

Abstract
During exocytosis, SNARE proteins of secretory vesicles interact with the corresponding SNARE proteins in the plasmalemma to initiate the fusion reaction. However, it is unknown whether SNAREs are uniformly distributed in the membrane or whether specialized fusion sites exist. Here we report that in the plasmalemma, syntaxins are concentrated in 200 nm large, cholesterol-dependent clusters at which secretory vesicles preferentially dock and fuse. The syntaxin clusters are distinct from cholesterol-dependent membrane rafts since they are Triton X-100-soluble and do not co-patch with raft markers. Synaptosomal-associated protein (SNAP)-25 is also clustered in spots, which partially overlap with syntaxin. Cholesterol depletion causes dispersion of these clusters, which is associated with a strong reduction in the rate of secretion, whereas the characteristics of individual exocytic events are unchanged. This suggests that high local concentrations of SNAREs are required for efficient fusion.
Keywords: cholesterol/exocytosis/PC12 cells/SNAREs
Introduction
SNAREs comprise a superfamily of small and mostly membrane-bound proteins that mediate membrane fusion. Eukaryotic cells possess different sets of SNAREs for the various intracellular fusion steps. For fusion to occur, corresponding SNAREs need to be localized on both of the membranes destined to fuse. Upon membrane contact, the SNAREs are thought to assemble into complexes that bridge the two membranes and thus facilitate bilayer mixing (reviewed by Jahn and Südhof, 1999; Lin and Scheller, 2000).
Most of our knowledge about the dynamics of SNARE assembly and disassembly is based on studies in solution using either soluble SNARE fragments, detergent extracts of biological membranes, or more recently, recombinant SNAREs reconstituted into proteoliposomes. However, little is known about how these proteins function in intact biological membranes. Most importantly, it remains to be clarified how many SNARE complexes need to cooperate in a fusion reaction. Membrane fusion mediated by viral fusion proteins is known to require multiple oligomers, which concentrate at the fusion site and may be involved in forming the initial fusion pore (Tse et al., 1993). If, as suggested, SNAREs function in a similar manner, it needs to be ascertained that a sufficient number of SNARE monomers is available at the fusion site (Hanson et al., 1997). These molecules may either be recruited from precursor pools that are freely diffusing in the plane of the membrane, or alternatively, they may be pre-assembled in clusters before contact between the fusing membranes is established. Moreover, SNARE function is regulated by other proteins that need to interact with individual SNAREs in a spatially and temporally controlled manner. For instance, more than a dozen proteins have been identified that bind specifically to syntaxin 1, suggesting the presence of complicated networks of interrelated protein–protein interactions (Jahn and Südhof, 1999; Lin and Scheller, 2000). It is not clear whether such complexes are evenly distributed over the membrane surface or how they relate to the ability of vesicles to dock and fuse.
In the present study we have investigated how SNAREs are spatially organized in intact biological membranes and how their spatial organization relates to the topology of vesicle docking and fusion. As a model we used PC12 cells, a neurosecretory cell line that releases neurotransmitter by Ca2+-dependent exocytosis (see e.g. Hay and Martin, 1992). Unstimulated PC12 cells contain numerous vesicles that are firmly attached to the plasma membrane. In our analysis we have concentrated on the SNAREs functioning in the regulated exocytosis of secretory vesicles. They include syntaxin 1 and synaptosomal-associated protein (SNAP)-25, which are predominantly localized in the plasma membrane, and synaptobrevin 1/2 (also referred to as VAMP, acronym for vesicle- associated membrane protein), which is predominantly associated with the membrane of secretory vesicles (Martin, 1997; Jahn and Südhof, 1999; Lin and Scheller, 2000). These proteins also operate in the exocytosis of synaptic vesicles in nerve terminals, and many features of exocytosis in neurons and PC12 cells are very similar (Burgoyne and Morgan, 1995; Martin, 1997). We found that in the plasma membrane, syntaxin and SNAP-25 are segregated in clusters of high local concentration that overlap only partially with each other and at which vesicles dock and fuse in a highly preferential manner. These clusters probably represent cholesterol-dependent microdomains that are distinct from rafts containing glyco- phosphatidyl inositol (GPI)-linked proteins. Cholesterol removal disintegrates these domains and concomitantly impairs exocytosis, suggesting that high local concentrations of SNAREs are required for efficient membrane fusion.
Results
Syntaxin and SNAP-25 are concentrated in clusters within the plasma membrane
To analyze the distribution of syntaxin and SNAP-25 in the plane of the membrane, we have taken advantage of a recently described procedure to produce flat, inside-out layers of plasma membrane that are devoid of all other cell constituents except bound secretory vesicles (Avery et al., 2000). We have shown previously that these vesicles can be stimulated to exocytose under appropriate conditions. Thus, this preparation is ideally suited for studying the topology of SNAREs with respect to docking sites of secretory vesicles. Also, as the thickness of the membrane sheets does not exceed 100 nm, there is no out-of-focus fluorescence to introduce noise and diminish lateral resolution.
PC12 cells were grown on coverslips and subjected to brief sonication in order to disrupt the cells, resulting in the generation of inside-out membrane layers. Membrane sheets were fixed with 4% paraformaldehyde (PFA), incubated with a syntaxin 1-specific monoclonal antibody, and subsequently labeled with a secondary fluorescence-labeled antibody. As shown in Figure 1A, the label was highly concentrated in numerous small dots that were scattered over the membrane surface, with only a low degree of staining in the surrounding membrane area. To confirm that the membrane sheets consist of intact phospholipid bilayers, they were also incubated with the lipophilic dye FM1-43. As judged from images such as Figure 1B, the membrane sheets were intact, with no holes, and flat. Furthermore, they were frequently decorated with fluorescent spots that presumably represent remnants of membranous organelles and generally did not correspond to the dots labeled for syntaxin.

Fig. 1. SNAREs are clustered in plasma membrane sheets derived from PC12 (A–H) or BHK cells (I and J). (A and B) Fixed sheets were immunostained for syntaxin 1 (A) followed by staining with FM1-43 (B) to visualize phospholipid membranes. Syntaxin is distributed in numerous brightly fluorescent dots (A). Fluorescence arising from stained syntaxin can not be seen through the brighter FM1-43 fluorescence (B). (C and D) Unfixed membrane sheets were stained for syntaxin 1 and imaged (C). They were then fixed and re-stained (D). Closed circles indicate syntaxin clusters on fixed membrane sheets that were already positive on the unfixed membrane sheet; dashed circles indicate syntaxin clusters that were visible only after fixation. Note that often a higher concentration of dots was observed at the rim of the membrane sheets (C and D), which is probably due to some membrane curling at the edges. (E and F) Imaging of an unfixed membrane sheet derived from a PC12 cell expressing syntaxin 1A–GFP (E), followed by immunostaining for syntaxin 1 (F). Arrows highlight spots that remained unchanged after antibody labeling. (G and H) Fixed membrane sheets double-stained for syntaxin 1 (G) and SNAP-25 (H). Spots positive for syntaxin 1 are positive (closed circles) or negative (dashed circles) for SNAP-25. (I and J) Unfixed membrane sheets immunostained for syntaxin 4 (I) followed by FM1-43 staining (J). Fluorescent spots on membranes derived from BHK cells were less numerous than syntaxin 1-positive spots on PC12-cell membranes.
Several control experiments were performed to ensure that the dots represent genuine accumulations of syntaxin and are not due to experimentally induced artefacts. First, the membranes were stained for syntaxin 1 without prior fixation (Figure 1C). After imaging, the sample was fixed, re-stained for syntaxin, and imaged again. As shown in Figure 1C and D, the staining patterns were largely identical, except that after fixation labeling intensity was higher and revealed additional spots that were not detectable before fixation. This increase is probably due to epitope masking in the native state, which may be caused by high local protein concentrations. Secondly, PC12 cells were transfected with syntaxin 1A C-terminally fused to green fluorescent protein (GFP) in order to visualize syntaxin by an independent approach. Membrane sheets were generated from transfected cells and analyzed directly by fluorescent microscopy without any further treatment. As shown in Figure 1E, the staining pattern was very similar to that observed after antibody labeling, with numerous brightly fluorescent dots. When these membranes were treated subsequently with primary and secondary antibodies, and again imaged by fluorescence microscopy, the staining patterns were very similar, indicating that clusters are not due to antigen patching (compare Figure 1E and F, see also below).
