

Abstract
Mitochondrial ADP-ribosylation leads to modification of two proteins of ∼26 and 53 kDa. The nature of these proteins and, hence, the physiological consequences of their modification have remained unknown. Here, a 55 kDa protein, glutamate dehydrogenase (GDH), was established as a specific acceptor for enzymatic, cysteine-specific ADP-ribosylation in mitochondria. The modified protein was isolated from the mitochondrial preparation and identified as GDH by N-terminal sequencing and mass spectrometric analyses of tryptic digests. Incubation of human hepatoma cells with [14C]adenine demonstrated the occurrence of the modification in vivo. Purified GDH was ADP-ribosylated in a cysteine residue in the presence of the mitochondrial activity that transferred the ADP-ribose from NAD+ onto the acceptor site. ADP- ribosylation of GDH led to substantial inhibition of its catalytic activity. The stoichiometry between incorporated ADP-ribose and GDH subunits suggests that modification of one subunit per catalytically active homohexamer causes the inactivation of the enzyme. Isolated, ADP-ribosylated GDH was reactivated by an Mg2+-dependent mitochondrial ADP-ribosylcysteine hydrolase. GDH, a highly regulated enzyme, is the first mitochondrial protein identified whose activity may be modulated by ADP-ribosylation.
Keywords: ADP-ribosylation/glutamate dehydrogenase/mitochondria/protein modification
Introduction
Besides its role in energy transduction, NAD+ serves an important function in the regulation of multiple cellular processes (Ziegler, 2000). This nucleotide may be utilized for post-translational protein modification known as poly- (Oei et al., 1997) or monoADP-ribosylation (Okazaki and Moss, 1996). It is also used as a substrate by NAD+ glycohydrolases, which may form cyclic ADP-ribose, a potent intracellular calcium-mobilizing agent (Lee, 1997; Ziegler et al., 1997; Galione et al., 1998). NAD+ glycohydrolases also generate free ADP-ribose from either NAD+ or cyclic ADP-ribose. Free ADP-ribose, in turn, can react non-enzymatically with protein lysine (Cervantes-Laurean et al., 1993; Jacobson et al., 1994) or cysteine (McDonald et al., 1992) residues, leading to protein glycation. However, the in vitro systems described so far suggest that this kind of protein modification occurs only at high concentrations of ADP-ribose (Frei and Richter, 1988; McDonald and Moss, 1993a).
In vivo, monoADP-ribosylation is catalyzed by monoADP-ribosyl transferases which have been found in eukaryotes and prokaryotes (Moss and Vaughan, 1988). Eukaryotic monoADP-ribosyl transferases modify specific amino acids of the acceptor proteins, including arginine (Moss et al., 1980; Zolkiewska et al., 1992), cysteine (Tanuma et al., 1987; Saxty and van Heyningen, 1995; Jorcke et al., 1998) and diphthamide (Lee and Iglewski, 1984) residues. Endogenous ADP-ribosylation also appears to occur in hydroxyl-containing amino acid residues (Cervantes-Laurean et al., 1995). Clostridial C3-like exoenzymes modify Rho proteins at an asparagine residue (Aktories, 1997). Several specific hydrolases have been described, which may remove the ADP-ribose from the modified protein and thereby regenerate the original amino acid residue (Okazaki and Moss, 1996).
MonoADP-ribosylation has been studied extensively in mitochondria (Kun et al., 1975; Richter et al., 1983; Hilz et al., 1984; Masmoudi and Mandel, 1987; Frei and Richter, 1988; Jorcke et al., 1998). Under well-defined conditions, the formation of free ADP-ribose from NAD+, and thus non-enzymatic ADP-ribosylation, could be virtually excluded (Masmoudi and Mandel, 1987; Jorcke et al., 1998). This permitted the detection of enzymatic, cysteine-specific ADP-ribosylation in bovine liver mitochondria (Jorcke et al., 1998), i.e. a direct transfer of ADP-ribose from NAD+ onto cysteine residues of the acceptor proteins. According to these observations, the mechanism of modification appears to resemble that of bacterial toxin ADP-ribosyl transferases, in particular pertussis toxin, which also modifies cysteine residues.
In a number of studies, mitochondrial proteins with approximate molecular masses of 30 and 50 kDa have been found to be modified specifically with ADP-ribose (Kun et al., 1975; Richter et al., 1983; Hilz et al., 1984; Masmoudi and Mandel, 1987; Jorcke et al., 1998). Here we report the identification of a modified ∼53 kDa protein as liver mitochondrial glutamate dehydrogenase (GDH; EC 1.4.1.3). The protein was also found to be ADP-ribosylated in vivo. ADP-ribosylation of GDH causes substantial inhibition of its enzymatic activity. Analysis of the chemical stability of the protein–ADP-ribose linkage confirmed that the modification occurs in a cysteine residue. A mitochondrial activity was detected that removed the ADP-ribose from the modified GDH and thereby restored its enzymatic activity. The discovery of an ADP-ribosylation cycle that regulates GDH activity provides important new insights into the role of ADP-ribosylation in higher eukaryotes.
