

Abstract
To investigate the relationship between chromatin dynamics and nucleotide excision repair (NER), we have examined the effect of chromatin structure on the formation of two major classes of UV-induced DNA lesions in reconstituted dinucleosomes. Furthermore, we have developed a model chromatin-NER system consisting of purified human NER factors and dinucleosome substrates that contain pyrimidine (6-4) pyrimidone photoproducts (6-4PPs) either at the center of the nucleosome or in the linker DNA. We have found that the two classes of UV-induced DNA lesions are formed efficiently at every location on dinucleosomes in a manner similar to that of naked DNA, even in the presence of histone H1. On the other hand, excision of 6-4PPs is strongly inhibited by dinucleosome assembly, even within the linker DNA region. These results provide direct evidence that the human NER machinery requires a space greater than the size of the linker DNA to excise UV lesions efficiently. Interestingly, NER dual incision in dinucleosomes is facilitated by recombinant ACF, an ATP-dependent chromatin remodeling factor. Our results indicate that there is a functional connection between chromatin remodeling and the initiation step of NER.
Keywords: ACF/chromatin remodeling/DNA damage/nucleosome/nucleotide excision repair
Introduction
DNA is frequently damaged by a variety of environmental and endogenous agents produced by physical or chemical reactions (Friedberg et al., 1995). Nucleotide excision repair (NER), one of the most important and well-studied pathways of DNA repair, is capable of eliminating a broad range of structurally unrelated lesions from DNA, including UV-induced damage (Sancar, 1996; de Laat et al., 1999; Batty and Wood, 2000). The NER process consists of four steps: (i) recognition of the damaged DNA; (ii) excision from the DNA of the 24–32 residues surrounding the damaged oligonucleotide; (iii) DNA synthesis to fill the gap; and (iv) ligation of the nick (Sancar, 1996; de Laat et al., 1999; Batty and Wood, 2000). Each process is carried out by multi-protein complexes and has been thoroughly analyzed using highly purified human proteins or recombinant polypeptides (Araki et al., 2000; Araujo et al., 2000). However, mechanisms of NER in the context of chromatin structure remain unclear, partly due to the lack of systematic analyses of NER in defined chromatin.
Packaging of eukaryotic DNA into chromatin affects many of the dynamic processes of DNA metabolism, including transcription, replication, recombination and repair. This is because the assembly of nucleosomes, the basic unit of chromatin, changes the structure of the DNA and restricts the access of DNA-binding factors to their recognition sites (Luger et al., 1997; Wolffe, 2000). Recent studies have led to the purification of >10 large protein complexes that locally disrupt or alter the association of histones with DNA depending on ATP hydrolysis (LeRoy et al., 2000; Shen et al., 2000; Vignali et al., 2000). All of these ATP-dependent chromatin remodeling complexes contain the ATPase subunit of the SNF2 superfamily and fall into one of three distinct families of remodeling complexes: SWI/SNF2-like (e.g. SWI/SNF, RSC and BRM), ISWI-like (e.g. NURF, CHRAC, ACF, yISWI and RSF) and Mi-2-like (e.g. NURD) (Kingston and Narlikar, 1999; Vignali et al., 2000). Although functional analysis of these complexes has been restricted mainly to transcription, it is conceivable that similar activities may also assist NER in chromatin (Thoma, 1999).
UV light induces two major classes of mutagenic DNA lesions: cyclobutane pyrimidine dimers (CPDs) and pyrimidine (6-4) pyrimidone photoproducts (6-4PPs). UV-induced damage formation has been shown to be affected by the chromatin environment (Pfeifer, 1997; Thoma, 1999). To investigate the relationship between chromatin dynamics and NER, we have used synthetic, physiologically spaced and positioned dinucleosome substrates that contain 6-4PP lesions at specific sites. The reconstituted dinucleosome system was previously established to analyze the effect of chromatin structure on transcription (Ura et al., 1995, 1997), and it demonstrated structural changes in chromatin depending on the incorporation of linker histones (Ura et al., 1995; Sato et al., 1999). Here, we have found that the excision of 6-4PP lesions by highly purified human NER factors (Araki et al., 2000) is inhibited when the DNA is folded into chromatin, irrespective of the position of 6-4PP within the chromatin. In addition, we demonstrate that ATP-dependent chromatin remodeling by ATP-utilizing chromatin assembly and remodeling factor (ACF) (Ito et al., 1997) facilitates the excision of 6-4PP lesions by the NER factors, in particular those situated in the linker DNA. Our results indicate that NER clearly requires chromatin reconfiguration, which probably involves alterations in histone–DNA interactions by ATP-dependent chromatin remodeling factors other than the minimal components of NER complexes.
Results
Effects of chromatin structure on acquisition of UV-induced DNA damage
CPDs and 6-4PPs are the two major classes of mutagenic DNA lesions induced by UV light, and they induce a DNA bend or kink of 7–9° and 44°, respectively (Wang and Taylor, 1991; Kim and Choi, 1995). Their frequency and distribution depend on the DNA sequence, the local DNA structure and the association of DNA with chromosomal proteins (Friedberg et al., 1995; Pfeifer, 1997; Thoma, 1999). To address the question of how the formation of UV-induced lesions is affected by histone octamers and linker histone H1, we used reconstituted dinucleosomes composed of two tandem 5S RNA genes (Figure 1A). The dinucleosome is the minimal unit of chromatin containing intact linker DNA. Purified 5′-end-labeled dinucleosome cores were incubated with histone H1, resulting in at least one H1 binding to each dinucleosome. The dinucleosomes with or without histone H1 were irradiated with a UV dose of 100 or 450 J/m2, as was the naked 5S DNA that served as a control. The CPD and 6-4PP UV-induced lesions were then mapped as described in Figure 1B. As shown in Figure 1C, the overall yields and positions of both CPDs (lanes 1–9) and 6-4PPs (lanes 10–18) were not significantly affected by the 5S nucleosome structure. On average, each dinucleosome irradiated with 450 J/m2 UV contained 0.50 CPD and 0.095 6-4PP lesions. Our previous atomic force microscopy images and biochemical analyses showed a structural change in the linker DNA of the dinucleosome, depending on the incorporation of histone H1 (Ura et al., 1995; Sato et al., 1999). However, we did not observe here that the association of H1 altered the formation of either of the two types of UV-induced lesion in dinucleosomes (Figure 1C, lanes 6 and 9 for CPD, 15 and 18 for 6-4PP). Careful comparison of CPD formation between naked DNA and nucleosomal DNA did, however, reveal inhibition of CPD formation at several sites, in particular where the minor groove faces the histone octamer surface (Figure 1C, arrowheads). This is in agreement with earlier work (Gale et al., 1987; Pehrson, 1995; Liu et al., 2000). In addition, CPDs in the dinucleosome were less frequent around the dyad axis of the nucleosome (Figure 1C, dots) compared with naked DNA. Our quantitative analysis shows that, compared with the naked DNA control, CPD formation was reduced up to 4-fold by nucleosome folding at specific sites in the nucleosome (Figure 1C).


