

Abstract
Tumor suppressor p53 induction in response to cellular stresses activates the mitogen-activated protein kinase (MAPK) cascade through pathways involving Ras and Raf. p53’s ability to activate this pathway is dependent on p53-mediated transcription. In order to investigate potential p53 target gene(s) involved, we utilized expression array analysis and identified heparin-binding epidermal growth factor-like growth factor (HB-EGF) as being markedly up-regulated by p53. In response to DNA damage, HB-EGF was induced in wild-type, but not in mutant p53-containing cells, implying its p53 dependence. HB-EGF neutralizing antibody and inhibitors of EGF receptor signaling abrogated p53-induced MAPK activation. Expression of HB-EGF was shown to protect cells from H2O2-induced apoptosis through MAPK activation. Additionally, the PI3K/Akt pathway was activated in response to p53 signaling through HB-EGF induction, and inhibition of MAPK and Akt activation after DNA damage decreased cell survival in wild-type p53-containing cells. All these findings point to a novel aspect of p53 function. Namely, p53-induced growth factors such as HB-EGF, which activate MAPK and Akt signaling, may be involved in a compensatory mechanism to alleviate adverse effects of cellular stresses.
Keywords: Akt/HB-EGF/MAPK/p53/tumor suppressor
Introduction
The p53 tumor suppressor gene is mutated in >50% of human tumors (Ko and Prives, 1996; Levine, 1997). The wild-type p53 protein functions as a transcription factor, capable of binding in a sequence-specific manner to well-defined DNA elements and inducing transcription of genes that contain these elements. p53 can also suppress transcription of other genes (Murphy et al., 1996, 1999; MacLachlan et al., 2000; Zhai and Comai, 2000; Zhang et al., 2000). p53 induces either cell cycle arrest, apoptosis or permanent growth arrest/senescence, depending on the cell type (Ko and Prives, 1996; Levine, 1997). p53-induced cell cycle arrest is mediated mainly by the p53 target gene, p21, an inhibitor of cyclin-dependent kinases (El-Deiry et al., 1993; Brugarolas et al., 1995). Bax, IGF-BP3 and DR5, which are also transcriptional targets of p53, are thought to be mediators of p53-induced apoptosis (Buckbinder et al., 1995; Miyashita and Reed, 1995; Wu et al., 1997). More recently, other p53 targets including PERP and noxa have been identified and are also believed to contribute to p53-induced apoptosis (Attardi et al., 2000; Oda et al., 2000).
The mitogen-activated protein kinase (MAPK) cascade is a highly conserved signaling system, through which cells respond to a wide variety of extracellular stimuli (Marshall, 1995). We have shown previously that the Ras/Raf/MAPK cascade can be activated in a sustained manner by p53. This ability of p53 depends on its transcriptional activity, suggesting that p53 transcriptional target gene(s) are involved in this response (Lee et al., 2000).
The Akt serine/threonine kinases are critical mediators of cell survival in response to growth factor stimulation. A number of pro-apoptotic proteins including Bad, caspase-9 and GSK-3 have been identified as direct substrates, which are suppressed upon phosphorylation by Akt (Datta et al., 1999). It is now clear that activation of phosphatidylinositol 3-kinase (PI3K)/Akt pathways and resulting inhibition of the apoptosis are critical events in tumorigenesis.
The identification of transcriptional targets of p53 is critical in understanding pathways by which p53 affects cellular outcomes. To date, the list of transcription targets of p53 is far from exhaustive. In an effort to identify downstream target genes of p53, and in particular those that might be involved in p53-mediated MAPK activation, we performed expression array analysis using tetracycline-regulatable p53-expressing EJ tumor cells that have lost p53 function (Sugrue et al., 1997). We report here that a p53 target gene, heparin-binding epidermal growth factor-like growth factor (HB-EGF), identified through this screening, contributes to p53-mediated MAPK and Akt activation. Blocking the function of HB-EGF by either neutralizing antibody or inhibitors of the EGF receptor, a known receptor for HB-EGF, significantly diminished MAPK activation induced by p53. In addition, exogenous expression of HB-EGF protected cells from H2O2-induced apoptosis. We show further that in wild-type p53-containing cells where HB-EGF is induced in response to DNA damage, blocking the function of HB-EGF by inhibiting MAPK or Akt activation resulted in decreased cell survival. These results demonstrate that p53 activates MAPK and Akt through HB-EGF up-regulation to promote cell survival, suggesting that in addition to p53 targets, which function as either cell cycle inhibitors or apoptosis promoters, other p53 targets such as HB-EGF function as survival factors.
Results
Induction of the HB-EGF transcript by p53
We have shown that the Ras/Raf/MAPK signaling pathway is activated in response to p53 (Lee et al., 2000). Moreover, the ability of p53 to activate this pathway is dependent on its transcriptional activity since tumor-derived p53 mutants fail to do so (Lee et al., 2000). In order to identify potential p53 transcriptional target genes that might be involved in this signaling response, we utilized the tetracycline (tet)-regulatable EJ-p53 system. EJ-p53 cells grow normally in the presence of tet but, following tet removal, p53 is induced and triggers a rapid permanent growth arrest/senescence program in these cells (Sugrue et al., 1997). Thus, we utilized a high-density cDNA array to compare genes expressed in the presence or absence of p53. Poly(A) RNAs from EJ-p53 cells grown in the presence or absence of tet were used to generated cDNAs, which were labeled with 33P and then hybridized to the filters containing 1176 genes (Atlas Human 1.2 array, Clontech). Results were analyzed using Imagequant software (Molecular Dynamics).
