

Abstract
Glucocorticoid hormones were found to regulate DNA demethylation within a key enhancer of the rat liver-specific tyrosine aminotransferase (Tat) gene. Genomic footprinting analysis shows that the glucocorticoid receptor uses local DNA demethylation as one of several steps to recruit transcription factors in hepatoma cells. Demethylation occurs within 2–3 days following rapid (<1 h) chromatin remodeling and recruitment of a first transcription factor, HNF-3. Upon demethylation, two additional transcription factors are recruited when chromatin is remodeled. In contrast to chromatin remodeling, the demethylation is stable following hormone withdrawal. As a stronger subsequent glucocorticoid response is observed, demethylation appears to provide memory of the first stimulation. During development, this demethylation occurs before birth, at a stage where the Tat gene is not yet inducible, and it could thus prepare the enhancer for subsequent stimulation by hypoglycemia at birth. In vitro cultures of fetal hepatocytes recapitulate the regulation analyzed in hepatoma cells. There fore, demethylation appears to contribute to the fine-tuning of the enhancer and to the memorization of a regulatory event during development.
Keywords: chromatin/DNA methylation/liver/nuclear receptor/transcription
Introduction
Cytosine methylation at CpG dinucleotides contributes to the transcriptional silencing of gene expression both by interfering with the action of activators and by allowing the recruitment of repressors that promote the formation of inactive chromatin structures (Bird and Wolffe, 1999). The generality of the repressive nature of cytosine methylation is widely admitted now, but the role of this DNA modification is still controversial. It could be involved in the reduction of transcriptional noise in organisms with a large number of genes (Bird, 1995), in the suppression of the activity of parasitic mobile elements (Walsh et al., 1998), in the memorization of developmental decisions (Regev et al., 1998) or in a combination of these roles, being a flexible evolutionary device allowing acquisitions of various functions involving DNA marking (Colot and Rossignol, 1999). It was proposed long ago that DNA methylation could contribute to the regulation of gene expression during development (reviewed in Russo et al., 1996). Nowadays, one of the arguments in favor of this hypothesis is the correlation between the modification of methylation patterns and gene expression during mammalian development. Methylation patterns are usually transmitted by clonal inheritance but can be modified selectively during development. A genome-wide resetting of the methylation patterns is observed in the early stages, followed by a selective gene-specific demethylation occurring mostly in tissues and at developmental stages where the gene is expressed (Razin and Kafri, 1994). The developmental defects that result from the inactivation of several cytosine methyltransferase genes (Dnmt) in both plants and vertebrates are also in favor of an important role for DNA methylation in development (Okano et al., 1999; Finnegan et al., 2000; Stancheva and Meehan, 2000). In contrast, there are arguments that do not favor this hypothesis. First, the developmental defects caused by the Dnmt gene inactivation could be indirect. They could result from the failure of other methylation-dependent mechanisms, e.g. proper X chromosome inactivation (Panning and Jaenisch, 1996), parental imprinting (Li et al., 1993), mobile element repression (Walsh et al., 1998), prevention of recombination between repeated sequences (Maloisel and Rossignol, 1998) or other functions needing the DNMT protein but not its methyltransferase activity. Secondly, a number of invertebrates, including model organisms like Drosophila melanogaster and Caenorhabditis elegans, execute sophisticated developmental programs in the absence of extensive DNA methylation (Regev et al., 1998). It suggests that, at least in certain organisms, the genome marking function of DNA methylation can be taken up by other chromatin modifications. Thirdly, the correlation between expression and DNA methylation is not absolute, and DNA demethylation is not sufficient to induce tissue-specific genes in non-expressing tissue (Walsh and Bestor, 1999). In the light of the contradictory data accumulated over years, a likely hypothesis would be that, in most cases, DNA methylation only participates in the control of gene expression during development, rather than being its sole determinant. Such a limited participation could be sufficient for methylation to contribute to cell determination during development by providing a memory of transient regulatory events. Despite the accumulation of data showing that some transcriptional activators are able to promote DNA demethylation (Saluz et al., 1988; Brandeis et al., 1994; Macleod et al., 1994; Kirillov et al., 1996; Matsuo et al., 1998; Hsieh, 1999), there is no evidence that DNA demethylation provides memory of a regulatory event. We describe here the glucocorticoid-regulated DNA demethylation of a regulatory sequence that is associated with a modification of its subsequent behavior.
In mammals, glucocorticoid hormones participate, in the adult, in the control of the response of the organism to various stresses, including starvation; whereas during development, they prepare various organs for the metabolic adaptations allowing autonomous life after birth (Greengard, 1970; Sassi et al., 1998; Tronche et al., 1998). The glucocorticoid receptor (GR) and many other members of the nuclear receptor superfamily are ligand-activated transcriptional regulators that make use of coactivator and corepressor complexes to modulate both chromatin structure and the activity of the basal transcription machinery (Glass and Rosenfeld, 2000). The GR acts cooperatively with other DNA-binding proteins, altogether interacting with regulatory sequences termed glucocorticoid responsive units (GRUs), which can integrate the hormonal response to other regulatory pathways (Schüle et al., 1988). In the liver, one of the genes whose transcription is induced by the activated GR in postnatal life is the tyrosine aminotransferase gene (Tat) (Granner and Hargrove, 1983). This induction is mediated through cooperative interaction of two GRUs located at –2.5 and –5.5 kb. The Tat GRUs consist of numerous contiguous and overlapping binding sites for the GR and other transcription factors, including members of the C/EBP, HNF-3 and Ets families (Grange et al., 1991; Espinás et al., 1994; Roux et al., 1995). This arrangement confers tissue specificity to the glucocorticoid response, and allows it to synergize positively with glucagon and negatively with insulin (Sassi et al., 1998 and references therein). The GRUs are thus activated in response to hypoglycemia, which is the developmental trigger that induces, at birth, a number of genes in the liver, including Tat (Greengard, 1970; Sassi et al., 1998). The GR is essential for neonatal induction of the Tat gene, as seen in the GR knockout mice (Tronche et al., 1998). However, it is not sufficient since a dimerized glucocorticoid responsive element (GRE) does not convey gene induction in the liver at birth, in contrast to the Tat GRUs (Sassi et al., 1998). The GR recruits HNF-3 at the –2.5 Tat GRU, presumably through chromatin remodeling (Rigaud et al., 1991). Thus, it is tempting to speculate that the GR is needed for neonatal induction through the Tat GRUs because it allows the recruitment of the transcription factors mediating the response to hypoglycemia (Sassi et al., 1998).