Results
All in vitro ADP-ribosylation assays were conducted in the presence of EDTA and dithiothreitol (DTT). Under these conditions, any significant contribution from non-enzymatic modification is excluded, because the mitochondrial NAD+ glycohydrolase is strongly inhibited (Jorcke et al., 1998). Consequently, there is virtually no liberation of ADP-ribose from NAD+.
Since chromatographic procedures led to substantial loss of incorporated label, blue native gel electrophoresis (Schägger and von Jagow, 1991) was performed as the first step. This procedure permitted efficient separation from non-covalently bound nucleotide and was sufficiently mild. Three bands carrying radioactivity with Rf values of 0.13, 0.31 and 0.64 were detected. The majority of the label resided in the band with an Rf of 0.31. This band was excised from the gel and applied to an SDS– polyacrylamide gel. As shown in Figure 1, left panel, the band excised from the non-denaturing gel still contained several proteins. However, only the protein migrating as an ∼53 kDa band contained the radioactive label (Figure 1, right panel). This band was excised and subjected to ‘in-gel’ digestion with trypsin (Eckerskorn and Lottspeich, 1990). The resulting peptides were analyzed by mass spectrometry utilizing the matrix-assisted laser desorption/ionization time of flight mass spectrometry (MALDI-TOF) technique (Figure 2). Searches with the identified tryptic peptide masses in several databases (SwissProt, EMBL and GenBank) returned by far the highest score for mitochondrial GDH. The majority of mass peaks, namely 14 (Figure 2), corresponded to peptide masses expected after tryptic digestion of this enzyme, assuming a maximum of one missed cleavage site. If more than one missed cleavage site and oxidized methionine residues were also taken into consideration, several additional mass peaks (marked with an asterisk in Figure 2) could be assigned to GDH.

Fig. 1. Separation of the ADP-ribosylated ∼53 kDa protein. Specific modification of solubilized mitochondrial proteins in the presence of 100 µM [32P]NAD+ (0.6 µCi/nmol) was performed. After incubation, the suspension was separated in a non-denaturing polyacrylamide gel. The band containing the majority of the radioactive label was excised from the gel and subjected to SDS–PAGE. The proteins were then blotted onto a PVDF membrane and subsequently revealed by staining with Coomassie Blue (left panel). The right panel shows the auto radiogram of the blot. Numbers (in daltons) on the right indicate the mobility of marker proteins.

Fig. 2. Mass spectrometric analysis of a tryptic digest obtained from the ADP-ribosylated ∼53 kDa mitochondrial protein. The incubation and separation of the modified protein were conducted as described in the legend to Figure 1. Following SDS–PAGE, the protein band containing the radioactive label was subjected to ‘in-gel’ digestion with trypsin. The mass spectrum of the resulting peptides was obtained by MALDI-TOF spectrometry. Only masses corresponding to internal peptides of GDH are indicated, assuming a maximum of one missed cleavage site per peptide. The asterisks indicate peaks corresponding to GDH peptide masses, considering more than one missed cleavage site or oxidized methionine residues.
Three masses were subjected to post-source decay analysis (Chaurand et al., 1999) to establish the amino acid sequences of the corresponding peptides: a 544.3 Da peptide, SNAPR; a 863.4 Da peptide, AQHSQHR; and a 1737.9 Da peptide, HGGTIPIVPTAEFQDR (cf. Figure 2). All three peptides were identical to tryptic peptides deduced from the known sequence of bovine liver mitochondrial GDH, except for the 863 Da peptide in which two C-terminal amino acids (QH) were found to be in the opposite order as compared with the known sequence. Since this result was obtained using two independent mitochondrial preparations, it seems likely that there is an error in the sequence deposited in the SwissProt database.
The identification of the ∼53 kDa ADP-ribosylated protein as GDH was confirmed further by N-terminal sequencing of the corresponding band (Figure 1). The sequence was XEAXADREDDPNFFKMV. The X indicates that the respective amino acid residue could not be clearly identified. A reported N-terminal sequence of the bovine enzyme (Julliard and Smith, 1979) lacks the first four amino acids and starts only at position five (ADR…) (Table I). However, the highly similar sequence of the mature (i.e. after cleavage of the mitochondrial targeting sequence) human enzyme does contain four additional amino acids at the N-terminus (cf. Table I) and has been confirmed for the bovine GDH (McCarthy et al., 1980). The N-terminal sequence of the highly purified bovine enzyme (twice recrystallized, from Roche) did not contain the first four amino acids (Table I), confirming that cleavage had occurred during extensive purification of the enzyme (McCarthy et al., 1980). Taken together, the results of these molecular analyses provide conclusive evidence that GDH (55 561 Da) represents the radioactively labeled protein separated by the two electrophoretic steps (Figure 1).