Fig. 1. The effect of chromatin structure on CPD and 6-4PP formation in 5S dinucleosome DNA. (A) Structure of the 5S dinucleosome DNA (Ura et al., 1995). The 5S dinucleosome DNA (418 bp) contains two 197 bp tandem repeats of the Xenopus borealis somatic 5S RNA gene and associated upstream sequences, including the entire nucleosome positioning element. Thick arrows indicate the location and orientation of the 120 bp 5S RNA genes. The major nucleosome positions in the absence (core particle position) or presence (chromatosome position) of linker histone are based upon our previous micrococcal nuclease mapping results and are indicated by ovals. All positions are relative to the transcription start site of the 5S RNA gene, which is denoted by +1. (B) Flow diagram showing how the UV-induced CPD and 6-4PP sites on UV-irradiated dinucleosomes were mapped. (C) Mapping of CPD and 6-4PP sites on UV-irradiated dinucleosomes. Naked DNA (lanes 1–3 and 10–12) and dinucleosomes without histone H1 (lanes 4–6 and 13–15) or with histone H1 (lanes 7–9 and 16–18) were irradiated with increasing UV doses (0, 100 or 450 J/m2) as shown at the top of the lanes. Purified DNA was digested with T4 endonuclease V to map CPD sites (lanes 1–9), or incubated with CPD photolyase followed by digestion with UVDE to map 6-4PP sites (lanes 10–18). The amount of DNA in each lane was adjusted according to the total radioactivity before performing denaturing PAGE. The position at which CPD formation is clearly inhibited by the nucleosome folding is shown by arrowheads. The rotational positioning of the 5S nucleosome was demonstrated by the hydroxyl radical cleavage pattern of dinucleosomes in lane 19. M, Maxam–Gilbert G tracks as markers. Dots indicate the the axis of dyad symmetry of the nucleosome. Vertical arrows show the location and orientation of the 5S RNA gene. Ovals indicate the predominant regions contacted by the nucleosome cores as shown in (A) (core particle position). Asterisks show the sites where synthetic 6-4PPs are introduced into the 5S dinucleosome for use as templates in the NER assay.
In contrast to CPD formation, dinucleosome assembly did not seem to affect the distribution of 6-4PPs (Figure 1C). We found that the 6-4PP distribution in the dinucleosome was almost the same as that of naked DNA, even in the linker DNA, where preferential formation of 6-4PPs in chromatin was reported with mixed-sequence nucleosomes from UV-irradiated cells (Mitchell et al., 1990). On the whole, our results demonstrate that the chromatin structure of DNA does not restrict its acquisition of UV-induced lesions.
5S dinucleosome substrates containing specifically positioned synthetic 6-4PPs
The chromatin structure restricts the access of proteins or protein complexes to their respective DNA binding sites (Wolffe, 2000). As we have shown above, however, UV-induced DNA lesions occur throughout chromatin DNA (Figure 1C). Thus, there must be molecular mechanisms that facilitate DNA repair within chromatin. To examine the initiation mechanisms of NER at the chromatin level, we made 5S dinucleosomes that contained 6-4PPs at specific positions on the non-transcribed strand of the 5S DNA: either at the center of two nucleosome cores (center-dinucleosome) or in the linker DNA (linker-dinucleosome). We chose 6-4PP as the model UV lesion because this lesion is removed rapidly by global genome repair (GG-NER), which is the transcription-independent NER subpathway. In contrast to 6-4PPs, CPDs are repaired only very slowly by GG-NER. The other NER subpathway, transcription-coupled repair (TC-NER), is instead responsible for the efficient removal of CPDs from the transcribed strand of expressed genes (de Laat et al., 1999; Thoma, 1999). In order to maintain 5S nucleosome positioning in damaged 5S dinucleosomes, the 6-4PP at the center of the nucleosome was generated at the 5′-TC (–2) site of the 5S gene. The TC sequence that faces out from the nucleosome surface at this site allowed efficient UV-induced 6-4PP formation (Figure 1C, asterisks). The 6-4PP site in the linker DNA was positioned at 5′-TC (+80). The DNA fragments bearing 6-4PPs at either of the two sites were prepared using chemically synthesized 6-4PP-containing oligonucleotides (Figure 2A). 5′-end-radiolabeled dinucleosomes were reconstituted by salt dialysis. The efficiency with which the dinucleosomes were reconstituted was not significantly affected by the presence of either or both of the 6-4PPs.


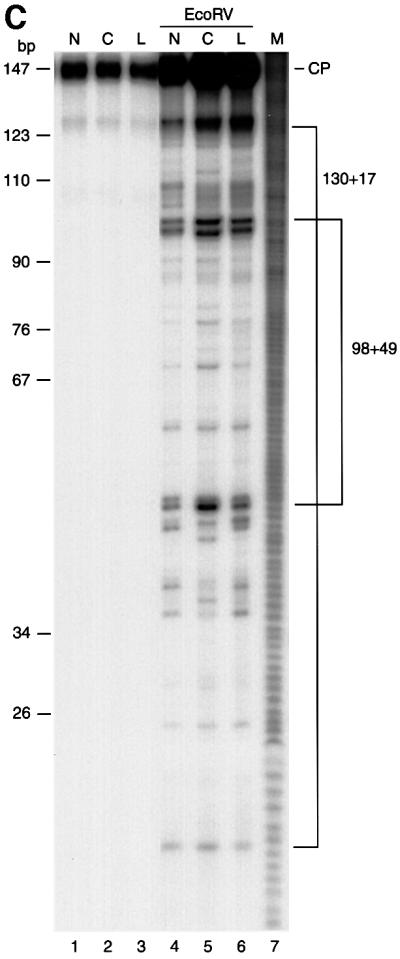
Fig. 2. Preparation and characterization of dinucleosome substrates containing synthetic 6-4PPs. (A) Schematic representation of dinucleosomes that contain 6-4PPs either in the center of the nucleosome (center-dinucleosome) or in the linker DNA (linker-dinucleosome). Asterisks indicate 6-4PPs. (B) DNase I footprinting of 6-4PP-containing dinucleosomes. Lanes 1–3, undigested DNA; lanes 4–6, digested naked DNA; lanes 7–9, digested dinucleosomes. Lane 10 shows a G-specific cleavage reaction as a marker. N, undamaged DNA (lanes 1, 4 and 7); C, center-dinucleosome DNA (lanes 2, 5 and 8); L, linker-dinucleosome DNA (lanes 3, 6 and 9). Dots indicate the axis of dyad symmetry of the nucleosome. The 10 bp repeat pattern, common to nucleosomal footprinting, is shown by arrowheads. As a control, we show that DNA fragments containing 6-4PPs are frequently slightly nicked at damage sites even without DNase I treatment (lanes 2 and 3) (Franklin et al., 1982). Dots, vertical arrows, ovals and asterisks indicate the same items described in Figure 1A. (C) Micrococcal nuclease mapping of core positions on dinucleosomes containing 6-4PPs. Dinucleosomes were digested with micrococcal nuclease. 5′-end-labeled core particle DNA (147 bp) was digested with EcoRV. There is no EcoRV site in the non-specific DNA that was used for nucleosome reconstitution with dinucleosome DNA. Lanes 1–3, undigested core particle DNA; lanes 4–6, DNA digested with EcoRV. Lane 7 shows a DNA maker produced by the hydroxyl radical cleavage reaction. N, undamaged DNA; C, center-dinucleosome DNA; L, linker-dinucleosome DNA; CP, core particle DNA products of digestion.
