order to understand the mechanism by which c-Abl neutralizes the inhibitory effects of Mdm2 on p53, we investigated whether c-Abl and human Mdm2 (Hdm2) can engage in a protein–protein interaction. To address this question, 293 cells were transfected with expression plasmids for hdm2 alone or together with c-abl. Twenty-four hours post-transfection, cells were harvested and the extracts were subjected to immunoprecipitation using anti-c-Abl antibody (ABL-148; Sigma), followed by western blot analysis using anti-Hdm2 (SMP14). Hdm2 was brought down efficiently with anti-c-Abl antibody when both Hdm2 and c-Abl were expressed, but not in the absence of c-Abl (Figure 1AI, lanes 2 and 3), confirming the existence of an interaction between the two proteins. This interaction was also observed when the co-immunoprecipitation was carried out via Hdm2 (data not shown). To examine whether the interaction between the two proteins is direct, the binding was tested in vitro. c-Abl and Mdm2 were synthesized and radioactively labeled with [35S]methio nine in vitro. Following incubation of the two proteins together, Mdm2 was immunoprecipitated using anti-Mdm2 antibody (2A10), and the amount of bound c-Abl was detected by exposure to film. A co-immunoprecipitation of the two proteins was clearly observed with radiolabeled c-Abl (Figure 1B, lane 4), but not with non-radioactive c-Abl (c-Abl*; Figure 1B, lane 5). Thus, c-Abl and Mdm2 interact both in vivo and in vitro.
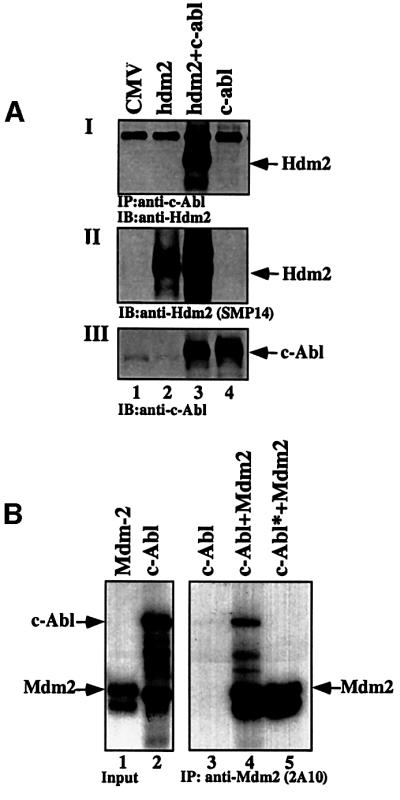
Fig. 1. Interaction between c-Abl and Hdm2 in vitro and in vivo. (A) 293 cells were transfected with the indicated expression plasmids (5 µg each). Twenty-four hours post-transfection, cell extracts were subjected to immunoprecipitation using anti-c-Abl antibody (ABL-148) followed by western blotting using anti-Hdm2 (SMP14) (I). The expression level of Hdm2 in the transfected cells was monitored by blotting with anti-Hdm2 antibody (SMP14) (II), and that of c-Abl using anti-c-Abl antibody (ABL-148) (III). (B) Radioactively labeled ([35S]methionine) Mdm2 and c-Abl were incubated in vitro before being subjected to immunoprecipitation using anti-Hdm2 antibody (2A10). The amount of bound c-Abl was detected by exposure to film. As a control, unlabeled c-Abl was used (lane 5). The amount of input for each protein is shown (lanes 1 and 2).
To define the interaction region of c-Abl within Hdm2, a series of Hdm2 mutants lacking different regions of the protein were tested in an in vivo binding assay. These deletions cover the entire Hdm2 protein (Figure 2A). 293 cells were transfected with expression plasmids for His-tagged c-abl alone, or together with wild-type hdm2 or deletion mutants of hdm2, as indicated in Figure 2A. His-c-Abl was isolated from the cell extracts using a nickel resin, and the amount of Hdm2 bound was determined by immunoblotting with anti-Hdm2 antibodies, SMP14 and Hdm2-323 (Sigma; M7815), which together recognize all the Hdm2 mutants (Figure 2B). All the Hdm2 deletion mutants tested were co-immunoprecipitated with c-Abl, but to varying degrees. A marked reduction in the level of interaction was observed with the Hdm2 6–339 mutant lacking the C-terminus (lane 10), while the Hdm2Δ222– 437 mutant bound with higher affinity (lane 9). This supports a role for the ring-finger domain of Hdm2 in this interaction. However, other Hdm2 mutants lacking the ring-finger domain (Hdm2Δ441–491 and Hdm2 1–200) still bind c-Abl well (lanes 8 and 11), supporting the presence of an additional binding site within the N-terminal half of Hdm2. The reduced binding of Hdm2Δ1–50 mutant to c-Abl (lane 4), relative to the other deletions at the N-terminus (lanes 5 and 6), may support a role for the first 50 residues of Hdm2 in this interaction. Furthermore, the stronger interaction of c-Abl with the Hdm2 1–200 mutant than with Hdm2 6–339 (lane 11 versus 10) may support the involvement of the first five residues of Hdm2 in this interaction. Thus, it appears that the extreme N- and C-termini of Hdm2 are both important for binding c-Abl.

Fig. 2. c-Abl binds Hdm2 at multiple sites. (A) A schematic representation of the Hdm2 protein and the different deletion mutants of Hdm2 used in this study. Solid lines represent the encoded cDNA of the mutants, while the broken lines denote internal deletions. (B) 293 cells were transfected with expression plasmids for His-tagged c-Abl alone (5 µg, lane 1), Hdm2 alone (5 µg, lane 2), or His-c-Abl together with Hdm2 wild-type or deletion mutants as indicated (5 µg each, lanes 3–11). His-c-Abl was isolated from the cell extracts using nickel resin, and the protein complex was subjected to western blotting using SMP14 and 323 anti-Hdm2 antibodies (B). (C) The levels of Hdm2 in the transfected cells were determined prior to immunoprecipitation using SMP14 and 323. (D) The level of c-Abl was monitored with anti-c-Abl antibody (ABL-148).
Both c-Abl and Mdm2 have nuclear localization and nuclear export signals (NLS and NES, respectively). Since both Mdm2 and c-Abl affect the subcellular distribution of p53 (Roth et al., 1998; Boyd et al., 2000; Geyer et al., 2000; Vogt Sionov et al., 2001a), it was of interest to define whether the interaction between Mdm2 and c-Abl is confined to a particular compartment within the cell. For this purpose, the effect of the subcellular localization of c-Abl on its interaction with Hdm2 was tested by comparing the binding of Hdm2 to NLS-defective c-Abl (mutant ‘K’) versus c-Abl that is distributed both in the nucleus and the cytoplasm (mutant ‘A’) (Wen et al., 1996). The distribution of the latter mutant ‘A’ is similar to that of wild-type c-Abl. 293 cells were transfected with an expression plasmid for hdm2, either alone or together with c-abl ‘A’ or ‘K’. Hdm2 bound efficiently to c-Abl mutant ‘A’, but only weakly to c-Abl mutant ‘K’ (Figure 3AI, lanes 2 and 3), suggesting that the interaction occurs primarily, but not exclusively, in the nucleus.
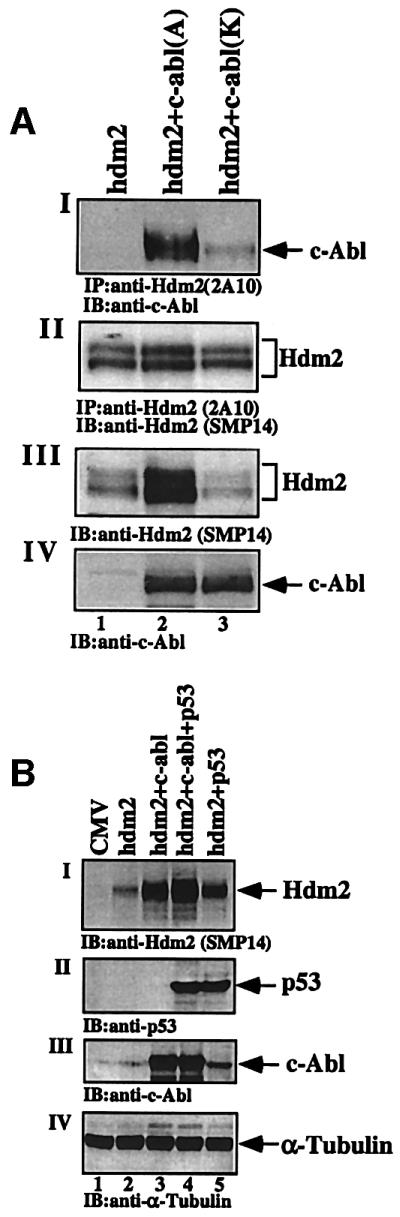
Fig. 3. c-Abl binds Hdm2 in the nucleus and enhances its accumulation in a p53-independent manner. (A) 293 cells were transfected with the expression plasmid for Hdm2 alone (5 µg, lane 1), or together with c-abl mutants (5 µg) that are expressed in the cytoplasm (c-abl K; lane 3) or in both the nucleus and cytoplasm (mutant ‘A’; lane 2). Cell extracts were subjected to immunoprecipitation using anti-Hdm2 antibody (2A10), followed by blotting with anti-c-Abl antibody (ABL-148) (I) or with anti-Hdm2 antibody (SMP14) (II). The levels of Hdm2 and c-Abl in the transfected cells were determined by western blotting with anti-Hdm2 (SMP14) (III) or anti-c-Abl (IV), respectively. (B) H1299 cells were transfected with the indicated expression plasmids Hdm2 (3 µg), c-Abl (4 µg) and p53 (1 µg). Twenty hours post-transfection, cell extracts were subjected to western blotting using anti-Hdm2 (I), anti-p53 (II) and anti-c-Abl (ABL-148) (III). The amount of extracts loaded was monitored with anti-α-tubulin (IV).