Table I. Comparison of the N-terminal sequences of the ∼53 kDa ADP-ribosylated mitochondrial protein with those of commercially available bovine liver GDH and published bovine and human GDH sequences.
| Source | N-terminal sequence | Reference |
|---|---|---|
| Bovine liver mitochondria | ADREDDPNFFKMV | Julliard and Smith (1979) |
| SEAVADREDDPNFFKMV | McCarthy et al. (1980) | |
| XEAXADREDDPNFFKMV | this study | |
| Bovine liver (Roche) | ADREDDPNFFKMV | this study |
| Human liver | SEAVADREDDPNFFKMV | Julliard and Smith (1979) |
The N-terminal sequences of the ADP-ribosylated ∼53 kDa mitochondrial protein and the purified bovine liver GDH were determined as described in Materials and methods. The N-terminal sequence of the labeled ∼53 kDa protein was obtained from blots similar to that shown in Figure 1. X indicates that the respective amino acid residue was not identified.
To verify the ability of GDH to serve as specific acceptor of a cysteine-specific ADP-ribosyl transferase, the following set of experiments was carried out. Use was made of the highly purified commercially available bovine liver GDH. First, this enzyme was incubated in the presence of a small amount of the mitochondrial preparation and [32P]NAD+. As shown in Figure 3A, GDH was labeled efficiently and co-migrated with the ∼53 kDa band detected in the mitochondria alone. Note that for lane 2 of Figure 3A, the amount of mitochondrial protein was 10 times higher than for lane 1. Lane 2 also exemplifies the high specificity of mitochondrial ADP-ribosylation for GDH and an unidentified ∼26 kDa protein. As for the endogenous mitochondrial protein (Jorcke et al., 1998), the modification was detected if [14C]adenine-labeled NAD+ was used (Figure 3B, lane 1), but not if the nicotinamide moiety carried the radioactive label (Figure 3B, lane 2). Importantly, the amount of radioactivity co-migrating with the purified GDH was negligible if incubation with [32P]NAD+ was carried out in the absence of mitochondria (Figure 3C). These observations rule out the possibility that the entire molecule, NAD+, was attached to GDH, as has been observed for glyceraldehyde-3-phosphate dehydrogenase (McDonald and Moss, 1993b).
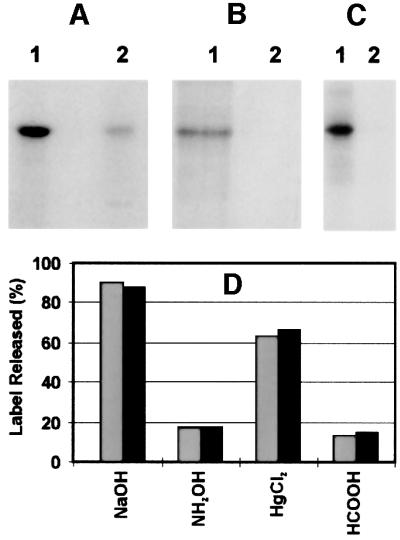
Fig. 3. Cysteine-specific ADP-ribosylation of purified bovine liver GDH and its co-migration with the modified mitochondrial ∼53 kDa protein. (A) Purified bovine liver GDH (10 µg) was ADP-ribosylated in the presence of the mitochondrial preparation (10 µg) and 100 µM [32P]NAD+ (1 µCi) for 30 min (lane 1). For lane 2, 100 µg of the mitochondrial preparation were treated as described for lane 1, except that no GDH was added. (B) The experiment was conducted as described for (A), lane 1, except that instead of [32P]NAD+, [14C]NAD+ labeled in either the adenine (lane 1) or the nicotinamide moiety (lane 2) was used (0.25 µCi each). (C) The sample used for lane 1 was prepared as described for (A), lane 1. For lane 2, purified GDH was subjected to a similar incubation with [32P]NAD+ in the absence of the mitochondrial suspension. Following the incubations, samples were precipitated with acetone and the pellets subjected to SDS–PAGE. The autoradiograms of the gels are shown. (D) Specific [32P]ADP-ribosylation of mitochondrial proteins or purified GDH was conducted as described in Materials and methods. Proteins of several identical samples were separated by SDS–PAGE and subsequently blotted onto PVDF membranes. The blots were washed twice in a solution containing 50 mM MOPS–KOH pH 7.0, 100 mM NaCl and 1 mM EDTA for 15 min. The regions of the ∼53 kDa protein (gray bars) or purified GDH (black bars) were excised and the incorporated ADP-ribose determined by Cerenkov counting. The individual pieces of the blots were then subjected to treatment at 37°C for 2 h with the reagents indicated below. Thereafter, the pieces were washed twice as detailed above and the remaining radioactivity determined. Subsequent staining with Coomassie Blue gave no indication that the amount of protein adsorbed to the blot membranes was affected by any of these treatments. The additions indicated in the figure were 10 mM HgCl2 (in wash buffer without EDTA), 2 M NH2OH (adjusted to pH 7.0) in wash buffer, 0.2 M NaOH plus 1 mM EDTA, or 44% HCOOH. Note that NaOH releases bound ADP-ribose non-specifically from modified amino acids. The data represent the average of two independent experiments.