Next, we analyzed the distribution of SNAP-25 in the membrane sheets. SNAP-25, which is anchored to the membrane via palmitoyl side chains, operates alongside syntaxin 1 in the exocytosis of secretory vesicles (Rothman, 1994; Jahn and Südhof, 1999). Staining for SNAP-25 revealed a dot-staining pattern that was more diffuse and widespread than that of syntaxin (Figure 1H, see also Figure 2D). Double-labeling for syntaxin and SNAP-25 revealed that a majority of the syntaxin clusters colocalized more or less precisely with corresponding domains labeled for SNAP-25 (compare Figure 1G and H). In contrast, many SNAP-25-positive areas were devoid of syntaxin, rendering it unlikely that the majority of SNAP-25 forms binary complexes with syntaxin in the plasma membrane. Probably, SNAP-25 is more mobile within the plane of the membrane due to the lack of a transmembrane domain.

Fig. 2. Secretory vesicles are preferentially docked to sites on membrane sheets that contain clusters of syntaxin 1 and SNAP-25. (A–D) Secretory vesicles colocalize with syntaxin 1 or SNAP-25. Membrane sheets were generated from cells expressing the secretory granule marker NPY–GFP, fixed and immunostained for either syntaxin 1 (A and B) or SNAP-25 (C and D). Circles were super imposed on spots of GFP fluorescence (A and C) and transferred to identical pixel locations of the corresponding immunofluorescence picture (B and D). Closed circles, vesicles with a corresponding signal; dotted circles, vesicles with an only partially corresponding overlap; dashed circles, vesicles with no detectable corresponding immuno reactivity. (E) Criteria for rating the association of granules with clusters. Left, cartoon of a granule (green, 120 nm-diameter) touching with its tip the edge of a cluster (red, 200 nm-diameter). Right, as above but after considering the diffraction-induced spreading of the signal by the objective lens (granule 340 nm, cluster 370 nm). Granules were rated to be associated with a cluster (positive) when their signals were at least overlapped to the degree shown; less-overlapping granules were rated as negative; in some cases, a clear assessment was not possible (rated as neutral).
We then asked the question of whether syntaxin clusters are confined to neurosecretory cells or whether they are also present in other cell types, for example cells that do not possess a regulated secretory pathway. Membrane sheets were generated from fibroblasts [baby hamster kidney (BHK) cells] and labeled for syntaxin 4, a syntaxin family member known to be expressed at the plasmalemma of many cells (Bennett and Scheller, 1993). As shown in Figure 1I, syntaxin 4 was found to be concentrated in dots that were similar in appearance to, but somewhat less dense than those observed for syntaxin 1 in PC12 cells. FM1-43 staining of the same membrane sheet (Figure 1J) confirmed that the plasma membrane remained intact after sonication.
To obtain an estimate of the size of the clusters, unfixed membrane sheets of PC12 cells were stained for syntaxin 1 and imaged at maximum magnification. Line scans were then performed through the center of fluorescent spots using a line width of one pixel (68 nm). Brightness exceeded half its maximum value over a 370 ± 7 nm distance (30 scans from five cells). Spots appear enlarged due to the diffraction-limited resolution of the objective. To obtain an upper limit for their size, the point spread function (PSF) was determined by measuring the size of 100-nm fluorescent beads {PSF = [(beadssize measured)2 – (beadsreal size)2]1/2}, resulting in 319 nm. The real size of the spots can then be calculated by subtraction of the PSF {spotreal size = [(spotmeasured size)2 – (PSF)2]1/2}, resulting in 189 ± 14 nm (median 198 nm).
SNARE clusters represent docking and fusion sites for secretory granules
As outlined above, exocytosis requires the interaction between plasma membrane-bound syntaxin 1A and SNAP-25 with granule-bound synaptobrevin. Similar to other non-polarized cells, there is no evidence that exocytosis occurs at preferential sites in undifferentiated PC12 cells. Thus, in principle, vesicles should be able to dock and fuse everywhere, and there are many examples of cells in which fusion events appear to be randomly scattered over the cell surface (Steyer et al., 1997; Angleson et al., 1999; Toomre et al., 2000). However, it it unknown whether each site of the plasma membrane is fusion competent. Rather, the uneven distribution of syntaxin described above suggests that docking and fusion may be confined to sites containing syntaxin clusters. We therefore used a combination of methods to investigate whether the syntaxin clusters correspond to sites where granules dock and fuse.
To analyze the association of secretory granules with syntaxin and SNAP-25 clusters, PC12 cells were transfected with neuropeptide Y fused to green fluorescent protein (NPY–GFP). This construct was previously shown to be sorted and packaged into secretory granules (Lang et al., 1997) and thus provides a convenient fluorescent marker for secretory granules. When membranes containing labeled granules were immunostained for syntaxin 1, a high degree of colocalization was observed (Figure 2A and B). Quantitation revealed that 62% of all labeled granules corresponded to a syntaxin cluster (Table I, for rating criteria see Figure 2E), whereas in ∼27%, no correspondence was observable (rated as ‘negative’, Table I). In ∼11%, a clear assessment was not possible, and these granules were rated as ‘neutral’ (Table I, examples in Figure 2A and B). To differentiate between specific association and accidential colocalization, we randomized the syntaxin clusters (by horizonal or vertical mirroring of the image) and then determined the degree of colocalization by the same procedure. Only 23% of the granules corresponded to syntaxin clusters by this procedure, whereas 72% were rated as negative (Table I), demonstrating that granules are associated with syntaxin clusters in a highly preferential manner.
Table I. Colocalization of secretory granules with clusters.
| Secretory granules (NPY–GFP) | Positive (%) | Negative (%) | Neutral (%) |
|---|---|---|---|
| Syntaxin 1 (n = 10) | 62.3 ± 4.9 | 26.8 ± 5.3 | 11.1 ± 1.7 |
| Syntaxin 1 (background) | 23.3 ± 3 | 72.0 ± 3.5 | 4.8 ± 1.3 |
| SNAP-25 (n = 6) | 74.2 ± 2.4 | 15.2 ± 3.4 | 10.8 ± 2.5 |
| SNAP-25 (background) | 45.5 ± 6.9 | 45.3 ± 7.9 | 9.0 ± 3.3 |
Colocalization was rated as described in Figure 2E where n is the number of membrane sheets (with at least 10 secretory granules per membrane sheet) analyzed for each condition. Values are given as mean ± SEM.
Staining of membranes for SNAP-25 again revealed a high degree of colocalization with GFP-labeled granules (Figure 2C and D) that exceeded 74%. However, due to the high density of SNAP-25-labeling it is difficult to differentiate between specific and accidental colocalization (Figure 2C and D). Indeed, when the same randomization procedure as for syntaxin was carried out, a ‘background’ association of 46% was found.
The presence of syntaxin patches and their association with granule docking sites was confirmed by immuno-gold electron microscopy using two different procedures. First, ultrathin cryosections were prepared from intact PC12 cells and labeled for syntaxin 1. As shown in Figure 3A–C, labeling was frequently observed at granule docking sites and was generally confined to the plasma membrane. Despite the low labeling intensity, >80% of all gold grains (n = 192) were separated by <100 nm, in agreement with the view that syntaxin is locally concentrated. Secondly, granule-containing membrane sheets were analyzed by pre-embedding immuno-gold electron microscopy. Figure 3D–F shows vertical sections through membrane sheets. Gold grains formed carpets of 100–200 nm on the inner leaflet of the plasma membrane (Figure 3D and F). Granules were usually found to be docked on such gold-labeled segments. Virtually no labeling was observed in the areas between the densely labeled carpets.

Fig. 3. Syntaxin clusters are visible after immuno-gold electron microscopy of PC12 cells (A–C) or of membrane sheets (D–F). (A–C) Ultrathin frozen sections were immuno-gold labeled for syntaxin 1 and viewed by electron microscopy. Gold grains (arrows) are frequently clustered and label specifically the plasma membrane. Gold clusters are found at contact sites between vesicles and the plasma membrane (A and B), but also at sites where apparently no granules reside (C). Membrane sheets were immuno-gold labeled for syntaxin 1, fixed and embedded for electron microscopy. (D–F) Gold grains form 100–200 nm-large carpets that are associated with secretory vesicles. (E) Control, where the primary antibody has been omitted. Scale bars, 250 nm.
Next we investigated whether vesicles attached to syntaxin clusters can undergo exocytosis at these sites. Membrane sheets were generated from NPY–GFP-expressing PC12 cells, which were subsequently immunolabeled for syntaxin to visualize the clusters. Cytosol containing Mg-ATP and 100 µM Ca2+ was then added to prime and trigger exocytosis. GFP-labeled granules disappeared in a Ca2+-dependent manner [43 ± 3% (n = 9 membrane sheets) versus 13 ± 2% (n = 5) when Ca2+ was omitted over an incubation period of 30 min], in agreement with our previous report (Avery et al., 2000). As shown in Figure 4, exocytosis often occurred directly on top of syntaxin clusters. In most cases, no obvious visible changes in the shape of the clusters occurred after exocytosis (not shown). Together, we conclude from these data that SNARE clusters represent functional sites for vesicle docking and fusion.

