

Abstract
SNF2/SWI2-related ATPases employ ATP hydrolysis to disrupt protein–DNA interactions, but how ATP hydrolysis is coupled to disruption is not understood. Here we examine the mechanism of action of MOT1, a yeast SNF2/SWI2-related ATPase that uses ATP hydrolysis to remove TATA binding protein (TBP) from DNA. MOT1 function requires a 17 bp DNA ‘handle’ upstream of the TATA box, which must be double stranded. Remarkably, MOT1-catalyzed disruption of TBP–DNA does not appear to require DNA strand separation, DNA bending or twisting of the DNA helix. Thus, TBP–DNA disruption is accomplished in a reaction apparently not driven by a change in DNA structure. MOT1 action is supported by DNA templates in which the handle is connected to the TATA box via single-stranded DNA, indicating that the upstream duplex DNA can be conformationally uncoupled from the TATA box. Combining these results with proposed similarities between SNF2/SWI2 ATPases and helicases, we suggest that MOT1 uses ATP hydrolysis to translocate along the handle and thereby disrupt interactions between TBP and DNA.
Keywords: ATPase/MOT1/TBP/transcription
Introduction
The ATPase activities of SNF2/SWI2-related proteins are of at least two types (Pazin and Kadonaga, 1997; Vignali et al., 2000). Many proteins in this family possess DNA-stimulated ATPase activity. ATPases with DNA-stimulated ATPase activity have been shown to alter histone–DNA contacts and to rearrange or displace nucleosomes from DNA in vitro (Kingston and Narlikar, 1999; Vignali et al., 2000). ATP-dependent remodeling of the extensive protein–DNA interface in the nucleosome might require movement of the enzyme along DNA coupled with multiple rounds of ATP hydrolysis (Logie and Peterson, 1997; Whitehouse et al., 1999; Havas et al., 2000). A second kind of ATPase activity is exemplified by MOT1, an essential transcriptional regulator in Saccharo myces cerevisiae (Davis et al., 1992; Piatti et al., 1992; Collart, 1996; Madison and Winston, 1997; Prelich, 1997). MOT1 interacts specifically with TATA binding protein (TBP) in vitro and in vivo (Auble and Hahn, 1993; Poon et al., 1994), and MOT1’s ATPase activity is stimulated by TBP in vitro (Auble et al., 1997; Adamkewicz et al., 2000). In vitro, interaction of MOT1 with TBP–DNA complexes leads to TBP–DNA complex disruption in the presence of ATP (Auble and Hahn, 1993; Auble et al., 1994). Previous work has shown that MOT1 acts locally and transiently at TBP–DNA complexes, and that MOT1 has no detectable processivity along DNA (Auble and Steggerda, 1999).
How is ATP hydrolysis coupled to disruption of protein–DNA complexes? SNF2/SWI2-related proteins have been widely suggested to fall within a helicase superfamily (Gorbalenya et al., 1989), but proteins in the SNF2/SWI2 family that have been tested do not display helicase activity in vitro (Pazin and Kadonaga, 1997). Interestingly, one member of this family, INO80, is found in a complex with proteins related to the bacterial RuvB branch migration protein, and the INO80 complex has helicase activity that depends on functional ATPase activity of the INO80 polypeptide (Shen et al., 2000). SNF2/SWI2 ATPase activity may therefore be coupled to helicase activity in some assemblies. Since the sequences of the ATPase domains of different SNF2/SWI2-related proteins are similar (Eisen et al., 1995), the catalytic portions of these enzymes are likely to have a similar overall fold. Structures of four helicases have recently been reported (Korolev et al., 1997; Yao et al., 1997; Benz et al., 1999; Johnson and McKay, 1999; Velankar et al., 1999) and all of the conserved ‘helicase’ motifs (Gorbalenya et al., 1989; Gorbalenya and Koonin, 1993) fall within domains 1A and 2A, comprising the ATPase motor (Korolev et al., 1998). These domains are coupled to less well conserved domains that confer specificity for different DNA substrates (Bird et al., 1998). Thus, many enzymes that have been defined as helicases based on conservation of the ATPase motor probably do not unwind DNA, but rather couple ATP hydrolysis to DNA translocation in other ways (Bird et al., 1998; Egelman, 1998; Hall and Matson, 1999; Soultanas et al., 2000).
One possibility is that SNF/SWI ATPases drive changes in protein–DNA interactions by using ATP hydrolysis to induce changes in DNA structure that propagate through protein–DNA interfaces, but that these changes in DNA structure do not represent classical helicase strand unwinding activity. Indeed, a persistent change in DNA topology that is induced by the SNF/SWI complex has been reported (Imbalzano et al., 1996; Schnitzler et al., 1998; Guyon et al., 1999). Furthermore, yeast SWI/SNF complex, Xenopus Mi-2 complex, recombinant ISWI and recombinant BRG1 have all been shown to generate negative superhelical torsion in an ATP-dependent manner, suggesting that DNA conformational change is involved in disruption of protein–DNA contacts by these enzymes (Havas et al., 2000). Propagation of a change in DNA structure by the CHRAC and ISWI complexes (Langst et al., 1999) and by the SNF2/SWI2 complex (Boyer et al., 2000) has also been suggested to explain how these ATPases induce nucleosome mobility. Evidence for a role for a SNF2/SWI2-related ATPase in driving conformational changes in DNA also derives from an analysis of RAD54 function (Petukhova et al., 1998). RAD54 has been shown to use ATP hydrolysis to drive homologous pairing between DNA strands in a reaction that requires the yeast RecA homolog RAD51 (Petukhova et al., 1998). Here we explicitly test the idea that MOT1 disrupts TBP–DNA complexes by using ATP hydrolysis to alter DNA structure through the TATA box. The data strongly suggest that MOT1 does not displace TBP from DNA via an induced change in DNA structure that involves DNA twisting, bending or strand separation propagating from the MOT1 DNA binding site. We suggest instead that MOT1 functions as a molecular plow that displaces TBP from DNA by translocation through the TATA box in an ATP-dependent manner. This model fits well with a predicted underlying similarity between helicases and SNF/SWI ATPases, which had previously been generally unsupported by performing conventional helicase assays. This function of the MOT1 ATPase also highlights the diversity associated with the utilization of a conserved ATPase motor.
Results
Structural organization of the MOT1–TBP–DNA complex
To determine the overall organization of MOT1 and TBP in the ternary complex, DNase I footprinting was performed. As shown in Figure 1, binding of TBP led to protection from DNase I digestion of ∼20 bp of DNA centered on the TATA box (lanes 3 and 8). Incubation of the DNA probe with MOT1 alone led to no significant changes in DNase I digestion (lanes 2 and 7), indicating that MOT1 alone does not specifically bind to the DNA. Incubation of MOT1 with TBP and DNA led to an extension of protection from DNase I digestion upstream of the TATA box (lanes 4 and 9). On the top strand, the upstream protection extends ∼20 bases to position –57; this protection is punctuated by a hypersensitive site at –50. A qualitatively similar result was obtained on the bottom strand with upstream protection punctuated by a hypersensitive site at –43, although for unknown reasons the upstream boundary of the footprint could not be determined. Thus, MOT1 appears to occupy a position adjacent to TBP and upstream of the TATA box, perhaps in contact with upstream DNA. Four other hypersensitive sites were also observed at –21 and –22 on the top strand and –21 and –23 on the bottom strand. These four hypersensitive sites encircle the DNA in a region that is protected from DNase I by TBP alone, suggesting that incubation with MOT1 causes a change in the contacts between TBP and DNA. The possible significance of this observation is discussed below. In the presence of ATP, MOT1 induces the removal of TBP from DNA, as expected (lanes 5 and 10).

Fig. 1. DNase I footprinting of TBP–DNA and MOT1–TBP–DNA complexes. A radiolabeled 110 bp DNA probe (∼0.15 nM) containing a consensus TATA sequence labeled on either the top (A) or the bottom strand (B) was incubated with 5 nM TBP and/or 20 nM MOT1 and 10 µM ATP as indicated. The reactions were treated with DNase I and reaction products were resolved on a denaturing high resolution polyacrylamide gel and visualized by autoradiography as described in Materials and methods.
The DNA substrate requirements for the formation of a MOT1–TBP–DNA ternary complex were analyzed by two kinds of experiment. First, hydroxyl radical treatment of DNA was used in ‘missing nucleoside’ experiments to determine which sugars and bases in the DNA substrate are required for ternary complex formation (Tullius et al., 1987). To do this, radiolabeled DNA was treated with hydroxyl radical to randomly remove one nucleoside on average per template molecule. This population of modified DNAs was then incubated with TBP or TBP and MOT1, and protein–DNA complexes were separated from one another and from free DNA by non-denaturing gel electrophoresis (Figure 2A). DNA in each of the complexes was purified, and analyzed by high resolution gel electrophoresis as described (Tullius et al., 1987; Auble and Steggerda, 1999). As shown in Figure 2B and C (lanes 4 and 9), incubation of modified DNA with TBP revealed 8 bp of protection centered on the TATA box (brackets) and DNAs missing nucleosides in the TATA box were enriched in the free DNA population (lanes 5 and 10). The 8 bp protection is in good agreement with the region of DNA protected by TBP observed in the co-crystal structure of TBP–DNA (Kim et al., 1993a,b). Suprisingly, only loss of nucleosides within the TATA box impairs formation of the MOT1–TBP–DNA complex (Figure 2B and C, compare lanes 2 with 4 and 7 with 9). Therefore, no nucleosides outside of the TATA box are absolutely required for formation of the MOT1–TBP– DNA ternary complex. In principle, it is possible to treat protein–DNA complexes with hydroxyl radical to map close contacts between proteins and DNA (Tullius et al., 1987), but the solution conditions required for hydroxyl radical treatment were found to be incompatible with MOT1 activity (data not shown).