Secondly, the chemical stability of the protein–ADP- ribose linkage found for the purified mitochondrial GDH coincided with that detected previously in the mitochondrial preparation (Jorcke et al., 1998) and, specifically, with that of the modified ∼53 kDa protein (Figure 3D). The observed sensitivity towards mercury ions clearly points to modification at a cysteine residue (Jacobson et al., 1994). The radioactive compounds released by Hg2+ were analyzed by anion-exchange HPLC. At least 60% of the liberated radioactivity was recovered as ADP-ribose, as indicated by the co-elution with unlabeled ADP-ribose which was added to the samples before the separation. This finding confirms that the modifying group was indeed ADP-ribose.
The possible effect of ADP-ribosylation on GDH’s enzymatic activity was investigated both in the mitochondrial preparation and using the pure enzyme. Regardless of whether endogenous GDH of the mitochondrial preparation or added purified GDH was measured, after 60 min glutamate formation was inhibited by ∼60–65% following incubation in the presence of 100 µM NAD+ (Figure 4). The reverse reaction of GDH (deamination of glutamate) was similarly affected (not shown). NAD+ alone or heat-treated mitochondria had only little effect (<10% inhibition after 60 min), if any, on the isolated enzyme (not shown). As a further control, endogenous malate dehydrogenase was measured in parallel to GDH in the mitochondrial preparation. Only a slight decrease (<15%) in endogenous malate dehydrogenase activity was observed after a 60 min incubation of the solubilized mitochondrial preparation in the presence of 100 µM NAD+. If incubations of solubilized mitochondria with NAD+ were performed for longer time periods, the modification and concomitant inhibition of GDH activity did not increase further. Obviously, this phenomenon is related to the reversibility of ADP-ribosylation in mitochondria (Richter et al., 1983; Frei and Richter, 1988; Jorcke et al., 1998; see below).

Fig. 4. Inhibition of GDH activity by ADP-ribosylation. Mitochondrial proteins or purified GDH were ADP-ribosylated in the presence of 100 µM NAD+. Samples containing 0.25 µg of GDH (squares) or ∼15 µg of mitochondrial protein (triangles) were withdrawn at the times indicated and their GDH activity determined. The initial specific activity of the GDH (100%) was 186 µmol/min/mg. The uninhibited activity of the solubilized mitochondrial preparation was ∼1.7 µmol/min/mg. In the absence of added NAD+, the GDH activity decreased only by ∼5–8% after 2 h. The inset shows the modification with ADP-ribose of endogenous GDH during a parallel experiment conducted in the presence of [32P]NAD+. Aliquots were withdrawn at the times indicated (in minutes) and precipitated with acetone. The autoradiogram of the SDS–PAGE of the samples is presented. The data represent five independent experiments.
GDH is catalytically active as a homohexamer (Fisher, 1985). It was of interest, therefore, to establish the stoichiometry between the enzyme and the ADP-ribose incorporated, which affords the inhibition. To address this point, the extent of ADP-ribosylation of purified GDH was varied by continuing the modification reaction for 0–60 min (cf. Figure 4) and the remaining GDH activity was determined. As may be inferred from Figure 5, a linear relationship between modification of hexamers and inhibition would extrapolate to a ratio of roughly one ADP-ribose incorporated per enzyme molecule (hexamer) to achieve complete inhibition.

Fig. 5. Correlation of inhibition and ADP-ribosylation of GDH. Purified GDH was inhibited by ADP-ribosylation to various extents by incubating the enzyme for different time intervals (0–60 min) in the presence of [32P]NAD+ and mitochondrial preparation (cf. Figure 4). Parallel samples (20 µl) were withdrawn and used for analysis of enzyme activity or subjected to gel filtration by HPLC. Prior to gel filtration, the samples were made 5 mM in unlabeled NAD+ to minimize co-migration with GDH of non-covalently bound radioactive NAD+. The eluting fractions containing GDH were pooled and both their protein content and bound radioactivity determined. From these data, the stoichiometry of ADP-ribose per enzyme hexamer was calculated assuming a subunit molecular mass of 55 561 Da. The data were corrected for the values obtained using mitochondria in the absence of added GDH or purified GDH without mitochondria. The straight line indicates the result of a linear regression analysis of the data (R2 = 0.92). The values represent two independent experiments using two different mitochondrial preparations.