Fig. 4. Secretory granules undergo exocytosis at syntaxin clusters. Unfixed, syntaxin-stained (red) membrane sheet produced from a cell expressing the secretory granule marker NPY–GFP (green). Syntaxin 1 (A) and NPY–GFP (B–D), imaged at various times after stimulation of exocytosis with elevated concentrations of free calcium in the presence of Mg-ATP and rat brain cytosol (times are indicated). Arrows indicate granules that display exocytic activity during the stimulation period, resulting in their disappearance or dimming. Circles mark identical regions in (A) and (B). Continuous circles, granules that are associated with a cluster; dashed circles, granules that lack a corresponding signal. Note that the pattern of syntaxin clusters did not change during the experiment.
The integrity of syntaxin clusters depends on cholesterol
In the following experiments we investigated which factors are responsible for syntaxin clustering. Syntaxin is known to homo-oligomerize via both its cytoplasmic domain (Lerman et al., 2000) and its transmembrane domain (Laage et al., 2000). Furthermore, one or several of the many hetero-oligomeric protein–protein interactions that syntaxin is known to undergo may be responsible for clustering. Alternatively, clustering may be due to phase separation within the bilayer, which could be caused by specific protein–lipid interactions. The best characterized of such lipidic ‘rafts’ are enriched in cholesterol and sphingomyelin and contain specific proteins such as caveolin and proteins with GPI anchors. They are also referred to as DRMs (detergent-resistant membranes) (Simons and Ikonen, 1997; Brown and London, 2000). These rafts are insoluble in Triton X-100, but disintegrate upon cholesterol removal by specific compounds such as methyl-β-cyclodextrin (Scheiffele et al., 1997; Keller and Simons, 1998).
Membrane sheets were treated with various concentrations of methyl-β-cyclodextrin in order to examine whether syntaxin clusters are sensitive to cholesterol depletion. Methyl-β-cyclodextrin is known to complex cholesterol and to deplete it from membranes with high selectivity, leaving phospholipids unaffected (Ohtani et al., 1989). As shown in Figure 5A, treatment led to a dramatic change in the staining pattern. First, the number of spots increased and became less separate, probably due to the break-up of existing clusters. In addition, the staining intensity increased in a similar manner as to that observed after fixation (see above), suggesting that cholesterol depletion, like fixation, increases accessibility of syntaxin to the antibody. When the concentration of methyl-β-cyclodextrin was increased or the incubation time was extended, the clusters disappeared and syntaxin was distributed almost uniformly over the membrane surface. Frequently, different states of cluster disintegration were observed on the same coverslip. A very similar behavior was observed for SNAP-25 (not shown). When treatment with methyl-β-cyclodextrin was extended further, the membrane integrity was progressively disrupted (Figure 5A). To examine whether syntaxin clusters of other cell types are also dependent on cholesterol, membrane sheets prepared from BHK cells were depleted of cholesterol. A similar disintegration of syntaxin 4-containing clusters was observed upon treatment with methyl-β-cyclodextrin (data not shown).

Fig. 5. The integrity of syntaxin clusters depends on cholesterol. (A) Clusters disintegate upon cholesterol depletion.Unfixed membrane sheets were treated for 30 min at 37°C with 15 mM methyl-β-cyclodextrin and then immunostained for syntaxin 1. Upper row, membranes displaying increasing degrees of cyclodextrin-induced changes. Spots become less separated until a uniform distribution of the label is observed. Finally, membranes disintegrate. Boxes in the images indicate regions that are magnified below. (B and C) In vivo labeling of syntaxin 1 with photocholesterol. PC12 cells were photoaffinity labeled with either [3H]photocholesterol (B) or 10-azistearic acid/[3H]choline (C). Syntaxin was immunoprecipitated from detergent-extracted cell lysates using a monoclonal antibody against syntaxin 1. Aliquots of starting extracts (start), the supernatants after immunoprecipitation (unbound) and the immunoprecipitates (bound) were subjected to SDS–PAGE followed by blotting on nitrocellulose membrane. Left, autoradiographs (detected by phosphoimaging); right, immunoblots for syntaxin 1(using a polyclonal antibody) of the same membranes. For comparison, the starting material was adjusted to approximately equal amounts of radioactivity.
The dependence of syntaxin clusters on membrane cholesterol prompted us to investigate whether syntaxin is directly associated with cholesterol in the membrane. A recently developed procedure was used that involves loading of intact PC12 cells with [3H]photocholesterol, a radiolabeled cholesterol derivative containing a diazirine group. Photoactivation generates a short-lived and highly reactive carbene intermediate that inserts itself into C–H bonds of neighboring molecules, such as the transmembrane domains of membrane proteins. A multitude of proteins are labeled under these conditions (Figure 5B, left lane), in agreement with earlier observations (Thiele et al., 2000). To investigate whether syntaxin was labeled, the protein was immunoprecipitated from labeled PC12 cell extracts. As shown in Figure 5B (right lane), syntaxin was radiolabeled. As a control, we photolabeled membrane proteins with a diazirino-derivative of phosphatidyl choline. No labeling of syntaxin was found, although a comparable amount of radioactivity was incorporated into protein. These data demonstrate that syntaxin is directly associated with cholesterol and indicate that the syntaxin clusters are enriched in cholesterol.
The dependence of syntaxin clustering on membrane cholesterol prompted us to investigate whether these clusters represent microdomains identical to DRMs. After solubilization of membranes in Triton X-100, DRMs float to a low density during density gradient centrifugation because of their high lipid content (Brown and Rose, 1992). However, they become Triton-soluble when cholesterol is depleted by methyl-β-cyclodextrin prior to Triton X-100 treatment (Scheiffele et al., 1997; Keller and Simons, 1998). Therefore we tested whether, after extraction with non-ionic detergents, syntaxin would co-fractionate with cholesterol, sphingomyelin and GPI-anchored proteins on a density gradient. PC12-cells were extracted with 1% Triton X-100 for 30 min at 4°C, adjusted to 40% OptiprepTM and layered on the bottom of a density gradient. After centrifugation, the distribution of lipids and proteins on the gradient was analyzed by immunoblotting and high-performance thin layer chromatography (HP-TLC). As a marker for GPI-linked proteins we used Thy-1 (Harder et al., 1998). As expected, Thy-1 was found floating together with sphingomyelin and cholesterol on the top of the gradient (Figure 6A, B and D), indicating that our protocol was appropriate for the separation of these rafts. In contrast, syntaxin and SNAP-25 remained at the bottom of the gradient together with the majority of phospholipids, well separated from the fractions enriched in Thy-1 and sphingomyelin (Figure 6B–D). In addition to Triton X-100, varying concentrations (up to 2%) of Lubrol (C12E9), Lubrol WX, Triton X-102, CHAPS and cholate were used. In all detergents, syntaxin and SNAP-25 remained at the bottom of the gradient unless the detergent concentrations were too low for complete micellarization of phospholipids.
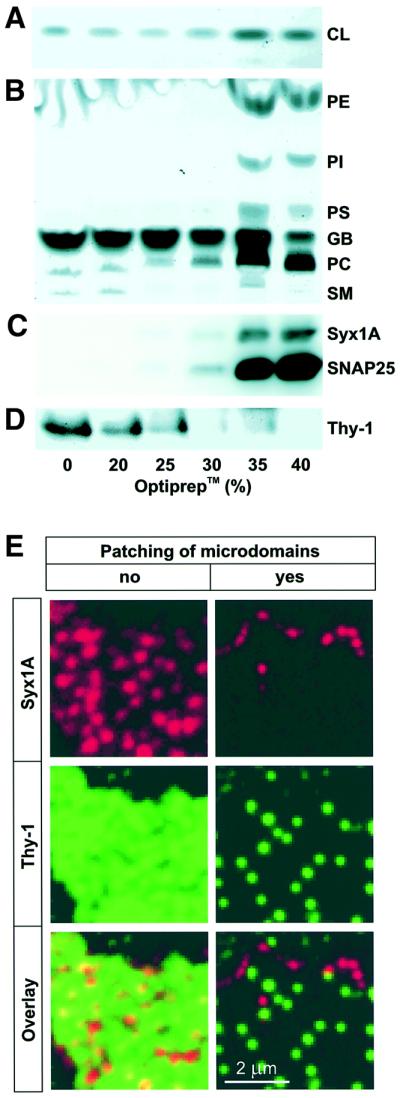
Fig. 6. Syntaxin clusters are distinct from DRMs. (A–D) Syntaxin clusters do not float on a density gradient after extraction with Triton X-100. Fractions of an OptiprepTM-sucrose flotation step gradient from cells extracted with 1% Triton X-100 were analyzed by HP-TLC (A and B) and immunoblotting using monoclonal antibodies against syntaxin 1 and SNAP-25 (C) or Thy-1 (D). (A) CL, cholesterol. (B) PE, phosphatidylethanolamine; PI, phosphatidylinositol; PS, phosphatidylserine; GB, gradient buffer; PC, phosphatidylcholine; SM, sphingomyelin, all as judged by comparison with standard markers or gradient buffer. (E) Syntaxin- and Thy-1-microdomains do not co-patch upon antibody-induced cross-linking. Left, numerous spots of syntaxin 1 (red) and Thy-1 (green) are visualized by indirect immunofluorescence on unfixed membrane sheets when excess amounts of primary and secondary antibodies were used. Right, same protocol, but primary and secondary antibodies were diluted to maximize cross-linking of microdomains, resulting in clear segregation of the Thy-1 and syntaxin 1 signal to different patches.