Fig. 2. Identification of nucleosides required for TBP and MOT1 interaction with DNA by hydroxyl radical treatment. The same radiolabeled DNA probes as used in Figure 1 were pre-treated with hydroxyl radical to remove random nucleosides. The collection of damaged DNAs (∼3 nM) was then incubated with 5 nM TBP and/or 20 nM MOT1 as indicated, and the free DNA and protein–DNA complexes were resolved by non-denaturing PAGE (A). The indicated bands were then excised and resolved on high resolution denaturing polyacrylamide gels to identify the nucleosides critical for interaction with TBP and MOT1 (B and C). Note the depletion of DNA molecules missing TATA box nucleosides in the reactions containing TBP and TBP plus MOT1.
DNA substrate requirements for MOT1 activity were also analyzed using DNA templates of different lengths. As shown in Figure 3, MOT1–TBP–DNA ternary complex formation required a 17 bp extension of DNA upstream of the TATA box, whereas templates with shorter upstream extensions did not support ternary complex formation or ATP-dependent TBP–DNA disruption very well or at all. In contrast, MOT1 binding was relatively unaffected by deletion of DNA downstream of the TATA box, with the only significant impairment in ternary complex formation occurring with a template truncated at position –24, which did not support binding of TBP alone under these conditions. Importantly, the requirement for DNA upstream of the TATA box for ternary complex formation indicates that MOT1 contacts the DNA in this region. Furthermore, there is no sequence conservation among templates that support MOT1 binding in vitro (Auble and Hahn, 1993) so the contacts made by MOT1 in this region do not appear to be sequence specific. Below, we refer to the DNA upstream of the TATA box required for MOT1 function as the ‘handle’.

Fig. 3. Formation of the MOT1–TBP–DNA complex requires 17 bp of DNA upstream of the TATA box. Radiolabeled DNA probes (0.15 nM) of the lengths indicated were incubated with 5 nM TBP, TBP plus 1 or 4 U (∼6 or 24 nM) of MOT1, and TBP plus MOT1 and 10 µM ATP. Protein–DNA complexes were detected by non-denaturing gel electrophoresis. The results are summarized in (A); electrophoretic mobility shift results using probes that bracket the boundary between functional and non-functional DNAs are shown in (B).
MOT1-catalyzed TBP–DNA disruption requires duplex DNA in the handle
Three general classes of models were considered to explain how MOT1 might catalyze disruption of TBP– DNA complexes. First, MOT1 could use ATP hydrolysis to drive a conformational change in DNA through a protein–DNA complex, or secondly, MOT1 might use ATP hydrolysis to translocate along DNA using DNA as a track. A third possibility is that MOT1 might induce a conformational change in TBP that alters TBP’s affinity for DNA, or that MOT1 induces a transient change in DNA structure that is localized to the TATA box. To test directly whether MOT1 uses ATP hydrolysis to induce changes in DNA structure proximal to or through the TATA box, DNA templates were constructed that either constrain or alter one or more of the properties of linear duplex DNA. The requirement for a 17 bp DNA handle that extends upstream of the TBP–DNA complex suggests that MOT1 may grip the handle and use ATP hydrolysis to twist the DNA duplex to rotate TBP away from its interaction with the minor groove. To test this idea, DNA constructs containing single-stranded tails and gaps were constructed (Figures 4A and 5A). We reasoned that twisting of the DNA duplex in the upstream region would require intact duplex DNA to provide the torque for driving TBP–DNA disruption downstream, whereas single-stranded DNA would allow rotation about the single bonds in the sugar–phosphate backbone. The twisting seemed possible because in addition to contacting DNA in the handle, MOT1 also contacts TBP in the absence of ATP (see below and Poon et al., 1994; Cang et al., 1999), so the enzyme bridges between the upstream DNA and TBP.

Fig. 4. DNA containing single base pair gaps is competent for MOT1 binding and ATP-driven disruption of TBP–DNA. (A) Diagram of DNA constructs. The control construct was based upon the adenovirus major late promoter (numbers indicate the position in the promoter), with most AT base pairs being changed to GC for the cross-linking experiment (below; see Table I for oligonucleotide sequences). Gray shading indicates the position of the sequence TATAAAAG. Eleven constructs with 1 bp gaps in the upstream DNA were made. Label was placed on the TATA-less upstream strand, so no shift is seen unless the construct anneals fully. (B) Gel mobility shift results using fully duplex control DNA (lanes 1–5) and nine of the constructs made. TBP core domain was used at 5 nM. The reaction in lane 1 contained no TBP. ATP, added where indicated, was used at 5 µM. Units of MOT1 were added as indicated. One unit of MOT1 is estimated to be 6 nM. The DNA concentration is ∼0.05 nM. Positions of ternary complex (‘3°’), TBP–DNA complex (‘TBP–DNA’) and unbound DNA probe (‘Probe’) are indicated. Results with 5G3 and 5G5 (not shown) are identical. On construct 3G3, two discrete TBP–DNA complexes were detected; the reason for this is unknown but was specific for a template with a gap at this position.

Fig. 5. Effects of large gaps in the upstream DNA on MOT1 binding and ATP-driven disruption of TBP–DNA. (A) Diagram of DNA constructs. The control is the same as in Figure 4; other constructs are derived from control, missing sequences at the positions indicated. Upstream segments are labeled. (B) Gel mobility shift results using fully duplex control DNA and the 5C7 template, which has an 18 base single-stranded tail. The positions of the MOT1–TBP–DNA complex (‘3°’) and TBP–DNA complex (‘TBP’) are indicated; the free DNA band is not shown. Reactions in lanes 1 contained no MOT1, 1 U of MOT1 (∼6 nM) was added to the reactions in lane 2 and the amount of MOT1 was doubled in successive lanes. (C) Gel mobility shift results obtained with each of the DNA constructs. Conditions were as described for Figure 4B, except that 4 U of MOT1 (∼24 nM) were used in each MOT1-containing reaction. Constructs 5C7 and 3C7 support ternary complex but MOT1-catalyzed disruption is severely impaired. Positions of ternary complex (‘3°’), TBP–DNA complex (‘TBP’) and free DNA probe (‘DNA’) are indicated. In (B) and (C), DNA was present at 0.15 nM and TBP was added to 5 nM concentration.
These constructs were tested for TBP binding, ternary complex formation and ATP-dependent disruption by gel shift assay. To account for potential differences in specific radioactivity or TBP binding by these templates, the relative abilities of the DNAs to support ternary complex formation were determined by measuring the ratio of the amount of ternary complexes formed to the amount of TBP–DNA complexes formed in reactions with different amounts of MOT1. The effects of single base pair gaps placed upstream of the TATA box are shown (Figure 4B). Construct 5G1 contains a gap immediately upstream of the TATA box and supports TBP binding, formation of the ternary complex and ATP-dependent disruption as well as the fully duplex control (Figure 4B, compare lanes 7–11 with lanes 2–6). Construct 3G1, containing the same gap but on the other strand, bound TBP poorly and could not be assayed (data not shown). However, constructs containing 1 bp gaps farther away from the TATA box in the 3′ strand did support TBP binding, ternary complex formation and ATP-dependent disruption (Figure 4B, lanes 32–46). Aside from 3G1, all constructs with 1 bp gaps were fully functional (Figure 4B and data not shown for 5G3 and 5G5), and these gaps were placed from 1 to 7 bp upstream of the TATA box, indicating that MOT1 does not require its upstream DNA binding site to be conformationally linked to the TATA box. Conformational flexibility imparted by the 1 bp gaps suggests that MOT1 action does not require a force generated by bending the DNA handle, a conclusion supported by data described below. Additionally, four constructs were made with 3 bp gaps >7 bp upstream of the TATA box (Figure 5A). Among these, three were functional in our assay (5T10, 3T7 and 3T10; Figure 5C). The fourth, 5T7, was defective for ternary complex formation (Figure 5C, lane 8) and possibly for TBP disruption (lane 9).
In comparison, two constructs were made in which the 18 bases at the upstream end of the DNA handle are single stranded (Figure 5A, 5C7 and 3C7). Both ‘tail’ constructs displayed reduced levels of ternary complex formation (titration for 5C7 shown in Figure 5B). By approximation from gel shift, TBP–3C7 binds MOT1 with half the affinity of the control TBP–DNA complex; the affinity of MOT1 for TBP–5C7 is 8-fold reduced. However, even with a significant amount of ternary complex formation, TBP–DNA disruption was almost undetectable in the presence of ATP (Figure 5C, lanes 5 and 7 versus lane 3).