In order to assess the binding region of the modification and the potential mechanism of inhibition, ADP-ribosylation of isolated GDH was conducted in the presence of purine and pyridine nucleotides. Besides the catalytic pyridine nucleotide-binding sites, GDH contains allosteric sites for purine nucleoside di- or triphosphates. In the presence of ADP or GDP, the activity of GDH is stimulated, while ATP and GTP are inhibitory (Fisher, 1985). It is shown in Figure 6 that NADP, NADPH and NADH strongly suppressed the ADP-ribosylation of GDH, whereas ATP and ADP were much less effective. Since GTP had no effect on the extent of modification, it would appear that the pyridine nucleotide-binding site may exhibit some affinity for the adenylate moiety of the adenine nucleotides, i.e. ADP-ribosylation is unlikely to occur in an allosteric purine nucleotide-binding site. Rather, these results suggest that the mode of inhibition of GDH by ADP-ribosylation includes the modification at a catalytic site that is prevented when this site is occupied by pyridine nucleotides. Consistent with the lack of modification of GDH in the presence of pyridine nucleotides, the enzyme was also protected from inhibition (not shown).
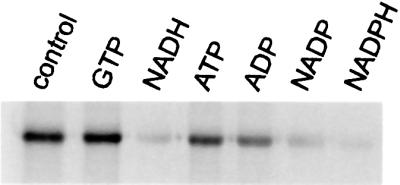
Fig. 6. Pyridine nucleotides prevent ADP-ribosylation of GDH. ADP-ribosylation assays were conducted under standard conditions in the presence of 25 µM [32P]NAD+, 10 µg of GDH, 10 µg of the mitochondrial suspension and the nucleotide (1 mM) indicated in the figure. Incubations were stopped after 15 min and the samples processed for SDS–PAGE. The autoradiogram of the gel is shown.
The potential physiological significance of the ADP-ribosylation of GDH was assessed in experiments aimed at visualizing the modification in vivo. For this purpose, a human hepatoma cell line (Hep-G2) was grown in the presence of 14C-labeled adenine. As shown in Figure 7A, GDH immunoprecipitated from the cell extracts was labeled by [14C]adenine. If only glutamate, but no glutamine, was added to the cell culture medium (lane 1), modification of GDH was significantly higher as compared with the conditions when glutamate and glutamine (lane 2) or glutamine alone (lane 3) were present in the medium. As controls, an unrelated rabbit IgG or just protein A–Sepharose was used for the immunoprecipitation. No labeled proteins were detected in these eluates, verifying the specificity of the assay (not shown). The specificity of the GDH antibody, which was affinity purified against bovine liver GDH, was tested by western blot analysis of bovine liver mitochondria and Hep-G2 cells (Figure 7B). Autoradiography to visualize the in vivo modified, immunoprecipitated GDH required an exposure time of ∼3 weeks. A period of even 3 months was required to detect endogenous ADP-ribosylation of Gβ using 3H-labeled adenine (Lupi et al., 2000). In principle, in these experiments, proteins could be labeled that were either ADP-ribosylated or adenylated. However, GDH was not adenylated in the presence of [α-32P]ATP and mitochondria (not shown), and the modification using NAD+ as substrate was clearly ADP-ribosylation (Figures 3 and 8). Therefore, the results presented in Figure 7 strongly support the regulatory significance of ADP-ribosylation for the activity of GDH in vivo.
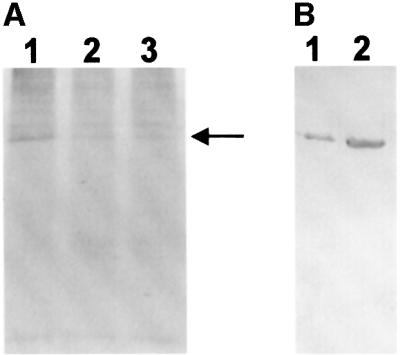
Fig. 7. ADP-ribosylation of GDH in vivo. Hep-G2 cells were grown in the presence of [14C]adenine and extracts immunoprecipitated with purified rabbit anti-GDH IgG. (A) The growth medium contained either 2 mM glutamate (lane 1), 1 mM glutamate and 1 mM glutamine (lane 2), or 2 mM glutamine (lane 3). Immunoprecipitated proteins were separated by 12% SDS–PAGE and their modification revealed by autoradiography. The film was exposed for 25 days. The arrow indicates the position at which GDH migrated. (B) Immunoblot of Hep-G2 cells (lane 1) and bovine liver mitochondria (lane 2) developed with the purified anti-GDH IgG used for the immunoprecipitation experiment shown in (A).