As 6-4PPs can cause marked DNA distortions, it is still possible that nucleosome positioning in 5S DNA is altered by the introduction of 6-4PPs. We thus investigated nucleosome positioning in reconstituted 6-4PP-containing dinucleosomes by using two independent nuclease mapping methodologies, namely, DNase I footprinting and micrococcal nuclease mapping. The DNase I cleavage pattern of chromatin shows the rotational positioning of nucleosome cores. Well-positioned nucleosome cores like undamaged 5S dinucleosomes give typical 10 nucleotide cleavage ladders because the nuclease cuts preferentially where the DNA is maximally exposed on the nucleosome surface (Ura et al., 1995). DNase I footprinting patterns of dinucleosomes that contain 6-4PPs at the center of nucleosomes (center-dinucleosomes) or in linker DNA (linker-dinucleosomes) were exactly the same as that of undamaged dinucleosomes, indicating that rotational positioning of 5S dinucleosomes was not changed by the introduction of 6-4PPs (Figure 2B). Furthermore, we performed micrococcal nuclease mapping of nucleosome core positions on each 5S dinucleosomes. Each dinucleosome was trimmed with micrococcal nuclease and digestion products were end labeled. Nucleosome core particle fragments (147 bp) were isolated and subjected to EcoRV restriction enzyme digestion, which gave cuts around the middle of the positioned 5S nucleosome cores (Figure 1A). Carrier DNA that was used for reconstitution of dinucleosomes had no EcoRV site and remained at the position of core particles (Figure 2C, CP). The EcoRV restriction enzyme cleavage patterns of core particle fragments derived from each dinucleosome containing 6-4PPs at the center of nucleosomes or in linker DNA were almost the same as that from undamaged dinucleosomes (Figure 2C). These results revealed that not only rotational positioning but also two major translational positions of 5S nucleosome cores (Figure 1A) remained unchanged after the introduction of positioned 6-4PPs.
Excision of 6-4PP from dinucleosomes by purified human NER factors
We assessed the ability of NER to remove the 6-4PP lesions from our dinucleosome substrates by adding the purified human NER factors RPA, XPA, XPC-hHR23B, XPG, XPF-ERCC1 and TFIIH. Excision reactions were monitored by following the appearance of internally labeled dual incision products. The predicted products generated by dual or uncoupled incision of the substrates are shown in Figure 3A. When the naked dinucleosome DNA template was used, the mixture of purified NER factors efficiently excised 6-4 PPs as 24–28 nucleotide oligomers. Approximately 30% of the total substrate signal was thus processed in 30 min (Figure 3B, lanes 2 and 5). Dual incision of both positioned 6-4PPs was repressed by increasing the number of histone octamers on both types of 5S dinucleosome DNA. Surprisingly, strong repression of NER in physiologically spaced dinucleosome templates was observed even when the 6-4PP lesion was located in the linker DNA (Figure 3B, lane 7). The excision activity in dinucleosomes, either at the center of nucleosomes or in the linker DNA, was reduced to <20% of that of naked DNA (Figure 3C). These results suggest that the space that human NER complex requires to excise UV lesions in chromatin is greater than the general length of linker DNA. In the mononucleosome, a histone octamer is located at one of the two 5S nucleosome positioning elements of the dinucleosome DNA. The 50% loss of repression on center-mononucleosomes compared with naked DNA suggests that displacement of only one histone octamer from a damaged DNA site facilitates NER in chromatin (Figure 3C). We concluded that six human NER factors: RPA, XPA, XPC-hHR23B, XPG, ERCC1-XPF and TFIIH, are insufficient to overcome the structural barriers that chromatin poses to the removal of DNA damage, even when the lesion is located within the linker DNA region.


Fig. 3. Effects of chromatin structure on NER. (A) Schematic representation of the predicted cleavage products after incision of the two internally labeled 6-4PP substrates, center-dinucleosome DNA (a) and linker-dinucleosome DNA (b). The labeled products of the excision assay are represented by thin bidirectional arrows along with their lengths in nucleotides (nt). Thick arrows indicate the location and orientation of the 120 bp 5S RNA gene. The 5′-internal promoter of two repeats of the 5S RNA gene was mutated by base substitution, and is indicated by the white box in plasmid pXSP5S. Internally radiolabeled sites of center- and linker-dinucleosome DNA are indicated by black and white asterisks, respectively. The major nucleosome core positions are shown by ovals. (B) Excision activity of NER factors on synthetic chromatin templates. Chromatin templates were assembled on internally labeled DNA fragments containing either no 6-4PP (lane 1), 6-4PPs at the nucleosome centers (lanes 2–4) or a 6-4PP in the linker DNA (lanes 5–7). These were used as substrates in the excision assay. Naked DNA (lanes 1, 2 and 5), mononucleosomes (lanes 3 and 6) or dinucleosomes (lanes 4 and 7) were incubated with purified NER factors for 30 min at 30°C. Cleavage products were analyzed by 12% denaturing PAGE. Each value obtained from the excision products is expressed as a percentage of the total signal in each lane, which is shown at the bottom of the lane. This figure is representative of data obtained from repeated trials of independently conducted experiments. (C) The quantitative data from (B) shown as a bar graph. The relative excision activity of dinucleosomes containing 6-4PP at the center of nucleosomes (black bars) or in linker DNA (white bars) was calculated by taking the value of excision on each naked DNA template as 100%.
ATP-dependent chromatin remodeling by ACF facilitates NER of damage in linker DNA
The strong inhibition of NER in the reconstituted dinucleosome demonstrates the dependence of repair on chromatin dynamics, as can be inferred from previous in vitro studies using bacterial repair enzymes (Schieferstein and Thoma, 1998). Recent studies indicate that chromatin modification, including ATP-dependent chromatin remodeling and histone modification, facilitates transcription of chromatinized genes (Kingston and Narlikar, 1999; Kornberg and Lorch, 1999; Vignali et al., 2000). To unravel the possible molecular mechanisms of NER in chromatin, we investigated the effect of ATP-dependent chromatin remodeling on NER of the 5S dinucleosomes using recombinant ACF. ACF is an ATP-dependent chromatin remodeling complex derived from Drosophila, consisting of ISWI, a member of the SWI2/SNF2 family containing an ATPase/helicase domain, and Acf1, in addition to two other polypeptides (Ito et al., 1997, 1999). Recently, purified recombinant ACF (rACF) that consists only of Acf1 and ISWI has been shown to function in ATP-dependent chromatin assembly and remodeling (Ito et al., 1999, 2000). To examine whether ACF can influence NER of damaged chromatin, we compared the excision activity of purified NER factors in the presence or absence of rACF. Recombinant ACF complex (Acf1 and ISWI) was expressed in insect cells and purified. As expected, there was no detectable change in NER activity on naked DNA templates upon the addition of ACF (Figure 4A, lanes 1, 2, 5 and 6). In contrast, in the presence of ACF, significant activation of NER was observed in linker-dinucleosome templates (Figure 4A, lanes 3 and 4). Interestingly, ACF had no effect on NER in 6-4PP-containing dinucleosomes when the lesions were located at the center of the nucleosomes (Figure 4A, lanes 7 and 8). Specific activation of NER in the linker DNA of dinucleosomes by ACF indicates that the addition of rACF did not cause artificial displacement or disruption of histone octamers from the short chromatin templates. In order to confirm the effect of ACF on NER in chromatin, we added a polyclonal antiserum against the N-terminal region of ISWI to the NER reaction mixtures. NER of the naked DNA template was not influenced by the addition of the antiserum (Figure 4A, lanes 1 and 10). In contrast to the naked DNA template, ACF-dependent activation of NER on linker-dinucleosome templates was strongly inhibited by the antiserum against ISWI (Figure 4A, lanes 3, 4 and 9). Curiously, NER activity on the linker-dinucleosome in the presence of ACF and anti-ISWI serum was only 7.1% of that of the naked DNA control. This is lower than the relative excision activities observed in dinucleosomes in the absence of ACF (Figure 4B). This effect of ISWI antiserum may be due to contamination of our partially purified TFIIH complex with ISWI-containing chromatin remodeling complexes. Indeed, the relative excision activities in dinucleosomes compared with control naked templates varied depending on the preparation of the TFIIH complex (data not shown). Our results demonstrate that ISWI complexes enhance NER dual incision activity by 3.8-fold, and that this can specifically occur in the linker DNA of structures as small as a dinucleosome (Figure 4A, lanes 4 and 9). Although the repressive effect of chromatin was not completely relieved by ACF, these results demonstrate for the first time a functional connection between ATP-dependent chromatin remodeling and NER.