Comparison of the effects of MOT1 on the 3C7 tailed template and the gapped template (3T7), which has a three base gap on the same strand as the strand truncation in 3T7, is also informative. Ternary complex formation occurred with similar concentrations of MOT1 on both of these templates, but the tailed template was defective for ATP-driven disruption, whereas the gapped template was not. Conformational changes in DNA can not be transmitted through the gapped DNA, so these data suggest that MOT1 requires ATP hydrolysis to move along the duplex DNA upstream of the 3T7 gap. The results of these experiments show that while the upstream DNA binding site need not be conformationally linked to the TATA box, both DNA strands upstream of the TATA box are involved in formation of the MOT1–TBP–DNA complex and may play a role in MOT1 catalysis.
MOT1 does not separate DNA strands between the handle and the TATA box
To test directly the idea that MOT1 uses ATP hydrolysis to separate the DNA strands, and that strand separation is required to dislodge TBP from the TATA box, DNA templates were constructed that contained site-specific psoralen cross-links either upstream or both upstream and downstream of the TATA box. As shown in Figure 6A, introduction of a psoralen cross-link upstream of the TATA box generated a DNA template that supported both TBP and MOT1 binding (lanes 6 and 7). In the presence of ATP, MOT1 efficiently removed TBP from this cross-linked template (lane 8). Thus, the upstream DNA sequence required for MOT1 loading onto TBP–DNA is not used to propagate a region of melted DNA through the TATA box, as would be expected if MOT1 functioned as a site-specific helicase. This result was expected because a similar observation was published while these experiments were in progress (Adamkewicz et al., 2000). To test the idea that MOT1 reaches into the TATA box and unwinds DNA specifically in this sequence, a template with cross-links both upstream and downstream of the TATA box was constructed. As shown in Figure 6B (lanes 4–6), the doubly cross-linked template supported TBP binding, MOT1–TBP–DNA ternary complex formation and MOT1- catalyzed TBP–DNA disruption in the presence of ATP. Quantitation of the band intensities by PhosphorImager demonstrated that under these conditions, MOT1 directed the disruption of ∼70% of the TBP–DNA complexes formed on the wild-type DNA probe and ∼60% of the TBP–DNA complexes formed on the psoralen-cross-linked probe. There may be a very small decrease in MOT1-catalyzed TBP–DNA disruption on the doubly cross-linked DNA template, but these results demonstrate that DNA strand separation through the TATA box is not required for ATP-dependent TBP–DNA disruption by MOT1.
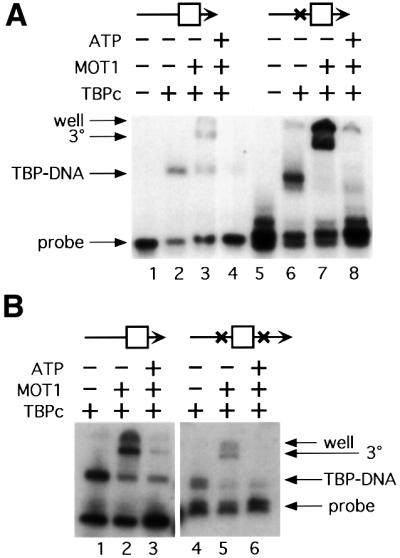
Fig. 6. Psoralen cross-links flanking the TATA box do not impair MOT1 function. (A) Singly cross-linked DNA compared with control. The psoralen cross-linking site is indicated (X), and the TATA box is schematized by the square. Where indicated, 5 nM TBP core domain, 1 U of MOT1 (∼6 nM) and 5 µM ATP were incubated with radiolabeled DNA probes (∼0.15 nM) constructed as described in Materials and methods. Results in lanes 1–4 are from reactions using duplex DNA with no cross-links. Lanes 5–8 contain DNA probe with a single psoralen cross-link between the upstream DNA handle and the TATA box. The positions of the free DNA, TBP–DNA complexes (‘TBP–DNA’) and MOT1–TBP–DNA complex (‘3°’) are indicated. ‘Well’ indicates the top of the gel. (B) Doubly cross-linked DNA compared with control. The procedure was the same as for (A).
MOT1 removes TBP bound to a highly constrained minicircle
Binding of TBP to the minor groove of the TATA sequence induces helix unwinding and bending of the DNA helix by ∼90° (Kim et al., 1993a,b). One possibility is that MOT1 uses ATP hydrolysis to straighten the TATA sequence and thereby induce dissociation of TBP. This idea was tested by constructing 156 bp minicircles containing a TATA sequence. The orientation of the TATA box was fixed in the minicircles by introducing phased A tracts either 31 or 37 bp away from the TATA sequence. The phased A tracts introduce a static bend in the DNA, which then orients the TATA sequence with either a bend towards the minor groove (31 bp spacer) or a bend towards the major groove (37 bp spacer) (Parvin et al., 1995; Kahn, 2000). As shown in Figure 7 (top panel), while binding of TBP to linear probes with 31 or 37 bp spacers was similar, pre-bending the TATA sequence towards the major groove substantially improved the binding of TBP to DNA (top panel, compare circular 37 bp spacer with 31 bp spacer). This result is in good agreement with previous results (Parvin et al., 1995), which demonstrated a 100-fold increase in the affinity of TBP for DNA when the TATA sequence was pre-bent towards the major groove compared with unbent DNA and a 300-fold increase in affinity when binding was compared with a minicircle with the TATA box pre-bent towards the minor groove.

Fig. 7. MOT1 disrupts TBP–DNA complexes formed on minicircle DNA. Gel shift experiments were performed with radiolabeled minicircle DNA (∼0.15 nM) or the identical linear sequence incubated with TBP (top panel) or TBP ± MOT1 ± ATP as indicated (bottom panel). Two sets of probes were used: circular and linear DNAs with a 31 bp spacer between the TATA box and phased A tracts, and circular and linear probes with a 37 bp spacer between the TATA box and phased A tracts. In the minicircle constructs, the 31 bp spacer pre-bends the DNA towards the minor groove of the TATA box (inhibiting TBP binding), whereas the minicircle with the 37 bp spacer pre-bends the TATA box towards the major groove, greatly enhancing the binding of TBP to this probe. The protein–DNA complexes are indicated. Reactions contained 5 nM TBP, 10 µM ATP and ∼1 or 4 U (6 or 24 nM) of MOT1 as indicated. In the top panel, note that a very small amount of residual circular DNA co-migrates with TBP–DNA complexes formed on the linear probes.
The effect of MOT1 addition to TBP–minicircle complexes is shown in Figure 7 (lower panel). Addition of MOT1 resulted in the formation of MOT1–TBP–DNA complexes and disruption of TBP–DNA in the presence of ATP. The effect of MOT1 on TBP–DNA complexes formed on the linear template of identical sequence is shown for comparison. In the absence of ATP, comparing the linear and circular templates, MOT1 stabilizes binding of TBP to linear DNA, which would otherwise be much less stable in non-denaturing gels (Hoopes et al., 1992); TBP–DNA complexes formed on pre-bent minicircle DNA are much more stable than those formed on linear DNA and stabilization of TBP–minicircle DNA complexes by MOT1 was not observed. Despite greater TATA box occupancy by TBP on the minicircle template, substantial disruption by equivalent amounts of MOT1 was detected in the presence of ATP, indicating that the efficiency of TBP–DNA disruption on circular and linear templates is similar. Since the minicircle is extremely small and constrained, these results suggest that MOT1 is unlikely to function by a mechanism involving straightening of the bent DNA in complex with TBP. Furthermore, the 156 bp minicircle would be expected to provide a substantial barrier for induction of topological stress by other mechanisms [helix twisting, strand melting (Kahn, 2000)], and these results therefore reinforce data described above indicating that MOT1 does not induce TBP–DNA disruption by driving transient conformational changes in DNA.
Interaction of MOT1 with TBP off DNA inhibits DNA binding by TBP
Previous results indicated that the major groove of the TATA box plays a role in the efficiency of TBP–DNA dissociation by MOT1 (Auble et al., 1997). MOT1 action could be accomplished by insertion of a portion of the protein into the TATA box on the ‘underside’ of the TBP–DNA complex; ATP-driven TBP–DNA dissociation could then occur by pushing TBP away from DNA. In the absence of ATP and DNA, MOT1 interacts with TBP, but the properties of the MOT1–TBP complex have not been analyzed. As shown in Figure 8, pre-incubation of MOT1 and TBP eliminated the ability to detect TBP–DNA or MOT1–TBP–DNA complexes when DNA was subsequently added (compare lanes 2 and 4). To demonstrate that this inhibition is due to the association of MOT1 with TBP, reactions were performed in parallel using affinity purified material from a yeast strain in which MOT1 is not epitope tagged. TBP binding to DNA was not inhibited by pre-incubation with the mock-purified material, indicating that MOT1 was required for this effect (Figure 8, lanes 6 and 8). In reactions containing MOT1 and ATP, TBP– DNA complexes were not detected either because ATP does not induce the dissociation of the MOT1–TBP complex or because TBP that is liberated from the complex was then removed from the DNA template by MOT1 once it had bound.