Fig. 8. Reversal of ADP-ribosylation and concomitant reactivation of GDH catalyzed by a mitochondrial hydrolase activity. (A and B) Purified GDH was [32P]ADP-ribosylated and then freed of non-covalently bound nucleotide by HPLC as described in the legend to Figure 5. The modified enzyme (10 µg) was then incubated in the presence of 5 mM MgCl2 (lanes 1), 5 mM MgCl2 and 10 µg of the mitochondrial preparation (lanes 2), or 10 µg of the mitochondrial preparation alone (lanes 3). After 30 min at 30°C, the protein was subjected to SDS–PAGE and the remaining modification visualized by autoradiography (A). The liberated nucleotides were analyzed by thin-layer chromatography (B). The autoradiogram is shown. On the right, the migration of standard compounds is indicated (ADPR, ADP-ribose). A control sample (no addition during incubation) was indistinguishable from the sample represented by lanes 1. (C) Purified GDH or endogenous GDH of the mitochondrial preparation was ADP-ribosylated to achieve ∼50% inhibition by incubating mitochondria and purified GDH (squares and circles) or mitochondria alone (triangles) in the presence of 100 µM NAD+ for ∼45 min (cf. Figure 4). The sample represented by the circles was then centrifuged to remove the added mitochondria. Thereafter (time zero in this figure), 5 mM MgCl2 was added to all samples and incubation continued. At the times indicated, aliquots received 10 mM EDTA and their GDH activity was determined. The arrow indicates re-addition of mitochondria (3 µg per µg of GDH, after 30 min of incubation) to the sample previously freed of mitochondria. The values given are related to the activities of purified or endogenous GDH, respectively, prior to the incubation with NAD+. The data represent 2–4 independent experiments using two different mitochondrial preparations.
Finally, if ADP-ribosylation of GDH were a regulatory mechanism, the modification should be reversible. Indeed, both ADP-ribosylation (Figure 8A and B) and the concomitant inhibition of GDH (Figure 8C) were reversed by a mitochondrial activity. This activity was stimulated greatly by Mg2+. Figure 8B demonstrates that the entire modifying group, ADP-ribose, was released from GDH in the presence of mitochondria and Mg2+. Therefore, the recovery of GDH’s enzymatic activity is most likely due to the restoration of the sulfhydryl group of the modified cysteine. The reactivation of ADP-ribosylated GDH exhibited the same requirements, namely the presence of Mg2+ and mitochondria (Figure 8C). The enzymatic nature of this process has been verified in several control experiments (in addition to those presented in Figure 8), including, for example, heat inactivation of the added mitochondrial preparation (not shown). According to the observed reaction, this activity can be designated as ADP-ribosylcysteine hydrolase.
The specificity of the detected hydrolase activity for modified cysteines was supported in experiments using [32P]ADP-ribosylated proteins modified in either a cysteine or an arginine residue. These potential hydrolase substrates were generated using known transferase/ acceptor systems. Thus, cysteine-modified Gαi1 was obtained by incubating the protein in the presence of [32P]NAD+ and pertussis toxin (Moss and Vaughan, 1988). Arginine-specific ADP-ribosylation was conducted using recombinant mouse RT6.2, a known arginine-specific ADP-ribosyl transferase (Koch-Nolte et al., 1996) carrying a FLAG tag (Hopp et al., 1988). In the presence of specific antibodies directed against the FLAG epitope and [32P]NAD+, recombinant RT6.2 ADP-ribosylates an arginine residue of the antibody’s light chain (Koch-Nolte et al., 1996). As can be inferred from Figure 9, the mitochondrial hydrolase activity catalyzed release of the modification from the protein labeled at a cysteine residue (Figure 9A), but not from the antibody modified at an arginine residue (Figure 9B).
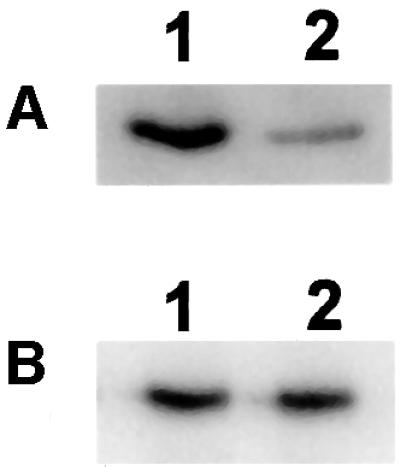
Fig. 9. Specificity of the mitochondrial hydrolase activity for ADP-ribosylcysteine. (A) A 5 µg aliquot of Gαi1 (Calbiochem) was ADP-ribosylated in a cysteine residue by 0.5 µg of activated pertussis toxin (Sigma) with 25 µM [32P]NAD+ for 30 min in a medium consisting of 50 mM MOPS–KOH pH 7.5, 10 mM DTT, 1 mM EDTA, 1 mM ATP and 1 mM GTP. Thereafter, 10 mM MgCl2 was added and incubation continued for 20 min in the absence (lane 1) or presence (lane 2) of 20 µg of the mitochondrial preparation. (B) FLAG-tagged arginine-specific ADP-ribosyltransferase (recombinant mouse RT6.2; kind gift of Dr Koch-Nolte, Hamburg, Germany) was immobilized on M2-Sepharose (Kodak/IBI) carrying FLAG-specific antibodies. The complex (20 µl of the resin) was incubated with 25 µM [32P]NAD+ for 30 min in a medium consisting of 10 mM potassium phosphate pH 7.0 and 150 mM NaCl. Under these conditions, the light chain of the antibody is ADP-ribosylated (Koch-Nolte et al., 1996). Thereafter, the resin was washed twice with 50 mM MOPS–KOH pH 7.5, 10 mM DTT, 1 mM EDTA. Incubation was then continued for 20 min in this medium containing in addition 10 mM MgCl2 in the absence (lane 1) or presence (lane 2) of 20 µg of the mitochondrial preparation. All samples were then subjected to SDS–PAGE and subsequent autoradiography.