Fig. 4. Effects of rACF on NER in vitro. (A) Excision in synthetic chromatin templates. The excision reaction of NER was carried out either in the presence or absence of ACF (300 fmol) as indicated at the top of each lane. DNA fragments containing 6-4PPs in linker DNA (lanes 1–4, 9 and 10) or at the nucleosome centers (lanes 5–8) were used as substrates for the excision assay. Naked DNA (lanes 1, 2, 5, 6 and 10) or dinucleosomes (lanes 3, 4, 7, 8 and 9) were incubated with purified NER proteins in the absence (lanes 1–8) or presence (lanes 9 and 10) of antiserum against ISWI for 45 min at 30°C. Each value obtained from the excision products is expressed as a percentage of the total signal in each lane, which is shown at the bottom of the lane. This figure is representative of data obtained from repeated trials of independently conducted experiments. (B) The quantitative data from (A) shown as a bar graph. Relative excision activity of center-dinucleosomes (black bars) or linker-dinucleosomes (white bars) was calculated by taking the value of excision from each naked DNA template as 100%.
To investigate the mechanism by which ACF stimulates NER activity on the 5S dinucleosomes, we compared the accessibility of dinucleosomal DNA to cleavage by restriction enzymes under conditions similar to those employed for the NER reaction. Both 6-4PP-bearing and undamaged 5′-end-labeled dinucleosomes were subjected to digestion by either EaeI or RsaI. Based on the restriction enzyme sites in 5S dinucleosome DNA (Figure 5A), EaeI digestion should cut the DNA 6 bp downstream from the site where the 6-4PP lesion is placed at the nucleosomal center, while RsaI should cleave dinucleosome DNA 2 bp upstream from the site where the 6-4PP lesion in the linker DNA is situated. In the absence of ACF, dinucleosomes that are undamaged or have the 6-4PP lesion in the linker DNA are digested by EaeI much more poorly than by RsaI. In contrast, the center-dinucleosomes are digested by EaeI more efficiently, indicating that the 6-4PP lesion at the nucleosomal centers may cause local DNA distortion that improves the accessibility of nucleosomal DNA to EaeI (Figure 5B, top, lanes 7–9). When ACF and ATP were present, the cleavage of both damaged and undamaged dinucleosomes at the 5′ RsaI site in the linker DNA was improved by ∼1.5-fold. In contrast, ACF did not affect EaeI digestion of any of the dinucleosomes (Figure 5B, lanes 5, 8 and 11). Thus, ACF seems to stimulate nucleosome movement that extends the nucleosome-free linker DNA region, rather than acting by displacing the nucleosomes. Supporting this notion is the observation of ACF-dependent nucleosome movement of the 5S dinucleosomes (H.Saeki, unpublished data). These results demonstrate that ATP-dependent chromatin remodeling by ACF facilitates NER of the linker DNA by increasing the accessibility of nucleosomal DNA to the NER factors.

Fig. 5. Effect of rACF on accessibility of damaged DNA in dinucleosomes. (A) The locations of the EaeI and RsaI sites on 5S dinucleosome DNA. Thick arrows show the location of the 5S RNA gene. Asterisks show the 6-4PP positions. (B) Restriction enzyme digestion of dinucleosomes. 5′-end-radiolabeled naked DNA (lanes 2 and 3) and dinucleosomes (lanes 4–12) were digested with either EaeI (top) or RsaI (bottom). The restriction digestion was carried out in the presence or absence of ACF (200 fmol) and 3 mM ATP as indicated at the top of each lane. Non, undamaged DNA; Center, center-dinucleosome DNA; Linker, linker-dinucleosome DNA. The percentage digestion of the 5′ EaeI and 5′ RsaI restriction sites is shown at the bottom of each lane.
Discussion
In this study, we have established a model chromatin-NER system using reconstituted 5S dinucleosomes and purified human NER factors. Systematic analyses of how NER can repair UV damage at different sites in such dinucleosomes should help elucidate the molecular mechanisms involved in NER of damaged chromatin.
Chromatin structure does not significantly affect the acquisition of UV-induced damage
The formation of two major classes of UV-induced DNA lesions, CPDs and 6-4PPs, was demonstrated at the nucleotide level in defined, reconstituted dinucleosomes by enzymic detection. CPD formation was somewhat reduced at sites where the minor groove faced the histone octamer and around the pseudo-center of the nucleosome (Figure 1C, arrowheads). These results are consistent with earlier work in mixed-sequence chromatin (Gale et al., 1987; Pehrson, 1995). However, the distribution of CPDs did not show the 10.3 bp periodicity observed with mixed-sequence nucleosome cores, which is consistent with other studies that used rotationally positioned mononucleosomes (Schieferstein and Thoma, 1996; Liu et al., 2000). The specific reduction of CPD formation across the pseudo-center of the nucleosome may reflect the relatively straight structure of nucleosomal DNA, which is supported by the tight interactions between histone H3 and H4 in the histone octamer around this region (Luger et al., 1997; Liu et al., 2000). Apart from these mild differences, however, the frequency and distribution of CPD lesions were generally equivalent between naked 5S DNA and the dinucleosome.
The yield and distribution of 6-4PP lesions were not affected by the dinucleosome structure of the 5S DNA (Figure 1C). It should be noted that the preferential formation of 6-4PP in linker DNA that was reported for bulk chromatin from UV-irradiated cells (Mitchell et al., 1990) was not observed with our 5S dinucleosomes. One possible explanation for this discrepancy is that nucleosome positioning may be altered after DNA damage in living cells. However, we could not see any significant changes in DNase I cleavage patterns between UV-irradiated and UV-untreated 5S dinucleosomes (data not shown). This is in agreement with other reports using defined, reconstituted nucleosome cores (Schieferstein and Thoma, 1996; Liu et al., 2000). Another possibility is that in living cells, chromatin binding proteins or chromatin remodeling factors may alter nucleosome positioning around the lesion sites after damage formation.