Fig. 8. The MOT1–TBP complex is not competent to bind DNA, but TBP is released from MOT1 in the presence of ATP. Gel mobility shift results obtained with a 40mer DNA probe that contains the upstream handle (lanes 1–9) and a 30mer DNA probe that does not support MOT1 function (lanes 10 and 11). Lane 1 shows the position of the TBP–DNA complex (also indicated by the leftward arrow adjacent to lane 11). TBP and DNA or TBP and MOT1 were pre-incubated for 20 min followed by the addition of MOT1 or DNA ± ATP for 5 min as indicated in the reaction schemes labeled 1 and 2. The reactions in lanes 2–5 and 10 and 11 contained wild-type MOT1, whereas the reactions in lanes 6–9 contained eluate obtained from a mock affinity purification using extract in which MOT1 was not epitope tagged. Note that pre-incubation of TBP and MOT1 prevents formation of protein–DNA complexes, whether or not ATP is present in the reaction (lanes 4 and 5); the effect depends on MOT1 in the reaction (lanes 8 and 9), and TBP binding to DNA can be recovered in the presence of ATP if a DNA probe is used that sequesters TBP from MOT1 action (lane 11). Reactions contained 5 nM TBP, 10 µM ATP and ∼30 nM MOT1 as indicated.
To determine whether ATP induces disruption of the MOT1–TBP complex, a radiolabeled DNA probe that is too short to support MOT1 binding was added with or without ATP to reactions containing pre-incubated MOT1 and TBP. As shown in Figure 8 (lane 11 versus lane 10), TBP–DNA complexes were detected under these conditions using the short DNA probe, suggesting that MOT1 blocks the DNA binding surface of TBP in the MOT1–TBP complex and that addition of ATP leads to dissociation of MOT1 and TBP. Since co-occupancy of MOT1 and TBP could be detected on DNA but the MOT1–TBP complex is incompetent to bind DNA, the MOT1 machinery may be inserted under or adjacent to the TBP–DNA complex. It is also possible that MOT1 binding to TBP does not occlude the TBP DNA binding surface, but that MOT1 induces a structural change in TBP that renders it incompetent to bind DNA.
Discussion
Models for MOT1 function
Three general models might explain how MOT1 functions. First, MOT1 might use ATP hydrolysis to induce a conformational change in the upstream DNA handle that is propagated through the TBP–DNA interface to cause disruption of the TBP–DNA complex. Secondly, MOT1 might use ATP hydrolysis to translocate along DNA and thereby remove TBP from DNA by pushing or pulling it off. Thirdly, MOT1 might function by using ATP hydrolysis to introduce a local change in TATA DNA or TBP structure that reduces the affinity of TBP for DNA. The experiments described here explicitly test the role of DNA structural change in driving TBP–DNA complex dissociation. Each of these models is discussed in the context of our findings below.
Structure of the MOT1–TBP–DNA complex
DNase I footprinting of MOT1–TBP–DNA complexes demonstrates a stereospecific interaction of MOT1 with TBP–DNA. The extension of DNase I protection upstream of the TATA box could be due to direct contacts between MOT1 and DNA or simple proximity of the large MOT1 protein to the upstream DNA. Deletion analysis demonstrates that DNA upstream of the TATA box is required for the formation of a stable ternary complex, indicating that MOT1 contacts upstream DNA (Figure 3). The observation that the 17 bp upstream DNA handle supports ternary complex formation, whereas shorter templates were greatly impaired, suggests that MOT1 contacts DNA at the upstream end of the minimum functional DNA template. However, no specific nucleosides upstream of the TATA box were found to be required for MOT1– TBP–DNA complex formation. Previous data demonstrated that MOT1 can remove TBP from DNA templates with no sequence similarity upstream of the TATA box (Auble and Hahn, 1993), so we suggest that MOT1 makes primarily electrostatic interactions with upstream DNA phosphates that are not perturbed in the missing nucleoside experiments (Tullius et al., 1987). As discussed below, recognition of DNA in a non-sequence-specific manner would be advantageous for a protein that translocates along DNA using the phosphate backbone as a track.
DNase I footprinting also revealed several DNase I-hypersensitive sites. These include sites at –43 (bottom strand) and –50 (top strand). Modeling standard B-form DNA extending upstream from the TBP–DNA complex locates these two hypersensitive sites on the ‘upper’ surface of the double helix with the –50 site just upstream of the boundary of the MOT1-induced footprint (not shown). The hypersensitive site at –43 is within the upstream MOT1 footprint, suggesting that MOT1 contacts two separate regions of the upstream DNA (punctuated by a DNase I-accessible site) or that the MOT1–DNA interface wraps around the helix. Interestingly, introduction of a three base gap just downstream of this hypersensitive site (5T7; Figure 5) strongly impairs the interaction of MOT1 with TBP–DNA. Since the extreme upstream end of the ‘handle’ also appears to stabilize MOT1’s interaction with TBP and DNA (discussed above), MOT1 interactions with the upstream DNA are apparently extended along the length of the upstream DNA.
Altered TBP–DNA contacts in the MOT1–TBP–DNA complex?
DNase I-hypersensitive sites were also located immediately downstream of the TATA box at positions –21, –22 (top strand) and –21 and –23 (bottom strand). These sites form a ring of DNase I hypersensitivity, which is induced by MOT1 in a region of DNA that is completely protected from nuclease digestion by TBP alone. The simplest interpretation of these hypersensitive sites is that binding of MOT1 to TBP–DNA alters contacts between TBP and DNA even though no ATP is present in the reaction. This might reflect partial dissociation of TBP from DNA in the ternary complex and could be analogous to formation of the ‘primed’ complex between PcrA and DNA in which duplex DNA is distorted prior to ATP hydrolysis (Velankar et al., 1999). As discussed above, one model for MOT1 function posits that MOT1 induces a conformational change in TBP and that this conformational change decreases the affinity of TBP for DNA; the DNase I footprinting data are consistent with this idea (Figure 9D). Likewise, the inability of the MOT1–TBP complex to bind DNA could reflect a conformational change in TBP induced by MOT1 that alters TBP’s DNA binding surface. MOT1 has been proposed to function by altering the structure of TBP (Adamkewicz et al., 2000), although there is as yet no direct experimental support for this. We favor instead the idea that MOT1 physically blocks access to the TBP DNA binding surface when it interacts with TBP. TBP–DNA complexes formed on DNAs with <17 bp of upstream DNA do not support formation of the MOT1–TBP–DNA ternary complex (Figure 3) despite the fact that MOT1–TBP complexes form in the absence of DNA (Figure 8). Small recombinant fragments of the MOT1 N-terminus can also specifically recognize TBP and prevent binding of TBP to DNA (our unpublished data). Thus, occupancy of the TBP DNA binding surface by a DNA molecule and binding of MOT1 to TBP appear to be mutually exclusive, consistent with the idea that MOT1 binding to TBP blocks access to TBP’s DNA binding surface.

Fig. 9. Model for MOT1-catalyzed ATP-dependent disruption of TBP–DNA. A cartoon of the MOT1–TBP–DNA ternary complex is shown in (A). Three mechanisms are possible. MOT1 could be anchored to the DNA handle and ATP hydrolysis is used to drive a wedge to dislodge TBP (B). Alternatively, ATP hydrolysis powers translocation of MOT1 along DNA to disrupt the association of TBP with DNA (C). In these cases, duplex DNA in the handle is employed as a track to direct movement of MOT1 or a domain of MOT1. In (C), movement of MOT1 could be in either direction: movement of MOT1 to the left would cause TBP dissociation by ‘pulling’, whereas movement to the right would cause disruption of TBP–DNA by ‘pushing’. See text for discussion. (D) The upstream DNA handle might function as an allosteric activator for MOT1, which is required for MOT1 to effect a change in TBP conformation or a local change in TATA DNA structure.
DNA templates that constrain bending and strand separation
Previous data demonstrated that MOT1 does not track along DNA over distances as short as ∼40 bp (Auble and Steggerda, 1999), but the observation that the interaction of MOT1 with TBP–DNA complexes is local and transient did not rule out the possibility that MOT1 induces local changes in DNA structure in the immediate vicinity of TBP–DNA complexes. Introduction of psoralen cross-links flanking the TATA box indicates that MOT1 does not function via propagation of DNA strand separation through the TATA box to dissociate TBP. These results support and extend those obtained by Adamkewicz et al. (2000), in which a DNA construct with a psoralen cross-link upstream of the TATA box also supports MOT1 activity in vitro. It has so far not been possible to either engineer a template with a cross-link within the TATA box that still binds TBP or to move the psoralen cross-links closer to the TATA box. It is formally possible, therefore, that MOT1 induces DNA denaturation only within the TATA box, but such a mechanism can not explain the effects observed with gapped DNA templates (see below). Similarly, TBP–DNA complexes formed on highly constrained minicircle DNA also function as efficient substrates for MOT1-catalyzed disruption. The efficiency of MOT1 function on the linear and circular probes is difficult to compare directly due to the substantially increased occupancy by TBP of the TATA box on the circular versus the linear probe, but over the range of MOT1 concentrations used, disruption of TBP–DNA complexes formed on the two templates is similar. Since the pre-bent minicircle DNA has ∼100-fold higher affinity for TBP than the linear probe (Parvin et al., 1995), and the increased affinity is due largely to a decrease in the TBP–DNA dissociation rate (Parvin et al., 1995; Kahn, 2000), the failure to detect any difference in MOT1 activity directed towards complexes formed on the linear versus circular probes provides a strong argument that DNA straightening is not required for MOT1 action. The covalently closed minicircle is also likely to provide a barrier to enzymes that induce other changes in DNA structure, although the magnitude of the barrier is difficult to estimate (Kahn, 2000).