This binding assay also revealed that c-Abl increases the steady-state level of Hdm2 (Figure 1AII, lane 2 versus 3). Since c-Abl stabilizes and activates p53 by neutralizing the inhibitory effects of Mdm2 (Vogt Sionov et al., 1999, 2001a), it is plausible that this action of c-Abl on Mdm2 expression is mediated through the induction of p53 transcriptional activation of the mdm2 gene. Therefore, the requirement for p53 for the elevation of Mdm2 expression by c-Abl was evaluated in H1299 cells lacking p53 expression. H1299 cells were transfected with expression plasmids for hdm2, either alone or together with c-abl or p53, or all three plasmids together. The steady-state level of Hdm2 was determined by western blot analysis using the anti-Hdm2 antibody SMP14 (Figure 3BI). The data show that accumulation of Hdm2 in response to c-Abl occurs efficiently also in the absence of p53 (Figure 3B, compare lanes 2 and 3). This effect is further enhanced in the presence of p53 (lane 4). Therefore, p53 contributes to, but is not essential for, the elevation of Hdm2 by c-Abl.
c-Abl kinase activity is important for the stabilization of p53 under normal conditions and in response to DNA damage
We have recently shown that the presence of endogenous c-Abl is important for maintaining the basal levels of p53, and for the maximal and efficient stabilization of p53 in response to DNA damage (Vogt Sionov et al., 2001a). To evaluate the role of c-Abl kinase activity in this process, the expression of p53 was compared between wild-type fibroblasts and c-Abl-deficient fibroblasts, either non-manipulated or reconstituted with a kinase-defective c-Abl mutant (K290R; cell lines K8 and K10). To evaluate the role of the c-Abl kinase activity in the basal expression levels of p53, equal numbers of untreated cells of each fibroblastic cell type were harvested and p53 protein levels were determined by western blot analysis. Consistent with our recent findings (Vogt Sionov et al., 2001a), the basal levels of p53 expression in c-Abl-deficient fibroblasts were significantly lower than those in normal cells expressing physiological levels of c-Abl (Figure 4A). Interestingly, the expression levels of p53 in cells expressing kinase-defective c-Abl were significantly lower than in wild-type cells, but slightly higher than in the c-Abl-negative cells (Figure 4A). Identical results were obtained when c-Abl-deficient fibroblasts reconstituted with wild-type c-Abl at physiological levels (Vogt Sionov et al., 1999) were used instead of wild-type fibroblasts (data not shown). Thus, the kinase activity of c-Abl is important for maintaining the basal levels of p53 expression.

Fig. 4. The kinase activity of c-Abl is important for the stabilization of p53 under basal and stress conditions. (A) Cell extracts were prepared from normal primary fibroblasts (c-Abl positive, lane 1), from c-Abl null fibroblasts (c-Abl negative, lane 2), or from two fibroblastic cell lines that were reconstituted with a c-Abl kinase-defective mutant (K8, lane 3; K10, lane 4). Extracts were subjected to western blotting using anti-p53 antibody (CM5; I) or anti-c-Abl (ABL-148; II). Long exposure of the film was required in order to detect p53 in the c-Abl-negative cells (lane 2), which resulted in an apparent high expression level of p53 in normal cells (lane 1) (I). The amounts of protein loaded were monitored by blotting with anti-α-tubulin (III). (B) Fibroblastic cell lines, either negative for c-Abl (lanes 5–8), or reconstituted with either wild-type c-Abl (c-Abl positive; lanes 1–4) or kinase-defective c-Abl (c-AblK– K8; lanes 9–12), were either untreated (lanes 1, 5, 9) or exposed to γ-radiation (10 Gy) and harvested at the times indicated. The expression levels of p53 were monitored by blotting with anti-p53 antibody (CM5). The amount of protein loaded was determined with anti-histone 2B (II). (C) The three fibroblastic cell lines were treated with mitomycin C (1 µg/ml; lanes 1–3) or doxorubicin (10 µg/ml; lanes 4–6) for 6 h. The expression levels of p53 were monitored as in (A)(I), and the amount of protein loaded was determined with anti-α-tubulin (II). (D) Northern blot analysis of c-Abl null primary fibroblasts (c-Abl negative; lanes 1–4) or normal primary fibroblasts (c-Abl positive; lanes 5–8). Cells were either untreated (NT) or exposed to the proteasome inhibitor ALLN (150 µM for 2 h), mitomycin C (Mito-C; 3 µg/ml for 6 h) or doxorubicin (3 µg/ml for 6 h). Total mRNA was prepared from each sample and subjected to northern blot analysis. The levels of p53 mRNA were determined by probing with 32P-labeled p53 cDNA, and the amount of mRNA loaded was monitored by reprobing with 32P-labeled cDNA for GAPDH.
Next, we asked whether the kinase activity of c-Abl is also important for the accumulation of p53 in response to DNA damage. For this purpose, fibroblasts lacking c-Abl and fibroblastic cell lines reconstituted with wild-type or kinase-defective c-Abl were exposed to different types of DNA-damaging agents. Cells were exposed to γ-radiation (10 Gy) and harvested at the indicated times. Further, cells were incubated with mitomycin C (1 µg/ml) or doxorubicin (10 µg/ml) for 6 h. At the indicated times after treatment, cells were harvested and p53 expression levels were determined. As shown in Figure 4, the stabilization of p53 in response to γ-radiation (B), mitomycin C or doxorubicin (C) was greater in cells expressing wild-type c-Abl than in cells lacking c-Abl or expressing a kinase-defective mutant. Taken together, these results demonstrate a clear role for the kinase activity of c-Abl in the stabilization of p53 in response to exposure to various DNA damaging agents, and in the maintenance of p53 expression under non-stressed conditions.
Since c-Abl has been implicated in transcription through the phosphorylation of RNA polymerase II (Baskaran et al., 1999), we examined whether the basal or stress-induced elevation of p53 expression levels by wild-type c-Abl results from enhanced transcription of endogenous p53 mRNA. c-Abl-negative and c-Abl-reconstituted fibroblasts were either not treated or exposed to the proteasome inhibitor ALLN (150 µM for 2 h), mitomycin C (3 µg/ml for 6 h) or doxorubicin (3 µg/ml for 6 h). Total mRNA was prepared from treated and untreated cells, and the expression of endogenous p53 was measured by northern blot analysis using a p53-specific cDNA probe. The amounts of mRNAs loaded were monitored by the expression levels of GAPDH. As shown in Figure 4D, the levels of p53 mRNAs were similar in cells lacking or expressing wild-type c-Abl irrespective of the treatment. Therefore, the presence of c-Abl does not increase the amount of p53 mRNA in untreated cells or in cells exposed to DNA damage. This strongly suggests that the effect of c-Abl on p53 occurs at the protein level.
c-Abl phosphorylates Mdm2 in vivo
The physical and functional link between c-Abl and Mdm2 encouraged us to examine whether Mdm2 is a direct target for phosphorylation by c-Abl. This possibility is attractive, particularly since c-Abl tyrosine kinase activity is induced in response to DNA damage (Kharbanda et al., 1995), and the kinase activity of c-Abl is required for the maximal accumulation of p53 in response to stress (Figure 4). The ability of c-Abl to phosphorylate Hdm2 in vivo was examined in 293 cells transfected with expression plasmids for hdm2, either alone or together with wild-type c-abl or kinase-defective c-abl. Twenty-four hours post-transfection, cells were harvested and Hdm2 was immunoprecipitated using anti-Hdm2 antibody (SMP14), followed by immunoblot analysis using anti-phosphotyrosine antibody (4G10; Upstate Biotechnology). Tyrosine phosphorylation of Hdm2 was observed in the presence of wild-type c-Abl but not with the c-Abl kinase-defective mutant (Figure 5AI, lanes 3 and 4). Similar results were obtained in 293 cells transfected with expression plasmids for glutathione S-transferase (GST)– c-Abl fusion proteins. The kinase-defective mutant (GST–c-AblK–) did not phosphorylate Hdm2, while an SH3-deficient mutant (GST–c-AblΔSH3) enhanced the phosphorylation of Hdm2 at least to the same extent as wild-type c-Abl (GST–c-Abl) (Figure 5B). These results demonstrate that c-Abl can phosphorylate Hdm2 on tyrosine in vivo.