Discussion
The present study establishes GDH as a specific target for enzymatic, cysteine-specific ADP-ribosylation. GDH is the first mitochondrial protein reported to be a substrate for this kind of post-translational modification. Modification and concomitant inhibition of GDH were reversed enzymatically by an ADP-ribosylcysteine hydrolase. The occurrence of ADP-ribosylated GDH in living cells suggests the physiological regulation of the enzyme in an ADP-ribosylation cycle (Figure 10). ADP-ribosylation cycles have been postulated for eukaryotic systems (Okazaki and Moss, 1996). However, only for dinitrogenase reductase of the photosynthetic bacterium Rhodospirillum rubrum and related prokaryotes has an ADP-ribosylation cycle been demonstrated conclusively (Ludden, 1994). Interestingly, glutamine synthetase of several bacteria has also been reported to be a target of ADP-ribosylation (Ludden, 1994), suggesting that ADP-ribosylation plays an important role in nitrogen metabolism. The identification of mitochondrial GDH as a target for this modification may indicate that the cellular nitrogen balance of eukaryotes might also be regulated by ADP-ribosylation. Given the central position of GDH in metabolism, at the crossroads of several important pathways (Figure 10), tight control of its catalytic activity is essential. In line with this suggestion, the results presented here (Figure 7A) indicate an enhanced ADP-ribosylation of GDH if glutamate, but no glutamine, is added to the cell culture medium. Inhibition of excessive glutamate utilization by GDH would prevent a condition that is toxic for the cells, i.e. accumulation of ammonia. Moreover, in the absence of added glutamine, this amino acid has to be synthesized from glutamate by glutamine synthetase. This enzyme is also central to the cellular nitrogen metabolism and is tightly controlled by a variety of mechanisms, including adenylation, feedback inhibition and uridylation of a regulatory protein. Thus, ADP-ribosylation of GDH may be part of the complex regulatory systems controlling the cellular nitrogen balance.

Fig. 10. Schematic representation of the mitochondrial ADP-ribosylation cycle regulating GDH activity. Metabolic pathways that may be affected by the inhibition of GDH are indicated. mART, mitochondrial ADP-ribosyl transferase.
ADP-ribosylation might also complement the well-documented allosteric regulation of the enzyme by purine nucleotides (GDP and ADP are stimulatory, while GTP and ATP are inhibitory) (Fisher, 1985). Under conditions of sufficiently high energy levels, glutamate would be used for anabolic reactions (GDH is inhibited), whereas energy depletion activates GDH, providing α-ketoglutarate for the citric acid cycle. Moreover, glutamate is known as a major excitatory neurotransmitter (Fonnum, 1984) with neurotoxic potential (McGeer and McGeer, 1976), which further illustrates the necessity to control its metabolism. The exact function of ADP-ribosylation of GDH and the underlying regulatory mechanisms will have to be addressed in future studies.
Analyses of the ADP-ribosylation reaction of pure GDH in the presence of small amounts of mitochondria (to provide the enzymatic activity for the modification) have confirmed the characteristics found for the previously unidentified ∼53 kDa protein (Jorcke et al., 1998). First, ADP-ribose, but not NAD+, is the modifying group. Secondly, the substantial release of label from the modified protein following treatment with mercury ions strongly supports the conclusion that a cysteine residue is ADP-ribosylated. This notion has been verified further in the present study by demonstrating that the hydrolytic activity that removes ADP-ribose from GDH is specific for modified cysteines. Thirdly, the modification occurs under conditions that exclude any significant contribution from non-enzymatic ADP-ribosylation. Consequently, the reaction is catalyzed by a cysteine-specific ADP-ribosyl transferase present in the mitochondrial preparation. The enzymatic nature of the reaction is supported further by the observation that modification of the endogenous ∼53 kDa mitochondrial protein, here identified as GDH, with 100 µM [32P]ADP-ribose was immeasurably low as compared with [32P]NAD+ as substrate (Jorcke et al., 1998).