To confirm the differences between syntaxin clusters and DRMs by an independent approach, we performed double-labeling of membrane sheets for syntaxin and Thy-1 (Figure 6E). The staining patterns were clearly different, but due to the labeling density for Thy-1 it could not be excluded that syntaxin is associated with a subpopulation of DRMs. Therefore, we performed an antibody-patching experiment in which antibodies against both Thy-1 and syntaxin were added to unfixed membrane sheets. The concentrations of both primary and secondary antibodies were adjusted to maximize patching. As shown in Figure 6E (right panels), Thy-1 and syntaxin segregated into separate sets of patches. Since the labeling was lower due to the reduced antibody concentrations, we also fixed the samples after patching and re-stained, yielding virtually identical results (not shown). We conclude that plasma membrane-associated SNAREs form clusters that are dependent on cholesterol, but are different from DRMs.
Dispersion of syntaxin clusters by cholesterol removal is associated with an inhibition of exocytosis
The data described so far show that plasma membranes contain microdomains with clustered SNARE proteins, which disintegrate upon removal of cholesterol. We have also shown that secretory granules preferentially dock and fuse at these sites, suggesting that SNARE clustering promotes, or even perhaps is required for exocytosis. Therefore, we investigated whether removal of cholesterol affects exocytosis.
The following experiments were performed on intact PC12 cells. First, we used confocal microscopy to confirm that treatment by methyl-β-cyclodextrin disperses syntaxin clusters in intact cells. Batches of PC12 cells were treated with cyclodextrin (15 mM, 30 min). Under these conditions, the cholesterol content was reduced by ∼40% as determined by quantitative HP-TLC. The cells were then fixed and immunostained for syntaxin. As shown in Figure 7A, untreated cells showed punctate staining of the plasma membrane. The resolution of the dots was usually not as clear as on the membrane sheets, which is due to the limited resolution of the confocal microscope in the z-axis. Cyclodextrin treatment led to a significant dispersal of these dots. In order to assess this effect in more detail, intensity profiles were obtained by placing a line scan through the center of the plasma membrane. Examples of such scans are shown in Figure 7A and B. To reduce instrument noise, low-pass filtering was performed (Figure 7C). For a quantitative evaluation of many cells, the ratio of peak amplitude to basal fluorescence was determined for each peak, and the histograms of these values were plotted. As shown in Figure 7 (right panels), a significant shift to smaller values was observed in cyclodextrin-treated cells.

Fig. 7. Disintegration of syntaxin clusters upon cholesterol depletion of intact PC12 cells. Confocal micrographs taken from the periphery of aldehyde-fixed PC12 cells stained for syntaxin 1. Prior to immuno staining, cells were either treated for 60 min with Ringer solution (A), or for 30 min with Ringer solution followed by 30 min 15 mM methyl-β-cyclodextrin in Ringer solution (B). For quantitative analysis of the differences between control and cyclodextrin-treated cells, a 600-nm-wide line scan (not shown for clarity) was put through the center of the peripheral fluorescence, and the fluorescence intensity was recorded (lower panels). Intensity traces were low-pass filtered to remove noise [(C), compare traces shown in (C) and (A)]. For each condition, the ratios of individual peak amplitudes to the basal fluorescence signal [see gray box in (C)] was determined from filtered traces and plotted as histograms [right panels in (A) and (B)]. (D) Histograms shown in (A) and (B) presented as cumulative plots.
Next we studied whether cyclodextrin influences exocytosis using amperometric measurements of dopamine release from PC12 cells. For this, PC12 cells were treated with methyl-β-cyclodextrin as described above. Depolarization-induced exocytosis was triggered by superfusion with a high potassium-containing solution. In untreated cells, depolarization led to robust exocytotic response. In contrast, the response of cells pretreated with methyl-β-cyclodextrin was reduced by ∼60% (Figure 8A and B). Furthermore, some exocytotic activity was observed after the end of the stimulus (Figure 8, and data not shown), suggesting that the decline of intracellular Ca2+ concentrations to baseline levels was retarded. To rule out differences in Ca2+ influx during depolarization, we also triggered exocytosis by the Ca2+-ionophore ionomycin. Again, a reduction by >50% in the frequency of exocytotic events was observed after cyclodextrin treatment (Figure 8C and D). In contrast, the properties of individual exocytotic events with respect to charge and kinetics remained unchanged (Table II).

Fig. 8. Cholesterol depletion inhibits exocytosis. (A) Superfusion of cells with a depolarizing solution (containing 80 mM KCl; bar) triggers an increase in the frequency of amperometric spikes (left trace) that is substantially reduced in methyl-β-cyclodextrin-treated cells (right trace). (B) Analysis of traces as exemplified in (A). In control cells, 32.6 ± 7.8 events (n = 9 cells) were detected in a stimulation period of 20 s versus 9.46 ± 3.1 events (n = 13 cells) in cyclodextrin-treated cells. (C) Superfusion of cells with ionomycin (2.5 µM; bar) triggers an increase in the frequency of amperometric spikes (left trace) that is substantially reduced in methyl-β-cyclodextrin-treated cells (right trace). (D) Quantitative analysis of traces as exemplified in (C). In control cells, 34 ± 7.2 events (n = 12 cells) were detected in a stimulation period of 60 s versus 15.2 ± 5.4 events (n = 11 cells) in cyclodextrin-treated cells. (E) Inhibition of exocytosis after a short time application of methyl-β-cyclodextrin. Cells were stimulated for 20 s with a depolarizing solution [as in (A)], superfused for 2 min with Ringer solution (control) or Ringer solution containing 15 mM methyl-β-cyclodextrin, and then superfused again for 20 s with a depolarizing solution for a second stimulation. The number of events of the first response (R1) was normalized to 100%. The following values were obtained for the second stimulation (R2): control cells, 78.1 ± 19% (n = 9 cells); cyclodextrin-treated cells, 28.7 ± 11% (n = 13). (F and G) Neurite from a NGF-differentiated PC12 cell expressing the secretory granule marker NPY–GFP before (F) and after (G) stimulation. The neurite was stimulated to secrete NPY–GFP into the buffer by permeabilization of the plasma membrane with digitonin in the presence of 50 µM free calcium, Mg-ATP and rat brain cytosol. Exocytosis is measured by the loss of GFP fluorescence from the neurite. (H) Quantitative determination (by video microscopy) of neurite fluorescence. In the presence of calcium, neurites lost 35.7 ± 5% (n = 18) of their GFP fluorescence; when calcium was omitted, a decrease of 8.2 ± 4% (n = 11) was observed. Pretreatment of neurites with 15 mM methyl-β-cyclodextrin for 30 min led to a decrease of only 13.3 ± 3.4% (n = 19), although calcium was present. Values are given as mean ± SEM.
Table II. Properties of transmitter release from secretory granules in PC12 cells.
| Event parameters | Control (eight cells, 273 events) | Cyclodextrin (15 mM) (13 cells, 276 events) |
|---|---|---|
| Charge (fC) | 30.0 ± 33 | 27.1 ± 29.6 |
| Amplitude (pA) | 9.3 ± 13.2 | 11.1 ± 17.2 |
| 50–90% rise-time (µs) | 1050 ± 1270 | 780 ± 1460 |
| Halfwidth (µs) | 3480 ± 3000 | 3145 ± 3100 |
Events with an amplitude >1.5 pA were analyzed. The 50–90% rise-time reflects the time span between 50 and 90% amplitude. The half-width gives the duration of the current transient at half height of its peak amplitude. Measurements are given as mean ± SD.