Fig. 5. c-Abl phosphorylates Hdm2 in vivo. (A) 293 cells were transfected with the indicated expression plasmids (5 µg each). Twenty-four hours later, cell extracts were subjected to immunoprecipitation using SMP14, followed by western blotting with 4G10 (I) or SMP14 (II). The levels of c-Abl in the transfected cells were monitored with anti-c-Abl antibody (ABL-148) prior to immunoprecipitation (III). (B) 293 cells were transfected with expression plasmids for Hdm2 together with GST fused to wild-type c-Abl (lane 1) or to SH3-deletion mutant (lane 2) or to a kinase-defective mutant (lane 3). The phosphorylation of Hdm2 (I) and the expression levels of Hdm2 (II) and c-Abl (III) were monitored as described in (A).
c-Abl phosphorylates Hdm2 on Y394
Hdm2 has 14 tyrosine residues that are dispersed along the protein. A search for the relevant tyrosine phosphorylation sites suggested that Tyr394 (Y394) was an attractive candidate. The sequence encompassing Y394 shares the highest homology with the phosphorylation consensus site of c-Abl (Till et al., 1999), and is highly conserved among mice, humans and hamsters (Figure 6A; Momand et al., 2000). Intriguingly, the Y394 site is very similar to the phosphorylation site of c-Abl in p73 (Figure 6A; Yuan et al., 1999). To test whether Y394 is a phosphorylation site, this tyrosine was substituted by phenylalanine (F), to prevent phosphorylation. The effect of this substitution on the phosphorylation of Hdm2 by c-Abl was measured in vivo and in vitro. For the in vivo assay, 293 cells were transfected with expression plasmids for wild-type hdm2 or hdm2 F394 alone, or together with an expression plasmid for c-abl. Hdm2 phosphorylation was examined by immunoprecipitation of Hdm2 followed by immunoblotting with anti-phosphotyrosine antibody (4G10). The Y394F substitution reduced significantly the extent of Hdm2 phosphorylation, as compared with wild-type Hdm2 (Figure 6BI, lanes 3 and 5). This reduction supports the notion that Y394 is a major phosphorylation site for c-Abl. However, the data leave open the possibility that c-Abl may phosphorylate Hdm2 on additional site(s). One potential site is Y489 (LTYFP). However, unlike F394, the Hdm2 F489 substitution mutant was effectively phosphorylated by c-Abl in vivo (Figure 6BI, lanes 3 and 7), arguing that Y489 is not a relevant target site of c-Abl.

Fig. 6. c-Abl phosphorylates Hdm2 on Tyr394 in vivo and in vitro. (A) Alignment of the amino acid sequence around Y394 showing the sequence homology in the Mdm2 sequence among three species and to the c-Abl phosphorylation site in p73 (Y99). The c-Abl consensus phosphorylation site (Till et al., 1999) is shown at the bottom. Gray boxes represent conserved amino acids and lines point to position substitution of conserved residues. (B) 293 cells were transfected with the indicated expression plasmids (5 µg each). Phosphorylation of Hdm2 (I), the levels of Hdm2 after (II) and before immunoprecipitation (III), and the levels of c-Abl (IV) were determined as described in Figure 5A. (C) 293 cells were transfected with expression plasmid for c-abl (5 µg) together with Hdm2 F394 (lane 2) or wild-type Hdm2 (lane 3). Hdm2 was immunoprecipitated using a mixture of antibodies (SMP14, 2A9 and 2A10), followed by immunoblotting with anti-phospho-Y394 antibodies (I) or anti-Hdm2 antibody (SMP14) (II). The position of phosphorylated Hdm2 and total Hdm2 is indicated. (D) SJSA cells were either non-treated (NT) or treated with the tyrosine phosphatase inhibitor sodium orthovanadate (Na3VO4) alone (lane 3) or together with H2O2 (lane 2) for 20 min. The detection of Hdm2 phosphorylated on Y394 was as described in (C). (E) An in vitro phosphorylation assay of Hdm2 by c-Abl. GST fused to full-length wild-type Hdm2 (lanes 1–4) or to N-terminally truncated mutants (Hdm2ΔN, lanes 5–8) bearing a Y394 (lanes 1, 2, 5 and 6) or an F394 substitution (lanes 3, 4, 7 and 8) were incubated with either wild-type c-Abl (lanes 1, 3, 5 and 7) or a kinase-defective mutant (lanes 2, 4, 6 and 8). The phosphorylation of Hdm2 on Y394 was determined by western blot analysis using anti-phospho-Y394 polyclonal antibodies (I). The amount of GST–Hdm2 proteins was determined with anti-GST antibody (II) and the levels of c-Abl were monitored using anti-c-Abl antibody (ABL-148) (III). Note, the phosphorylated form of c-Abl migrates slower on SDS–PAGE.
To substantiate the phosphorylation of Hdm2 on Y394, we applied an anti-phospho-Y394 polyclonal antibody raised against a phosphorylated Y394-bearing peptide. For the in vivo assay, two experimental approaches were used. In the first, 293 cells were transfected with expression plasmids for c-Abl together with either wild-type Hdm2 or Hdm2 F394. Twenty-four hours after transfection, Hdm2 was immunoprecipitated from the cell extracts using a mixture of anti-Hdm2 antibodies (SMP14, 2A9 and 2A10), followed by western blot analysis using anti-phospho-Y394 polyclonal antibodies. As shown in Figure 6C, specific phosphorylation was detected with anti-phospho-Y394 only in cells expressing wild-type Hdm2 (lane 3) but not Hdm2 F394 (lane 2). Furthermore, to examine whether endogenous Hdm2 is also phosphorylated on Y394, SJSA-1 osteosarcoma cells were either non-treated, or treated with the tyrosine phosphatase inhibitor sodium orthovanadate alone or together with H2O2, which prevents dephosphorylation of Hdm2 and activates c-Abl (Sun et al., 2000). Endogenous Hdm2 was immunoprecipitated as in 293 cells, and the immune complex was subjected to western blot analysis using anti-phospho-Y394 antibodies. Specific phosphorylation of endogenous Hdm2 was detected only in cells treated with orthovanadate and H2O2 (Figure 6D, lane 2). These results support the phosphorylation of Hdm2 on Y394 in vivo, and further demonstrate that phosphorylation of endogenous Hdm2 occurs at physiological levels of c-Abl under conditions that trigger its kinase activity (Sun et al., 2000).
For the in vitro assay, wild-type or a kinase-defective c-Abl was isolated by immunoprecipitation from 293 cells transfected with the respective expression plasmids. The kinase activity of c-Abl was verified using bacterially derived GST–c-Crk (120–225), or GST–c-Crk (120–212), the latter lacking Tyr221 (Shafman et al., 1997). Wild-type c-Abl, but not the kinase-defective mutant, phosphorylated c-Crk (120–225) but not c-Crk (120–212) (data not shown). To test the in vitro phosphorylation of Hdm2 on Y394, GST was fused to full-length Hdm2 or to the C-terminus of Hdm2 (Hdm2ΔN; amino acids 358–493), with either Y or F at position 394. The proteins were expressed and purified from bacteria, and subjected to a phosphorylation assay using wild-type or a kinase-defective c-Abl immunoprecipitated from transfected 293 cells. Reaction products were subjected to immunoblotting using anti-phospho-Y394 antibodies. A significant and specific phosphorylation of wild-type Hdm2, both full length and ΔN, but not their equivalent F394 mutants, was observed (Figure 6E, lanes 1 and 5 versus lanes 3 and 7). Taken together, these results demonstrate the phosphorylation of Hdm2 on Y394 by c-Abl both in vivo and in vitro.
Modifications of the ATM phosphorylation site at Ser395 do not affect Y394 phosphorylation by c-Abl
Our finding of Hdm2 phosphorylation by c-Abl at Y394 is particularly striking as this site is adjacent to the ATM-mediated phosphorylation site, Ser395 (S395) (Khosravi et al., 1999; Maya et al., 2001). This raised the question whether phosphorylation of S395 by ATM affects the phosphorylation of Y394 by c-Abl. To address this question, the effect of modifications of S395 on the phosphorylation of Y394 was measured in vitro and in vivo.
The in vitro assay was carried out with purified His-tagged c-Abl, comparing wild-type with the kinase-defective c-Abl mutant. The phosphorylation of four different GST-fused Hdm2 proteins by c-Abl was compared. These include wild-type Hdm2 and Hdm2 F394, and two Hdm2 mutants bearing substitutions at S395: serine to alanine substitution (Hdm2 S395A) to prevent phosphorylation at S395, and serine to aspartate (Hdm2 S395D) to mimic the phosphorylated state of S395 (Maya et al., 2001). Following the in vitro phosphorylation assay, the reaction products were subjected to immunoblot analysis using anti-phospho-Y394 antibodies. A strong and specific phosphorylation of wild-type Hdm2, Hdm2 S395A and Hdm2 S395D was observed in the presence of wild-type c-Abl, but not with the kinase-defective mutant (Figure 7A). Therefore, the phosphorylation at Y394 by c-Abl is independent of the modifications at S395, and neither the lack nor the mimicking of S395 phosphorylation prevented phosphorylation at Y394 (lanes 1, 3 and 4). If anything, the S395A substitution slightly enhanced Y394 phosphorylation (lanes 1 and 4). Nevertheless, neither substitution at S395 affected the reactivity of the anti-Y394 antibody with Hdm2 in the absence of phosphorylation by c-Abl (lanes 5–8).