Among highly up-regulated genes detected, the transcript for HB-EGF, a member of the EGF family, was found to be increased in response to p53 induction. To quantitate the level of HB-EGF induction, we performed northern blot analysis. As shown in Figure 1A, at 24 h after tet removal in EJ-p53 cells, the transcripts of p53 as well as of its immediate target gene, p21, were readily detectable (lane 4). The HB-EGF transcript was induced >10-fold with kinetics similar to p21. In contrast, HB-EGF was not induced in EJ-CAT control cells after CAT induction by tet removal (lanes 7–10). These results demonstrated that the HB-EGF transcript was up-regulated by p53, consistent with expression array analysis.

Fig. 1. p53-dependent induction of the HB-EGF transcript. (A) Induction in EJ-p53 cells after tetracycline withdrawal. Total RNA was prepared from EJ-p53 or EJ-CAT cells grown in the presence or absence of tet for 6, 12, 24 and 48 h. Northern blots were performed sequentially using 32P-labeled probes against p53, HB-EGF, p21 and 36B4. (B) Induction in VhD and Vm10 cells after temperature downshift. Total RNA was prepared from 10.1, VhD and Vm10 cells grown at 32 and 38°C for 24 and 48 h. Northern blots were performed sequentially using 32P-labeled probes against HB-EGF, p21, mdm2 and 36B4. (C) Induction in p53+/+ fibroblasts after mitomycin C (MMC, left) or actinomycin D (Act. D, right) treatment. Total RNA was prepared from cells after mitomycin C treatment at 0 and 24 h or from cells after actinomycin D treatment at 0, 12 and 24 h. Northern blots were performed sequentially with 32P-labeled probes against HB-EGF, p21 and 36B4.
To confirm p53-mediated up-regulation of HB-EGF further, we analyzed mouse fibroblast cell lines which contain a temperature-sensitive p53 mutant (ts-p53). Both VhD and Vm10 cells are derivatives of the mouse fibroblast cell line 10.1 (Wu et al., 1993). After temperature downshift from 38 to 32°C, VhD cells undergo growth arrest while Vm10 cells, which contain the myc oncogene in addition to the ts-p53 mutant, undergo apoptosis (Wu et al., 1993). Total RNA from VhD, Vm10 and parental 10.1 cells grown at permissive (32°C) and non-permissive (38°C) temperatures was isolated, and northern blot analysis was performed. As shown in Figure 1B, two p53 transcriptional targets, p21 and mdm2, were markedly induced at 24 and 48 h after temperature downshift in both VhD and Vm10 cells (lanes 2, 4, 8 and 10). The HB-EGF transcript was also induced in these cells under the same conditions (Figure 1B, lanes 2, 4, 8 and 10). As a control, the transcript level of 36B4 remained constant. Thus, the induction of HB-EGF by p53 was not dependent on the cellular outcomes induced by p53, since HB-EGF was induced in both VhD (growth arrest) and Vm10 cells (apoptosis). When p53+/+ and p53–/– mouse embryonic fibroblasts were exposure to mitomycin C or actino mycin D, p21 as well as HB-EGF transcripts were up-regulated in p53+/+ but not in p53–/– cells (Figure 1C). These results demonstrate that the HB-EGF transcript could be induced by p53 activated under DNA-damaging stress conditions and that this induction requires wild- type p53.
Inhibition of HB-EGF function abrogates MAPK activation by p53
Previous studies have shown that signaling by the EGF receptor in response to EGF leads to MAPK activation through Ras and Raf (Ullrich and Schlessinger, 1990). HB-EGF is known to activate both the EGF receptor and ErbB4 (Higashiyama et al., 1991, 1992; Elenius et al., 1997). When full-length HB-EGF cDNA under the cytomegalovirus (CMV) promoter was transfected into 293T cells, increasing levels of HB-EGF expression resulted in increased levels of phospho-specific MAPK while the total MAPK level remained relatively constant (Figure 2A, compare lanes 2–4 with lane 1). Dominant-negative (DN) Raf or DN Ras substantially decreased the level of HB-EGF-activated MAPK (Figure 2B, compare lanes 3 and 4 with lane 2). Under these conditions, the level of HB-EGF was not affected by co-expression of either DN Raf or DN Ras (compare lanes 3 and 4 with lane 2). Moreover, DN Ras blocked MAPK activation induced by HB-EGF expression to a similar extent to that induced by soluble EGF (Figure 2C, compare lanes 5 and 6 with lanes 2 and 3). These findings demonstrated that exogenous expression of HB-EGF activates MAPK through Ras and Raf, as has been demonstrated for p53-induced MAPK activation (Lee et al., 2000). It is of note that we detected several species of the membrane-anchored form of HB-EGF with mol. wts ranging between 21 and 32 kDa (Figure 2A and B), consistent with previous studies (Gechtman et al., 1999).

Fig. 2. HB-EGF activates MAPK through Ras and Raf. (A) 293T cells were transfected with vector or increasing amount of HB-EGF as indicated at the top of each lane. Lysates were prepared at 48 h post-transfection. Immunoblots were performed using antibodies against phospho-MAPK (p-MAPK), MAPK and HB-EGF, respectively. (B) 293T cells were transfected with vector, HB-EGF, HB-EGF plus DN Raf, or HB-EGF plus DN Ras as indicated at the top of each lane. Lysates were prepared at 48 h post-transfection. Immunoblots were performed using antibodies against p-MAPK, MAPK and HB-EGF, respectively. (C) 293T cells were transfected with vector or DN Ras as indicated at the top of each lane. Cells were serum starved and 10 ng/ml EGF was added. Cell lysates were collected at 0, 20 and 60 s after EGF treatment as indicated. Immunoblots were performed using antibodies against phospho-MAPK and MAPK.