We also did not observe any effects of the linker histone H1 on the acquisition of UV-induced DNA lesions in the 5S dinucleosomes (Figure 1C). Physiologically spaced dinucleosome templates incorporate histone H1 without aggregation (Ura et al., 1995). We demonstrated previously that the inclusion of linker histones into dinucleosomal templates causes histone–DNA contacts to be established over the linker DNA regions (Ura et al., 1995, 1996; Sato et al., 1999). Regardless of these effects of histone H1 on the chromatin structure, the formation of either CPDs or 6-4PPs in the 5S dinucleosomes was not significantly affected by the incorporation of histone H1.
Thus, our results reveal that the effect of chromatin structure on UV damage formation is insignificant compared with that of other processes, such as binding of factors to their recognition sites or DNA damage formation that is generated by other agents such as chemicals. In general, the induction of CPDs and 6-4PPs by UV light seems to depend more on the DNA sequence than on the chromatin environment. As will be discussed below, however, the NER activity that can correct such DNA lesions is strongly affected by chromatin structure.
NER is inhibited by the chromatin structure of DNA
To investigate whether the chromatin structure of DNA affects NER, we developed 5S dinucleosomes that contained 6-4PPs either at the centers of nucleosomes or in the linker DNA. Using these dinucleosomes as substrates, we performed dual incision reactions with a mixture of six purified human NER factors (Araki et al., 2000). DNase I footprinting patterns and micrococcal nuclease mapping results revealed that 5S nucleosome positioning was not changed by the introduction of one or two 6-4PPs in a 418 bp dinucleosome DNA fragment (Figure 2). It thus appears that the severe DNA kink induced by the 6-4PP lesion does not influence the intrinsic DNA structure of the 5S RNA gene, which is required for nucleosome positioning, although enzymic digestion studies did show that it affects local histone–DNA contact around the nucleosome center (Figure 5B).
Using these defined, damaged dinucleosome templates, we demonstrated that nucleosome assembly drastically inhibited dual incision, an early step of NER (Figure 3). This inhibitory effect of chromatin structure on NER was observed even after a 15 min incubation (data not shown). That DNA repair can be strongly and rapidly inhibited by nucleosome structures was demonstrated previously in vitro by using UV-irradiated mononucleosome cores as substrates for direct repair by bacterial DNA photolyase or T4 endonuclease (Schieferstein and Thoma, 1998). However, that the folding of DNA into nucleosomes can also inhibit NER had at the time of our studies not been demonstrated clearly in vitro, although earlier NER studies had suggested that it is an important factor (Pfeifer, 1997; Thoma, 1999). Very recently, a study using 136 bp nucleosomal DNA has now also shown that the nucleosome structure inhibits NER (Hara et al., 2000), thus confirming our observations.
We found that damage excision was repressed drastically by increasing the number of histone octamers associated with the 5S dinucleosome DNA. Surprisingly, the inhibition of NER activity was equivalent whether the 6-4PP lesion was present at the center of the nucleosome or in the linker DNA between the nucleosomes (Figure 3). One might have expected that the lesion on the linker DNA might be more accessible to the NER factors. However, our 5S dinucleosome templates have 50–80 bp of linker DNA. This is shorter than the ∼100 bp stretch apparently required for excision of a lesion from naked DNA by the human NER complex (Huang and Sancar, 1994). Our results demonstrate that human NER complexes need a nucleosome-free DNA space that is longer than 80 bp to excise 6-4PPs in chromatin.
Our data show that although the chromatin structure of DNA does not inhibit its acquisition of UV-induced lesions, excision of this damage with the six human NER factors are severely inhibited by this structure. Therefore, we suggest that a dynamic change in the histone–DNA interaction by protein(s) other than the six human NER factors, e.g. chromatin modification complexes, is involved in the NER process in chromatin.
ATP-dependent chromatin remodeling promotes NER of damaged chromatin
Chromatin modification complexes can be classified into two broad classes: ATP-dependent chromatin remodeling complexes and histone modification complexes (Kingston and Narlikar, 1999; Vignali et al., 2000). In this study, we have investigated the effect of the former complexes on NER in our defined chromatin-NER system, using recombinant Drosophila ACF. Our biochemical analyses demonstrated that rACF facilitates NER dual incision reactions specifically in the linker DNA of dinucleosomes but not at the center of dinucleosome cores (Figure 4). In agreement with our in vitro data, NER rates for CPDs and 6-4PPs on the non-transcribed strand are influenced by the chromatin environment and are removed more efficiently in linker DNA than in nucleosomal DNA in yeast cells (Wellinger and Thoma, 1997; Tijsterman et al., 1999). Our results suggest that NER in linker DNA is mediated preferentially by chromatin remodeling. ATP-dependent chromatin remodeling has been considered to facilitate NER in chromatin (Paetkau et al., 1994; Thoma, 1999), and a recent yeast study indicates that there exists genetically a connection between chromatin remodeling and DNA repair (Shen et al., 2000). We have presented here novel biochemical evidence that there is indeed a direct connection between the ATP-dependent chromatin remodeling complex and the early step of NER.
One possible mechanism by which ACF promotes NER of damaged dinucleosomes is by mediating nucleosome movement (Hamiche et al., 1999; Langst et al., 1999; Brehm et al., 2000; Guschin et al., 2000). In this model, ACF assists histone octamer sliding, which extends the nucleosome-free DNA region around 6-4PPs in linker DNA and thereby permits access of NER factors to the damaged sites. ACF has strong ATP-dependent histone octamer sliding activity, even on 5S DNA (H.Saeki, unpublished data). With respect to our dinucleosomes, this model suggests that ACF could induce the movement of two histone octamers to opposite ends of the dinucleosome together with NER factors, which would increase the length of linker DNA to a maximum of ∼120 bp. This would facilitate the dual incision reaction by ∼2- to 3-fold. On the other hand, center 6-4PPs would still be associated with nucleosome cores and would be less efficiently removed than lesions in the linker DNA. We examined this hypothesis by assessing the activity of two restriction enzymes. One of these enzymes digests a site close to the 6-4PP lesion at the nucleosome center, while the other cuts at a site close to the 6-4PP on the linker DNA. When ACF was present, digestion of the site on the linker DNA was improved, while digestion of the site within the nucleosome center was unaltered (Figure 5B). These results suggest that ACF promotes NER of damaged linker DNA regions by stretching these regions and thus allowing the NER complex to access the damage.
That the improvement of NER dual incision by ACF is not complete suggests that the increased length of linker DNA in dinucleosomes is still insufficient for efficient recognition and excision of the lesion by the NER complex within the chromatin context, although the length required for excision on naked DNA is ∼100 bp (Huang and Sancar, 1994). Alternatively, auxiliary factors other than ISWI and Acf1 in the ACF complex, or other ATP-dependent chromatin remodeling complexes, may be required for full activation of NER in chromatin. Nucleosome sliding in the nucleus may be more pronounced than in dinucleosomes and might actually provide enough space for NER of DNA lesions in linker DNA and at the center of nucleosomes. Once the initiation complex of NER is formed on damaged DNA, at least one nucleosome would be disrupted during damage excision. Reassembly of the original chromatin structure could be achieved by coupling components of the NER complex to chromatin assembly factor 1 (CAF-1) and proliferating cell nuclear antigen (PCNA) (Gaillard et al., 1996; Moggs et al., 2000; Verreault, 2000). ACF can mediate chromatin assembly with CAF-1 in vitro (Ito et al., 1997). It might also function for chromatin assembly during NER together with CAF-1.