Fig. 7. Modifications at S395 do not affect phosphorylation of Y394 by c-Abl. (A) An in vitro phosphorylation of Hdm2 mutants by c-Abl. GST fusions to wild-type Hdm2, Hdm2 F394, Hdm2 395D and Hdm2 395A were incubated with either His-tagged wild-type c-Abl (lanes 1–4) or a kinase-defective mutant (lanes 5–8). The phosphorylation of Y394 was determined by western blot analysis using anti-phospho-Y394 antibodies (I). The amount of GST–Hdm2 proteins loaded was monitored with anti-Hdm2 antibody (2A9). (B) An in vivo phosphorylation assay in 293 cells transfected with expression plasmids for c-abl alone (5 µg; lane 5) or together with either wild-type Hdm2 (lane 1) or Hdm2 F394 (lane 2), or Hdm2 S395A (lane 3) or Hdm2 S395D (lane 4) (6 µg of each mutant). Twenty hours post- transfection, cells were treated with sodium orthovanadate for 1 h. From triplicates of each plasmid combination, Hdm2 was immunoprecipitated with a mixture of anti-Hdm2 antibodies (SMP14, 2A9, 2A10 and 4B11), followed by immunoblotting with anti-phospho-Y394 (I). The amount of Hdm2 loaded was determined with SMP14 (II). The amount of Hdm2 and c-Abl proteins expressed in the transfected cells was also determined before the immunoprecipitation by western blotting using anti-Hdm2 (2A9; III) or anti-c-Abl (ABL-148; IV) antibodies, respectively.
The effect of S395 modifications on the phosphorylation of Y394 was also examined in vivo. 293 cells were transfected with expression plasmids for wild-type hdm2, hdm2 F394, hdm2 S395A and hdm2 S395D together with an expression plasmid for c-abl. Twenty-four hours after transfection, cells were treated with sodium orthovanadate for 1 h. The phosphorylation of Hdm2 on Y394 was examined by immunoprecipitation of Hdm2 followed by immunoblotting with anti-phospho-Y394 antibodies. As in the in vitro phosphorylation assay, Y394 phosphorylation was specific for Y394 and was unaffected to any significant level by the modifications at S395 (Figure 7B). Taken together, these results demonstrate that phosphorylation of Hdm2 at Y394 by c-Abl occurs independently of ATM-mediated phosphorylation at S395. Furthermore, modifications at S395 do not affect the reactivity or the specifity of the anti-phospho-Y394 for the phosphorylated Y394 site.
Substitution of Tyr by Phe at position 394 enhances the ability of Hdm2 to promote p53 degradation
The demonstration that the kinase activity of c-Abl contributes to the accumulation of p53, and that Hdm2 is phosphorylated by c-Abl on Y394, begged the question as to whether phosphorylation of this site modulates the regulation of p53 by Hdm2. To address this question, the effect of substitution of Tyr by Phe at position 394 on the ability of Hdm2 to promote p53 degradation was assessed. For this purpose, p53/mdm2 double knockout (2KO) fibroblasts were transfected with the expression plasmid for wild-type p53, either alone or together with expression plasmids for either wild-type Hdm2 or Hdm2 F394. As seen in Figure 8A, Hdm2 F394 was significantly more efficient than wild-type Hdm2 in promoting the degradation of p53. This difference was observed despite equal expression levels of Hdm2 F394 as compared with wild-type Hdm2 (Figure 8B). These results strongly support the notion that the Y394 substitution, which prevents the phosphorylation of Hdm2 on Y394, enhances the ability of Hdm2 to promote p53 degradation.
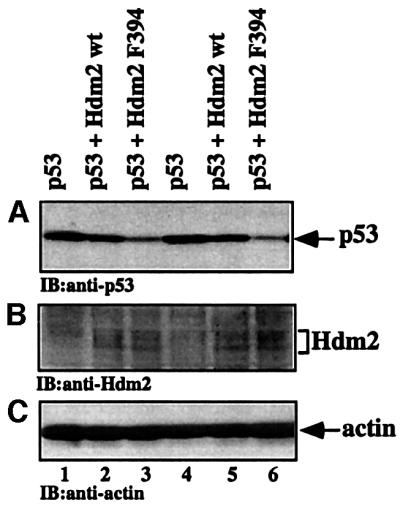
Fig. 8. Enhanced degradation of p53 by the Hdm2 F394 mutant. p53/mdm2 2KO fibroblasts were transfected with expression plasmids for wild-type p53 (25 ng), either alone (lanes 1 and 4), or together with expression plasmids for either wild-type Hdm2 (lanes 2 and 5) or Hdm2 F394A (lanes 3 and 6; 500 ng each). The experiment was carried out in replicate, from which a representative result is shown. The expression levels of p53 (A) and Hdm2 (B) were monitored by western blot analysis using anti-p53 (PAb1801 and DO1) and anti-Hdm2 (SMP14, SMP15, 2A9) antibodies, respectively. (C) The amount of protein loaded was monitored with anti-actin.
Substitution of Tyr by Phe at position 394 enhances the ability of Hdm2 to inhibit the transcriptional activity of p53
The effect of the substitution of Tyr by Phe at position 394 on the enhanced degradation of p53 encouraged us to examine the effect of the substitution on the inhibition of p53 transcriptional activity by Hdm2 (Chen et al., 1996). For this purpose, H1299 cells were transfected with wild-type p53 expression plasmid, either alone or together with expression plasmids for either wild-type Hdm2 or Hdm2 F394. The transcriptional activity of p53 was monitored by measuring the activity of the luciferase gene under the control of the cyclin G promoter. The inhibition of p53 transcriptional activity by Hdm2 F394 was more efficient than that by wild-type Hdm2. While the difference was moderate, it was highly reproducible in three independent experiments using three to seven replicate samples for each plasmid combination. Figure 9 represents one such experiment, carried out in seven replicate samples. These results demonstrate that the Y394 substitution enhances the ability of Hdm2 to inhibit p53 transcriptional activity.

Fig. 9. Enhanced inhibition of p53 transcriptional activity by Hdm2 F394. H1299 cells were transfected with expression plasmids for wild-type p53 (30 ng; black bar), either alone or together with expression plasmids for wild-type Hdm2 (400 ng; dark gray bar) or Hdm2 F394 (400 ng; light gray bar). The transcriptional activity of p53 was monitored by measuring the activity of the luciferase gene under the control of the cyclin G promoter (white bar). The results from three independent experiments are shown, each of which represents an average of three to seven independent transfections for each combination of expression plasmids. The standard error is given.
Substitution of Tyr by Phe at position 394 enhances the ability of Hdm2 to inhibit p53-mediated apoptosis
Hdm2 inhibits the apoptotic activity of p53 (Chen et al., 1996; Haupt et al., 1996), even under conditions where cells are matched for equal expression levels of p53 (Haupt et al., 1996; Unger et al., 1999). To examine whether the Tyr to Phe substitution at position 394 affects this inhibitory activity of Hdm2, H1299 cells were transfected with a wild-type p53 expression plasmid, either alone or together with increasing amounts of expression plasmid for wild-type Hdm2 or Hdm2 F394. Apoptosis was measured 55 h later (Haupt et al., 1996). As with the transcriptional activity, here too one could observe a mild but consistent increase in the inhibitory effect of Hdm2 F394 relative to wild-type Hdm2, at both the low and high levels of Hdm2 expression. The results of a representative experiment, carried out in triplicate, are summarized in Figure 10A. This difference in the inhibitory activity between wild-type Hdm2 and Hdm2 F394 was maintained at different ratios of Hdm2 and p53 (data not shown). A more robust effect of the Y394 to Phe substitution on the inhibitory effect of Hdm2 was revealed in Saos-2 cells (Figure 10B) transfected with the expression plasmid for wild-type p53, either alone or together with expression plasmids for wild-type Hdm2 or Hdm2 F394. While wild-type Hdm2 reduced the apoptotic activity of p53 by 42%, the same amount of Hdm2 F394 reduced this activity by 65% (Figure 10B). Taken together, these results demonstrate that the substitution of Y394 by F394 increases the inhibitory activity of Hdm2 on p53 apoptotic function.

Fig. 10. Hdm2 F394 inhibits p53-mediated apoptosis more efficiently than wild-type Hdm2. (A) H1299 cells were transfected with expression plasmids for wild-type p53 (3 µg, black bar), either alone or together with increasing amounts (4 µg, dark gray bar; 8 µg light gray bar) of expression plasmids for wild-type Hdm2 or Hdm2 F394. At 55 h post-transfection, floating and adherent cells were collected, fixed and stained for p53 expression. Cells were simultaneously stained for DNA with propidium iodide. Stained cells were subjected to flow cytometric analysis as described previously (Haupt et al., 1996). Cells transfected with p53 were collected and their cell cycle distribution was determined. The sub-G1 region represents apoptotic subpopulation (Haupt et al., 1996). The results of one representative experiment, in triplicate, are summarized. (B) The same assay as in (A), but in Saos-2 cells. In this experiment, 3 µg of expression plasmid for p53 and 5 µg of expression plasmid for Hdm2 were used. The result of one representative experiment, in triplicate, is shown. The standard error is also given.