Chemicals such as PD153035 (Fry et al., 1994; Bridges et al., 1996) and PD158780 (Cunnick et al., 1998; Rewcastle et al., 1998) have been identified as potent and specific inhibitors of the tyrosine kinase activity of the EGF receptor. As shown in Figure 3A, both inhibitors completely inhibited MAPK activation by HB-EGF (compare lanes 10–13 with lane 9), implying that HB-EGF signaling was mediated by the EGF receptor in the HB-EGF-transfected 293T cells. To investigate the contribution of HB-EGF to p53-mediated MAPK activation, we exposed p53-transfected 293T cells to the same inhibitors. As shown in Figure 3A, while both PD153035 and PD158780 inhibited p53-mediated MAPK activation, they did so to a lesser extent than what was observed with the HB-EGF transfectants (compare lanes 5–8 with lane 4). Treatment with either inhibitor had no detectable effect on cell growth, and the levels of total MAPK remained unchanged. Moreover, these inhibitors had no (10 µM) or little (30 µM) effect on the expression levels of p53 and HB-EGF (compare Figure 3B, lanes 3–6 with lane 2 for p53, and C, lanes 3–6 with lane 2 for HB-EGF).

Fig. 3. Inhibition of p53-mediated MAPK activation by inhibitors of the EGF receptor. 293T cells were transfected with vector, p53 or HB-EGF. PD153035 or PD158780 (10 or 30 µM) was added at the indicated lanes. Lysates were made and immunoblots were performed using antibodies against phospho-MAPK, MAPK (A), p53 (B) and HB-EGF (C), respectively.
As an independent approach to implicate HB-EGF in p53-mediated MAPK activation, we next tested whether the HB-EGF neutralizing antibody had any effect on p53-induced phospho-MAPK. As shown in Figure 4A, the HB-EGF neutralizing antibody decreased the phospho-MAPK level by ∼50% (lane 3) compared with transfection of p53 in the presence (lane 4) or absence (lane 2) of control IgG (compare lane 3 with lanes 2 and 4). Under the same conditions, this antibody was more effective in inhibiting induction of phospho-MAPK in HB-EGF-transfected 293T cells (Figure 4B, lane 3). All of these findings indicated that HB-EGF was not the only effector, but accounted for a significant component of p53-induced MAPK activation.

Fig. 4. Inhibition of p53-mediated MAPK activation by HB-EGF neutralizing antibody. 293T cells were transfected with the indicated plasmid. HB-EGF neutralizing antibody (10 µg/ml) or control goat IgG was added to the indicated lanes. Lysates were made and immunoblots were performed using antibodies against phospho-MAPK, MAPK and HB-EGF, respectively. 293T cells were transfected with vector or p53 (A) and with vector or HB-EGF (B).
Expression of HB-EGF increases cell survival in response to oxidative stress through activation of MAPK signaling
In order to investigate HB-EGF function in p53-mediated cellular responses, we generated a chronic HB-EGF-expressing human mammary epithelial cell line by retrovirus-mediated gene transfer. Following retrovirus infection and marker selection, HB-EGF- or LacZ-expressing 184B5 cell lines were obtained. A high level of HB-EGF expression as well as a significantly increased and sustained level of phospho-MAPK was observed by immunoblot analysis, as shown in Figure 5A (lane 2). The expression and localization of HB-EGF were confirmed further by immunostaining (Figure 5B). Whereas 184B5-LacZ control cells lacked detectable HB-EGF immunoreactivity, HB-EGF was readily detectable and was localized to the cell membrane in 184B5-HB-EGF cells.

Fig. 5. Chronic expression of HB-EGF activates MAPK. (A) Immunoblot analysis of HB-EGF expression and MAPK activation. Lysates were prepared from 184B5-HB-EGF and control 184B5-LacZ cells followed by immunoblot using antibodies against phospho-MAPK, MAPK and HB-EGF. (B) Cellular localization of HB-EGF in 184B5-HB-EGF cells. Immunostaining was carried out as described in Materials and methods. Cells were photographed using a Nikon Eclipse TE200 microscope (×400). Top, 184B5-LacZ cells; bottom, 184B5-HB-EGF cells.
It has been reported that sustained activation of the MAPK cascade induces permanent growth arrest/senescence in normal human fibroblasts (Serrano et al., 1997; Lin et al., 1998; Zhu et al., 1998). When we tested whether expression of HB-EGF caused any inhibitory effects on 184B5 cell proliferation, no differences in cell cycle profiles compared with control cells were observed by fluorescence activated cell sorting (FACS; data not shown). Conversely, it has been suggested that expression of the membrane-anchored form of HB-EGF is involved in promoting survival of cultured cells (Miyoshi et al., 1997; Takemura et al., 1997). As shown in Figure 6B, HB-EGF increased the percentage of surviving cells in response to H2O2 treatment. Survival increased from 65% in 184B5-LacZ cells to 83.9% in 184B5-HB-EGF cells treated with 0.5 mM H2O2 and from 15.5 to 38.6% when treated with 1 mM H2O2. These results demonstrated that HB-EGF exerts a protective effect on 184B5 cells exposed to oxidative stress.