We show here that the GR induces stable DNA demethylation at the –2.5 Tat GRU. This event is associated with the recruitment of additional factors that increase the subsequent glucocorticoid responses, hence providing a memory of the first stimulation. In vitro, DNA methylation directly affects the binding of one of these factors, suggesting that it is directly responsible for the effect observed in living cells. During development, the GR induces this demethylation before birth, at a stage where the Tat gene is not yet inducible. This indicates that demethylation prepares the Tat GRU for later stimulation by the hypoglycemia occurring at birth. Therefore, regulation of local DNA methylation status by transcription factors could indeed provide a way to modulate gene expression during development.
Results
Prolonged glucocorticoid treatment induces stable DNA demethylation at the Tat GRU
In rat hepatoma cells, a short-term glucocorticoid treatment (15 min to 1 h) induces chromatin remodeling at the –2.5 Tat GRU, as well as HNF-3 recruitment (Rigaud et al., 1991). Both of these events are reversible in ∼1 h following glucocorticoid withdrawal (Reik et al., 1991; Espinás et al., 1995). As the GRU contains several CpGs, we have analyzed whether glucocorticoid treatment also affects cytosine methylation. This was studied using ligation-mediated PCR (LM–PCR) analysis of genomic DNA treated with hydrazine and piperidine in conditions where DNA is cleaved only at unmethylated cytosines (Thomassin et al., 1999). Figure 1 shows that the methyl ation status of three CpGs contained within the –2.5 GRU is modified over the course of a 3 day dexamethasone treatment in the H4II hepatoma cell line. In untreated cells, there are three neighboring cytosines, all contained within the dinucleotide CpG, which do not react, indicating that they are methylated (lane 1). After 1 h of glucocorticoid treatment, whereas the chromatin has been remodeled and HNF-3 recruited, there is no modification of this cytosine methylation pattern (lane 2). Prolonged glucocorticoid treatment induces a demethylation of these cytosines, as shown by the appearance of the corresponding bands in the genomic sequencing ladder (lanes 3–7). Six hours after dexamethasone treatment, two out of the three Cs begin to react (lane 3) and the third C becomes reactive after ∼1 day (lane 4). The demethylation proceeds over 3 days, after which the three cytosines appear fully demethylated (lanes 6–7). A similar kinetic of DNA demethylation is observed on the other strand (data not shown).
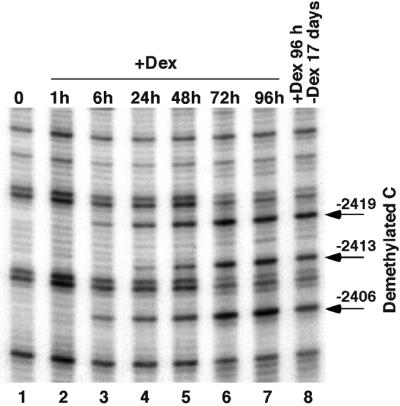
Fig. 1. Prolonged glucocorticoid treatment induces a stable DNA demethylation at the –2.5 Tat GRU. Rat hepatoma cells (H4II) were grown with 10–7 M dexamethasone (+Dex) for the indicated time (0, no hormone addition) and the corresponding genomic DNA was treated with hydrazine and piperidine. The upper strand of the –2.5 Tat GRU was analyzed by LM–PCR.
Glucocorticoids induce chromatin remodeling over a 350 bp region encompassing the Tat gene GRU (indicated on Figure 2; Reik et al., 1991; M.Flavin, L.Cappabianca, H.Thomassin and T.Grange, in preparation). Within this area, there is a fourth CpG dinucleotide (–2341) that is also demethylated during a 3 day glucocorticoid treatment (Figure 2 and data not shown). The CpGs located outside, notably directly upstream of the GRU, are not affected (data not shown).

Fig. 2. Schematic representation of the –2.5 Tat GRU region. The relative location of the transcription factor binding sites, the dinucleotide CpGs with their methylation status, and the extent of the region where chromatin is remodeled following glucocorticoid stimulation are indicated. The data not described in the present study originate from other sources (Grange et al., 1991; Reik et al., 1991; Espinás et al., 1994; Roux et al., 1995; M.Flavin, L.Cappabianca, H.Thomassin and T.Grange, in preparation). DR0-TF, transcription factor(s) interacting with the DR0 site; MeS-TF, methylation-sensitive transcription factor.
As chromatin remodeling, HNF-3 recruitment and transcriptional activation require the continuous presence of glucocorticoids, we wondered whether DNA demethylation was stable following glucocorticoid withdrawal (Figure 1, lane 8). The study shows that the CpGs, once demethylated, are not remethylated following culture in the absence of glucocorticoids for 3 weeks (lane 8) and even 3 months (data not shown). Thus, a stable mark of the prolonged glucocorticoid stimulation has remained at the regulatory sequence.
DNA demethylation is associated with a memorization of the first glucocorticoid stimulation
We sought to determine whether the stable mark left after a first glucocorticoid stimulation affects subsequent hormonal responses of the gene. We analyzed on a northern blot the transcriptional response of the Tat gene over the course of a prolonged glucocorticoid treatment of both naive cells and cells that have been in contact once with glucocorticoids for 4 days. These later cells were grown for 1 month in the absence of glucocorticoids before their response to a second hormonal stimulation was analyzed. This study reveals that the transcription induction is faster and stronger in cells that have been previously demethylated by a first glucocorticoid treatment (Figure 3). This is particularly notable in the first 2–4 h of stimulation where the response in demethylated cells is, respectively, five to three times stronger than in naive cells. The induction levels achieved in naive cells become similar to those achieved in demethylated cells only after 1–2 days of stimulation at a time when the GRU is largely demethylated. Thus, demethylation of the Tat gene GRU appears to improve its activity and to provide a memory of the first glucocorticoid stimulation.

Fig. 3. The first glucocorticoid stimulation enhances the subsequent hormonal response. Glucocorticoid-induced variations in TAT mRNA levels in H4II cells were analyzed by northern blotting using a TAT cDNA probe (Grange et al., 1985). After stripping of the probe, the blots were reprobed for an internal control with a cDNA of a housekeeping gene, that coding for poly(A) binding protein (PABP) (Grange et al., 1987). The graph shows the variation with time of the corrected TAT mRNA levels following quantitative analysis of the blots using a phosphoimager.