Discussion
The p53 protein is subject to tight positive and negative regulation. Under normal conditions and during recovery from stress, p53 is very labile and presumably inactive. This regulation is mediated primarily by the Mdm2 protein. By binding to p53, Mdm2 overcomes its transcriptional activity and catalyzes its ubiquitylation and subsequent degradation. Through these combined effects, Mdm2 achieves the efficient and rapid inhibition of p53-mediated signaling. However, under stress conditions, p53 has to be freed from these restrictive effects of Mdm2 in order to exert its growth-inhibitory functions. This relief of p53 from Mdm2-dependent inhibition involves multiple mechanisms depending on the type of stress signal imposed. For example, in response to oncogenic stimuli, the ubiquitylation and subsequent degradation of p53 by Mdm2 are inhibited by ARF (reviewed in Sherr and Weber, 2000).
In response to DNA damage, the inhibitory effect of Mdm2 is relieved in part by phosphorylation of p53. For instance, the phosphorylation of p53 on Ser20 renders it more resistant to the destabilizing activity of Mdm2 (Chehab et al., 1999; Shieh et al., 1999; Unger et al., 1999) and also inhibits its nuclear export (Zhang and Xiong, 2001). However, under certain conditions, the phosphorylation of p53 does not appear to exert a pronounced effect on its stability (Ashcroft et al., 1999; Blattner et al., 1999; Dumaz and Meek, 1999), lending support to the notion that additional mechanisms are involved in this regulation. An attractive candidate for DNA damage-induced modification is Mdm2 itself. Indeed, Mdm2 is phosphorylated on multiple sites (Fakharzadeh et al., 1991; Hay and Meek, 2000). Mdm2 is phosphorylated by casein kinase-2 (CK2) (Guerra et al., 1997; Gotz et al., 1999; Hjerrild et al., 2001) and by DNA-dependent protein kinase (DNA-PK); the phosphorylation by the latter prevents p53–Mdm2 interaction (Mayo et al., 1997). The in vivo relevance of these phosphorylation sites awaits future studies. Mdm2 is also phosphorylated by the PI3-kinase/Akt pathway, and this phosphorylation promotes the nuclear entry of Mdm2 and subsequently the inhibition and degradation of p53 (Mayo and Donner, 2001; Zhou et al., 2001; Gottlieb et al., 2002). Of note, the exposure of cells to DNA damage triggers the in vivo phosphorylation of Hdm2 by ATM (Khosravi et al., 1999). Phosphorylation of Hdm2 on S395 by ATM impairs the ability of Hdm2 to promote the degradation of p53 and to promote its nuclear export (Maya et al., 2001). This is consistent with the role of ATM in the stabilization of p53 in response to ionizing radiation (Kastan et al., 1992). Thus, phosphorylation of Hdm2 can affect the autoregulatory feedback loop between Hdm2 and p53.
A pivotal phosphorylation target of ATM is the c-Abl tyrosine kinase (Baskaran et al., 1997; Shafman et al., 1997). We have recently demonstrated an important role for c-Abl in the accumulation of p53 in response to DNA damage (Vogt Sionov et al., 2001a). Taken together with the ability of c-Abl to abrogate the inhibitory effects of Mdm2 (Vogt Sionov et al., 1999, 2001a), this raised the question to what degree this action of c-Abl is mediated by direct physical and functional interaction with Hdm2. We found that c-Abl and Hdm2 physically interact in vivo and in vitro (Figure 1). Importantly, the kinase activity of c-Abl contributes to the maximal accumulation of p53 in response to DNA damage. Moreover, we show that this activity of c-Abl is also required for the regulation of p53 expression at the basal level.
The kinase activity of c-Abl is essential for the induction of growth arrest and apoptosis by c-Abl in response to different stress signals (Sawyers et al., 1994; Goga et al., 1995; Wen et al., 1996; Yuan et al., 1996b, 1997; Huang et al., 1997). These biological activities of c-Abl, at least to some extent, are p53 dependent (Goga et al., 1995; Wen et al., 1996; Yuan et al., 1997). Our findings implicating a role for the kinase activity of c-Abl in the cooperation with p53 were surprising in view of previous studies suggesting that this activity of c-Abl is not essential for this cooperation (Goga et al., 1995; Yuan et al., 1996a,b; Vogt Sionov et al., 1999).
How can this apparent contradiction be resolved? One possible explanation is based on differences between the experimental systems used. In previous studies, the c-Abl kinase-defective mutant was either transiently overexpressed (Goga et al., 1995; Vogt Sionov et al., 1999), or constitutively expressed in stable cell lines (Yuan et al., 1996b, 1997). In both cases, this mutant was introduced into cells that express endogenous wild-type c-Abl. In these systems, it was assumed that the kinase-defective mutant of c-Abl neutralizes the kinase activity of the endogenous protein in a dominant-negative fashion (Sawyers et al., 1994). However, it remains possible that in these experimental systems the p53 protein is kept active by residual kinase activity of endogenous c-Abl. On the other hand, in our experimental system, the c-Abl kinase-defective mutant was reconstituted in c-Abl null cells. Another possibility is that c-Abl activates p53 by multiple mechanisms. It is possible that when c-Abl is expressed at high levels, a single pathway may suffice to achieve the maximum effect of c-Abl on p53 activation. For example, c-Abl interacts with the C-terminus of p53 and stabilizes its interaction with DNA (Nie et al., 2000), and this interaction is independent of the kinase activity (Yuan et al., 1996a). This interaction is also likely to stabilize the tetrameric form of p53 and consequently hinder nuclear export and/or interfere with the ubiquitylation of p53 on lysines residing within the C-terminus (Nakamura et al., 2000; Rodriguez et al., 2000). These two explanations are not mutually exclusive.
Importantly, c-Abl phosphorylates Hdm2 on multiple tyrosine residues both in vivo and in vitro. We propose that this phosphorylation underlies an important mechanism by which c-Abl neutralizes the inhibitory effect of Hdm2 on p53. We characterized the phosphorylation of Hdm2 on Y394, which fits the c-Abl consensus site (Till et al., 1999) and shares high homology with the c-Abl phosphorylation site (Y99) in p73α (Yuan et al., 1999). To evaluate the functional role of Y394 phosphorylation, we substituted tyrosine by phenylalanine at position 394 in order to prevent phosphorylation while imposing minimal structural impact on the Hdm2 protein. This substitution enhanced the ability of Hdm2 to promote p53 degradation, as well as its ability to inhibit the transcriptional activity of p53 and its apoptotic activity. Therefore, the prevention of Hdm2 phosphorylation on Y394 increases the inhibitory activities of Hdm2 on p53 in multiple experimental systems. Our results support the model whereby phosphorylation of Hdm2 on Y394 is important for the regulation of Hdm2 function by c-Abl. The characterization of other c-Abl phosphorylation site(s) in Hdm2 and their contribution to the regulation of Hdm2 activities require further study.
The enhanced ability of the Hdm2 F394 mutant to promote the degradation of p53 is consistent with our finding that c-Abl impairs the ability of Hdm2 to promote the ubiquitylation of p53 and inhibit its nuclear export (Vogt Sionov et al., 2001a). These two effects of Hdm2 on p53 may be linked, where ubiquitylation precedes nuclear export (Boyd et al., 2000; Geyer et al., 2000). However, ubiquitylation assays of p53 in vivo and in vitro did not reveal major differences between wild-type Hdm2 and Hdm2 F394 (data not shown). Interestingly, similar effects were seen when the adjacent phosphorylation site, S395, was substituted by alanine (Maya et al., 2001). Thus, Hdm2 F394 may enhance the degradation of p53 without affecting the extent of its ubiquitylation. It is of note that Argentini et al. (2001) described Hdm2 mutants that are impaired in the degradation of p53, but not in its ubiquitylation. Such mutants may be defective in their ability to promote the nuclear export of p53 and/or directing p53 to the 26S proteasome.
The finding of two adjacent phosphorylation sites (Y394 and S395) being targeted by two types of protein kinases (Maya et al., 2001; this study) is intriguing. Since ATM acts upstream of c-Abl in the cellular response to DNA damage (Baskaran et al., 1997; Shafman et al., 1997), we asked whether the phosphorylation of Y394 by c-Abl requires the prior phosphorylation of Hdm2 on S395 by ATM. Phosphorylation assays, both in vivo and in vitro, revealed that the phosphorylation of Hdm2 on Y394 by c-Abl occurs independently of the phosphorylation of S395 by ATM (Figure 7). Nevertheless, it is tempting to speculate that ATM and c-Abl cooperate in the regulation of Hdm2 in response to DNA damage. This speculation is particularly attractive because, in response to double-strand DNA breaks, ATM binds, phosphorylates and activates c-Abl (Baskaran et al., 1997; Shafman et al., 1997). Furthermore, in response to ionizing radiation, both ATM and c-Abl play important roles in the accumulation of p53 (Kastan et al., 1992; Vogt Sionov et al., 2001a). The phosphorylation of Hdm2 by these two kinases may occur simultaneously in a single complex, or sequentially. Either way, the phosphorylation of these two adjacent residues has an inhibitory effect on Hdm2 activity, thereby contributing to the rapid accumulation of p53 in response to DNA damage.