Fig. 6. Protective effects of HB-EGF expression on H2O2-induced apoptosis through activation of MAPK. (A) Immunoblot analysis of HB-EGF expression and effects of inhibitors PD98059 and PD158780 on MAPK activation. Lysates were prepared from 184B5-LacZ and 184B5-HB-EGF cells treated with 20 µM PD98059 or 10 µM PD158780, followed by immunoblot using antibodies against phospho-MAPK and MAPK. (B) FACS analysis of 184B5-LacZ and 184B5-HB-EGF cells treated with the indicated concentrations of H2O2 in the presence or absence of the indicated inhibitors. Cells were pre-incubated with the indicated inhibitors before exposure to H2O2. FACS analysis was performed 5 h later. The effect of the inhibitors was normalized and the percentages of live cells were compared. Error bars = means ± SD of three independent experiments.
In order to investigate whether these protective effects correlated with up-regulation of MAPK signaling, 184B5-HB-EGF cells were treated with the MEK1 inhibitor, PD98059, or the inhibitor of EGF receptor signaling, PD158780. Inhibition of MAPK activity was confirmed by immunoblot analysis using an antibody against the phospho-specific MAPK. As shown in Figure 6A, the elevated level of phospho-MAPK in 184B5-HB-EGF cells (lane 2) was decreased to a level (lanes 3 and 4) comparable to that in control 184B5-LacZ cells (lane 1) by treatment with the inhibitors. Exposure of 184B5-HB-EGF cells to these inhibitors also decreased the protective effects of HB-EGF (Figure 6B). These results imply that HB-EGF protects cells from H2O2-induced oxidative stress through activation of MAPK signaling.
p53 activates PI3K/Akt signaling through induction of HB-EGF
In addition to activating MAPK, growth factors are capable of activating the PI3K/Akt pathway (Datta et al., 1999). When serum-starved MCF7 cells were treated with either insulin or soluble HB-EGF, the level of phospho-Akt was increased (Figure 7A, compare lanes 2 and 4 with lane 1). The activation of Akt was blocked when insulin- or HB-EGF-treated cells were pre-incubated with the PI3K inhibitor, LY294002 (compare lane 3 with 2, and lane 5 with 4), indicating that HB-EGF activates Akt through PI3K.
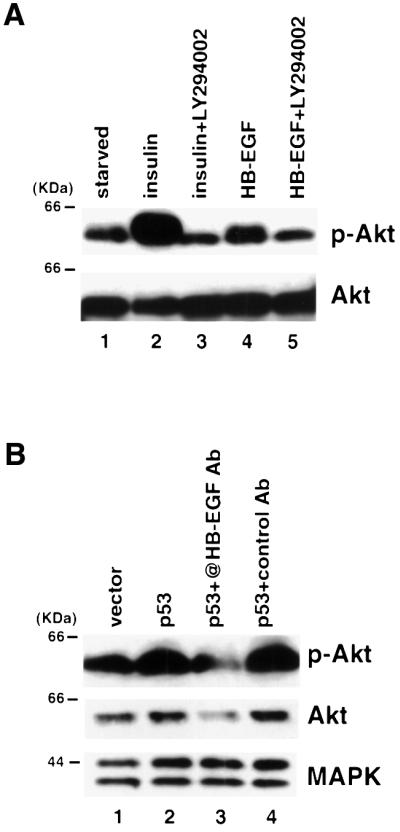
Fig. 7. p53 activates Akt through induction of HB-EGF. (A) MCF7 cells were serum starved and pre-treated with 10 µM LY294002 as indicated. Cell lysates were collected after 15 min treatment with either 200 ng/ml insulin or 10 ng/ml HB-EGF. Immunoblots were performed using antibodies against phospho-Akt (p-Akt) and Akt. (B) 293T cells were transfected with vector or p53. HB-EGF neutralizing antibody (10 µg/ml) or control goat IgG was added to the indicated lanes. Lysates were made and immunoblots were performed using antibodies against p-Akt, Akt and MAPK, respectively.
In order to test whether p53 activates Akt, 293T cells were transiently transfected with vector or p53, and the levels of phospho-Akt and total Akt were analyzed by immunoblot. As shown in Figure 7B, p53 expression increased the level of phospho-Akt (compare lane 2 with lane 1). Next we tested whether p53 activated Akt through induction of HB-EGF by using the HB-EGF neutralizing antibody. As shown in Figure 7B, the HB-EGF neutralizing antibody substantially decreased the level of phospho-Akt (lane 3) induced by p53 com pared with transfection of p53 in the presence (lane 4) or absence (lane 2) of control IgG (compare lane 3 with lanes 2 and 4). These results demonstrated that expression of p53 activated Akt through induction of HB-EGF.
p53-mediated induction of HB-EGF promotes cell survival through activation of MAPK and Akt in response to DNA damage
In order to investigate directly the function of HB-EGF as a p53 target gene, we utilized MCF7 human mammary epithelial carcinoma cells, which contain wild-type p53. Mitomycin C, a DNA-damaging/apoptosis-inducing agent, is known to trigger apoptosis in human breast cancer cells including MCF7 (Xu et al., 1995). When MCF7 cells were exposed to mitomycin C, p21 expression was dramatically induced (Figure 8A, lanes 2 and 3), and the HB-EGF transcript was also increased several fold (lanes 2 and 3). Moreover, activation of MAPK was observed in response to p53 induction (Figure 8B, lanes 2), as previously described (Lee et al., 2000). Pre-incubation with the MEK1 inhibitor PD98059 markedly inhibited MAPK activation (lanes 4). At concentrations of mitomycin C of 2–5 µg/ml, treatment in the presence of this inhibitor resulted in a marked increase in cell death. The percentage of dead cells increased from 18.2 to 43.6% with 2 µg/ml mitomycin C and from 58.5 to 78.8% with 5 µg/ml mitomycin C treatment (Figure 8D). Treatment of the inhibitor alone had no detectable effect. These results are consistent with the concept that p53-induced HB-EGF promotes cell survival in response to DNA-damaging stress through activation of MAPK.