In a separate set of experiments, cells were superfused only briefly (2 min) with methyl-β-cyclodextrin, and the response to a brief depolarizing pulse was recorded before and after superfusion. This protocol was chosen to minimize nonspecific membrane damage (including intracellular membranes). In cells superfused only with buffer, the second stimulus resulted in an exocytotic frequency of ∼75% of the first pulse, which is typical for this type of repetitive stimulation. In cyclodextrin-treated cells, a significant reduction of event frequency was observed (28% of the first stimulus) (Figure 8E). These data show that inhibition of exocytosis is observable before the dispersal of the SNARE clusters can be resolved with the microscope (see Discussion).
The effect of cyclodextrin treatment on exocytosis was also monitored using an independent approach. PC12 cells transfected with NPY–GFP were grown for three days in the presence of nerve growth factor (NGF) to promote neurite outgrowth. In agreement with earlier observations, GFP-containing secretory vesicles accumulated at the tips of the neurites (Figure 8F). The cells were then permeabilized with digitonin and incubated in cytosol, Mg-ATP and Ca2+ to stimulate exocytosis. Exocytosis is monitored by the release of fluorescent NPY–GFP, which is quantified by video microscopy (compare Figure 8F and G). As shown in Figure 8H, addition of Ca2+ to the buffer resulted in a 35% reduction of fluorescence during 10 min, whereas <9% reduction was observed when EGTA was added instead of Ca2+. When cells were pretreated with cyclodextrin, fluorescence decreased only by 13%, again documenting that exocytosis is inhibited under these conditions.
Discussion
In the present study we have shown that SNAREs form clusters in the plasma membrane that define sites at which secretory vesicles dock and fuse with high preference. The clusters contain cholesterol but are distinct from DRMs. Cholesterol removal causes dispersion of these clusters, which is associated with an inhibition of exocytosis.
The syntaxin clusters are characterized by the following features. First, they form circumscribed areas of high local protein concentration, whereas in the surrounding areas syntaxin was only detectable after overexpression. Secondly, their integrity depends on the presence of cholesterol. Cholesterol is probably enriched in these domains because syntaxin can be cross-linked with cholesterol but not with phosphatidylcholine. Thirdly, the domains are clearly different from DRMs, since (i) their distributions in the membrane are not identical to DRMs; (ii) they do not co-patch with DRMs upon antibody cross-linking; and (iii) they are soluble in all detergents examined. Solubility in Lubrol WX also distinguishes these clusters from prominin-containing microdomains that are associated with microvilli (Röper et al., 2000). Fourthly, the stability of the clusters does not depend on the presence of attached vesicles since only a subset of domains had vesicles bound to them.
Unfortunately, the clusters can not be isolated due to their sensitivity to detergents, precluding a comprehensive biochemical analysis. However, some conclusions can be drawn concerning the nature of these clusters and the mechanisms by which the proteins are kept together. Since cholesterol removal suffices to disperse evenly syntaxin in the plane of the membrane, a major contribution of cross-linking proteins (e.g. elements of the submembrane cytoskeleton) is unlikely. Rather, the domains are probably stabilized by cholesterol–protein interactions and by phase-partitioning of cholesterol and other membrane lipids. Indeed it is possible that lateral phase segregation of lipids may contribute to a micro-patterning of the membrane that promotes the formation of protein clusters for not only SNAREs but also other membrane proteins. The extent and composition of individual clusters may then be determined by specific protein–protein interactions that, for a given cluster, may incorporate some proteins and exclude others. The interacting forces, however, are probably relatively weak and primarily hydrophobic, since the clusters are dependent on the presence of cholesterol for stabilization. Indeed, preliminary results suggest that other membrane proteins (such as the Na,K-ATPase) are also clustered (although less conspicuously than the SNAREs) and that these clusters are segregated from clusters containing SNARE proteins. DRMs may represent a special case of such domains that are of unusual stability.
How and where are the clusters formed? DRMs and the recently described prominin rafts (Röper et al., 2000) are assembled in the trans-Golgi network and may retain their integrity during membrane trafficking (Brown and Rose, 1992; Scheiffele et al., 1997). The origin of syntaxin clusters remains to be established. Preliminary experiments indicate that cyclodextrin-mediated disassembly of syntaxin is reverted when the membranes are re-loaded with cholesterol. It is currently under investigation whether such re-assembly depends on membrane traffic, is mediated by the cytoskeleton, or is only driven by cholesterol-dependent interactions, such as phase-partitioning and the formation of cholesterol–protein complexes, in the plane of the plasma membrane.
The striking correlation between bound vesicles and syntaxin clusters documents that the clusters represent preferred docking sites for secretory vesicles. Further more, our data show that vesicles exocytose at these sites, showing that they represent functional entities for membrane fusion. Although the concentration of syntaxin in the clusters still needs to be determined, it is most probably very high. This results in densely packed carpets at the fusion site that may facilitate exocytosis. Interestingly, we observed a minor population of vesicles that underwent exocytosis, although they were not associated with detectable syntaxin clusters (our unpublished observations). Thus, it appears that some freely mobile syntaxin may be present in membrane areas outside of clusters, which allows for the formation of fusion-competent SNARE complexes (albeit less efficiently) away from such domains.
Cholesterol depletion is known to inhibit endocytosis (Rodal et al., 1999; Subtil et al., 1999). Our data shows clearly that removal of cholesterol not only disassembles syntaxin clusters but also reduces exocytosis. For the following reasons we believe that a lack of cholesterol does not cause an unspecific disintegration of the plasma membrane.
First, depolarization-induced, Ca2+-dependent exocytosis is not abolished, showing that cyclodextrin treatment does not lead to a collapse of the membrane potential. Secondly, the shape and kinetics of the amperometric signals are unchanged, showing that neither the fusion process nor the integrity of the secretory organelle is altered. Thirdly, a similar inhibition is already observed after 2 min superfusion with cyclodextrin. Under such conditions it is unlikely that intracellular membranes are affected, indicating that inhibition is due to a specific process that is sensitive to even moderate cholesterol depletion of the plasma membrane. Finally, the number and localization of secretory granules is not changed as judged by comparing electron micrographs of treated and untreated cells (our unpublished observations). Although alternative explanations can not be excluded, we believe that the inhibitory effect on exocytosis caused by cholesterol depletion is due to the dispersal of SNARE proteins.
In summary, our data show that vesicles dock and fuse with the plasma membrane preferentially at sites of high local concentrations of SNAREs. Since it is generally believed that multiple trans-SNARE complexes need to form for fusion to occur, such local clusters would greatly facilitate the fusion reaction. Moreover, recent evidence has shown that formation of trans-complexes is reversible and initially of low affinity (Xu et al., 1999). Thus, SNAREs would be able to diffuse out of the contact site between the fusing membranes if they were freely mobile in the plane of the membrane, and it would be more difficult to achieve coordinated assembly of enough trans-complexes. The need for high local SNARE concentrations may also explain, at least in part, why proteoliposomes reconstituted with appropriate sets of SNAREs only fuse with a low rate and efficiency unless abnormally high SNARE concentrations are used (Weber et al., 1998; our unpublished observations). Clustering of SNAREs may thus be a prerequisite for efficient function of SNAREs in membrane fusion.
Materials and methods
Antibodies
Mouse monoclonal antibodies were used for the following proteins: syntaxin 1A/B (HPC-1; Barnstable et al., 1985), SNAP-25 (clone Cl 71.1; Bruns et al., 1997; available from Synaptic Systems, Göttingen, Germany, Cat#111001), and Thy-1 (obtained from Serotec Ltd, Oxford, UK, Cat#MCA47R). Rabbit polyclonal sera were used for SNAP-25 (Aguado et al., 1996) and syntaxin 4 (affinity purified; Hansen et al., 1999). A rabbit anti-serum for syntaxin (R31) was generated using recombinant syntaxin 1A as antigen that lacked the transmembrane region and contained a His6-tag (Fasshauer et al., 1997).
Plasmids
The cDNAs encoding chimerae of either rat syntaxin 1A (provided by R.H.Scheller) or human neuropeptide Y (Lang et al., 1997) with enhanced GFP (EGFP) were constructed by inserting the corresponding proteins in the multi-cloning site of the EGFP-containing mammalian expression vector pEGFP-N1 (Clonetech Lab., Heidelberg, Germany). We modified the syntaxin 1A and neuropeptide Y cDNAs by PCR to introduce restriction sites that allowed subsequent cloning in-frame with the N-terminal end of EGFP. Both constructs were verified by restriction analysis and DNA sequencing.