Materials and methods
Cell culture and transfection assays
H1299 lung adenocarcinoma cells, Saos-2 and SJSA-1 osteosarcoma cells were cultivated in RPMI-1640 medium supplemented with 10% FCS. 293 kidney epithelial cells, the three fibroblast cell lines [c-Abl negative (Abl–/– fibroblasts reconstituted with LacZ); c-Abl positive (Abl–/– fibroblasts reconstituted with c-Abl); and c-AblK– (c-Abl fibroblasts reconstituted with the c-Abl kinase-defective mutant K290R)] (Vogt Sionov et al., 1999), primary fibroblasts from normal and c-abl null mice, and fibroblasts (174-2) derived from p53/mdm2 2KO mice (McMasters et al., 1996) were grown in DMEM supplemented with 10% heat-inactivated FCS. The c-Abl-positive fibroblasts expressed physiological levels of c-Abl (Vogt Sionov et al., 1999). Transfections by the calcium phosphate precipitation method, and western blot analyses, were carried out as described previously (Haupt et al., 1996). A constant amount of plasmid DNA in each sample was maintained by supplementing with empty plasmid.
The antibodies used were: anti-human p53 monoclonal antibodies PAb1801 and DO1; anti-mouse p53 polyclonal antibody CM5 (Novocastra); anti-Hdm2 SMP14, SMP15, 2A9 and 2A10, 4B11, Hdm2-323 (Sigma; M7815), anti-c-Abl ABL-148 (Sigma), anti-α-tubulin DM1A (Sigma), anti-histone H2B (LG2-2; kindly provided by Dr D.Eilat), anti-phosphotyrosine 4G10 (Upstate Biotechnology, NY), anti-GST polyclonal rabbit immunosera (purified on GST column), HRP-conjugated affinity-purified goat anti-mouse IgG (Jackson Immuno Research Laboratories), HRP-conjugated protein A (Jackson ImmunoResearch Laboratories), EnVision peroxidase anti-mouse or anti-rabbit IgG (DAKO Corporation), and FITC-conjugated affinipure F(ab′)2 fragment goat anti-mouse IgG (Jackson ImmunoResearch Laboratories). The anti-phosphoY394 polyclonal antibodies were raised against a phosphorylated peptide, ESEDY(PO3H2)SQPSTC, as described previously for anti-phospho-Ser15 antibody against human p53 (Shieh et al., 1997).
Plasmids
Expression plasmids were: human wild-type p53 (pCMV-Neo-Bam-p53), human wild-type p53 (pRC-CMV-p53), human wild-type mdm2 (pCMV-Neo-Bam-hdm2), human wild-type mdm2 (pCHDM1A-hdm2; kindly provided by Dr A.Levine), hdm2 1–200, hdm2ΔRing, mouse wild-type c-abl (pCMV-c-abl, type IV), mouse kinase-defective c-abl (pCMV-c-ablK290H, type IV), mouse c-ablk– mutant ‘A’ [pcDNA- K290RC4 1NLSQ (Q5) (Wen et al., 1996)], mouse cytoplasmic c-Ablk– mutant ‘K’ [(pcDNA-K290RC4 1NLSQ2NLSQ3NLSQ, type IV) (Wen et al., 1996), mouse GST-c-Abl (EBG-Abl, type IV), mouse c-Abl kinase-defective GST-c-AblK290M, mouse GST-c-AblΔSH3 (the GST–c-Abl fusion constructs were kindly provided by Dr B.Mayer), p53-responsive Cyclin G-luciferase (Haupt et al., 1996), and empty vectors (pCMV-Neo-Bam, pCDNA3 and pCOC). His-tagged c-Abl was generated by fusing six His residues to mouse c-Abl using PCR. The following truncated forms of hdm2 were kindly provided by Dr A.Levine: pCHDM Δ1–50, pCHDM Δ58–89, pCHDM Δ90–150, pCHDM Δ1–200, pCHDM Δ150–230, pCHDM Δ222–437, pCHDM Δ441–491 and pCHDM 6–339 (Freedman et al., 1997). The Tyr to Phe substitutions at 394 and 489 of Hdm2 were generated by using site-directed mutagenesis of wild-type pCMV-Neo-Bam-hdm2 (Stratagene). The generation of Hdm2S395A and Hdm2S395D was described previously (Maya et al., 2001). The following plasmids were used for in vitro translation– transcription (TNT): mouse wild-type c-abl (SP6) and mouse mdm2 (T7 pGEM-X2); for in vitro GST-conjugated protein production: pGEX-c-CRK 120–225 and pGEX-cCRK 120–212 (lacking Tyr221) (kindly provided by Dr K.K.Khanna, Australia), C-terminal mouse mdm2-GST (amino acids 356–482) and pGEX (encoding GST alone). The C-termini (amino acids 356–493) of wild-type hdm2, hdm2Y394F and hdm2Y389F were generated by PCR and fused to pGEX. Fragments of full-length wild-type hdm2, hdm2Y394F, hdm2S395A and hdm2S395D were generated by PCR and fused to pGEX. pGEX fusion plasmids were expressed in bacteria and the GST fusion proteins were isolated from the bacterial extract on glutathione beads (Sigma).
Immunoprecipitation and nickel pull down
Immunoprecipitation was carried out essentially as described previously (Haupt et al., 1996), where the lysis buffer contained 50 mM Tris pH 8.0, 150–300 mM NaCl, 5 mM EDTA, 0.5% NP-40, 2 mM PMSF, 20 µg/ml aprotinin, 25 mM NaF and 0.2 mM sodium orthovanadate. Samples were taken from the cell extracts prior to immunoprecipitation for analysis of the steady-state levels of the relevant proteins by western blotting. Proteins were synthesized in vitro by the TNT transcription– translation-coupled reticulocyte lysate system (Promega), either non-radiolabeled or labeled with [35S]methionine. Immunoprecipitation of the in vitro synthesized proteins was carried out in the same way.
For nickel pull down, 293 cells were transfected with wild-type or mutant Hdm2 together with His-tagged c-Abl. Cell extracts were prepared in the above lysis buffer but in the absence of EDTA. Binding to nickel resin was carried out according to the manufacturer’s instructions (CytoSignal) in the presence of 15 mM imidazole for 90 min, followed by three washes in washing buffer containing increasing concentrations of imidazole (20–60 mM). His-c-Abl was eluted from the resin in elution buffer (20 mM Tris pH 8, 500 mm NaCl and 300 mM imidazole).
In vivo kinase assay
SJSA-1 cells were treated with 3 mM H2O2 (Merck, Germany) and/or 1 mM sodium orthovanadate (tyrosine phosphatase inhibitor; Sigma) for the indicated period. Endogenous Hdm2 was immunoprecipitated by using a combination of SMP14, 2A9 and 2A10 anti-Hdm2 antibodies, followed by immunoblotting using the anti-phosphotyrosine monoclonal antibody (4G10) or anti-phospho-394Y polyclonal antibodies.
293 cells were transfected with 5 µg of hdm2 and/or 5 µg of c-abl or c-ablK–. Twenty-four hours post-transfection, cells were either harvested directly or when indicated cells were treated with sodium orthovanadate before harvest. Cell extracts were immunoprecipitated with SMP14 or a combination of SMP14, 2A9 and 2A10, followed by immunoblotting using 4G10 or anti-phospho-Y394 antibodies as indicated.
In vitro kinase assay
The in vitro kinase assay was carried out with c-Abl isolated from 293 cells transfected with 5 µg of expression plasmids, for either His-tagged or non-tagged wild-type c-Abl or a kinase-defective c-Abl (c-ablK–). Non-tagged c-Abl was isolated by immunoprecipitation, while the His-tagged c-Abl was isolated by a nickel pull down. Purified His-c-Abl was incubated with 30 µl of substrate in kinase buffer (25 mM Tris pH 7.4, 10 mM MgCl2, 1 mM MnCl2, 0.5 mM DTT, 10 µM ATP) for 15 min at room temperature. The incubation mix was subjected to western blot analysis using 4G10 or anti-phospho-Y394 antibodies as indicated.
Luciferase assay
H1299 cells were transfected with 30 ng of expression plasmid for wild-type p53, either alone or together with 400 ng of expression plasmids for wild-type hdm2 or hdm2Y394F. All the samples contained 1 µg of the reporter plasmid encoding the luciferase gene under the control of the Cyclin G promoter. Post-transfection (17–24 h), the luciferase activity was measured in the cell extract with the Promega luciferase kit using the Lumat luminometer (LB9507; Berthold).
Apoptosis assay
H1299 cells and Saos-2 cells were transfected with the indicated amount of expression plasmid for wild-type hp53, either alone or together with the indicated amount of expression plasmids for hdm2 or hdm2Y394F. At the indicated time post-transfection, cells were collected, fixed, stained for p53, and analysed by flow cytometry as described previously (Haupt et al., 1996) using the FACSCalibur (Becton Dickinson).
Northern blot analysis
Northern blot analysis of fibroblasts, either non-treated or treated as indicated in the text, was carried out essentially as described previously (Haupt et al., 1997).
Acknowledgments
Acknowledgements
We thank Y.Ben-Neriah, M.Dobbelstein and G.Lozano for the generous gift of the fibroblastic cell lines; K.K.Khanna, B.Mayer, A.Levine and R.Maya for the generous gift of plasmids; and D.Lane, S.Picksley, A.Levine and D.Eilat for the generous gift of antibodies. We are grateful to S.Haupt for critical comment on the manuscript. This work was supported by a grant of the Israel Science Foundation, by the Research Career Development Award from the Israel Cancer Research Fund, by the Lejwa Fund for Biochemistry, and supported in part by research grant No.1-FY01-177 from the March of Dimes Birth Defects Foundation. In addition, the work contributed by R.A.V.E. was supported by NIH grant CA72465.