Fig. 8. HB-EGF promotes cell survival in response to DNA damage in wild-type p53-containing cells. (A) Northern blot analysis. Total RNA was prepared from MCF7 cells after mitomycin C (MMC) treatment for 0, 12 and 24 h. Northern blots were performed sequentially using 32P-labeled probes against HB-EGF, p21 and 36B4. (B) Immunoblot analysis. Lysates were prepared from MCF7 cells after mitomycin C treatment for 0 and 24 h with or without the MEK1 inhibitor, PD98059. Immunoblot analysis was performed using antibodies against p53, phospho-MAPK and MAPK. (C) Immunoblot analysis. Lysates were prepared from MCF7 cells after mitomycin C treatment for 0 and 24 h with or without the PI3K inhibitor, LY294002. Immunoblot analysis was performed using antibodies against p53, phospho-Akt and Akt. (D) Effects of inhibitors on cell survival after DNA damage. Cells were treated with the indicated concentration of mitomycin C for 48 h in the presence or absence of the indicated inhibitors followed by trypan blue staining. The percentages of dead cells were calculated and compared. Error bars = means ± SD of two independent experiments with duplicate plates.
To investigate the role of the PI3K/Akt pathway in the p53-mediated response, western blot analysis was performed after mitomycin C treatment of MCF7 cells. As shown in Figure 8C, treatment with mitomycin C increased the level of phospho-Akt, which was blocked by pre-incubation of the PI3K inhibitor LY294002 (compare lane 4 with lane 2), indicating that Akt activation in response to p53 accumulation was mediated through PI3K. More importantly, blocking activation of Akt resulted in a marked decrease in surviving cells. The percentage of dead cells increased from 18.2 to 45.5% with 2 µg/ml mitomycin C and from 58.5 to 76.8% with 5 µg/ml mitomycin C treatment (Figure 8D). These results demonstrated that activation of the PI3K/Akt pathway in response to p53 also contributes to cell survival.
Discussion
Recent studies have established a biochemical link between p53 signaling and activation of the Ras/Raf/MAPK cascade. Findings that MAPK activation occurs in a p53-dependent manner in response to DNA-damaging stress in multiple cell types, but is delayed, suggested that this signaling occurred in response to induction of a primary p53 target gene(s) (Lee et al., 2000). In the present study, we identified a novel p53 target gene, HB-EGF, a growth factor of the multimember EGF family, as being up-regulated in response to p53 induction. The kinetics of this response, which were comparable to those of p21, a known primary p53 transcriptional target, are consistent with HB-EGF being transcriptionally activated by p53. Moreover, HB-EGF induction was shown to be p53 dependent, since it was induced by DNA damage in wild-type but not p53–/– mouse embryo fibroblasts. However, whether HB-EGF is a direct p53 target or its expression is increased by some indirect mechanism in response to p53 remains to be elucidated.
HB-EGF is known to activate the EGF receptor (Higashiyama et al., 1991, 1992; Elenius et al., 1997). EGF triggering of the EGF receptor has been shown previously to act through Ras to cause MAPK activation (Ullrich and Schlessinger, 1990). In the present report, we established that HB-EGF activates Ras/Raf/MAPK through the EGF receptor, since MAPK activation in response to HB-EGF could be inhibited by EGF receptor antagonists as well as DN Ras or Raf. The ability of both HB-EGF neutralizing antibody and EGF receptor antagonists partially to inhibit p53-induced MAPK activation argues that increased HB-EGF expression in response to p53 is mechanistically involved in this p53 signaling response.
HB-EGF is a heparin-binding member of the EGF family, which was identified initially in the conditional medium of human macrophages. It is a potent mitogen and chemotactic factor for fibroblasts and smooth muscle cells, but not endothelial cells (Raab and Klagsbrun, 1997). HB-EGF exists as a membrane-anchored form and a secreted form, the latter of which results from proteolytic cleavage. The transmembrane form of HB-EGF is a juxtocrine growth factor, which is immobilized on the surface of the cell and interacts with neighboring cells. It has been shown that transmembrane HB-EGF synthesized by one type of cell can stimulate tyrosine phosphorylation of the EGF receptor in another type of cell in co-culture (Raab and Klagsbrun, 1997). In the present studies, we detected the membrane-anchored form of HB-EGF induced in response to p53, implying that up-regulation of transmembrane HB-EGF may primarily influence neighboring cells.
The expression of HB-EGF protected cells from H2O2-induced apoptosis, consistent with previous reports (Miyoshi et al., 1997; Takemura et al., 1997). We showed further that this protective effect was mediated by MAPK activation in response to this growth factor since MAPK inhibitors reversed this cyto-protective effect.