Cell culture and transfection
PC12 cells (clone 251; Heumann et al., 1983) were maintained and propagated as described (Lang et al., 1997). Transfection of PC12 cells was performed essentially as described previously (Lang et al., 1997).
BHK cells were maintained and propagated in 15-cm-diameter culture dishes in Dulbecco’s modified Eagle’s medium [DMEM, with 4.5 g/l glucose, 5% fetal calf serum (FCS), 10% tryptose phosphate, 4 mM glutamine and 60 U/ml each of penicillin and streptomycin] at 37°C in 5% CO2. For immunofluorescence experiments, cells were scraped from their substrate with a rubber policeman, titurated, diluted in growth medium and plated onto 20 mm-glass coverslips coated with poly-l-lysine (Lang et al., 1997). PC12 cells (Greene and Tischler, 1976) for electrophysiological measurements were cultured as previously described (Greene et al., 1991).
Immunofluorescence on membrane sheets
For preparation of membrane sheets, cells were grown on poly-l-lysine-coated coverslips and disrupted as previously described (Avery et al., 2000) using a 100 ms ultrasound treatment in ice-cold sonication buffer [20 mM HEPES pH 7.2, 120 mM potassium glutamate, 20 mM potassium acetate, 10 mM EGTA, 2 mM Mg-ATP and 0.5 mM dithiothreitol (DTT)]. Before immunostaining of the membrane sheets, all antibodies were diluted 100-fold in phosphate-buffered saline (PBS) (137 mM NaCl, 2.7 mM KCl, 0.9 mM CaCl2, 0.5 mM MgCl2, 8.1 mM Na2HPO4 pH 7.3) containing 1% bovine serum albumin (BSA), incubated for 45 min at room temperature and centrifuged for 10 min at 16 000 g. Freshly prepared membrane sheets were fixed for 30 min at room temperature in 4% PFA in PBS, quenched for 20 min in PBS containing 50 mM NH4Cl and washed three times in PBS for 10 min each. They were then incubated for 30 min with the primary antibodies. Subsequently, membranes were washed three times in PBS for 10 min each, followed by a 30 min incubation with the secondary antibodies (Cy3-coupled goat-anti-mouse and Cy2-coupled goat-anti-rabbit; Dianova, Hamburg, Germany) diluted 1:200 in PBS containing 1% BSA. Before imaging, membrane sheets were washed twice for 20 min and once for 10–40 min in PBS.
For immunostaining of unfixed membranes, the same protocol was used except that instead of PBS, Kglu buffer (20 mM HEPES pH 7.2, 120 mM potassium glutamate, 20 mM potassium acetate, 2 mM Mg-ATP, 0.5 mM DTT) was used. For cholesterol depletion, membrane sheets were treated in a humid chamber for 30 min at 37°C with 15 mM methyl-β-cyclodextrin (Sigma) in Kglu buffer. After two washing steps in Kglu buffer, membrane sheets were stained for syntaxin 1 using the HPC-1 monoclonal antibody.
Fluorescence microscopy
Membrane sheets were analyzed using either a Zeiss Axiophot 2 or a Zeiss Axiovert 100 TV fluorescence microscope with a 100× 1.4 NA plan achromate objective. For imaging we used either a front-illuminated slow-scan CCD-camera (1317 × 1035 Kodak chip, 6.8 × 6.8 µm pixel size; Princeton Instruments Inc.) or a back-illuminated frame transfer CCD-camera (2 × 512 × 512-EEV chip, 13 × 13 µm pixel size; Princeton Instruments Inc.) with a magnifying lens (1.6× Optovar) to avoid spatial undersampling by the large pixels. GFP-, Cy2- and FM1-43-fluorescence were detected using Zeiss filter set 10 (excitation filter BP 450–490, BS 510, emission filter BP 515–565); for Cy3-fluorescence Zeiss filter set 15 was used (excitation filter BP 540–552, BS 580, emission LP 590). Images were analyzed with Metamorph (Universal Imaging Corporation, West Chester, PA).
Electron microscopy of ultrathin cryo-sections
Ultrathin cryo-sections were prepared following Tokuyasu (1973). PC12 cells were fixed with 2% PFA and 0.1% glutaraldehyde in 0.1 M sodium phosphate pH 7.4 for 30 min at room temperature directly on cell culture dishes. After removal of the fixation solution, cells were postfixed with 4% PFA and 0.1% glutaraldehyde for 2 h on ice. Cells were removed with a cell scraper from the dishes. After washing with phosphate buffer, cells were embedded in 10% gelatine, cooled on ice and cut into small blocks. The blocks were infused with 2.3 M sucrose at 4°C overnight, mounted on small metal pins, frozen and stored in liquid nitrogen until cryo-ultramicrotomy. Ultrathin cryo-sections were cut at approximately –110°C using a diamond knife (Diatome) in an ultracryomicrotome (Leica), collected using a 1:1 mixture of 1.8% methyl-cellulose and 2.3 M sucrose, and deposited on formvar and carbon-coated nickel grids. For immunolabeling, sections were incubated with anti-serum for syntaxin 1A (diluted 1:30) for 1 h followed by a 25 min incubation with 5 nm colloidal gold-labeled goat-anti-rabbit Fabs (British BioCell). After washing, sections were incubated with uranyl acetate/oxalate, embedded in a mixture of methyl-cellulose, uranyl acetate (Tokuyasu, 1978) and examined with a Phillips CM120 electron microscope.
Electron microscopy on membrane sheets
Coverslips were coated with a thin layer of EM bed 812 (Electron Microscopy Science), polymerized for 8 h at 80°C, coated with poly-l-lysine as given above and glow discharged in air. Cells were grown on the pre-treated coverslips. Membrane sheets were prepared by sonication as described above. For immuno-gold labeling, unfixed membrane sheets were blocked with 0.5% BSA in PBS for 15 min and then incubated for 1 h with HPC-1 monoclonal antibody (ascites diluted 1:100 in PBS containing 0.5% BSA). After three washing steps, membrane sheets were incubated with 6 nm protein A-gold conjugate diluted 1:100 (obtained from G.Posthuma, Utrecht University, The Netherlands) in 0.5% BSA in PBS plus 0.5 M NaCl. As a control, labeling was carried out without the first antibody. After five washes in PBS, the samples were fixed for 10 min with 2% glutaraldehyde (Electron Microscopy Science) in 0.1 M cacodylate buffer pH 7.4 for 2 h at room temperature. Coverslips were washed and transferred into 1% OsO4 in cacodylate buffer. After a pre-embedding stain with 1% aqueous uranyl acetate, the samples were dehydrated in EtOH and propylenoxid and embedded in EM bed 812. After polymerization for 48 h at 60°C the coverslips were removed by etching with hydrofluoric acid. Ultrathin sections were cut perpendicular to the monolayer and stained with uranyl acetate and lead citrate. The sections were mounted on unfilmed 200 mesh copper grids and evaluated in a Phillips CM 120 Bio Filter.
Cell-free exocytosis on immunostained membrane sheets
Membrane sheets were prepared from PC12 cells transfected with NPY–EGFP as described above. Membrane sheets with ≥10 brightly fluorescent granules were located, and immunostained as for other unfixed preparations with the following modifications to preserve biological activity. Incubations with primary (anti-syntaxin 1 monoclonal antibody) and secondary (Cy3-coupled goat-anti-mouse) antibodies were reduced to 1 min. Subsequent to the incubation with antibody, membranes were rinsed once, followed by two 10 min washes with Kglu buffer. Exocytosis was stimulated as described (Avery et al., 2000) with images of both red and green channels captured every minute for 30 min.
Photoaffinity labeling of cultured PC12 cells
Experiments were performed as described (Thiele et al., 2000). In brief, for labeling with [3H]photocholesterol, cells were supplemented with delipidized FCS and [3H]photocholesterol-methyl-β-cyclodextrin complex was added. After 16 h, cells were washed three times with Ca/Mg-PBS (PBS containing 0.1 mM CaCl2 and 1 mM MgCl2) and irradiated for 30 min in Ca/Mg-PBS with a 100 W mercury lamp. For photoaffinity labeling with 10-azistearic acid/[3H]choline, cells were grown for 16 h in delipidized, 10-azistearic acid-loaded FCS and choline-free DMEM supplemented with [3H]choline.