References
- Agami R., Blandino,G., Oren,M. and Shaul,Y. (1999) Interaction of c-Abl and p73α and their collaboration to induce apoptosis. Nature, 399, 809–813. [DOI] [PubMed] [Google Scholar]
- Argentini M., Barboule,N. and Wasylyk,B. (2001) The contribution of the acidic domain of MDM2 to p53 and MDM2 stability. Oncogene, 20, 1267–1275. [DOI] [PubMed] [Google Scholar]
- Ashcroft M., Kubbutat,M.H. and Vousden,K.H. (1999) Regulation of p53 function and stability by phosphorylation. Mol. Cell. Biol., 19, 1751–1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baskaran R. et al. (1997) Ataxia telangiectasia mutant protein activates c-Abl tyrosine kinase in response to ionizing radiation. Nature, 387, 516–519. [DOI] [PubMed] [Google Scholar]
- Baskaran R., Escobar,S.R. and Wang,J.Y. (1999) Nuclear c-Abl is a COOH-terminal repeated domain (CTD)-tyrosine (CTD)-tyrosine kinase-specific for the mammalian RNA polymerase II: possible role in transcription elongation. Cell Growth Differ., 10, 387–396. [PubMed] [Google Scholar]
- Blattner C., Tobiasch,E., Litfen,M., Rahmsdorf,H.J. and Herrlich,P. (1999) DNA damage induced p53 stabilization: no indication for an involvement of p53 phosphorylation. Oncogene, 18, 1723–1732. [DOI] [PubMed] [Google Scholar]
- Boyd S.C., Tsai,K.Y. and Jacks,T. (2000) An intact Hdm2 RING-finger domain is required for nuclear exclusion of p53. Nat. Cell Biol., 2, 563–568. [DOI] [PubMed] [Google Scholar]
- Chehab N.H., Malikzay,A., Stavridi,E.S. and Halazonetis,T.D. (1999) Phosphorylation of Ser-20 mediates stabilization of human p53 in response to DNA damage. Proc. Natl Acad. Sci. USA, 96, 13777–13782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J., Wu,X., Lin,J. and Levine,A.J. (1996) Mdm-2 inhibits the G1 arrest and apoptosis functions of the p53 tumor suppressor protein. Mol. Cell. Biol., 16, 2445–2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumaz N. and Meek,D.W. (1999) Serine15 phosphorylation stimulates p53 transactivation but does not directly influence interaction with HDM2. EMBO J., 18, 7002–7010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fakharzadeh S.S., Trusko,S.P. and George,D.L. (1991) Tumorigenic potential associated with enhanced expression of a gene that is amplified in a mouse tumor cell line. EMBO J., 10, 1565–1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang S., Jensen,J.P., Ludwig,R.L., Vousden,K.H. and Weissman,A.M. (2000) Mdm2 is a RING finger-dependent ubiquitin protein ligase for itself and p53. J. Biol. Chem., 275, 8945–8951. [DOI] [PubMed] [Google Scholar]
- Freedman D.A., Epstein,C.B., Roth,J.C. and Levine,A.J. (1997) A genetic approach to mapping the p53 binding site in the Mdm2 protein. Mol. Med., 3, 248–259. [PMC free article] [PubMed] [Google Scholar]
- Geyer R.K., Yu,Z.K. and Maki,C.G. (2000) The Mdm2 RING-finger domain is required to promote p53 nuclear export. Nat. Cell Biol., 2, 569–573. [DOI] [PubMed] [Google Scholar]
- Giaccia A.J. and Kastan,M.B. (1998) The complexity of p53 modulation: emerging patterns from divergent signals. Genes Dev., 12, 2973–2983. [DOI] [PubMed] [Google Scholar]
- Goga A., Liu,X., Hambuch,T.M., Senechal,K., Major,E., Berk,A.J., Witte,O.N. and Sawyers,C.L. (1995) p53 dependent growth suppression by the c-Abl nuclear tyrosine kinase. Oncogene, 11, 791–799. [PubMed] [Google Scholar]
- Gong J.G., Costanzo,A., Yang,H.Q., Melino,G., Kaelin,W.G.,Jr, Levrero,M. and Wang,J.Y. (1999) The tyrosine kinase c-Abl regulates p73 in apoptotic response to cisplatin-induced DNA damage. Nature, 399, 806–809. [DOI] [PubMed] [Google Scholar]
- Gottifredi V. and Prives,C. (2001) Getting p53 out of the nucleus. Science, 292, 1851–1852. [DOI] [PubMed] [Google Scholar]
- Gottlieb T.M., Martinez Leal,J.F., Seger,R., Taya,Y. and Oren,M. (2002) Cross-talk between Akt, p53 and Mdm2: possible implications for the regulation of apoptosis. Oncogene, 21, 1299–1303. [DOI] [PubMed] [Google Scholar]
- Gotz C., Scholtes,P., Prowald,A., Schuster,N., Nastainczyk,W. and Montenarh,M. (1999) Protein kinase CK2 interacts with a multi-protein binding domain of p53. Mol. Cell. Biochem., 191, 111–120. [PubMed] [Google Scholar]
- Guerra B., Gotz,C., Wagner,P., Montenarh,M. and Issinger,O.G. (1997) The carboxy terminus of p53 mimics the polylysine effect of protein kinase CK2-catalyzed MDM2 phosphorylation. Oncogene, 14, 2683–2688. [DOI] [PubMed] [Google Scholar]
- Haupt Y., Barak,Y. and Oren,M. (1996) Cell type-specific inhibition of p53-mediated apoptosis by Mdm2. EMBO J., 15, 1596–1606. [PMC free article] [PubMed] [Google Scholar]
- Haupt Y., Maya,R., Kazaz,A. and Oren,M. (1997) Mdm2 promotes the rapid degradation of p53. Nature, 387, 296–299. [DOI] [PubMed] [Google Scholar]
- Hay T.J. and Meek,D.W. (2000) Multiple sites of in vivo phosphorylation in the MDM2 oncoprotein cluster within two important functional domains. FEBS Lett., 478, 183–186. [DOI] [PubMed] [Google Scholar]
- Hjerrild M., Milne,D., Dumaz,N., Hay,T., Issinger,O.G. and Meek,D. (2001) Phosphorylation of murine double minute clone 2 (MDM2) protein at serine-267 by protein kinase CK2 in vitro and in cultured cells. Biochem. J., 355, 347–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda R. and Yasuda,H. (1999) Association of p19ARF with Mdm2 inhibits ubiquitin ligase activity of Mdm2 for tumor suppressor p53. EMBO J., 18, 22–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y., Yuan,Z.M., Ishiko,T., Nakada,S., Utsugisawa,T., Kato,T., Kharbanda,S. and Kufe,D.W. (1997) Pro-apoptotic effect of the c-Abl tyrosine kinase in the cellular response to 1-β-d-arabinofuranosyl cytosine. Oncogene, 15, 1947–1952. [DOI] [PubMed] [Google Scholar]
- Jimenez G.S., Khan,S.H., Stommel,J.M. and Wahl,G.M. (1999) p53 regulation by post-translational modification and nuclear retention in response to diverse stresses. Oncogene, 18, 7656–7665. [DOI] [PubMed] [Google Scholar]
- Kastan M.B. and Lim,D.S. (2000) The many substrates and functions of ATM. Nat. Rev. Mol. Cell Biol., 1, 179–186. [DOI] [PubMed] [Google Scholar]
- Kastan M.B., Zhan,Q., el Deiry,W.S., Carrier,F., Jacks,T., Walsh,W.V., Plunkett,B.S., Vogelstein,B. and Fornace,A.J.,Jr (1992) A mammalian cell cycle checkpoint pathway utilizing p53 and GADD45 is defective in ataxia-telangiectasia. Cell, 71, 587–597. [DOI] [PubMed] [Google Scholar]
- Kharbanda S., Ren,R., Pandey,P., Shafman,T.D., Feller,S.M., Weichselbaum,R.R. and Kufe,D.W. (1995) Activation of the c-Abl tyrosine kinase in the stress response to DNA-damaging agents. Nature, 376, 785–788. [DOI] [PubMed] [Google Scholar]
- Kharbanda S., Yuan,Z.M., Weichselbaum,R. and Kufe,D.W. (1998) Determination of cell fate by c-Abl activation in the response to DNA damage. Oncogene, 17, 3309–3318. [DOI] [PubMed] [Google Scholar]
- Khosravi R., Maya,R., Gottlieb,T., Oren,M., Shiloh,Y. and Shkedy,D. (1999) Rapid ATM-dependent phosphorylation of MDM2 precedes p53 accumulation in response to DNA damage. Proc. Natl Acad. Sci. USA, 96, 14973–14977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubbutat M.H.G., Jones,S.N. and Vousden,K.H. (1997) Regulation of p53 stability by Mdm2. Nature, 387, 299–303. [DOI] [PubMed] [Google Scholar]
- Lain S., Midgley,C., Sparks,A., Lane,E.B. and Lane,D.P. (1999) An inhibitor of nuclear export activates the p53 response and induces the localization of HDM2 and p53 to U1A-positive nuclear bodies associated with the PODs. Exp. Cell Res., 248, 457–472. [DOI] [PubMed] [Google Scholar]