Many templates with gaps between the TATA box and the handle support MOT1 function
As shown in Figure 4, single base gaps can be introduced at 11 different positions on either strand of the DNA linking the TATA box and the upstream handle without affecting MOT1–TBP–DNA ternary complex formation or MOT1-driven ATP-dependent disruption. Duplex DNA in the handle is required for MOT1 activity, but these results demonstrate that MOT1’s grip on the handle is not used as a lever to force TBP off DNA by twisting or bending, since the single-stranded DNA connecting the TATA box and the handle allows the two duplex regions to be rotated or bent with respect to one another. Likewise, we suggest that templates containing a single-stranded swivel between the TATA and the handle would be unlikely to support MOT1 function if MOT1 worked via a localized change in TATA DNA structure or by inducing a conformational change in TBP. It is possible that the upstream DNA functions as an allosteric activator of MOT1, which induces a conformational change in MOT1 required for catalysis (Figure 9D). However, this model is not easily reconciled with the requirement for duplex DNA in the handle and the failure of template 3C7 to support MOT1-catalyzed disruption while displaying only a modest defect in ternary complex formation. Also, the ATPase activity of MOT1 is activated by TBP and not by DNA, implicating TBP and not DNA as an allosteric effector of MOT1 (Auble et al., 1997; Adamkewicz et al., 2000). Therefore, while the experiments presented here do not disprove models for MOT1 function that involve either conformational changes in TBP or a very local change in TATA structure, we believe that this type of model is less likely. Future work using probes for transient changes in TATA DNA and TBP structure should afford a more direct test of these ideas.
A proposed MOT1 mechanism involving translocation along DNA
Whereas cross-linked and minicircle DNA templates do not impair MOT1 function, loss of duplex DNA in the upstream DNA handle does inhibit ATP-dependent disruption of TBP–DNA (Figure 5, 5C7 and 3C7). Are these defects due to poor binding by MOT1 or a requirement for duplex DNA for disruption, or both? Template 5C7–TBP complexes have an 8-fold lower affinity for MOT1 than those formed on wild-type DNA, and the consequences of this defect for catalysis of TBP–DNA disruption are unclear. On the other hand, TBP–DNA complexes formed on the 3C7 template display a very modest decrease (∼2-fold) in affinity for MOT1, and yet this template supports virtually no ATP-dependent disruption by MOT1. Additionally, comparison of results obtained with probes 3C7 and 3T7 (Figure 5) is informative. These probes support roughly equivalent binding of MOT1, but the gapped template functions equivalently to wild-type DNA whereas the tailed template is defective. These results further define a clear function for duplex DNA in the upstream end of the handle even when this upstream duplex DNA is linked to the TBP–DNA complex via a single-stranded gap. These results suggest, therefore, that duplex DNA in the handle is important for the formation of MOT1–TBP–DNA complexes and, independently, duplex DNA in the handle is required for ATP-dependent TBP–DNA dissociation. The requirement for duplex DNA for catalysis can not be due to induction of conformational change in the DNA handle that propagates through the TATA box, because a gap in either strand has no effect on disruption (Figure 4; except 5T7, discussed above). The property of double-stranded DNA that is required by MOT1, then, is not its rigid linkage to TBP–DNA, but only that it is attached to the TATA box. The simplest interpretation of these results is that the MOT1 ATPase motor translocates along the duplex DNA handle and removes TBP from the TATA box by pulling or pushing.
Two possibilities are schematized in Figure 9. In Figure 9B, a domain of MOT1 remains anchored to the DNA handle during the ATP-driven power stroke. The tight grip of MOT1 on the handle allows extension of the effector portion of the protein to push TBP off DNA. The effector domain is schematized as a wedge, but many other possibilities for effector action are possible. Alternatively, in Figure 9C, ATP hydrolysis drives movement of MOT1 along duplex DNA; movement of the entire molecule would then cause dissociation of TBP either by pushing or pulling TBP as movement occurs.
Several SNF2/SWI2-related ATPases involved in chromatin remodeling have been shown to generate ATP-dependent superhelical torsion and it has been proposed that these ATPases remodel chromatin structure by DNA twisting and/or translocation (Havas et al., 2000). The translocation model is consistent with the findings reported here, but MOT1 appears to differ from these enzymes in not utilizing DNA distortion to alter protein–DNA contacts. Among members of the helicase superfamily (Gorbalenya et al., 1989; reviewed in Hall and Matson, 1999), the conserved ATP-driven motor has been shown to use ATP hydrolysis in many different ways; many proteins in this large family do not appear to catalyze DNA strand separation. Rather than a mis-classification of function, it has been argued that many proteins that have been defined as helicases do not unwind DNA, but instead translocate along DNA using the same structurally and functionally conserved motor domain that is used in bona fide helicases to unwind DNA (Egelman, 1998; Hall and Matson, 1999). In fact, even enzymes with demonstrated helicase activity can use ATP hydrolysis to translocate along DNA without unwinding it (Kaplan, 2000), and the bacterial RuvB protein appears to facilitate branch migration by ‘DNA pumping’ rather than by strand unwinding (Egelman, 1998; George et al., 2000). Strand-specific effects of single-stranded gaps on RecBC helicase have been interpreted to mean that the enzyme uses ATP hydrolysis to translocate along one strand of duplex DNA (Blanco and Kowalczykowski, 2000). MOT1 requires duplex DNA for catalysis, but in contrast to helicases, the polarity of MOT1 movement is likely to be determined by its asymmetric and stereospecific association with TBP.
Further tests of the translocation model will require trapping intermediates of the ATPase reaction cycle. While this has not been possible for MOT1 so far using non-hydrolyzable analogs of ATP and other biochemical approaches, we have begun to isolate and characterize mutant MOT1 proteins with defects in the conserved ATPase domain. By analogy with helicases, such mutations may help to define the region of the SNF2/SWI2 motor involved in transmission of the ATP-driven conformational change to the effector portion of the enzyme (Velankar et al., 1999).
Materials and methods
Recombinant proteins
Recombinant yeast TBP core domain (Geiger et al., 1996) was obtained from an Escherichia coli expression system in which amino acids 61–240 of yeast TBP were expressed under the control of the T7 promoter as a fusion with an N-terminal His6 tag (Novagen). One liter cultures of E.coli BL21 cells harboring the T7–yeast TBP expression plasmid were grown in YT medium plus ampicillin to an OD600 of 0.7–1.0 and expression was induced by the addition of 0.4 mM imidazole for 2 h at 37°C. Cells were harvested by centrifugation and resuspended in buffer I [30 mM Tris–HCl pH 7.5, 500 mM KCl, 10% glycerol, 1 mM dithiothreitol (DTT), 2 mM EDTA, 1 mM phenylmethylsulfonyl fluoride (PMSF)]. Cells were lysed by sonication (3 × 30 s using a Branson Sonifier microtip set at maximum output), Triton X-100 was added to 0.1% and the cellular debris was removed by centrifugation (20 000 r.p.m. for 30 min in a Sorvall SS34 rotor at 4°C). The supernatant was then added to 1 ml of nickel–agarose beads (Qiagen) pre-equilibrated in buffer I. The agarose beads were washed with 10 column volumes of buffer I plus 5 mM imidazole, then with buffer I plus 20 mM imidazole, and finally TBP was eluted in buffer I containing 250 mM imidazole with an adjusted pH of 6.5. The TBP was then purified further on a 1 ml Resource S column (Pharmacia) by diluting the pooled fractions to achieve a conductivity equivalent to that of buffer containing 10 mM HEPES pH 7.5, 100 mM KCl, 10% glycerol, 1 mM EDTA, 1 mM DTT, 1 mM PMSF; TBP that bound to the Resource S column was eluted using a 10 ml linear gradient of KCl from 0.1 to 1.0 M contained in the same HEPES buffer above. TBP core domain eluted from the Resource S column in HEPES buffer containing ∼200 mM KCl. The TBP core domain used in these studies was a gift from Jim Geiger. Epitope-tagged MOT1 was immunopurified from yeast exactly as described previously (Auble et al., 1997). Briefly, wild-type yeast cells (YPH499; Sikorski and Hieter, 1989) containing a LEU2-marked 2µ plasmid that directs expression of the MOT1 allele of interest under GAL1 control were grown in synthetic medium without leucine and containing raffinose (Auble et al., 1997). MOT1 expression was induced in mid-log phase by the addition of galactose to 2%, and following 2 h incubation at 30°C, cells were harvested by centrifugation. Cells were then lysed by vortexing with glass beads, and MOT1 was purified using protein G–Sepharose beads coupled to a monoclonal antibody that recognizes the Py epitope, which is fused to the N-termini of MOT1 and derivatives (Schneider et al., 1994; Auble et al., 1997).