For further analysis, cells were lysed on ice for 1 h in lysis buffer [50 mM Tris–HCl pH 7.4, 140 mM NaCl, 5 mM EDTA, 0.5 mM phenylmethylsulfonyl fluoride (PMSF), 1% Triton X-100, 0.5% deoxycholate)] The lysate was centrifuged for 8 min at 1000 g, and the supernatant was subjected to an immunoprecipitation using an anti-syntaxin 1 monoclonal antibody. Aliquots of starting extract, the supernatants after immunoprecipitation and the immunoprecipitates were separated by SDS–PAGE (11%, Laemmli) followed by blotting of the proteins on nitrocellulose membrane. Radioactivity was detected by phosphoimaging (Fujifilm BAS-2500, Fuji Photo Film Co., Ltd, Japan) using a Tritium-sensitive BAS-TR2025 imaging plate (Fuji Photo Film Co., Ltd, Japan). From the same membrane, an immunoblot was performed using a rabbit serum for syntaxin (R31) followed by goat-anti-rabbit horse radish peroxidase-coupled secondary antibodies (Bio-Rad Laboratories) and chemiluminescence detection (NENTM; Life Science Products Inc., Boston, MA).
Analysis of syntaxin clusters by confocal microscopy
For controls, PC12 cells were treated for 60 min in Ringer solution (130 mM NaCl, 4 mM KCl, 1 mM CaCl2, 1 mM MgCl2, 48 mM glucose and 10 mM HEPES–NaOH pH 7.4); for cyclodextrin treatment they were incubated for 30 min in Ringer solution followed by 30 min in Ringer solution supplemented with 15 mM methyl-β-cyclodextrin. Cells were then washed twice in Ringer solution and fixed for 20 min with 3.7% PFA in PBS, quenched for 10 min in PBS containing 50 mM NH4Cl, and washed twice, for 5 min each time, first in PBS and then in Tris-buffered saline (TBS). Cells were treated for 5 min in TBS containing 0.1% Triton X-100, and then incubated three times for 10 min in BSA-TBS (TBS containing 0.5% BSA and 0.1% Triton X-100). Cells were then incubated for 1 h with an anti-syntaxin 1 monoclonal antibody diluted 100-fold in BSA-TBS and then washed three times, for 10 min each, with antibody-free BSA-TBS. The cells were incubated subsequently for 1 h with Cy3-coupled goat-anti-mouse diluted 1:500 in BSA-TBS, and washed three times, for 10 min each, in antibody-free BSA-TBS, and twice, for 10 min each, in TBS. Cells were imaged with a Leica confocal microscope at a resolution of 40 nm per pixel, using a 100× 1.4 NA plan achromate objective. The optical sections were scanned using the 568 nm laser line. The images were exported as TIF files and analyzed with Metamorph (Universal Imaging Corporation, West Chester, PA). Low-pass filtering (reducing 88% of the frequency spectrum) of intensity traces was performed using SigmaPlot (SPSS Inc., Richmond, CA).
Flotation density gradient
PC12 cells grown on 3-cm dishes were washed twice in ice-cold PBS, scraped and spun down at 2000 r.p.m. at 4°C in a tabletop centrifuge (Eppendorf). The cells were subsequently lysed at 4°C at a protein concentration of 1.2 mg/ml in 200 µl TNE (25 mM Tris–HCl pH 7.4, 150 mM NaCl, 5 mM EDTA, 1 mM DTT, 1 mg/ml antipain, 1 mg/ml leupeptin, and 1 mM PMSF), 10% sucrose, 1% Triton X-100. After 30 min the suspension was mixed with 400 µl of cold 60% OptiprepTM (Nycomed-Pharma, Oslo, Norway). The mix was split and transferred to two TLS-55 centrifuge tubes (Beckmann, München, Germany). The sample was overlaid with a 0.35 ml step each of 35, 30, 25, 20 and 0% OptiprepTM in TNE, 10% sucrose, 1% Triton X-100. The gradients were spun for 3 h at 50 000 r.p.m. in a TLS-55 rotor (Beckmann, München, Germany) at 4°C. Six fractions were collected from the top of the gradient. The fractions were precipitated with trichloroacetic acid and analyzed by either HP-TLC or immunoblotting followed by goat-anti-mouse horse radish peroxidase-coupled secondary antibodies (Bio-Rad Laboratories) and chemiluminescence detection (NENTM; Life Science Products Inc., Boston, MA).
HP-TLC
For extraction of lipids, 1.2 ml of chloroform:methanol (1:1) were added to 0.6 ml aliquots from the step gradient. The chloroform phase was evaporated with nitrogen. Evaporated lipids were resuspended in chloroform:methanol (3:1), spotted on Nano-Silicagel 60 plates (Merck, Darmstadt, Germany) and pre-developed (only Figure 7B) in chloroform:methanol:acetic acid:water (100:70:7:3). Plates were baked at 180°C for 10 min and then developed again in hexane:diethylether:acetic acid (50:50:0.5). Plates were dried at 180°C for 10 min, cooled down and then soaked in CuSO4 solution. Plates were than baked at 180°C until bands had been developed.
Co-patching of microdomains on unfixed membrane sheets
Membrane sheets were produced as described above. Incubations with primary antibodies [rabbit polyclonal anti-syntaxin 1 (R31) and mouse monoclonal anti-Thy-1] were performed in BSA-PBS (PBS containing 0.5% BSA) for 90 min at 37°C. Membranes were washed three times for 20 min each with BSA-PBS and then incubated for 90 min at 37°C with secondary antibodies (Cy2-coupled goat-anti-rabbit, Cy3-coupled goat-anti-mouse). Membranes were then washed three times for 10 min each and imaged. For controls (Figure 7E, left panel), dilution of antibodies was as follows: R31, 1:4; Thy-1, 1:50; Cy2-goat-anti-rabbit, 1:50; Cy3-goat-anti-mouse, 1:50. For antibody-induced microdomain patching, antibodies were diluted as follows: R31, 1:500; Thy-1, 1:32 000; Cy2-goat-anti-rabbit, 1:5000; Cy3-goat-anti-mouse, 1:10 000.
Electrophysiology
Electrochemical detection of dopamine release was performed with carbon fiber amperometry as described (Bruns et al., 2000). Oxidative current transients were recorded with a List EPC-7 (Heka Electronics, Lambrecht, Germany; electrode voltage set to +800 mV). Amperometric currents were filtered at 3 kHz, digitized gapfree at a rate of 25 kHz, and stored on a personal computer. Signals were again digitally filtered at 3 kHz (effective overall filtering at 2.1 kHz). External saline contained 130 mM NaCl, 4 mM KCl, 5 mM CaCl2, 1 mM MgCl2, 48 mM glucose, 10 mM HEPES–NaOH pH 7.3. For stimulation, KCl was elevated to 80 mM and NaCl reduced to 50 mM. Data shown in Table II were obtained from events recorded during a 20 s stimulation period and the following superfusion period (90 s) with external saline.
In the ionomycin experiments, the external calcium concentration was adjusted to 1 mM CaCl2. Ionomycin was obtained from Calbiochem (Germany). Drugs and external solutions were delivered from a multichannel-perfusion pipette placed ∼400 µm from the cell. Change of the perfusion solution was accomplished by triggered electromagnetic valves (Lee Co., Westbrook, CT).
GFP release from neurites
Cell culture, stimulation of neurites and analysis of GFP release from neurites was performed as previously described (Lang et al., 1997), except that the stimulation protocol was modified extensively. First, neurites were imaged in Ringer solution; permeabilization was performed for 1 min at room temperature with 15 µM digitonin in Kglu buffer containing 0.5 mg/ml rat brain cytosol and, where indicated, 50 µM free Ca2+. Digitonin was then removed and, after an additional 9 min, neurites were imaged for the second time. Prior to the first imaging, neurites were incubated for 30 min at 37°C in Ringer solution, or where indicated, Ringer solution containing 15 mM methyl-β-cyclodextrin.
Acknowledgments
Acknowledgements
The authors gratefully acknowledge the help of Ursel Ries in performing lipid extraction and HP-TLC, Wolfram Antonin for providing anti-syntaxin 4 antibodies, Silke Bruns-Engers for excellent technical assistance, K.Simons for stimulating discussions and W.B.Huttner for making available photocholesterol.