Cytosine demethylation permits glucocorticoid-dependent recruitment of a transcription factor through a direct effect on DNA binding
To clarify the molecular bases of the memorization event observed, we used genomic footprinting to analyze transcription factor interaction with the GRU over the course of a prolonged glucocorticoid treatment of both naive and demethylated cells. Dimethyl sulfate (DMS) footprinting of the region overlapping the three CpGs, all contained within a 15-bp region, revealed a glucocorticoid-dependent change of reactivity, indicating the recruitment of a transcription factor (Figure 4A). This is visible essentially as a hyperreactivity of the guanine –2420. In naive methylated cells, a 1 h glucocorticoid treatment does not induce detectable factor recruitment (compare lanes 1 and 2 in Figure 4A). The characteristic hyperreactivity steadily increases over prolonged hormonal stimulation (compare lanes 1–5 in Figure 4A) with a kinetic similar to that of DNA demethylation (compare with lanes 1–6 of Figure 1). In contrast, the kinetic of recruitment of this factor is much faster in cells where the –2.5 Tat GRU has been demethylated during a first glucocorticoid exposure (lanes 6–10 in Figure 4A). The interaction is not detected in the absence of hormone (lane 6) but maximal recruitment is observed after 1 h of dexamethasone treatment (lanes 7 and 8–10). Therefore, factor recruitment at this site requires both a stable event induced by prolonged glucocorticoid stimulation and an activated GR.

Fig. 4. DNA demethylation permits glucocorticoid-dependent loading of a transcription factor at the demethylated sites. (A) Genomic footprinting of hepatoma cells using DMS. Methylated cells, naive H4II cells; demethylated cells, H4II cells treated for 4 days with 10–7 M Dex and grown for 1 month in the absence of GR ligand. Cells were grown with Dex for the indicated time prior to treatment with DMS. The upper strand of the –2.5 Tat GRU was analyzed by LM–PCR. (B) Mobility shift analysis of factor binding as a function of DNA methylation. Crude nuclear extracts prepared from H4II cells treated or not with Dex for 24 h were incubated with double-stranded oligonucleotide probes (–2399/–2425) containing either only unmethylated cytosines (lanes 1 and 2) or methylated cytosines at all CpGs (lanes 3 and 4).
In view of this result, we tested whether cytosine methylation has a direct impact on transcription factor binding using electrophoretic mobility shift assay (EMSA; Figure 4B). Sequence-specific DNA-binding activities present in nuclear extracts of hepatoma cells treated or not with glucocorticoids for 1 day were analyzed using a 25-bp double-stranded oligonucleotide probe overlapping the three CpGs (see Materials and methods). All the cytosines within the CpGs were either unmethylated or methylated and the corresponding probes were analyzed side by side. The results show that several activities bind in vitro to this site and that the binding of some of these is sensitive to the methylation status of the probe. Among the four bands detected, the upper one is better seen with the unmethylated probe, whereas the two lower bands are better seen with the methylated probe. Furthermore, a 24 h glucocorticoid treatment does not modify the levels of DNA-binding activities interacting with this site. The results suggest that CpG methylation directly prevents factor interaction with this site in living cells during the early steps of the first glucocorticoid response.
DNA demethylation appears required for glucocorticoid-dependent recruitment of factors at a second site
We used genomic footprinting to determine whether methylation is also affecting transcription factor occupancy in the region of the fourth CpG of the GRU demethylated upon glucocorticoid treatment. A few bases 3′ to this CpG, there is a direct repeat of the AGGTCA motif characteristic of type II nuclear receptor DNA-binding sites. Several activities binding to that site are detected in vitro, among them COUP-TF and FTF (Galarneau et al., 1996) as assessed by supershift analysis (data not shown). When DNase I is used as the footprinting reagent, a clear protection of the direct repeat is observed following a 2 day glucocorticoid treatment of H4II cells (compare lane 3 with lanes 1–2 in Figure 5). Occupancy of this site requires both the presence of glucocorticoids and DNA demethylation. It is detected neither in the absence of glucocorticoids (lanes 1 and 5) nor following a 1 h glucocorticoid treatment of naive H4II cells (lane 2). Following this 1 h hormonal treatment, only the slight changes of reactivity characteristic of the chromatin remodeling event can be observed at this site (M.Flavin, L.Cappabianca, H.Thomassin and T.Grange, in preparation). However, the same short-term hormonal treatment is sufficient to observe protection of the site when demethylation of the GRU has been induced by a prior glucocorticoid treatment (lane 6). Thus, as for the transcription factor binding site containing the three CpGs, occupancy of this site is observed only when GR has induced chromatin remodeling and DNA demethylation.
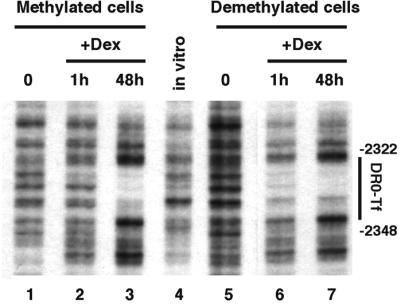
Fig. 5. DNA demethylation is associated to glucocorticoid-dependent loading of another transcription factor nearby a demethylated site. Genomic footprinting of hepatoma cells using DNase I. The lower strand of the –2.5 Tat GRU was analyzed by LM–PCR.
The effect of demethylation on transcription factor interaction with that site was studied using EMSA with both methylated and unmethylated probes encompassing the –2431 CpG and the direct repeat (Materials and methods). None of the activities detected in hepatoma cell nuclear extracts is affected by the methylation status of the CpG (data not shown). This is consistent with the location of this dinucleotide outside the DNA sequence contacted by type II nuclear receptors (Rastinejad et al., 1995). Thus, in contrast to the previously analyzed site, DNA methylation does not seem to directly affect transcription factor binding at that site.
The Tat gene GRU is demethylated during rat liver development in parallel to the prenatal peak of glucocorticoids
Many tissue-specific genes undergo a selective demethylation of their regulatory sequences at the appropriate stage of differentiation and in the tissues where they are expressed (Razin and Kafri, 1994). In the liver, extensive biochemical differentiation occurs during the last few days of gestation and in early postnatal life (Greengard, 1970). The Tat gene is rapidly turned on at birth in response to hypoglycemia (Greengard, 1970; Sassi et al., 1998). To assess whether the –2.5 GRU methylation profile is related to the Tat gene expression profile in vivo, we analyzed its methylation status in liver and other tissues during the perinatal period. As Tat gene transcription is restricted to parenchymal cells of the liver, determination of the degree of methylation in these cells may be obscured by the cellular heterogeneity of the liver, particularly at fetal stages. Indeed, hematopoietic cells represent ∼60% of the liver cells at embryonic day (e.d.) 15 and ∼30% just after birth (Greengard et al., 1972). To minimize interference by red cells, DNA was extracted from parenchymal nuclei purified by ultracentrifugation through sucrose layers. We found that the Tat GRU is essentially methylated in fetal liver at e.d.15 (Figure 6A, lane 1). Demethylation is partial 4 days later, at e.d.19 (lane 2), and the GRU is mostly demethylated at birth (lane 3), just when the gene is going to be induced. The GRU undergoes further demethylation in postnatal life (lanes 4–6). Demethyla tion of the GRU is liver-specific and does not occur in non-expressing tissues like spleen and kidney (lanes 7 and 8). As in H4II cells, demethylation occurs on both strands and is restricted to the four CpGs located within the GRU (data not shown).