- Maya R. et al. (2001) ATM-dependent phosphorylation of Mdm2 on serine 395: role in p53 activation by DNA damage. Genes Dev., 15, 1067–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayo L.D. and Donner,D.B. (2001) A phosphatidylinositol 3-kinase/Akt pathway promotes translocation of Mdm2 from the cytoplasm to the nucleus. Proc. Natl Acad. Sci. USA, 98, 11598–11603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayo L.D., Turchi,J.J. and Berberich,S.J. (1997) Mdm-2 phosphoryl ation by DNA-dependent protein kinase prevents interaction with p53. Cancer Res., 57, 5013–5016. [PubMed] [Google Scholar]
- McMasters K.M., Montes de Oca Luna,R., Pena,J.R. and Lozano,G. (1996) Mdm2 deletion does not alter growth characteristics of p53-deficient embryo fibroblasts. Oncogene, 13, 1731–1736. [PubMed] [Google Scholar]
- Meek D.W. (1999) Mechanisms of switching on p53: a role for covalent modification? Oncogene, 18, 7666–7675. [DOI] [PubMed] [Google Scholar]
- Michael D. and Oren,M. (2002) The p53 and Mdm2 families in cancer. Curr. Opin. Genet. Dev., 12, 53–59. [DOI] [PubMed] [Google Scholar]
- Momand J., Wu,H.H. and Dasgupta,G. (2000) MDM2-master regulator of the p53 tumor suppressor protein. Gene, 242, 15–29. [DOI] [PubMed] [Google Scholar]
- Nakamura S., Roth,J.A. and Mukhopadhyay,T. (2000) Multiple lysine mutations in the C-terminal domain of p53 interfere with MDM2-dependent protein degradation and ubiquitination. Mol. Cell. Biol., 20, 9391–9398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie Y., Li,H.H., Bula,C.M. and Liu,X. (2000) Stimulation of p53 DNA binding by c-Abl requires the p53 C-terminus and tetramerization. Mol. Cell. Biol., 20, 741–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oren M. (1999) Regulation of the p53 tumor suppressor protein. J. Biol. Chem., 274, 36031–36034. [DOI] [PubMed] [Google Scholar]
- Rodriguez M.S., Desterro,J.M., Lain,S., Lane,D.P. and Hay,R.T. (2000) Multiple C-terminal lysine residues target p53 for ubiquitin– proteasome-mediated degradation. Mol. Cell. Biol., 20, 8458–8467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth J., Dobbelstein,M., Freedman,D.A., Shenk,T. and Levine,A.J. (1998) Nucleo-cytoplasmic shuttling of the hdm2 oncoprotein regulates the levels of the p53 protein via a pathway used by the human immunodeficiency virus rev protein. EMBO J., 17, 554–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawyers C.L., McLaughlin,J., Goga,A., Havlik,M. and Witte,O. (1994) The nuclear tyrosine kinase c-Abl negatively regulates cell growth. Cell, 77, 121–131. [DOI] [PubMed] [Google Scholar]
- Shafman T. et al. (1997) Interaction between ATM protein and c-Abl in response to DNA damage. Nature, 387, 520–522. [DOI] [PubMed] [Google Scholar]
- Shaul Y. (2000) c-Abl: activation and nuclear targets. Cell Death Differ., 7, 10–16. [DOI] [PubMed] [Google Scholar]
- Sherr C.J. and Weber,J.D. (2000) The ARF/p53 pathway. Curr. Opin. Genet. Dev., 10, 94–99. [DOI] [PubMed] [Google Scholar]
- Shieh S.Y., Ikeda,M., Taya,Y. and Prives,C. (1997) DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell, 91, 325–334. [DOI] [PubMed] [Google Scholar]
- Shieh S.-Y., Taya,Y. and Prives,C. (1999) DNA damage-inducible phosphorylation of p53 at N-terminal sites including a novel site, Ser20, requires tetramerization. EMBO J., 18, 1815–1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiloh Y. (2001) ATM and ATR: networking cellular responses to DNA damage. Curr. Opin. Genet. Dev., 11, 71–77. [DOI] [PubMed] [Google Scholar]
- Stommel J.M., Marchenko,N.D., Jimenez,G.S., Moll,U.M., Hope,T.J. and Wahl,G.M. (1999) A leucine-rich nuclear export signal in the p53 tetramerization domain: regulation of subcellular localization and p53 activity by NES masking. EMBO J., 18, 1660–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, X., Majumbder,P., Shioya,H., Wu,F., Kumar,S., Weichselbaum,R., Kharbanda, and Kufe,D. (2000) Activation of the cytoplasmic c-Abl tyrosine kinase by reactive oxygen species. J. Biol. Chem., 275, 17237–17240. [DOI] [PubMed] [Google Scholar]
- Tao W. and Levine,A.J. (1999) Nucleocytoplasmic shuttling of oncoprotein Hdm2 is required for Hdm2-mediated degradation of p53. Proc. Natl Acad. Sci. USA, 96, 3077–3080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Till J.H., Chan,P.M. and Miller,W.T. (1999) Engineering the substrate specificity of the Abl tyrosine kinase. J. Biol. Chem., 274, 4995–5003. [DOI] [PubMed] [Google Scholar]
- Unger T., Juven-Gershon,T., Moallem,E., Berger,M., Vogt Sionov,R., Lozano,G., Oren,M. and Haupt,Y. (1999) Critical role for Ser20 of human p53 in the negative regulation of p53 by Mdm2. EMBO J., 18, 1805–1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Etten R.A. (1999) Cycling, stressed-out and nervous: cellular functions of c-Abl. Trends Cell Biol., 9, 179–186. [DOI] [PubMed] [Google Scholar]
- Vogt Sionov R.V. and Haupt,Y. (1999) The cellular response to p53: the decision between life and death. Oncogene, 18, 6145–6157. [DOI] [PubMed] [Google Scholar]
- Vogt Sionov R., Moallem,E., Berger,M., Kazaz,A., Gerlitz,O., Ben Neriah,Y., Oren,M. and Haupt,Y. (1999) c-Abl neutralizes the inhibitory effect of Mdm2 on p53. J. Biol. Chem., 274, 8371–8374. [DOI] [PubMed] [Google Scholar]
- Vogt Sionov R., Coen,S., Goldberg,Z., Berger,M., Bercovich,B., Ben Neriah,Y., Ciechanover,A. and Haupt,Y. (2001a) c-Abl regulates p53 levels under normal and stress conditions by preventing its nuclear export and ubiquitination. Mol. Cell. Biol., 21, 5869–5878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt Sionov R., Hayon,L.I. and Haupt,Y. (2001b) The regulation of p53 growth suppression. In Blagosklonny,M.V. (ed.), Cell Cycle Checkpoints and Cancer. Austin Landes Bioscience, pp. 106–125.
- Wen S.-T., Jackson,P.K. and Van Etten,R.A. (1996) The cytostatic function of c-Abl is controlled by multiple nuclear localization signals and requires the p53 and Rb tumor suppressor gene products. EMBO J., 15, 1583–1595. [PMC free article] [PubMed] [Google Scholar]
- Xirodimas D.P., Stephen,C.W. and Lane,D.P. (2001) Cocompart mentalization of p53 and Mdm2 is a major determinant for Mdm2-mediated degradation of p53. Exp. Cell Res., 270, 66–77. [DOI] [PubMed] [Google Scholar]
- Yu Z.K., Geyer,R.K. and Maki,C.G. (2000) MDM2-dependent ubiquitination of nuclear and cytoplasmic p53. Oncogene, 19, 5892–5897. [DOI] [PubMed] [Google Scholar]
- Yuan Z.-M., Huang,Y., Fan,M.M., Sawyers,C., Kharbanda,S. and Kufe,D. (1996a) Genotoxic drugs induce interaction of the c-Abl tyrosine kinase and the tumor suppressor protein p53. J. Biol. Chem., 271, 26457–26460. [DOI] [PubMed] [Google Scholar]
- Yuan Z.-M., Huang,Y., Whang,Y., Sawyers,C., Weichselbaum,R., Kharbanda,S. and Kufe,D. (1996b) Role for c-Abl tyrosine kinase in growth arrest response to DNA damage. Nature, 382, 272–274. [DOI] [PubMed] [Google Scholar]
- Yuan Z.-M., Huang,Y., Ishiko,T., Kharbanda,S., Weichselbaum,R. and Kufe,D. (1997) Regulation of DNA damage-induced apoptosis by the c-Abl tyrosine kinase. Proc. Natl Acad. Sci. USA, 94, 1437–1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Z.M. et al. (1999) p73 is regulated by tyrosine kinase c-Abl in the apoptotic response to DNA damage. Nature, 399, 814–817. [DOI] [PubMed] [Google Scholar]
- Zhang Y. and Xiong,Y. (2001) A p53 amino-terminal nuclear export signal inhibited by DNA damage-induced phosphorylation. Science, 292, 1910–1915. [DOI] [PubMed] [Google Scholar]
- Zhou B.P., Liao,Y., Xia,W., Zou,Y., Spohn,B. and Hung,M.-C. (2001) HER-2/neu induces p53 ubiquitination via Akt-mediated MDM2 phosphorylation. Nat. Cell Biol., 3, 973–982. [DOI] [PubMed] [Google Scholar]