NER is a highly conserved pathway among eukaryotes, and ISWI-containing chromatin remodeling complexes have also been found in various species from yeast to human. Importantly, ACF is a well-conserved and abundant remodeling complex (Ito et al., 1999; Guschin et al., 2000; LeRoy et al., 2000). Although the exact functions of ACF in the nucleus remain unknown and there exist many chromatin remodeling complexes other than ACF, our results indicate a possible role for ACF in DNA repair. At the very least, we have shown that an active chromatin reconfiguration is required during the NER process. Using our 6-4PP-containing dinucleosome sytem, we can now compare the NER activity and the structural changes induced by different chromatin remodeling complexes. This should help to determine which of the chromatin remodeling complex(es) contribute maximally to efficient NER of damaged chromatin. Acetylation of N-terminal tails of core histones may also facilitate the NER process coupled or uncoupled with ATP-dependent chromatin remodeling, although the removal of core histone tails does not stimulate DNA repair in 5S mononucleosomes (Liu and Smerdon, 2000). In order to reveal the chromatin dynamics associated with NER, further study is required. Our 5S dinucleosome-NER system provides a powerful tool for systematic analyses of NER in chromatin.
Materials and methods
Preparation of labeled DNA fragments for dinucleosome assembly
5′-end-labeled 418 bp XbaI–XhoI fragments derived from plasmid pX5S197-2 were used to assemble dinucleosomes for mapping of UV-induced damage sites (Ura et al., 1995).
In order to introduce a unique 6-4PP site into the linker DNA of the dinucleosome, the 5′ promoter of two tandem repeats of the 5S RNA gene in plasmid pX5S197-2 was mutated by substitution of 22 bp as follows. The promoter of the 5S RNA gene (+79 to +100) in plasmid pX5S197′ (Ura et al., 1995) was substituted with the SP6 promoter by PCR using oligonucleotides (5′-ATCGATTTAGGTGACTATAG-3′), giving rise to plasmid pXSP′. A 216 bp SalI–KpnI fragment derived from plasmid pX5S197 (Ura et al., 1995) was cloned into the XhoI–KpnI site of plasmid pXSP′ to generate plasmid pXSP5S. The 5′-end-labeled 418 bp DNA fragments (XhoI–XbaI) of pXSP5S were used for reconstitution of damaged 5S dinucleosome substrates. Micrococcal nuclease mapping and DNase I footprinting of the new dinucleosomes confirmed that the rotational and translational positioning of 5S nucleosome cores were not affected by the 22 bp substitution. Dinucleosome DNA fragments containing positioned 6-4PPs were prepared using the plasmid pXSP5S. Two 30mer oligonucleotides containing 6-4PPs were synthesized chemically (Mizukoshi et al., 1998). The sequences of the oligonucleotides were 5′-AAAGTCAGCCTTGTGCTCGCCTACGGCCAT-3′ (–18 to +12) for the center-dinucleosome and 5′-CCTGGTTAGTACATCGATTTAGGTGACACT-3′ (+67 to +96) for the linker-dinucleosome and contained 6-4PPs at the underlined sites. Each oligonucleotide was annealed to single-stranded circular DNA from pXSP5S and then treated with T4 DNA polymerase, T7 Sequenase DNA polymerase (Life Sciences) and T4 DNA ligase. Double-stranded closed circular plasmids containing positioned 6-4PPs were purified by cesium chloride–ethidium bromide density gradient centrifugation. DNA substrates lacking the lesion were prepared in the same way except that undamaged 30mer oligonucleotides for the center dinucleosome were used instead of 30mers containing 6-4PPs. The 418 bp DNA fragments (XhoI–XbaI) derived from each synthetic plasmid were used for assembly of 5S dinucleosomes. For the in vitro NER assay, 5S dinucleosome DNA fragments were internally radiolabeled using 5′-end-radiolabeled 30mer oligonucleotides.
Nucleosome reconstitution
Histone octamers were prepared from HeLa cells and histone H1 was prepared from calf thymus. Dinucleosomes were reconstituted onto radiolabeled 418 bp DNA fragments by salt dialysis as described previously (Sato et al., 1999). Briefly, radiolabeled dinucleosome DNA (500 ng) and unlabeled non-specific DNA (4.5 µg) were mixed with core histones (6 µg) in 2.0 M NaCl dialysis buffer at 4°C. Samples were dialyzed at 4°C against dialysis buffer containing NaCl as follows: 2.0 M, overnight; 1.5 M, 1 h; 1.0 M, 4 h; 0.75 M, 4 h; 0 M, overnight. The oligonucleosome cores were loaded onto 5–20% sucrose gradients and separated according to the number of associated histone octamers (Ura et al., 1995). The fractionated oligonucleosomes were concentrated to >5 µg/ml and stored on ice until use.
Histone H1 incorporation into dinucleosome cores
Dinucleosomes (50 ng DNA) were incubated with histone H1 (35 ng) in 50 µl of binding buffer (50 mM NaCl, 10 mM Tris–HCl pH 8.0, 0.1 mM EDTA) at room temperature for 20 min. Ten percent of the sample was loaded onto a 0.7% agarose gel to confirm the binding of histone H1 to dinucleosomes (Ura et al., 1995). The remaining sample was UV irradiated with increasing UV doses (100 or 450 J/m2).
Mapping of UV-induced CPD and 6-4PP sites
Dinucleosomes were placed on ice in a 12-well plate and irradiated with 254 nm germicidal UV light at a rate of 2.0 J/m2/s. Naked DNA served as a control and was also irradiated. UV fluence was measured using a Topcon UV radiometer. UV-irradiated dinucleosomes were treated with proteinase K. The DNA was then purified and 10 ng were digested at 37°C for 30 min with 4 ng of T4 endonuclease V (Epicenter Technologies) in 20 µl of T4 buffer [50 mM Tris–HCl pH 7.5, 5 mM EDTA, 0.005% bovine serum albumin (BSA)] to map CPD sites. The 6-4PP sites were mapped by first incubating 10 ng of the purified irradiated DNA with cyanobacterium Anacystis nidulans CPD photolyase in 10 µl of reaction buffer [100 mM NaCl, 50 mM Tris–HCl pH 7.0, 1 mM EDTA, 10 mM dithiothreitol (DTT)] at room temperature for 1 h under a fluorescent light. This caused all the CPDs that were present to be repaired. The DNA was then digested with 40 ng of Neurospora crassa UV-damage endonuclease (UVDE), which introduces an incision immediately 5′ to both CPDs and 6-4PPs, in reaction buffer (100 mM NaCl, 50 mM Tris–HCl pH 8.0, 10 mM MgCl2, 1 mM DTT) at 37°C for 30 min. CPD photolyase and UVDE were gifts of A.Yasui (Yasui and McCready, 1998). Digestion products were analyzed by 6% denaturing PAGE and a Fuji BAS imaging analyzer.