Fig. 6. Glucocorticoid-induced DNA demethylation during development. (A) Liver-specific demethylation of the Tat GRU occurs before birth and parallels the glucocorticoid peak. DNA methylation was analyzed at various time points in the perinatal period as described in Figure 1. The postnatal days (d) are indicated using birth (B) as the reference. The prenatal stages analyzed are e.d.15 and e.d.19. (B) Glucocorticoid-induced demethylation and chromatin remodeling in primary cultures of e.d.15 fetal rat hepatocytes. After isolation, fetal hepatocytes were cultured in vitro for the indicated time without (0) or with 10–7 M dexamethasone. (a) DNA methylation analysis as described in Figure 1. Similar demethylation was observed using bisulfite-PCR analysis (data not shown). (b) Chromatin remodeling analysis as assessed by restriction enzyme accessibility. Nuclei from fetal hepatocytes were incubated with XbaI for 1 h prior to DNA isolation and indirect-end labeling was performed as described in Materials and methods. The band corresponding to cleavage by XbaI at the –2.5 GRU (at position –2558) is indicated. (C) Liver-specific demethylation of the Tat promoter occurs several days after birth. Data were obtained as described in (A), except that the upper strand of the proximal promoter was analyzed (Rigaud et al., 1991).
Demethylation of the Tat GRU in the fetal liver follows the increase in the concentration of free corticosteroid in fetal rat plasma. Indeed, this concentration steadily increases from e.d.15 to e.d.19–20 and then decreases to reach low levels at birth and in the first 4 days of life (Martin et al., 1977). This suggests that glucocorticoids are responsible for Tat GRU demethylation during development as they are in H4II hepatoma cells. To assess whether glucocorticoids are able to promote a premature demethylation of the Tat GRU in fetal liver, we performed primary culture of hepatocytes isolated from e.d.15 fetuses, i.e. before the glucocorticoid peak. The method we used provides a homogeneous population of hepatocytes by elimination of hematopoietic cells (Plas et al., 1973). After an 18 h glucocorticoid treatment, demethylation of the GRU is visible (Figure 6Ba, lane 4). As in H4II cells, glucocorticoids are able to trigger a rapid chromatin remodeling at the Tat GRU as determined by restriction enzyme accessibility (Figure 6Bb). Concomitant with this remodeling, HNF-3 recruitment is observed (data not shown). The chromatin remodeling is visible soon after hormone addition (1 h; Figure 6Bb, lane 2) and takes place before DNA demethylation (Figure 6Ba, compare lanes 2 and 4). Therefore, both in H4II hepatoma cells and in fetal hepatocytes, glucocorticoids induce similar events at the Tat GRU: first a rapid chromatin remodeling followed by a slower demethylation of the CpGs located within the remodeled area.
In contrast to the situation in hepatoma cells, demethylation of the GRU in fetal rat liver is not accompanied by transcriptional activation of the Tat gene. Indeed, the gene is turned on as a result of hypoglycemia occurring at birth (Greengard, 1970). We suspect that the Tat gene could be unresponsive to the glucocorticoid peak preceding birth, at least in part because of regulatory sequences outside the GRU. Indeed, in transgenic mice harboring a reporter gene controlled by a housekeeping promoter regulated by the Tat GRUs (Sassi et al., 1995), glucocorticoid injection at e.d.20 can enhance expression of the transgene without inducing the endogenous Tat gene (H.Sassi and T.Grange, unpublished results). As all regulatory events occurring on the various enhancers must ultimately be integrated at the proximal promoter to be converted into transcriptional activation, we analyzed whether the proximal Tat promoter sequences were also subject to CpG demethylation in prenatal life. The promoter appears methylated at e.d.15 and remains methylated during the entire perinatal period (Figure 6C). Liver-specific demethylation occurs after birth, between day 5 and 22 (lanes 5–6 in Figure 6C). As for the GRU, no demethylation is observed in kidney and spleen (lanes 7–8). Therefore, demethylation of the –2.5 GRU and the proximal promoter is uncoupled during development. Demethylation of the GRU precedes the neonatal activation of the gene while demethylation of the proximal promoter occurs several days after birth, after the gene has been turned on.
Discussion
Glucocorticoid-regulated DNA demethylation of the Tat GRU in hepatoma cells as a model system
A critical aspect of the overall regulatory role of DNA methylation is the process of demethylation. The mechanisms that underlie the selective demethylation of tissue-specific genes, mostly in the cell type and at the developmental stage where they are expressed, are still largely unknown (Razin and Kafri, 1994). In addition, the contribution of DNA demethylation to developmental regulation of gene expression remains elusive. In most circumstances, selective demethylation of a given gene is part of a more general differentiation process where the dramatic changes occurring in the genetic expression program complicate the analysis of the role of DNA demethylation. One of the limits to our understanding of the mechanisms of demethylation, and its consequences on tissue-specific gene expression, is the lack of relevant models that can be easily manipulated. In this study, we describe a cultured cell system in which the activated GR triggers a cascade of events at the –2.5 Tat GRU, including chromatin remodeling and DNA demethylation. Genomic footprinting analysis allowed us to clarify the relationships among chromatin remodeling, demethylation, transcription factor recruitment and gene activation, and thus the contribution of DNA methylation to tissue-specific gene expression.