We also established that p53, through its target gene, HB-EGF, activated PI3K/Akt. Moreover, blocking p53-mediated MAPK or Akt activation in response to DNA damage resulted in a decreased percentage of live cells. Thus, it is possible that p53-induced growth factors such as HB-EGF may be involved in a cellular compensatory mechanism to alleviate adverse effects induced by various cellular stresses, a novel aspect of p53 function. Our findings that HB-EGF signaling accounts for a portion of p53-mediated MAPK and Akt activation suggest that other growth factors may also be involved in this response. In the context of the whole organism, p53 induction of autocrine or paracrine acting growth factors may play a role in the renewal of damaged tissues.
Materials and methods
Cell culture
EJ-p53 and EJ-CAT cells were cultured in the presence or absence of tetracycline (1 µg/ml) in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum (FBS) (Gibco) as described previously (Sugrue et al., 1997). Mouse fibroblast cell line 10.1 was maintained in DMEM plus 10% FBS. The 10.1 derivative cell lines were maintained in the same medium except that 100 µg/ml hygromycin was added for VhD cells, and 100 µg/ml hygromycin and 200 µg/ml geneticin were added for Vm10 cells, as described previously (Wu et al., 1993). Human MCF7 breast and EJ bladder carcinoma as well as 293T embryonic kidney cells were maintained in DMEM plus 10% FBS. The 184B5 human breast epithelial cell line was provided by M.Stampfer (Lawrence Berkeley National Laboratory, Berkeley, CA) (Stampfer et al., 1997). The MEK1 inhibitor PD98059 was purchased from New England Biolabs, and the EGF receptor inhibitors, PD153035 and PD158780, as well as the PI3K inhibitor, LY294002, were purchased from Calbiochem.
Plasmid construction, cell transfection and virus infection
Full-length cDNAs for HB-EGF as well as dominant-negative mutants of Ras (H-RasN17) (Lange-Carter and Johnson, 1994) and Raf (K375M) (Dent et al., 1995) were subcloned into pcDNA3 and used for transfection studies. 293T cells (∼50% confluent) were transfected with the indicated plasmids using Fugene 6 (Boehringer Mannheim). In transient co-transfection experiments, the total amount of plasmid DNA was kept constant with empty vector. cDNAs for HB-EGF and LacZ were also subcloned into a pBabe-derived retrovirus vector and were co-transfected with the pCL-ampho packaging plasmid into 293T cells for virus production. Retrovirus infection of 184B5 cells was performed in the presence of 2 µg/ml polybrene. Infected cells were selected with 2 µg/ml puromycin.
Expression array screening
The Clontech Atlas human array was hybridized with 33P-labeled cDNA probes derived from mRNAs extracted from EJ-p53 cells grown in the presence or absence of tetracycline, respectively. Quantitation of the hybridization intensity of each dot doublet on the array was performed using Imagequant software (Molecular Dynamics).
Northern blot analysis
Total RNA was extracted, denatured, electrophoresed through a 1% agarose–formaldehyde gel (20 µg total RNA per lane) and transferred to a nylon membrane (Bio-Rad). Hybridization was performed with 32P-labeled probes prepared by the randomly primed DNA labeling method for the indicated genes.
Immunoblot analysis
Cells were washed twice with ice-cold phosphate-buffered saline (PBS) with 2 mM sodium vanadate and lysed in EBC lysis buffer as described previously (Fang et al., 1999). Lysates were cleared by centrifugation at 14 000 r.p.m. for 20 min at 4°C. Protein concentrations were then determined using the BCA protein assay kit (Pierce). A 40 µg aliquot of total cellular protein per sample was subjected to SDS–PAGE and transferred to an Immobilon (Millipore) polyvinylidene difluoride filter. HB-EGF was detected using polyclonal antibody (R&D Systems) followed by the ECL detection system (Amersham). Antibodies included 1801 monoclonal for p53, the Ab-1 monoclonal for p21 (Oncogene Research Products), polyclonal for MAPK, E10 monoclonal for phospho-specific MAPK and polyclonal antibodies for phospho-Akt (Ser473) and Akt (New England Biolabs).
Immunofluorescent staining
184B5-HB-EGF and control 184B5-LacZ cells were seeded onto glass coverslips and cultured for 24 h. Coverslips were rinsed with PBS and fixed with methanol/acetone (1:1) for 2 min at room temperature. Cells were stained for HB-EGF using goat polyclonal antibody (R&D Systems) followed by Texas red-conjugated anti-goat immunoglobulin.
Treatment of cells
184B5-LacZ and 184B5-HB-EGF cells were grown to 50% confluence prior to the exposure to H2O2. Five hours later, both floating and adherent cells were collected, stained with the Annexin-V-Fluo staining kit (Boehringer Mannheim) according to the manufacturer’s instructions and analyzed with FACScan (Becton-Dickinson) using Cellquest software (Becton-Dickinson). MCF7 cells were treated with 20 µM PD98059 or 10 µM LY294002 prior to addition of mitomycin C. After 48 h, both floating and adherent cells were collected and stained with 2% trypan blue. The percentage of the blue cells was calculated by counting the total and blue cell number using a hemacytometer.
Acknowledgments
Acknowledgements
We thank Drs Arnold Levine and Xiangwei Wu for kindly providing VhD and Vm10 cells, Dr M.Klagsburn for HB-EGF cDNA and Dr M.Stampfer for 184B5 cells. This work was supported in part by National Institutes of Health Grants CA80058 and CA85214 and the T.J.Martell Foundation for Leukemia Cancer and AIDS Research (to S.A.A.), and National Institutes of Health Grants CA78356, CA82211 and AG13314 (to S.W.L.). L.F. is a recipient of a postdoctoral fellowship from the Forchheimer Foundation.