DNase I footprinting and micrococcal nuclease mapping
DNase I footprinting and micrococcal nuclease mapping were carried out as described previously (Ura et al., 1995, 1996).
Preparation of NER proteins and ACF
NER factors prepared for in vitro NER assays were purified as described previously (Araki et al., 2000), except for RPA and TFIIH. Human XPA–His protein was expressed in Escherichia coli. Recombinant human XPF–ERCC1–His complex and XPG protein were expressed in insect cells. Heterotrimeric human RPA complex (RPA70, RPA32 and RPA14) tagged with His6 on the C-terminus of the p32 subunit was expressed in E.coli and purified through three chromatographic steps (Ni-NTA agarose, single-stranded DNA cellulose and Mono Q). TFIIH was partially purified through five column chromatographic steps (phosphocellulose, single-stranded DNA cellulose, HiTrap SP, Resource Q and HiTrap heparin) from HeLa nuclear extracts.
Recombinant ACF complex (Acf1 and ISWI) was expressed in insect cells (Sf9) using recombinant baculovirus and purified using Flag-M2 resin as previously described (Ito et al., 1999, 2000).
NER dual incision assay
Chromatin was assembled onto internally labeled DNA fragments containing 6-4PPs and used as the substrate for the excision assay. Chromatin templates or naked DNA as a control (20 ng of DNA each) were incubated with purified NER factors at 30°C for the indicated times (15–60 min) in 15 µl of reaction buffer (20 mM HEPES pH 7.9, 50 mM KCl, 7.5 mM MgCl2, 40 mM phosphocreatine, 50 µg of creatine phosphokinase, 100 ng of cold non-specific DNA, 250 µg of BSA, 2 mM ATP and 10 mM DTT). Cleavage products were analyzed by 12% denaturing PAGE. The degree of excision was quantified by a Fuji BAS imaging analyzer.
Restriction enzyme digestion
5′-end-labeled dinucleosomes or naked DNA as a control (10 ng of DNA each) were incubated with 10 U of restriction enzyme at 30°C for 60 min in 15 µl of reaction buffer (10 mM Tris–HCl pH 7.9, 50 mM NaCl, 8 mM MgCl, 100 ng of cold non-specific DNA, 100 µg/ml BSA, 50 µM each dNTPs and 1 mM DTT). DNA was purified after proteinase K treatment and analyzed by 6% non-denaturing PAGE. The level of digestion was quantified by a Fuji BAS imaging analyzer.
Acknowledgments
Acknowledgements
We thank Drs A.Yasui for generously providing N.crassa UVDE and A.nidulan CPD photolyase, C.Wu for the ISWI cDNA, T.Ohta for the antiserum against ISWI and S.Kass, Y.Ohkuma and K.Sugasawa for critically reading the manuscript. This work was supported by grants from the Ministry of Education, Culture, Sports, Science and Technology of Japan, the Uehara Memorial Foundation and the Japanese Society for the Promotion of Science (JSPS). M.A. is a JSPS fellow.
References
- Araki M. et al. (2000) Reconstitution of damage DNA excision reaction from SV40 minichromosomes with purified nucleotide excision repair proteins. Mutat. Res., 459, 147–160. [DOI] [PubMed] [Google Scholar]
- Araujo S.J., Tirode,F., Coin,F., Pospiech,H., Syvaoja,J.E., Stucki,M., Hübscher,U., Egly,J.M. and Wood,R.D. (2000) Nucleotide excision repair of DNA with recombinant human proteins: definition of the minimal set of factors, active forms of TFIIH and modulation by CAK. Genes Dev., 14, 349–359. [PMC free article] [PubMed] [Google Scholar]
- Batty D.P. and Wood,R.D. (2000) Damage recognition in nucleotide excision repair of DNA. Gene, 241, 193–204. [DOI] [PubMed] [Google Scholar]
- Brehm A., Langst,G., Kehle,J., Clapier,C.R., Imhof,A., Eberharter,A., Muller,J. and Becker,P.B. (2000) dMi-2 and ISWI chromatin remodelling factors have distinct nucleosome binding and mobilization properties. EMBO J., 19, 4332–4341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Laat W.L., Jaspers,N.G. and Hoeijmakers,J.H. (1999) Molecular mechanism of nucleotide excision repair. Genes Dev., 13, 768–785. [DOI] [PubMed] [Google Scholar]
- Franklin W.A., Lo,K.M. and Haseltine,W.A. (1982) Alkaline lability of fluorescent photoproducts produced in ultraviolet light-irradiated DNA. J. Biol. Chem., 257, 13535–13543. [PubMed] [Google Scholar]
- Friedberg E.C., Walker,G.C. and Siede,W. (1995) DNA Repair and Mutagenesis. ASM Press, Washington, DC.
- Gaillard P.H., Martini,E.M., Kaufman,P.D., Stillman,B., Moustacchi,E. and Almouzni,G. (1996) Chromatin assembly coupled to DNA repair: a new role for chromatin assembly factor I. Cell, 86, 887–896. [DOI] [PubMed] [Google Scholar]
- Gale J.M., Nissen,K.A. and Smerdon,M.J. (1987) UV-induced formation of pyrimidine dimers in nucleosome core DNA is strongly modulated with a period of 10.3 bases. Proc. Natl Acad. Sci. USA, 84, 6644–6648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guschin D., Geiman,T.M., Kikyo,N., Tremethick,D.J., Wolffe,A.P. and Wade,P.A. (2000) Multiple ISWI ATPase complexes from Xenopus laevis: functional conservation of an ACF/CHRAC homolog. J. Biol. Chem., 275, 35248–35255. [DOI] [PubMed] [Google Scholar]
- Hamiche A., Sandaltzopoulos,R., Gdula,D.A. and Wu,C. (1999) ATP-dependent histone octamer sliding mediated by the chromatin remodeling complex NURF. Cell, 97, 833–842. [DOI] [PubMed] [Google Scholar]
- Hara R., Mo,J. and Sancar,A. (2000) DNA damage in the nucleosome core is refractory to repair by human excision nuclease. Mol. Cell. Biol., 20, 9173–9181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J.C. and Sancar,A. (1994) Determination of minimum substrate size for human excinuclease. J. Biol. Chem., 269, 19034–19040. [PubMed] [Google Scholar]
- Ito T., Bulger,M., Pazin,M.J., Kobayashi,R. and Kadonaga,J.T. (1997) ACF, an ISWI-containing and ATP-utilizing chromatin assembly and remodeling factor. Cell, 90, 145–155. [DOI] [PubMed] [Google Scholar]
- Ito T., Levenstein,M.E., Fyodorov,D.V., Kutach,A.K., Kobayashi,R. and Kadonaga,J.T. (1999) ACF consists of two subunits, Acf1 and ISWI, that function cooperatively in the ATP-dependent catalysis of chromatin assembly. Genes Dev., 13, 1529–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito T., Ikehara,T., Nakagawa,T., Kraus,W.L. and Muramatsu,M. (2000) p300-mediated acetylation facilitates the transfer of histone H2A-H2B dimers from nucleosomes to a histone chaperone. Genes Dev., 14, 1899–1907. [PMC free article] [PubMed] [Google Scholar]