Previous studies were performed using transfection of in vitro methylated DNA constructs into either a myoblast cell line (Paroush et al., 1990) or B cell lines (Kirillov et al., 1996), but demethylation of tissue-specific genes in their natural chromatin context was only rarely studied. Genomic sequencing and footprinting were used to analyze demethylation of the avian vitellogenin II gene upon estradiol treatment (Saluz et al., 1988). However, this model suffers several limitations: demethylation only occurs in the entire animal, and the hormonal injection affects several interacting tissues and induces extensive changes in the tissue where the demethylation is observed (Jost et al., 1973). The contribution of demethylation to transcription factor recruitment has been indirectly analyzed at the 3′ enhancer of the M lysozyme gene, but it relies on the comparison of different tumor cell lines that express different genetic programs (Ammerpohl et al., 1998). In contrast, the demethylation of the –2.5 Tat gene GRU, induced by glucocorticoid in the H4II hepatoma cell line, offers a powerful model system because: (i) the demethylation is observed in a well characterized cell line, thus rendering it more accessible to analysis than developing animals; (ii) the activator triggering the demethylation can be turned on or off at will by adding or removing its ligand. This allows monitoring of the kinetics and the reversibility of regulatory events such that various questions about the relationship between gene activation and demethylation can be addressed; (iii) the reversibility of all visible effects of the activator upon ligand withdrawal, but the demethylation of the GRU, allows comparison of two kinds of cells that differ minimally. Indeed, H4II cells that have been submitted to a 4 day glucocorticoid treatment do not exhibit any sign of changes in differentiation or proliferation rate; and (iv) the events triggered by the GR at the Tat GRU in hepatoma cells recapitulate the regulation of the GRU occurring during liver development. The GRU is demethylated during the 2–3 days preceding birth, following a peak of fetal plasma corticosterone. In vitro culture of e.d.15 fetal hepatocytes reveals that, indeed, glucocorticoids trigger similar events in H4II cells to during development.
Consequences of DNA demethylation on transcription factor recruitment and gene expression
It is generally assumed that DNA methylation is inversely related to transcriptional activity. Although selective demethylation of tissue-specific genes occurs in the territories where they are expressed, whether demethylation plays a primary regulatory role in transcription activation remains obscure. As shown here, cytosine methylation does not prevent chromatin remodeling, HNF-3 recruitment and subsequent transcription activation at the Tat gene GRU. Furthermore, once local demethylation has been established, the GRU does not remain active in the absence of glucocorticoids. Upon hormone withdrawal, transcription goes back down to basal level (Figure 3), chromatin remodeling, i.e. the DNase I hypersensitive site, disappears (data not shown), and the transcription factors that were recruited upon glucocorticoid stimulation dissociate from the GRU. As the GRU remains demethylated following hormone withdrawal, the data reveal that demethylation per se is not sufficient to induce GRU activation and transcription factor recruitment. Another event, presumably chromatin remodeling, must be triggered to reveal the effects of demethylation. This is consistent with experiments showing that demethylation is not sufficient to reactivate most tissue-specific genes in tissue where they are not expressed (Weih et al., 1991; Walsh and Bestor, 1999). However, our results indicate that demethylation contributes nevertheless to gene expression as, upon chromatin remodeling, recruitment of additional transcription factors and enhanced transcription activation occurs when the gene is demethylated.
How could demethylation achieve this permissive role in transcription factor recruitment at the GRU? Our results indicate that the mode of action might differ depending on the binding site considered. The binding site of the methylation-sensitive transcription factor (MeS-TF, Figure 2) contains three CpGs. At this site, direct inter ference of the 5-methyl group on factor binding is most likely responsible for the demethylation-dependent interaction observed in cells. Indeed, in vitro, we found a factor that binds preferentially to the demethylated DNA site (Figure 4). The interaction of the other factors detected in vitro is either unaffected or negatively affected by the methylation status of the probe, and these properties are not consistent with the pattern of interaction in living cells. Direct interference of DNA methylation with transcription factor interaction has been observed for many transcription factors (Razin and Kafri, 1994; Bird and Wolffe, 1999). It depends on the precise location of the methyl CpG within the binding site and thus cannot account for all effects of DNA methylation, but it is likely that it contributes to the fine-tuning of many regulatory sequences.
Within the same GRU, DNA demethylation appears to permit transcription factor recruitment at another site containing a CpG. This sequence contains a direct repeat (DR0) of the AGGTCA motif bound by nuclear receptors. None of the binding activities detected in vitro is sensitive to the methylation status of the probe, in agreement with the location of the CpG outside the region contacted in the major groove by nuclear receptors (Rastinejad et al., 1995). Thus, an indirect effect of DNA demethylation must be considered. For instance, differences in accessibility could be exerted through methylation-induced modification of the chromatin structure. Proteins harboring a methyl-CpG binding domain (MBD) are good candidates for mediating such modifications, particularly by promoting histone tail deacetylation. Three of these MBD proteins (MeCP2, MBD2, MBD3) are repressors that are in complexes containing histone deacetylases, and treatment with the deacetylase inhibitor trichostatin A (TSA) can relieve to a large extent their repressor activity (Bird and Wolffe, 1999). TSA treatment of H4II cells, in conditions that induce histone hyperacetylation as well as partial relaxation of the chromatin structure of the Tat gene, has not permitted any of the transcription factor recruitment induced by DNA demethylation (M.Flavin, L.Cappabianca, H.Thomassin and T.Grange, in preparation). Therefore, methylation-induced histone deacetylation does not seem to participate in the effects observed here. This is consistent with the observations that repression induced by the MBD proteins would be observed at a higher methylated CpG density than that of the Tat GRU (Boyes and Bird, 1991). Alternative possibilities must be considered, for example: (i) other components of the MBD repression complexes, like Sin 3, could exert a deacetylase-independent effect on accessibility (Bird and Wolffe, 1999); (ii) DNA methylation could affect the interaction of members of the linker histone H1 family with nucleosomes (Schwarz et al., 1997 and references therein); and (iii) the demethylation-dependent recruitment of the MeS-TF provides a nucleation site allowing or facilitating the recruitment of the other methylation-sensitive transcription factors.
Control of demethylation and regulation of Tat gene expression during development
The developmental relevance of the demethylation event we observed in an immortalized cell line is supported by its in vivo occurrence before birth, in parallel with fluctuations in the level of glucocorticoid hormones. It is further supported by the reproduction of this glucocorticoid-dependent demethylation using fetal hepatocytes isolated at a stage preceding demethylation (e.d.15).
How could this demethylation event contribute to the control of Tat gene expression during development? The GRU where this demethylation occurs is able to activate transcription not only in response to glucocorticoids, but also to hypoglycemia thanks to some of the transcription factors cooperating with GR within the GRU (Sassi et al., 1998). In the liver, hypoglycemia is the developmental stimuli that turns on the Tat gene at birth as well as a number of other genes from the so-called neonatal cluster (Greengard, 1970; Sassi et al., 1998). Before birth, the Tat gene is hardly expressed, and is responsive neither to a peak of glucocorticoids that precedes birth by 2–3 days, nor to experimental manipulation of the glucocorticoid levels (Greengard, 1970; Ruiz-Bravo and Ernest, 1982; H.Sassi and T.Grange, unpublished results). Glucocorti coids appear nevertheless important for Tat gene activation at birth as it is prevented by inactivation of the GR gene (Tronche et al., 1998). Since the prenatal glucocorticoid peak triggers demethylation of the Tat GRU without inducing transcription, and since demethylation appears to allow recruitment of additional transcription factors, we propose that the prenatal demethylation plays a preparatory role in permitting the optimal response of the GRU to the hypoglycemia occurring at birth.