References
- Attardi L.D., Reczek,E.E., Cosmas,C., Demicco,E.G., McCurrach,M.E., Lowe,S.W. and Jacks,T. (2000) PERP, an apoptosis-associated target of p53, is a novel member of the PMP-22/gas3 family. Genes Dev., 14, 704–718. [PMC free article] [PubMed] [Google Scholar]
- Bridges A.J., Zhou,H., Cody,D.R., Rewcastle,G.W., McMichael,A., Showalter,H.D., Fry,D.W., Kraker,A.J. and Denny,W.A. (1996) Tyrosine kinase inhibitors. 8. An unusually steep structure–activity relationship for analogues of 4-(3-bromoanilino)-6,7-dimethoxy quinazoline (PD 153035), a potent inhibitor of the epidermal growth factor receptor. J. Med. Chem., 39, 267–276. [DOI] [PubMed] [Google Scholar]
- Brugarolas J., Chandrasekaran,C., Gordon,J.I., Beach,D., Jacks,T. and Hannon,G.J. (1995) Radiation-induced cell cycle arrest compromised by p21 deficiency. Nature, 377, 552–557. [DOI] [PubMed] [Google Scholar]
- Buckbinder L., Talbott,R., Velasco-Miguel,S., Tanenaka,I., Faha,B., Seeizinger,B.R. and Kley,N. (1995) Induction of the growth inhibitor IGF-binding protein 3 by p53. Nature, 377, 646–649. [DOI] [PubMed] [Google Scholar]
- Cunnick J.M., Dorsey,J.F., Standley,T., Turkson,J., Kraker,A.J., Fry,D.W., Jove,R. and Wu,J. (1998) Role of tyrosine kinase activity of epidermal growth factor receptor in the lysophosphatidic acid-stimulated mitogen-activated protein kinase pathway. J. Biol. Chem., 273, 14468–14475. [DOI] [PubMed] [Google Scholar]
- Datta S.R., Brunet,A. and Greenberg,M.E. (1999) Cellular survival: a play in three Akts. Genes Dev., 13, 2905–2927. [DOI] [PubMed] [Google Scholar]
- Dent T., Reardon,D.B., Morrison,D.K. and Sturgill,T.W. (1995) Regulation of Raf-1 and Raf-1 mutants by Ras-dependent and Ras-independent mechanisms in vitro. Mol. Cell. Biol., 15, 4125–4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Deiry W.S. et al. (1993) WAF1, a potential mediator of p53 tumor suppression. Cell, 75, 817–825. [DOI] [PubMed] [Google Scholar]
- Elenius K., Paul,S., Allison,G., Sun,J. and Klagsbrun,M. (1997) Activation of HER4 by heparin-binding EGF-like growth factor stimulates chemotaxis but not proliferation. EMBO J., 16, 1268–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang L., Igarashi,M., Leung,J., Sugrue,M., Lee,S. and Aaronson,S. (1999) p21Waf1/Cip1/Sdi1 induces permanent growth arrest with markers of replicative senescence in tumor cells lacking functional p53. Oncogene, 18, 2789–2797. [DOI] [PubMed] [Google Scholar]
- Fry D.W., Kraker,A.J., McMichael,A., Ambroso,L.A., Nelson,J.M., Leopold,W.R., Connors,R.W. and Bridges,A.J. (1994) A specific inhibitor of the epidermal growth factor receptor tyrosine kinase. Science, 265, 1093–1095. [DOI] [PubMed] [Google Scholar]
- Gechtman Z., Alonso,J.L., Raab,G., Ingber,D.E. and Klagsbrun,M. (1999) The shedding of membrane-anchored heparin-binding epidermal-like growth factor is regulated by the Raf/mitogen-activated protein kinase cascade and by cell adhesion and spreading. J. Biol. Chem., 274, 28828–28835. [DOI] [PubMed] [Google Scholar]
- Higashiyama S., Abraham,J.A., Miller,J., Fiddes,J.C. and Klagsbrun,M. (1991) A heparin-binding growth factor secreted by macrophage-like cells that is related to EGF. Science, 251, 936–939. [DOI] [PubMed] [Google Scholar]
- Higashiyama S., Lau,K., Besner,G.E., Abraham,J.A. and Klagsbrun,M. (1992) Structure of heparin-binding EGF-like growth factor. Multiple forms, primary structure and glycosylation of the mature protein. J. Biol. Chem., 267, 6205–6212. [PubMed] [Google Scholar]
- Ko L.J. and Prives,C. (1996) p53: puzzle and paradigm. Genes Dev., 10, 1054–1072. [DOI] [PubMed] [Google Scholar]
- Lange-Carter C.A. and Johnson,G.L. (1994) Ras-dependent growth factor regulation of MEK kinase in PC12 cells. Science, 265, 1458–1461. [DOI] [PubMed] [Google Scholar]
- Lee S.W., Fang,L., Igarashi,M., Ouchi,T., Lu,K. and Aaronson,S.A. (2000) Sustained activation of Ras/Raf/MAPK cascade by tumor suppressor p53. Proc. Natl Acad. Sci. USA, 97, 8302–8305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine A.J. (1997) p53, the cellular gatekeeper for growth and division. Cell, 88, 323–331. [DOI] [PubMed] [Google Scholar]
- Lin A.W., Barradas,M., Stone,J.C., Aelst,L., Serrano,M. and Lowe,S.W. (1998) Premature senescence involving p53 and p16 is activated in response to constitutive MEK/MAPK mitogenic signaling. Genes Dev., 12, 3008–3019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLachlan T.K., Dash,B., Dicker,D.T. and El-Deiry,W. (2000) Repression of BRCA1 through a feedback loop involving p53. J. Biol. Chem., 275, 31869–31875. [DOI] [PubMed] [Google Scholar]