Detection of protein–DNA complexes
Gel mobility shift assays were performed using 6% polyacrylamide gels (6% acrylamide from a 20%:0.33% acrylamide:bis-acrylamide stock, 2.5% glycerol, 190 mM glycine, 10 mM MgOAc2, 2.5 mM Tris pH 8.3, 1 mM EDTA, 0.5 mM DTT), pre-run at 100 V and 4°C (in buffer containing 190 mM glycine, 5 mM MgOAc2, 2.5 mM Tris pH 8.3, 1 mM EDTA) for at least 1 h before loading. Samples were incubated for 30–60 min in 20 µl volumes containing 2 µg bovine serum albumin (BSA), 100 ng poly(dG–dC), 4% glycerol, 0.1% Brij 58, 60 mM KCl, 5 mM Tris pH 8, 5 mM MgOAc2, 1 mM DTT and 50 p.p.m. bromo phenol blue. Each reaction contained 1000 c.p.m. (∼0.25 nM) of DNA and the following as indicated: 2 ng of TBP, 5 µM ATP, 0.5–8.0 U of MOT1. One unit (U) of MOT1 is defined as the amount required to super-shift 50% of the TBP–DNA complexes formed under the conditions described above. The amount of MOT1 is defined in this way because the amounts of this large polypeptide that we obtain from the yeast overexpression system (<1 µg MOT1 per preparation) are not sufficient for routine quantitation of mass by protein assay or Coomassie Blue staining of gels (Auble et al., 1997). In general, under the conditions described above, we estimate 1 U of MOT1 activity to represent ∼30 ng of MOT1 polypeptide. Thus, the reactions described in this work contained 5 nM TBP core domain and between 3 and 48 nM MOT1. Gel mobility shift gels were loaded and run at 160 V and 4°C for 40–80 min before being dried and exposed to film or PhosphorImager screen (Molecular Dynamics) for 12–24 h. To compare the abilities of the modified DNA templates to support MOT1–TBP–DNA ternary complex formation, the band intensities representing the TBP–DNA and MOT1– TBP–DNA complexes were measured by PhosphorImager analysis and we compared the ratio of ternary complex abundance to TBP–DNA complex abundance on a test template to the ratio obtained with fully duplex, wild-type DNA. These ratios were measured and compared over an 8- to 16-fold range of MOT1 concentration.
DNase I footprinting was performed as described previously (Auble and Steggerda, 1999) and employed a radiolabeled 110 bp fragment of the adenovirus major late promoter uniquely labeled at the 5′ end of one strand (Auble and Hahn, 1993). Binding reactions were performed under the same conditions as for gel mobility shift assays except that ∼20 000 c.p.m. of labeled DNA were used per reaction; at the end of the 20–30 min incubation period, DNase I (Worthington) was added (amount determined empirically to achieve cleavage of ∼30% of the input DNA) for 1 min and the reaction was stopped by addition of an equal volume of 5 M ammonium acetate and 2 vols of ethanol. Following ethanol precipitation and washing with 80% ethanol, the DNA pellets were dried, resuspended in formamide loading solution, and the reaction products were resolved by electrophoresis on 8% polacrylamide sequencing gels (Sambrook et al., 1989). Missing nucleoside experiments were performed exactly as described (Tullius et al., 1987).
Oligonucleotide preparation
Oligonucleotides (Table I) were synthesized at the University of Virginia Biomolecular Research Facility and purifed by denaturing gel electrophoresis. [The gel contained 15% polyacrylamide (40:1 acrylamide:bis-acrylamide stock), 2.4 M urea, 91 mM boric acid, 90 mM Tris pH 8.3, 2.5 mM EDTA, and electrophoresis was performed in 1× TBE buffer (Sambrook et al., 1989).] DNA was excised from the gels and eluted into 0.5 ml of TE (10 mM Tris pH 8, 1 mM EDTA), bound to DEAE (CL-6B), eluted in 0.9 ml (1 M NaCl, 50 mM NaAc pH 5.5), ethanol precipitated and resuspended in 50 µl (10 mM Tris pH 8, 1 mM EDTA). For psoralen cross-linking studies, oligonucleotides X3L, X3M, X3R, X5L, X5M and X5R were designed such that a single specific psoralen cross-linking site lies 8 bp on either side of the TATA box (Table I; psoralen preferentially cross-links thymidines). Ten picomoles of each oligonucleotide marked with an asterisk in Table I were labeled with 20 µCi of [γ-32P]ATP (6000 Ci/mmol) using T4 polynucleotide kinase and the buffer provided (New England Biolabs) in 20 µl reactions. Labeled oligonucleotides were purified by the MERmaid spin kit (Bio101) according to the instructions provided by the manufacturer. Annealing reactions were performed in 10–20 µl of MERmaid elution buffer with 100 mM NaCl, boiled for 10 min and allowed to cool for at least 6 h. Oligonucleotides X5L and X3L were annealed to make construct XL; likewise, X5M and X3M form construct XM, and X5R and X3R form construct XR; 3-33 and 5-29 were annealed to make a control duplex used in one cross-linking experiment; 3-36 and 5-37 were annealed as a control duplex for the other cross-linking experiments and the gap and tail experiments. The tail constructs were made by annealing labeled 3-36 with X5M and labeled 5-37 with 3-T7. The gap constructs were made by annealing oligonucleotides in the combinations listed in Table I. Note that the radiolabel was placed so that no TBP shift would be observed unless all three oligonucleotides were properly annealed.
Table I. Oligonucleotides used for construction of gaps and single-stranded tails.
| Oligonucleotide | Sequence |
|---|---|
| 3-0T | 5′-CCCCCTTTTATA-3′ |
| 3-2T | 5′-CCCCCTTTTATAGC-3′ |
| 3-3T | 5′-CCCCCTTTTATAGCC-3′ |
| 3-5T | 5′-CCCCCTTTTATAGCCCC-3′ |
| 3-7T | 5′-CCCCTTTTATAGCCCCCC-3′ |
| 3-10T | 5′-CCCCCTTTTATAGCCCCCCATC-3′ |
| 3-33 | 5′-CGCCCCCTTTTATAGCCCCCCATCCGGGGCGCC-3′ |
| 3-36 | 5′-CCCCTTTTATAGCCCCCCATCCGGGGCGCCCGGCCG-3′ |
| 3-36N | 5′-CCCCCTTTTATAGCCCCCCTTCAGGAACACCCGGTC-3′ |
| 3-E12 | 5′-GGCGCCCGGCCG-3′ |
| 3-E15 | 5′-CGGGGCGCCCGGCCG-3′ |
| 3-E18 | 5′-CTTCAGGAACACCCGGTC-3′ |
| 3-E20 | 5′-CCCTTCAGGAACACCCGGTC-3′ |
| 3-E21 | 5′-CCCCTTCAGGAACACCCGGTC-3′ |
| 3-E23 | 5′-CCCCCCTTCAGGAACACCCGGTC-3′ |
| 5-0T | 5′-TATAAAAGGGGG-3′ |
| 5-1T | 5′-CTATAAAAGGGGG-3′ |
| 5-2T | 5′-GCTATAAAAGGGGG-3′ |
| 5-3T | 5′-GGCTATAAAAGGGGG-3′ |
| 5-4T | 5′-GGGCTATAAAAGGGGG-3′ |
| 5-5T | 5′-GGGGCTATAAAAGGGGG-3′ |
| 5-6T | 5′-GGGGGCTATAAAAGGGGG-3′ |
| 5-10T | 5′-GATGGGGGGCTATAAAAGGGG-3′ |
| 5-29 | 5′-GGCGCCCCGGATGGGGGGCTATAAAAGGG-3′ |
| 5-36 | 5′-GACCGGGTGTTCCTGAAGGGGGGCTATAAAAGGGGG-3′ |
| 5-37 | 5′-CGGCCGGGCGCCCCGGATGGGGGGCTATAAAAGGGGG-3′ |
| 5-E12 | 5′-CGGCCGGGCGCC-3′ |
| 5-E15 | 5′-CGGCCGGGCGCCCCG-3′ |
| 5-E17 | 5′-GACCGGGTGTTCCTGAA-3′ |
| 5-E18 | 5′-GACCGGGTGTTCCTGAAG-3′ |
| 5-E19 | 5′-GACCGGGTGTTCCTGAAGG-3′ |
| 5-E20 | 5′-GACCGGGTGTTCCTGAAGGG-3′ |
| 5-E21 | 5′-GACCGGGTGTTCCTGAAGGGG-3′ |
| 5-E22 | 5′-GACCGGGTGTTCCTGAAGGGGG-3′ |
| 5-E23 | 5′-GACCGGGTGTTCCTGAAGGGGGG-3′ |
| X3L | 5′-CCCCATCCGGGGCGCCCGGCCG-3′ |
| X3M | 5′-Pi-CGCCCCCTTTTATAGCC-3′ |
| X3R | 5′-CCGGGCGCGTACC-3′ |
| X5L | 5′-CGGCCGGGCGCCCCGCAT-3′ |
| X5M | 5′-Pi-GGGGGGCTATAAAAGGG-3′ |
| X5R |
5′-GGCGGGTACGCGCCCGG-3′ |
| Construct |
Components (asterisk indicates 32P-labeled strand) |
| 3C7 | 3-7T + 5-37* |
| 3G1 | 3-0T + 3-E23* + 5-36 |
| 3G3 | 3-2T + 3-E21* + 5-36 |
| 3G4 | 3-3T + 3-E20* + 5-36 |
| 3G6 | 3-5T + 3-E18* + 5-36 |
| 3T10 | 3-10T + 3-E12* + 5-37 |
| 3T7 | 3-7T + 3-E15* + 5-37 |
| 5C7 | 3-36* + 5-7T |
| 5G1 | 3-36N + 5-0T + 5-E23* |
| 5G2 | 3-36N + 5-1T + 5-E22* |
| 5G3 | 3-36N + 5-2T + 5-E21* |
| 5G4 | 3-36N + 5-3T + 5-E20* |
| 5G5 | 3-36N + 5-4T + 5-E19* |
| 5G6 | 3-36N + 5-5T + 5-E18* |
| 5G7 | 3-36N + 5-6T + 5-E17* |
| 5T10 | 3-36 + 5-10T + 5-E12* |
| 5T7 | 3-36 + 5-7T + 5-E15* |
| Control (Figures 4, 5 and 6B) | 3-36* + 5-37* |
| Control (Figure 6A) | 3-33 + 5-29* |
| XL | X3L* + X5L |
| XM | X3M + X5M |
| XR | X3R + X5R* |