The Tat GRU is not responsible for the lack of induction of the Tat gene before birth as the Tat GRU is partially responsive to glucocorticoid injection at this developmental stage when driving a housekeeping promoter (Sassi et al., 1995; H.Sassi and T.Grange, unpublished results). Interestingly, demethylation of the Tat proximal promoter is delayed by several days when compared with the GRU. This presumably reflects the asynchrony in the activation of the two regulatory sequences. The GRU appears to be activated first, at a stage when the proximal promoter is still inactive, and thus this activation does not result in transcriptional stimulation, even though it causes local demethylation. Activation of the proximal promoter at birth precedes its demethylation, just like activation of the GRU precedes its demethylation in hepatoma cells. In contrast to the demethylation taking place at the GRU, our data do not allow us to assess whether demethylation of the promoter also participates in Tat gene regulation or is a by-product of gene activation.
Despite the similarity of the demethylation of the GRU in hepatoma cells and during development, not all events affecting the Tat gene regulatory sequences are identical in the two conditions. In the naive hepatoma cells, the GRU is methylated but the proximal promoter is demethylated, its chromatin is remodeled and it is loaded with transcription factors independently of glucocorticoid presence (Rigaud et al., 1991 and data not shown). Thus, the proximal promoter appears poised, ready to respond to activation of the GRU, and this activation causes transcriptional stimulation that occurs before complete demethylation of the GRU. It is likely that the differences in relative behavior of the enhancer and promoter in the two experimental systems reflect the fact that the methylation of the GRU has been later regained during the immortalization or culture process of the hepatoma cell line. Indeed, this line originates from an adult rat liver where the two regulatory sequences were most likely demethylated. Such a deregulated de novo methylation of regulatory sequences appears common in cultured cells (Antequera et al., 1990). Nevertheless, it is a fortunate event here as it provides us with a cell line that recapitulates a regulated local DNA demethylation event occurring during development.
Twenty-five years have passed since the initial proposition that DNA demethylation could play a key role in the control of gene expression during development by providing a memory of regulatory events (reviewed in Russo et al., 1996). A large number of correlations between DNA methylation status of regulatory sequences and their activity have been observed but it could not be shown that changes in the methylation status provide memory during development. The ambiguous nature of the data has led to the proposal that methylation participates in highly specialized functions (allele-specific gene expression and silencing of parasitic sequences) and plays only a very minor role in the regulation of vertebrate development (Walsh and Bestor, 1999). In agreement with the observations of Walsh and Bestor, we observe here that DNA demethylation is not sufficient to activate a tissue-specific gene in the absence of the regulatory factor promoting chromatin remodeling, and that DNA demethylation is a consequence of transcriptional activation rather than a cause. Nevertheless, demethylation appears to be used for the fine-tuning of gene expression and to provide memory of a regulatory event during development. It is conceptually more satisfactory that DNA methylation plays such a role during development, in addition to its special ized functions. Indeed, it is likely that demethylation mechanisms have not been selected during evolution for allele-specific gene expression or silencing of parasitic sequences as it seems that these processes should remain irreversible.
Materials and methods
Cells, tissues and DNA preparation
Rat hepatoma cells H4IIEC3 (H4II herein) were cultured as described previously (Grange et al., 1991). Primary cultures of hepatocytes were obtained from 15-day-old Wistar rat fetuses as described (Plas et al., 1973). Seven hours after plating of the isolated liver cells on collagen-coated culture dishes, the non-adhering hematopoietic cells were removed. At this point, dexamethasone (10–7 M) was introduced or not in the fresh culture medium and the homogeneous population of hepatocytes was grown for the culture period chosen. DNA was purified from cell culture as described (Cappabianca et al., 1999).
Tissue DNA was prepared from liver, kidney and spleen of Wistar rats. To enrich the fetal liver DNA preparations in hepatocytes DNA and to minimize the contribution of hematopoietic cells, DNA was extracted from parenchymal nuclei purified according to Marshall and Burgoyne (1976). DNA was isolated from resuspended nuclei as described (Cappabianca et al., 1999). Genomic DNA of 22-day-old rat liver, kidney and spleen was directly extracted from powdered frozen tissues according to current procedures (Sambrook et al., 1989).
Methylation analysis, genomic footprinting and restriction enzyme accessibility
Cytosine methylation was analyzed by LM–PCR using hydrazine/piperidine treatment as described (Thomassin et al., 1999). Genomic footprinting using DNase I was performed according to Rigaud et al. (1991) with the modifications described (Grange et al., 1997; Cappabianca et al., 1999). Genomic footprinting using DMS was performed as described (Espinás et al., 1994). The –2500 GRU and the promoter of the rat Tat gene were analyzed using the sets of primers described by Rigaud et al. (1991).
Restriction enzyme (XbaI) accessibility analysis was performed according to Reik et al. (1991). DNA was then cleaved with HindIII (cleaving the Tat gene at –3337) and BamHI (–1300), separated on an agarose gel and transferred to a nylon membrane (Sambrook et al., 1989). Indirect end labeling was performed with an abutting probe (–1820/–1300).
Crude nuclear extract preparation and EMSA
Prior to extract preparation, cells were treated or not for 24 h with 10–7 M dexamethasone. Cells were recovered and lysed as described (Grange et al., 1991) except that buffer H was supplemented with 1 µg/ml each of leupeptin, antipain, chymostatin, pepstatin and aprotinin, 20 mM Na2HPO4 and 5 mM NaF. The nuclei were recovered by centrifugation onto a 0.5 M sucrose layer in buffer H. The nuclear extracts were then prepared as described (Roux et al., 1995).
EMSAs were performed as described (Roux et al., 1995) using as a probe a gel-purified, blunt-ended, double-stranded oligonucleotide generated by the annealing of two complementary 5′-32P-labeled oligonucleotides. The following oligonucleotides were used: (–2425/ –2399), 5′-GTCCTGCGTAGTCGCCTGTCGGTTTCT-3′ and its complementary partner, (–2346/–2322) 5′-GGATCGGGAGTTCAAGGTCAGCTTG-3′ and its complementary partner, and their modified counterparts in which the cytosines included in all the CpGs indicated in bold italic characters were replaced by 5-methylcytosines. The direct repeat of the AGGTCA motif characteristic of type II nuclear receptor binding sites is underlined.