- Marshall C.J. (1995) Specificity of receptor-tyrosine kinase signalling: transient versus sustained extracellular-signal regulated kinase activation. Cell, 80, 179–185. [DOI] [PubMed] [Google Scholar]
- Miyashita T. and Reed,J.C. (1995) Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell, 80, 293–299. [DOI] [PubMed] [Google Scholar]
- Miyoshi E., Higashiyama,S., Nakagawa,T., Hayashi,N. and Taniguchi,N. (1997) Membrane-anchored heparin-binding epidermal growth factor-like growth factor acts as a tumor survival factor in a hepatoma cell line. J. Biol. Chem., 272, 14349–14355. [DOI] [PubMed] [Google Scholar]
- Murphy M., Hinman,A. and Levine,A.J. (1996) Wild-type p53 negatively regulates the expression of a microtuble-associated protein. Genes Dev., 10, 2971–2980. [DOI] [PubMed] [Google Scholar]
- Murphy M., Ahn,J., Walker,K.K., Hoffman,W.H., Evans,R.M., Levine,A.J. and George,D.L. (1999) Transcriptional repression by wild-type p53 utilizes histone deacetylases, mediated by interaction with mSin3a. Genes Dev., 13, 2490–2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oda E., Ohki,R., Murasawa,H., Nemoto,J., Shibue,T., Yamashita,T., Tokino,T., Taniguchi,T. and Tanaka,N. (2000) Noxa, a BH3-only member of the bcl-2 family and candidate mediator of p53-induced apoptosis. Science, 288, 1053–1058. [DOI] [PubMed] [Google Scholar]
- Raab G. and Klagsbrun,M. (1997) Heparin-binding EGF-like growth factor. Biochim. Biophys. Acta, 1333, F179–F199. [DOI] [PubMed] [Google Scholar]
- Rewcastle G.W. et al. (1998) Tyrosine kinase inhibitors. 14. Structure– activity relationships for methylamino-substituted derivatives of 4-[(3-bromophenyl)amino]-6-(methylamino)-pyrido[3,4-d]pyrimidine (PD 158780), a potent and specific inhibitor of the tyrosine kinase activity of receptors for the EGF family of growth factors. J. Med. Chem., 41, 742–751. [DOI] [PubMed] [Google Scholar]
- Serrano M., Lin,A.W., McCurrach,M.E., Beach,D. and Lowe,S.W. (1997) Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell, 88, 593–602. [DOI] [PubMed] [Google Scholar]
- Stampfer M.R., Bodnar,A., Garbe,J., Wong,M., Pan,A., Villeponteau,B. and Yaswen,P. (1997) Gradual phenotypic conversion associated with immortalization of cultured human mammary epithelial cells. Mol. Biol. Cell, 8, 2391–2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugrue M.M., Shin,D.Y., Lee,S.W. and Aaronson,S.A. (1997) Wild-type p53 triggers a rapid senescence program in human tumor cells lacking functional p53. Proc. Natl Acad. Sci. USA, 94, 9648–9653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takemura T., Kondo,S., Homma,T., Sakai,M. and Harris,R.C. (1997) The membrane-bound form of heparin-binding epidermal growth factor-like growth factor promotes survival of cultured renal epithelial cells. J. Biol. Chem., 272, 31036–31042. [DOI] [PubMed] [Google Scholar]
- Ullrich A. and Schlessinger,J. (1990) Signal transduction by receptors with tyrosine kinase activity. Cell, 61, 203–212. [DOI] [PubMed] [Google Scholar]
- Wu G.S. et al. (1997) KILLER/DR5 is a DNA damage-inducible p53-regulated death receptor gene. Nature Genet., 17, 141–143. [DOI] [PubMed] [Google Scholar]
- Wu X., Bayle,J.H., Olson,D. and Levine,A.J. (1993) The p53–mdm2 autoregulatory feedback loop. Genes Dev., 7, 1126–1132. [DOI] [PubMed] [Google Scholar]
- Xu C., Meikrantz,W., Schlegel,R. and Sager,R. (1995) The human papilloma virus 16E6 gene sensitizes human mammary epithelia cells to apoptosis induced by DNA damage. Proc. Natl Acad. Sci. USA, 92, 7829–7833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai W. and Comai,L. (2000) Repression of RNA polymerase I transcription by the tumor suppressor p53. Mol. Cell. Biol., 20, 5930–5938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L., Yu,D., Hu,M., Xiong,S., Lang,A., Ellis,L.M. and Pollock,R.E. (2000) Wild-type p53 suppresses angiogenesis in human leiomyosarcoma and synovial sarcoma by transcriptional suppression of vascular endothelial growth factor expression. Cancer Res., 60, 3655–3661. [PubMed] [Google Scholar]
- Zhu J., Woods,D., McMahon,M. and Bishop,J.M. (1998) Senescence of human fibroblasts induced by oncogenic Raf. Genes Dev., 12, 2997–3007. [DOI] [PMC free article] [PubMed] [Google Scholar]