Construction of minicircle DNA
Construction of radiolabeled minicircle DNA was performed using the starting plasmids pBS-TATA-31 and pBS-TATA-37 (Parvin et al., 1995). First, PCR was performed with reactions containing 10 µl of 10× Taq buffer (Gibco-BRL), 2 µl of 1 mM ATP, 10 µl of 2 mM dTTP, 10 µl of 2 mM dGTP, 10 µl of 2 mM dCTP, 1 µl (0.1 mg/ml) of T7 sequencing primer, 1 µl (0.1 mg/ml) of T3 sequencing primer, 7.5 µl of [α-32P]dATP (10 µCi/µl, 3000 Ci/mmol), 1 µl of Taq DNA polymerase (Gibco-BRL), 66.5 µl of water and 50 ng of TATA-31 or TATA-37 plasmids. Cycling was performed as follows: reactions were incubated for 1 min at 94°C, 2 min at 92°C, 1 min at 55°C and then 2 min at 72°C. Steps 2–4 were repeated for 30 cycles. Following PCR, radiolabeled DNA was purified away from unincorporated nucleotides using Amersham probe quant spun columns, the reactions were extracted with phenol, ethanol precipitated and digested with ClaI as recommended by the manufacturer (NEB). The reaction products were then resolved on native 6% polyacrylamide gels in 1× TBE (Sambrook et al., 1989) and the linear, TATA-containing bands that co-migrated with the xylene cyanol marker dye were excised and purified by passive elution overnight into 0.5 M ammonium acetate, 2 mM EDTA. The eluted DNA was then ethanol precipitated and ligated overnight in a reaction volume of 300 µl using the buffer supplied by the manufacturer (NEB). The ligation was then ethanol precipitated and the ligated monomeric minicircles were separated from linear molecules (except linear dimers) and circular dimers by electrophoresis on non-denaturing 6% polyacrylamide gels as above. The circular monomer migrated slightly more slowly than the xylene cyanol marker dye. The minicircle DNA was excised from the gel and eluted as in the first purification step; circular monomers were then separated from contaminating linear dimers by electrophoresis on non-denaturing 4% polyacrylamide gels in 1× TBE buffer. On 4% gels, the circular monomers migrated more quickly than the linear dimers. Minicircles eluted from the 4% gels were ethanol precipitated, resuspended in 10 mM Tris–HCl pH 7.5, 1 mM EDTA and used in reactions for gel mobility shift analysis as described above. The minicircles were shown to be covalently closed circles by treatment with Bal31 exonuclease (Parvin et al., 1995 and data not shown).
Psoralen cross-linking
In separate reactions, constructs XL and XR were mixed with saturating 4, 5′, 8-trimethylpsoralen in the dark, then exposed to high-intensity UV light for 20–30 min using a portable UVGL-58 short wave lamp (UVP, Inc.) clamped directly over the reactions in open microcentrifuge tubes on ice. Reactions were boiled for 10 min in sample buffer (5% formamide, 1.5 mM EDTA, 20 p.p.m. bromophenol blue, 20 p.p.m. xylene cyanol) and loaded on a denaturing electrophoresis gel (15% polyacrylamide–2.4 M urea, 91 mM boric acid, 90 mM Tris pH 8.3, 2.5 mM EDTA). High molecular weight bands, running with xylene cyanol (∼40 bp), which did not appear in uncross-linked samples, were excised from the gel, eluted in 0.5 ml of 10 mM Tris pH 8, 1 mM EDTA, ethanol precipitated and eluted in 50 µl of 10 mM Tris pH 8. Cross-linked XL was ligated to XM at 16°C overnight using T4 DNA ligase (New England Biolabs) and the buffer provided in an 80 µl volume. The desired product was purified by denaturing PAGE (running above the position of cross-linked XL or XR and above the xylene cyanol); some was saved as ‘XLM’, and the rest was ligated to cross-linked XR, both procedures performed as above; the full-length constructs were purified by denaturing gel electrophoresis, as above (the position of the correct bands was determined by running samples of the previous products in the same gel). The full-length constructs were further purified by polyacrylamide gel (2.5% glycerol, 190 mM glycine, 10 mM MgOAc2, 2.5 mM Tris pH 8.3, 1 mM EDTA, 0.5 mM DTT), using an autoradiograph of a gel run with unpurified, full-length, doubly cross-linked construct as a guide, then eluted and ethanol precipitated as before. At the end of this procedure, between 300 and 2000 c.p.m. of full-length, doubly cross-linked construct were recovered for use in gel shift assays.
Acknowledgments
Acknowledgements
We are grateful to Ed Egelman, Jason Kahn, Fraydoon Rastinejad and Dale Wigley for discussion, to Ed Egelman for comments on the manuscript, to Chunming Zhu for purified MOT1, to Jim Geiger for purified TBP core domain, and to Jeff Parvin for reagents and advice on minicircle construction. This work was supported by NIH grant GM55763 to D.T.A.
References
- Adamkewicz J.I., Mueller,C.G.F., Hansen,K.E., Prud’homme,W.A. and Thorner,J. (2000) Purification and enzymic properties of Mot1 ATPase, a regulator of basal transcription in the yeast Saccharomyces cerevisiae. J. Biol. Chem., 275, 21158–21168. [DOI] [PubMed] [Google Scholar]
- Auble D.T. and Hahn,S. (1993) An ATP-dependent inhibitor of TBP binding to DNA. Genes Dev., 7, 844–856. [DOI] [PubMed] [Google Scholar]
- Auble D.T. and Steggerda,S.M. (1999) Testing for DNA tracking by MOT1, a SNF2/SWI2 protein family member. Mol. Cell. Biol., 19, 412–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auble D.T., Hansen,K.E., Mueller,C.G.F., Lane,W.S., Thorner,J. and Hahn,S. (1994) Mot1, a global repressor of RNA polymerase II transcription, inhibits TBP binding to DNA by an ATP-dependent mechanism. Genes Dev., 8, 1920–1934. [DOI] [PubMed] [Google Scholar]
- Auble D.T., Wang,D., Post,K.W. and Hahn,S. (1997) Molecular analysis of the SNF2/SWI2 protein family member MOT1, an ATP-driven enzyme that dissociates TATA-binding protein from DNA. Mol. Cell. Biol., 17, 4842–4851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benz J., Trachsel,H. and Baumann,U. (1999) Crystal structure of the ATPase domain of translation initiation factor 4A from Saccharomyces cerevisiae—the prototype of the DEAD box protein family. Structure, 7, 671–679. [DOI] [PubMed] [Google Scholar]
- Bird L.E., Subramanya,H.S. and Wigley,D.B. (1998) Helicases, a unifying structural theme? Curr. Opin. Struct. Biol., 8, 14–18. [DOI] [PubMed] [Google Scholar]
- Blanco P.R. and Kowalczykowski,S.C. (2000) Translocation step size and mechanism of the RecBC DNA helicase. Nature, 405, 368–372. [DOI] [PubMed] [Google Scholar]
- Boyer L.A., Shao,X., Ebright,R.H. and Peterson,C.L. (2000) Roles of the histone H2A–H2B dimers and the (H3–H4)2 tetramer in nucleosome remodeling by the SWI–SNF complex. J. Biol. Chem., 275, 11545–11552. [DOI] [PubMed] [Google Scholar]
- Cang Y., Auble,D.T. and Prelich,G. (1999) A new regulatory domain on the TATA-binding protein. EMBO J., 18, 6662–6671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collart M.A. (1996) The NOT, SPT3 and MOT1 genes functionally interact to regulate transcription at core promoters. Mol. Cell. Biol., 16, 6668–6676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis J.L., Kunisawa,R. and Thorner,J. (1992) A presumptive helicase (MOT1 gene product) affects gene expression and is required for viability in the yeast Saccharomyces cerevisiae. Mol. Cell. Biol., 12, 1879–1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egelman E.H. (1998) Bacterial helicases. J. Struct. Biol., 124, 123–128. [DOI] [PubMed] [Google Scholar]
- Eisen J.A., Sweder,K.S. and Hanawalt,P.C. (1995) Evolution of the SNF2 family of proteins: subfamilies with distinct sequences and functions. Nucleic Acids Res., 23, 2715–2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger J.H., Hahn,S., Lee,S. and Sigler,P.B. (1996) Crystal structure of the yeast TFIIA/TBP/DNA complex. Science, 272, 830–836. [DOI] [PubMed] [Google Scholar]