RNA preparation and analysis
Total RNA was prepared using the single-step guanidinium method, separated on formaldehyde denaturing agarose gel and then transferred to a nylon membrane according to current procedures (Sambrook et al., 1989).
Acknowledgments
Acknowledgements
We thank Jean-Luc Rossignol, Frédéric Pâques, Clémence Kress and Allyson Holmes for critical reading of the manuscript. This work was supported in part by the CNRS and grants from the Association de Recherche sur le Cancer, the Ligue Nationale contre le Cancer and the Association Française contre les Myopathies. M.-L.E. was supported by a fellowship from Fondation MEDIC.
References
- Ammerpohl O., Schmitz,A., Steinmüller,L. and Renkawitz,R. (1998) Repression of the mouse M-lysozyme gene involves both hindrance of enhancer factor binding to the methylated enhancer and histone deacetylation. Nucleic Acids Res., 26, 5256–5260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antequera F.J., Boyes,J. and Bird,A. (1990) High levels of de novo methylation and altered chromatin structure at CpG islands in cell lines. Cell, 6, 503–514. [DOI] [PubMed] [Google Scholar]
- Bird A.P. (1995) Gene number, noise reduction and biological complexity. Trends Genet., 11, 94–100. [DOI] [PubMed] [Google Scholar]
- Bird A.P. and Wolffe,A.P. (1999) Methylation-induced repression—belts, braces, and chromatin. Cell, 99, 451–454. [DOI] [PubMed] [Google Scholar]
- Boyes J. and Bird,A. (1991) DNA methylation inhibits transcription indirectly via a methyl-CpG binding protein. Cell, 64, 1123–1134. [DOI] [PubMed] [Google Scholar]
- Brandeis M., Frank,D., Keshet,I., Siegfried,Z., Mendelsohn,M., Nemes,A., Temper,V., Razin,A. and Cedar,H. (1994) Sp1 elements protect a CpG island from de novo methylation. Nature, 371, 435–438. [DOI] [PubMed] [Google Scholar]
- Cappabianca L., Thomassin,H., Pictet,R. and Grange,T. (1999) Genomic footprinting using nucleases. Methods Mol. Biol., 119, 427–442. [DOI] [PubMed] [Google Scholar]
- Colot V. and Rossignol,J.L. (1999) Eukaryotic DNA methylation as an evolutionary device. BioEssays, 21, 402–411. [DOI] [PubMed] [Google Scholar]
- Espinás M.L., Roux,J., Ghysdael,J., Pictet,R. and Grange,T. (1994) Participation of Ets transcription factor in the glucocorticoid response of rat tyrosine aminotransferase gene. Mol. Cell. Biol., 14, 4116–4125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espinás M.L., Roux,J., Pictet,R. and Grange,T. (1995) Glucocorticoids and protein kinase A coordinately modulate transcription factor recruitment at a glucocorticoid-responsive unit. Mol. Cell. Biol., 15, 5346–5354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finnegan E.J., Peacock,W.J. and Dennis,E.S. (2000) DNA methylation, a key regulator of plant development and other processes. Curr. Opin. Genet. Dev., 10, 217–223. [DOI] [PubMed] [Google Scholar]
- Galarneau L., Pare,J.F., Allard,D., Hamel,D., Levesque,L., Tugwood,J.D., Green,S. and Belanger,L. (1996) The α1-fetoprotein locus is activated by a nuclear receptor of the Drosophila FTZ-F1 family. Mol. Cell. Biol., 16, 3853–3865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass C.K. and Rosenfeld,M.G. (2000) The coregulator exchange in transcriptional functions of nuclear receptors. Genes Dev., 14, 121–141. [PubMed] [Google Scholar]
- Grange T., Guenet,C., Dietrich,J.B., Chasserot,S., Fromont,M., Befort,N., Jami,J., Beck,G. and Pictet,R. (1985) Complete complementary DNA of rat tyrosine aminotransferase messenger RNA. Deduction of the primary structure of the enzyme. J. Mol. Biol., 184, 347–350. [DOI] [PubMed] [Google Scholar]
- Grange T., Martins de Sa,C., Oddos,J. and Pictet,R. (1987) Human mRNA polyadenylate binding protein: evolutionary conservation of a nucleic acid binding motif. Nucleic Acids Res., 15, 4771–4787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grange T., Roux,J., Rigaud,G. and Pictet,R. (1991) Cell-type specific activity of two glucocorticoid responsive units of rat tyrosine aminotransferase gene is associated with multiple binding sites for C/EBP and a novel liver-specific nuclear factor. Nucleic Acids Res., 19, 131–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grange T., Bertrand,E., Rigaud,G., Espinás,M.L., Fromont-Racine,M., Roux,J. and Pictet,R. (1997) In vivo footprinting of the interaction of proteins with DNA and RNA. Methods, 11, 151–163. [DOI] [PubMed] [Google Scholar]
- Granner D.K. and Hargrove,J.L. (1983) Regulation of the synthesis of tyrosine aminotransferase: the relationship to mRNA-TAT. Mol. Cell. Biochem., 53/54, 113–128. [DOI] [PubMed] [Google Scholar]
- Greengard O. (1970) The developmental formation of enzymes in rat liver. In Litwack,G. (ed.), Biochemical Actions of Hormones. Vol. 1. Academic Press, New York, NY, pp. 53–87.