- Kim J.K. and Choi,B.S. (1995) The solution structure of DNA duplex-decamer containing the (6-4) photoproduct of thymidylyl (3′→5′)thymidine by NMR and relaxation matrix refinement. Eur. J. Biochem., 228, 849–854. [DOI] [PubMed] [Google Scholar]
- Kingston R.E. and Narlikar,G.J. (1999) ATP-dependent remodeling and acetylation as regulators of chromatin fluidity. Genes Dev., 13, 2339–2352. [DOI] [PubMed] [Google Scholar]
- Kornberg R.D. and Lorch,Y. (1999) Twenty-five years of the nucleosome, fundamental particle of the eukaryote chromosome. Cell, 98, 285–294. [DOI] [PubMed] [Google Scholar]
- Langst G., Bonte,E.J., Corona,D.F. and Becker,P.B. (1999) Nucleosome movement by CHRAC and ISWI without disruption or trans-displacement of the histone octamer. Cell, 97, 843–852. [DOI] [PubMed] [Google Scholar]
- LeRoy G., Loyola,A., Lane,W.S. and Reinberg,D. (2000) Purification and characterization of a human factor that assembles and remodels chromatin. J. Biol. Chem., 275, 14787–14790. [DOI] [PubMed] [Google Scholar]
- Liu X. and Smerdon,M.J. (2000) Nucleotide excision repair of the 5S ribosomal RNA gene assembled into a nucleosome. J. Biol. Chem., 275, 23729–23735. [DOI] [PubMed] [Google Scholar]
- Liu X., Mann,D.B., Suquet,C., Springer,D.L. and Smerdon,M.J. (2000) Ultraviolet damage and nucleosome folding of the 5S ribosomal RNA gene. Biochemistry, 39, 557–566. [DOI] [PubMed] [Google Scholar]
- Luger K., Mader,A.W., Richmond,R.K., Sargent,D.F. and Richmond,T.J. (1997) Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature, 389, 251–260. [DOI] [PubMed] [Google Scholar]
- Mitchell D.L., Nguyen,T.D. and Cleaver,J.E. (1990) Nonrandom induction of pyrimidine-pyrimidone (6-4) photoproducts in ultraviolet- irradiated human chromatin. J. Biol. Chem., 265, 5353–5356. [PubMed] [Google Scholar]
- Mizukoshi T., Hitomi,K., Todo,T. and Iwai,S. (1998) Studies on the chemical synthesis of oligonucleotides containing the (6-4) photo product of thymine-cytosine and its repair by (6-4) photolyase. J. Am. Chem. Soc., 120, 10634–10642. [Google Scholar]
- Moggs J.G., Grandi,P., Quivy,J.P., Jonsson,Z.O., Hubscher,U., Becker,P.B. and Almouzni,G. (2000) A CAF-1-PCNA-mediated chromatin assembly pathway triggered by sensing DNA damage. Mol. Cell. Biol., 20, 1206–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paetkau D.W., Riese,J.A., MacMorran,W.S., Woods,R.A. and Gietz,R.D. (1994) Interaction of the yeast RAD7 and SIR3 proteins: implications for DNA repair and chromatin structure. Genes Dev., 8, 2035–2045. [DOI] [PubMed] [Google Scholar]
- Pehrson J.R. (1995) Probing the conformation of nucleosome linker DNA in situ with pyrimidine dimer formation. J. Biol. Chem., 270, 22440–22444. [PubMed] [Google Scholar]
- Pfeifer G.P. (1997) Formation and processing of UV photoproducts: effects of DNA sequence and chromatin environment. Photochem. Photobiol., 65, 270–283. [DOI] [PubMed] [Google Scholar]
- Sancar A. (1996) DNA excision repair. Annu. Rev. Biochem., 65, 43–81. [DOI] [PubMed] [Google Scholar]
- Sato M.H., Ura,K., Hohmura,K.I., Tokumasu,F., Yoshimura,S.H., Hanaoka,F. and Takeyasu,K. (1999) Atomic force microscopy sees nucleosome positioning and histone H1-induced compaction in reconstituted chromatin. FEBS Lett., 452, 267–271. [DOI] [PubMed] [Google Scholar]
- Schieferstein U. and Thoma,F. (1996) Modulation of cyclobutane pyrimidine dimer formation in a positioned nucleosome containing poly(dA.dT) tracts. Biochemistry, 35, 7705–7714. [DOI] [PubMed] [Google Scholar]
- Schieferstein U. and Thoma,F. (1998) Site-specific repair of cyclobutane pyrimidine dimers in a positioned nucleosome by photolyase and T4 endonuclease V in vitro. EMBO J., 17, 306–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen X., Mizuguchi,G., Hamiche,A. and Wu,C. (2000) A chromatin remodelling complex involved in transcription and DNA processing. Nature, 406, 541–544. [DOI] [PubMed] [Google Scholar]
- Thoma F. (1999) Light and dark in chromatin repair: repair of UV-induced DNA lesions by photolyase and nucleotide excision repair. EMBO J., 18, 6585–6598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tijsterman M., de Pril,R., Tasseron-de Jong,J.G. and Brouwer,J. (1999) RNA polymerase II transcription suppresses nucleosomal modulation of UV-induced (6-4) photoproduct and cyclobutane pyrimidine dimer repair in yeast. Mol. Cell. Biol., 19, 934–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ura K., Hayes,J.J. and Wolffe,A.P. (1995) A positive role for nucleosome mobility in the transcriptional activity of chromatin templates: restriction by linker histones. EMBO J., 14, 3752–3765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ura K., Nightingale,K. and Wolffe,A.P. (1996) Differential association of HMG1 and linker histones B4 and H1 with dinucleosomal DNA: structural transitions and transcriptional repression. EMBO J., 15, 4959–4969. [PMC free article] [PubMed] [Google Scholar]
- Ura K., Kurumizaka,H., Dimitrov,S., Almouzni,G. and Wolffe,A.P. (1997) Histone acetylation: influence on transcription, nucleosome mobility and positioning and linker histone-dependent transcriptional repression. EMBO J., 16, 2096–2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verreault A. (2000) De novo nucleosome assembly: new pieces in an old puzzle. Genes Dev., 14, 1430–1438. [PubMed] [Google Scholar]
- Vignali M., Hassan,A.H., Neely,K.E. and Workman,J.L. (2000) ATP-dependent chromatin-remodeling complexes. Mol. Cell. Biol., 20, 1899–1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C.I. and Taylor,J.S. (1991) Site-specific effect of thymine dimer formation on dAn.dTn tract bending and its biological implications. Proc. Natl Acad. Sci. USA, 88, 9072–9076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellinger R.E. and Thoma,F. (1997) Nucleosome structure and positioning modulate nucleotide excision repair in the non-transcribed strand of an active gene. EMBO J., 16, 5046–5056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolffe A.P. (2000) Chromatin: Structure and Function. Academic Press, San Diego, CA.
- Yasui A. and McCready,S.J. (1998) Alternative repair pathways for UV-induced DNA damage. BioEssays, 20, 291–297. [DOI] [PubMed] [Google Scholar]