References
- Aguado F., Majo,G., Ruiz-Montasell,B., Canals,J.M., Casanova,A., Marsal,J. and Blasi,J. (1996) Expression of synaptosomal-associated protein SNAP-25 in endocrine anterior pituitary cells. Eur. J. Cell Biol., 69, 351–359. [PubMed] [Google Scholar]
- Angleson J.K., Cochilla,A.J., Kilic,G., Nussinovitch,I. and Betz,W.J. (1999) Regulation of dense core release from neuroendocrine cells revealed by imaging single exocytic events. Nature Neurosci., 2, 440–446. [DOI] [PubMed] [Google Scholar]
- Avery J., Ellis,D.J., Lang,T., Holroyd,P., Riedel,D., Henderson,R.M., Edwardson,J.M. and Jahn,R. (2000) A cell-free system for regulated exocytosis in PC12 cells. J. Cell Biol., 148, 317–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnstable C.J., Hofstein,R. and Akagawa,K. (1985) A marker of early amacrine cell development in rat retina. Brain Res., 352, 286–290. [DOI] [PubMed] [Google Scholar]
- Bennett M.K. and Scheller,R.H. (1993) The molecular machinery for secretion is conserved from yeast to neurons. Proc. Natl Acad. Sci. USA, 90, 2559–2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown D.A. and London,E. (2000) Structure and function of sphingolipid- and cholesterol-rich membrane rafts. J. Biol. Chem., 275, 17221–17224. [DOI] [PubMed] [Google Scholar]
- Brown D.A. and Rose,J.K. (1992) Sorting of GPI-anchored proteins to glycolipid-enriched membrane subdomains during transport to the apical cell surface. Cell, 68, 533–544. [DOI] [PubMed] [Google Scholar]
- Bruns D., Engers,S., Yang,C., Ossig,R., Jeromin,A. and Jahn,R. (1997) Inhibition of transmitter release correlates with the proteolytic activity of tetanus toxin and botulinus toxin A in individual cultured synapses of Hirudo medicinalis. J. Neurosci., 17, 1898–1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruns D., Riedel,D., Klingauf,J. and Jahn,R. (2000) Quantal release of serotonin. Neuron, 28, 205–220. [DOI] [PubMed] [Google Scholar]
- Burgoyne R.D. and Morgan,A. (1995) Ca2+ and secretory-vesicle dynamics. Trends Neurosci., 18, 191–196. [DOI] [PubMed] [Google Scholar]
- Fasshauer D., Bruns,D., Shen,B., Jahn,R. and Brunger,A.T. (1997) A structural change occurs upon binding of syntaxin to SNAP-25. J. Biol. Chem., 272, 4582–4590. [DOI] [PubMed] [Google Scholar]
- Greene L.A. and Tischler,A.S. (1976) Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proc. Natl Acad. Sci. USA, 73, 2424–2428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene L.A., Sobeih,M.M.and Teng,K.K. (1991) Methodologies for the culture and experimental use of the PC12 rat pheochromocytoma cell line. In Banker,G. and Goslin,K. (eds), Culturing Nerve Cells. The MIT Press, London, UK, pp. 207–226.
- Hansen N.J., Antonin,W. and Edwardson,J.M. (1999) Identification of SNAREs involved in regulated exocytosis in the pancreatic acinar cell. J. Biol. Chem., 274, 22871–22876. [DOI] [PubMed] [Google Scholar]
- Hanson P.I., Roth,R., Morisaki,H., Jahn,R. and Heuser,J.E. (1997) Structure and conformational changes in NSF and its membrane receptor complexes visualized by quick-freeze/deep-etch electron microscopy. Cell, 90, 523–535. [DOI] [PubMed] [Google Scholar]
- Harder T., Scheiffele,P., Verkade,P. and Simons,K. (1998) Lipid domain structure of the plasma membrane revealed by patching of membrane components. J. Cell Biol., 141, 929–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay J.C. and Martin,T.F. (1992) Resolution of regulated secretion into sequential MgATP-dependent and calcium-dependent stages mediated by distinct cytosolic proteins. J. Cell Biol., 119, 139–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heumann R., Kachel,V. and Thoenen,H. (1983) Relationship between NGF-mediated volume increase and ‘priming effect’ in fast and slow reacting clones of PC12 pheochromocytoma cells. Role of cAMP. Exp. Cell Res., 145, 179–190. [DOI] [PubMed] [Google Scholar]
- Jahn R. and Südhof,T.C. (1999) Membrane fusion and exocytosis. Annu. Rev. Biochem., 68, 863–911. [DOI] [PubMed] [Google Scholar]
- Keller P. and Simons,K. (1998) Cholesterol is required for surface transport of influenza virus hemagglutinin. J. Cell Biol., 140, 1357–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laage R., Rohde,J., Brosig,B. and Langosch,D. (2000) A conserved membrane-spanning amino acid motif drives homomeric and supports heteromeric assembly of presynaptic SNARE proteins. J. Biol. Chem., 275, 17481–17487. [DOI] [PubMed] [Google Scholar]
- Lang T., Wacker,I., Steyer,J., Kaether,C., Wunderlich,I., Soldati,T., Gerdes,H.H. and Almers,W. (1997) Ca2+-triggered peptide secretion in single cells imaged with green fluorescent protein and evanescent-wave microscopy. Neuron, 18, 857–863. [DOI] [PubMed] [Google Scholar]
- Lerman J.C., Robblee,J., Fairman,R. and Hughson,F.M. (2000) Structural analysis of the neuronal SNARE protein syntaxin 1A. Biochemistry, 39, 8470–8479. [DOI] [PubMed] [Google Scholar]
- Lin R.C. and Scheller,R.H. (2000) Mechanisms of synaptic vesicle exocytosis. Annu. Rev. Cell. Dev. Biol., 16, 19–49. [DOI] [PubMed] [Google Scholar]
- Martin T.F.J. (1997) Stages of regulated exocytosis. Trends Cell Biol., 7, 271–275. [DOI] [PubMed] [Google Scholar]
- Ohtani Y., Irie,T., Uekama,K., Fukunaga,K. and Pitha,J. (1989) Differential effects of α-, β- and γ-cyclodextrins on human erythrocytes. Eur. J. Biochem., 186, 17–22. [DOI] [PubMed] [Google Scholar]
- Rodal S.K., Skretting,G., Garred,O., Vilhardt,F., van Deurs,B. and Sandvig,K. (1999) Extraction of cholesterol with methyl-β-cyclodextrin perturbs formation of clathrin-coated endocytic vesicles. Mol. Biol. Cell, 10, 961–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Röper K., Corbeil,D. and Huttner,W.B. (2000) Retention of prominin in microvilli reveals distinct cholesterol-based lipid microdomains within the apical plasma membrane of epthelial cells. Nature Cell Biol., 2, 582–592. [DOI] [PubMed] [Google Scholar]
- Rothman J.E. (1994) Mechanisms of intracellular protein transport. Nature, 372, 55–63. [DOI] [PubMed] [Google Scholar]
- Scheiffele P., Roth,M.G. and Simons,K. (1997) Interaction of influenza virus haemagglutinin with sphingolipid–cholesterol membrane domains via its transmembrane domain. EMBO J., 16, 5501–5508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons K. and Ikonen,E. (1997) Functional rafts in cell membranes. Nature, 387, 569–572. [DOI] [PubMed] [Google Scholar]
- Steyer J.A., Horstmann,H. and Almers,W. (1997) Transport, docking and exocytosis of single secretory granules in live chromaffin cells. Nature, 388, 474–478. [DOI] [PubMed] [Google Scholar]
- Subtil A., Gaidarov,I., Kobylarz,K., Lampson,M.A., Keen,J.H. and McGraw,T.E. (1999) Acute cholesterol depletion inhibits clathrin-coated pit budding. Proc. Natl Acad. Sci. USA, 96, 6775–6780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiele C., Hannah,M.J., Fahrenholz,F. and Huttner,W.B. (2000) Cholesterol binds to synaptophysin and is required for biogenesis of synaptic vesicles. Nature Cell Biol., 2, 42–49. [DOI] [PubMed] [Google Scholar]
- Tokuyasu K.T. (1973) A technique for ultracryotomy of cell suspensions and tissues. J. Cell Biol., 57, 551–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokuyasu K.T. (1978) A study of positive staining of ultrathin frozen sections. J. Ultrastruct. Res., 63, 287–307. [DOI] [PubMed] [Google Scholar]
- Toomre D., Steyer,J.A., Keller,P., Almers,W. and Simons,K. (2000) Fusion of constitutive membrane traffic with the cell surface observed by evanescent wave microscopy. J. Cell Biol., 149, 33–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tse F.W., Iwata,A. and Almers,W. (1993) Membrane flux through the pore formed by a fusogenic viral envelope protein during cell fusion. J. Cell Biol., 121, 543–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber T., Zemelman,B.V., McNew,J.A., Westermann,B., Gmachl,M., Parlati,F., Sollner,T.H. and Rothman,J.E. (1998) SNAREpins: minimal machinery for membrane fusion. Cell, 92, 759–772. [DOI] [PubMed] [Google Scholar]
- Xu T., Rammner,B., Margittai,M., Artalejo,A.R., Neher,E. and Jahn,R. (1999) Inhibition of SNARE complex assembly differentially affects kinetic components of exocytosis. Cell, 99, 713–722. [DOI] [PubMed] [Google Scholar]