- Greengard O., Federman,M. and Knox,W.E. (1972) Cytomorphometry of developing rat liver and its application to enzymic differentiation. J. Cell Biol., 52, 261–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh C.L. (1999) Evidence that protein binding specifies sites of DNA demethylation. Mol. Cell. Biol., 19, 46–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jost J.P., Keller,R. and Dierks-Ventling,C. (1973) Deoxyribonucleic acid and ribonucleic acid synthesis during phosvitin induction by 17β-estradiol in immature chicks. J. Biol. Chem., 248, 5262–5266. [PubMed] [Google Scholar]
- Kirillov A., Kistler,B., Mostoslavsky,R., Cedar,H., Wirth,T. and Bergman,Y. (1996) A role for nuclear NF-κB in B-cell-specific demethylation of the Igκ locus. Nature Genet., 13, 435–441. [DOI] [PubMed] [Google Scholar]
- Li E., Beard,C. and Jaenisch,R. (1993) Role for DNA methylation in genomic imprinting. Nature, 366, 362–365. [DOI] [PubMed] [Google Scholar]
- Macleod D., Charlton,J., Mullins,J. and Bird,A.P. (1994) Sp1 sites in the mouse aprt gene promoter are required to prevent methylation of the CpG island. Genes Dev., 8, 2282–2292. [DOI] [PubMed] [Google Scholar]
- Maloisel L. and Rossignol,J.L. (1998) Suppression of crossing-over by DNA methylation in Ascobolus. Genes Dev., 12, 1381–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall A.J. and Burgoyne,L.A. (1976) Interpretation of the properties of chromatin extracts from mammalian nuclei. Nucleic Acids Res., 3, 1101–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin C.E., Cake,M.H., Hartmann,P.E. and Cook,I.F. (1977) Relationship between foetal corticosteroids, maternal progesterone and parturition in the rat. Acta Endocrinol. (Copenh.), 84, 167–176. [DOI] [PubMed] [Google Scholar]
- Matsuo K., Silke,J., Georgiev,O., Marti,P., Giovannini,N. and Rungger,D. (1998) An embryonic demethylation mechanism involving binding of transcription factors to replicating DNA. EMBO J., 17, 1446–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okano M., Bell,D.W., Haber,D.A. and Li,E. (1999) DNA methyl transferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell, 99, 247–257. [DOI] [PubMed] [Google Scholar]
- Panning B. and Jaenisch,R. (1996) DNA hypomethylation can activate Xist expression and silence X-linked genes. Genes Dev., 10, 1991–2002. [DOI] [PubMed] [Google Scholar]
- Paroush Z., Keshet,I., Yisraeli,J. and Cedar,H. (1990) Dynamics of demethylation and activation of the α-actin gene in myoblasts. Cell, 63, 1229–1237. [DOI] [PubMed] [Google Scholar]
- Plas C., Chapeville,F. and Jacquot,R. (1973) Development of glycogen storage ability under cortisol control in primary cultures of rat fetal hepatocytes. Dev. Biol., 32, 82–91. [DOI] [PubMed] [Google Scholar]
- Rastinejad F., Perlmann,T., Evans,R.M. and Sigler,P.B. (1995) Structural determinants of nuclear receptor assembly on DNA direct repeats. Nature, 375, 203–211. [DOI] [PubMed] [Google Scholar]
- Razin A. and Kafri,T. (1994) DNA methylation from embryo to adult. Prog. Nucleic Acid Res. Mol. Biol., 48, 53–81. [DOI] [PubMed] [Google Scholar]
- Regev A., Lamb,M.J. and Jablonka,E. (1998) The role of DNA methylation in invertebrates: developmental regulation or genome defense? Mol. Biol. Evol., 15, 880–891. [Google Scholar]
- Reik A., Schütz,G. and Stewart,A.F. (1991) Glucocorticoids are required for establishment and maintenance of an alteration in chromatin structure: induction leads to a reversible disruption of nucleosomes over an enhancer. EMBO J., 10, 2569–2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigaud G., Roux,J., Pictet,R. and Grange,T. (1991) In vivo footprinting of the rat TAT gene: dynamic interplay between the glucocorticoid receptor and a liver-specific factor. Cell, 67, 977–986. [DOI] [PubMed] [Google Scholar]
- Roux J., Pictet,R. and Grange,T. (1995) Hepatocyte nuclear factor 3 determines the amplitude of the glucocorticoid response of rat tyrosine aminotransferase gene. DNA Cell Biol., 14, 385–396. [DOI] [PubMed] [Google Scholar]
- Ruiz-Bravo N. and Ernest,M.J. (1982) Induction of tyrosine aminotransferase mRNA by glucocorticoids and cAMP in fetal rat liver. Proc. Natl Acad. Sci. USA, 79, 365–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo X., Martienssen,R.A. and Riggs,A.D. (1996) Epigenetic Mechanisms of Gene Regulation. Cold Spring Harbor Laboratory Press, Plainview, NY.
- Saluz H.P., Feavers,I.M., Jiricny,J. and Jost,J.P. (1988) Genomic sequencing and in vivo footprinting of an expression-specific DNase I-hypersensitive site of avian vitellogenin II promoter reveal a demethylation of a mCpG and a change in specific interactions of proteins with DNA. Proc. Natl Acad. Sci. USA, 85, 6697–6700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J., Fritsch,E.F. and Maniatis,T. (1989) Molecular Cloning: A Laboratory Manual. 2nd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Sassi H., Fromont-Racine,M., Grange,T. and Pictet,R. (1995) Tissue-specificity of a glucocorticoid-dependent enhancer in transgenic mice. Proc. Natl Acad. Sci. USA, 92, 7197–7201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sassi H., Pictet,R. and Grange,T. (1998) Glucocorticoids are insufficient for neonatal gene induction in the liver. Proc. Natl Acad. Sci. USA, 95, 5621–5625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schüle R., Muller,M., Kaltschmid,C. and Renkawitz,R. (1988) Many transcription factors interact synergistically with steroid receptors. Science, 242, 1418–1420. [DOI] [PubMed] [Google Scholar]
- Schwarz S., Hess,D. and Jost,J.P. (1997) The methylated DNA binding protein-2-H1 (MDBP-2-H1) consists of histone H1 subtypes which are truncated at the C-terminus. Nucleic Acids Res., 25, 5052–5056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stancheva I. and Meehan,R.R. (2000) Transient depletion of xDnmt1 leads to premature gene activation in Xenopus embryos. Genes Dev., 14, 313–327. [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Thomassin H., Oakeley,E.J. and Grange,T. (1999) Identification of 5-methylcytosine in complex genomes. Methods, 19, 465–475. [DOI] [PubMed] [Google Scholar]
- Tronche F., Kellendonk,C., Reichardt,H.M. and Schutz,G. (1998) Genetic dissection of glucocorticoid receptor function in mice. Curr. Opin. Genet. Dev., 8, 532–538. [DOI] [PubMed] [Google Scholar]
- Walsh C.P. and Bestor,T.H. (1999) Cytosine methylation and mammalian development. Genes Dev., 13, 26–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh C.P., Chaillet,J.R. and Bestor,T.H. (1998) Transcription of IAP endogenous retroviruses is constrained by cytosine methylation. Nature Genet., 20, 116–117. [DOI] [PubMed] [Google Scholar]
- Weih F., Nitsch,D., Reik,A., Schutz,G. and Becker,P.B. (1991) Analysis of CpG methylation and genomic footprinting at the tyrosine aminotransferase gene: DNA methylation alone is not sufficient to prevent protein binding in vivo. EMBO J., 10, 2559–2567. [DOI] [PMC free article] [PubMed] [Google Scholar]