- George H., Kuraoka,I., Nauman,D.A., Kobertz,W.R., Wood,R.D. and West,S.C. (2000) RuvAB-mediated branch migration does not involve extensive DNA opening within the RuvB hexamer. Curr. Biol., 10, 103–106. [DOI] [PubMed] [Google Scholar]
- Gorbalenya A.E. and Koonin,E.V. (1993) Helicases: amino acid sequence comparisons and structure–function relationships. Curr. Biol., 3, 419–429. [Google Scholar]
- Gorbalenya A.E., Koonin,E.V., Donchenko,A.P. and Blinov,V.M. (1989) Two related superfamilies of putative helicases involved in replication, recombination, repair and expression of DNA and RNA genomes. Nucleic Acids Res., 17, 4713–4730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guyon J.R., Narlikar,G.J., Sif,S. and Kingston,R.E. (1999) Stable remodeling of tailless nucleosomes by the human SWI–SNF complex. Mol. Cell. Biol., 19, 2088–2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall M.C. and Matson,S.W. (1999) Helicase motifs: the engine that powers DNA unwinding. Mol. Microbiol., 34, 867–877. [DOI] [PubMed] [Google Scholar]
- Havas K., Flaus,A., Phelan,M., Kingston,R., Wade,P.A., Lilley,D.M.J. and Owen-Hughes,T. (2000) Generation of superhelical torsion by ATP-dependent chromatin remodeling activities. Cell, 103, 1133–1142. [DOI] [PubMed] [Google Scholar]
- Hoopes B.C., LeBlanc,J.F. and Hawley,D.K. (1992) Kinetic analysis of yeast TFIID–TATA box complex formation suggests a multi-step pathway. J. Biol. Chem., 267, 11539–11547. [PubMed] [Google Scholar]
- Imbalzano A.N., Schnitzler,G.R. and Kingston,R.E. (1996) Nucleosome disruption by human SWI/SNF is maintained in the absence of continued ATP hydrolysis. J. Biol. Chem., 271, 20726–20733. [DOI] [PubMed] [Google Scholar]
- Johnson E.R. and McKay,D.B. (1999) Crystallographic structure of the amino terminal domain of yeast initiation factor 4A, a representative DEAD-box RNA helicase. RNA, 5, 1526–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahn J.D. (2000) Topological effects of the TATA box binding protein on minicircle DNA and a possible thermodynamic linkage to chromatin remodeling. Biochemistry, 39, 3520–3524. [DOI] [PubMed] [Google Scholar]
- Kaplan D.L. (2000) The 3′-tail of a forked-duplex sterically determines whether one of two DNA strands pass through the central channel of a replication-fork helicase. J. Mol. Biol., 301, 285–299. [DOI] [PubMed] [Google Scholar]
- Kim J.L., Nikolov,D.B. and Burley,S.K. (1993a) Co-crystal structure of TBP recognizing the minor groove of a TATA element. Nature, 365, 520–527. [DOI] [PubMed] [Google Scholar]
- Kim Y., Geiger,J.H., Hahn,S. and Sigler,P.B. (1993b) Crystal structure of a yeast TBP/TATA-box complex. Nature, 365, 512–520. [DOI] [PubMed] [Google Scholar]
- Kingston R.E. and Narlikar,G.J. (1999) ATP-dependent remodeling and acetylation as regulators of chromatin fluidity. Genes Dev., 13, 2339–2352. [DOI] [PubMed] [Google Scholar]
- Korolev S., Hsieh,J., Gauss,G.H., Lohman,T.M. and Waksman,G. (1997) Major domain swiveling revealed by the crystal structures of complexes of E. coli Rep helicase bound to single-stranded DNA and ADP. Cell, 90, 635–647. [DOI] [PubMed] [Google Scholar]
- Korolev S., Yao,N., Lohman,T.M. and Weber,P.C. (1998) Comparisons between the structures of HCV and Rep helicases reveal structural similarities between SF1 and SF2 super-families of helicases. Protein Sci., 7, 605–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langst G., Bonte,E.J., Corona,D.F.V. and Becker,P.B. (1999) Nucleosome movement by CHRAC and ISWI without disruption or trans-displacement of the histone octamer. Cell, 97, 843–852. [DOI] [PubMed] [Google Scholar]
- Logie C. and Peterson,C.L. (1997) Catalytic activity of the yeast SWI/SNF complex on reconstituted nucleosome arrays. EMBO J., 16, 6772–6782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madison J.M. and Winston,F. (1997) Evidence that Spt3 functionally interacts with Mot1, TFIIA and TBP to confer promoter-specific transcriptional control in Saccharomyces cerevisiae. Mol. Cell. Biol., 17, 287–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parvin J.D., McCormick,R.J., Sharp,P.A. and Fisher,D.E. (1995) Pre-bending of a promoter sequence enhances affinity for the TATA-binding factor. Nature, 373, 724–727. [DOI] [PubMed] [Google Scholar]
- Pazin M.J. and Kadonaga,J.T. (1997) SWI2/SNF2 and related proteins: ATP-driven motors that disrupt protein–DNA interactions? Cell, 88, 737–740. [DOI] [PubMed] [Google Scholar]
- Petukhova G., Stratton,S. and Sung,P. (1998) Catalysis of homologous DNA pairing by yeast Rad51 and Rad54 proteins. Nature, 393, 91–94. [DOI] [PubMed] [Google Scholar]
- Piatti S., Tazzi,R., Pizzagalli,A., Plevani,P. and Lucchini,G. (1992) Control of DNA synthesis genes in budding yeast: involvement of the transcriptional modulator MOT1 in the expression of the DNA polymerase α gene. Chromosoma, 102, S107–S113. [DOI] [PubMed] [Google Scholar]
- Poon D., Campbell,A.M., Bai,Y. and Weil,P.A. (1994) Yeast Taf170 is encoded by MOT1 and exists in a TBP–TAF complex distinct from TFIID. J. Biol. Chem., 269, 23135–23140. [PubMed] [Google Scholar]
- Prelich G. (1997) Saccharomyces cerevisiae BUR6 encodes a DRAP1/NC2α homolog that has both positive and negative roles in transcription in vivo. Mol. Cell. Biol., 17, 2057–2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J., Fritsch,E.F. and Maniatis,T. (1989) Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Schneider K.R., Smith,R.L. and O’Shea,E.K. (1994) Phosphate-regulated inactivation of the kinase PHO80–PHO85 by the CDK inhibitor PHO81. Science, 266, 122–126. [DOI] [PubMed] [Google Scholar]
- Schnitzler G., Sif,S. and Kingston,R.E. (1998) Human SWI/SNF interconverts a nucleosome between its base state and a stable remodeled state. Cell, 94, 17–27. [DOI] [PubMed] [Google Scholar]
- Shen X., Mizuguchi,G., Hamiche,A. and Wu,C. (2000) A chromatin remodelling complex involved in transcription and DNA processing. Nature, 406, 541–544. [DOI] [PubMed] [Google Scholar]
- Sikorski R.S. and Hieter,P. (1989) A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics, 122, 19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soultanas P., Dillingham,M.S., Wiley,P., Webb,M.R. and Wigley,D.B. (2000) Uncoupling DNA translocation and helicase activity in PcrA: direct evidence for an active mechanism. EMBO J., 19, 3799–3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tullius T.D., Dombroski,B.A., Churchill,M.E.A. and Kam,L. (1987) Hydroxyl radical footprinting: a high-resolution method for mapping protein–DNA contacts. Methods Enzymol., 155, 537–559. [DOI] [PubMed] [Google Scholar]
- Velankar S.S., Soultanas,P., Dillingham,M.S., Subramanya,H.S. and Wigley,D.B. (1999) Crystal structures of complexes of PcrA DNA helicase with a DNA substrate indicate an inchworm mechanism. Cell, 97, 75–84. [DOI] [PubMed] [Google Scholar]
- Vignali M., Hassan,A.H., Neely,K.E. and Workman,J.L. (2000) ATP-dependent chromatin-remodeling complexes. Mol. Cell. Biol., 20, 1899–1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitehouse I., Flaus,A., Cairns,B.R., White,M.F., Workman,J.L. and Owen-Hughes,T. (1999) Nucleosome mobilization catalyzed by the yeast SWI/SNF complex. Nature, 400, 784–787. [DOI] [PubMed] [Google Scholar]
- Yao N., Hesson,T., Cable,M., Hong,Z., Kwong,A.D. and Weber,P.C. (1997) Structure of the hepatitis C virus RNA helicase domain. Nature Struct. Biol., 4, 463–467. [DOI] [PubMed] [Google Scholar]