In the experiments presented here, the extent of ADP-ribosylation of GDH usually did not exceed ∼65% of the expected maximum. It has been established in this study that a Mg2+-stimulated ADP-ribosylcysteine hydrolase is present in the mitochondrial preparation that removes the modification from the GDH. Even under the conditions used (EDTA, no Mg2+ added), the hydrolase is sufficiently active to prevent complete modification. Therefore, it is conceivable that in previous attempts to identify the modified protein using, for example, chromatographic procedures, the initial separation of the hydrolase activity from the labeled protein was insufficient, resulting in a rapid loss of the label. Related to the transferase activity, the hydrolase activity is indeed substantial. When purified GDH was ADP-ribosylated and then isolated by immunoprecipitation, about one-third of the modification was released by the same amount of mitochondria and under the same conditions used for the modification reaction (not shown). Therefore, it would appear that during the ADP-ribosylation reaction, in the absence of Mg2+, an equilibrium between transferase and hydrolase activities is attained, precluding the complete modification of GDH. It is likely that regulatory mechanisms exist that adjust this equilibrium according to the needs of the cell.
The demonstration of a cysteine-specific hydrolase activity represents an important finding of the present study, because it establishes the existence of an ADP-ribosylation cycle in mitochondria. Previous reports support the occurrence of cysteine-specific ADP-ribosylation in vivo (Jacobson et al., 1990). Only very few eukaryotic cysteine-specific ADP-ribosyl transferases or hydrolases have been reported so far (Tanuma et al., 1987; Saxty and van Heyningen, 1995). Therefore, it will be important to isolate these enzymes, characterize them on a molecular level and identify potential further target proteins.
The observation that NADP(H) largely prevented modification of GDH points to a mechanism of inhibition that includes the occupation of a pyridine nucleotide-binding site with ADP-ribose at a cysteine residue. Consistent with this supposition is the loss of GDH activity following chemical modification of an active site cysteine (Cho et al., 1999). GDH is known to be catalytically active as a homohexamer (Fisher, 1985). The results presented here indicate that if one-sixth of the subunits of GDH were modified, complete inhibition of the enzyme would occur. This conclusion is consistent with the suggestion that ADP-ribosylation of one subunit of an active hexamer inactivates the molecule. Therefore, ADP-ribosylation represents a highly efficient means of GDH inhibition. The inactivation of the hexameric enzyme by modification of a single pyridine nucleotide site is probably attributable to the highly cooperative interactions between the subunits (Fisher, 1985). It indicates that catalysis in different catalytic sites proceeds sequentially rather than randomly, i.e. inactivation of one catalytic site disrupts the ordered cycle of catalysis. Such a mechanism has been established for other enzymes with cooperatively interacting catalytic sites, e.g. F1-ATPase (Boyer, 1997).
Under normal conditions, the ADP-ribosylation of GDH would be expected to be rather low owing to the sufficient amounts of NAD(P)H. Moreover, the prevalence of the reduced form of the pyridine nucleotides in mitochondria limits the substrate (NAD+) supply for the modification. Consequently, stimulation of ADP-ribosylation should take place under oxidizing conditions leading to an increase in the oxidized form of the pyridine nucleotides. Indeed, under these conditions, ADP-ribosylation of mitochondrial proteins is enhanced (Richter and Kass, 1991). The occurrence of ADP-ribosylated mitochondrial proteins in vivo has also been documented previously (Frei and Richter, 1988; Cervantes-Laurean et al., 1995). It was estimated that in freshly prepared rat liver mitochondria, up to ∼200 pmol of protein-bound ADP-ribose were present per milligram of protein.
In conclusion, the present study has established GDH as a target of mitochondrial ADP-ribosylation. This reversible modification may serve as an efficient device to control the activity of this enzyme. Therefore, it may have a severe impact on a variety of cellular functions, most prominently the regulation of nitrogen metabolism.
Materials and methods
Isolation of mitochondria
Bovine liver mitochondria were isolated and washed in 10 mM Tris–HCl pH 7.5, 250 mM sucrose, 0.5 mM EDTA as described (Ziegler et al., 1996) and stored frozen (–70°C). Owing to the freezing, the mitochondria were not coupled and respired upon addition of NADH (Jorcke et al., 1998). Therefore, the particles so prepared are termed ‘mitochondrial preparation’.
ADP-ribosylation assays
The mitochondrial preparations used for analysis of ADP-ribosylation (in the absence or presence of added GDH) were always pre-incubated for 15 min with 10 mM DTT and 2 mM EDTA to prevent cleavage of NAD+ by the NAD+ glycohydrolase (Jorcke et al., 1998). The standard incubation mixture (usually 50 µl) contained 50 mM MOPS–KOH pH 7.0, 10 mM DTT, 2 mM EDTA and [32P]NAD+ at the indicated specific radioactivity. Some experiments were conducted using [14C]adenine- or [14C]nicotinamide-labeled NAD+. Other additions were made as indicated in the figure legends. If mitochondrial proteins were to be modified in the absence of added GDH, 0.1 mg of the mitochondrial preparation was used. After incubation at 30°C for the time indicated, the reaction was stopped by acetone precipitation. The pellets were resuspended in sample buffer used for gel electrophoresis.
ADP-ribosylation of purified GDH (10 µg) was investigated by incubating the enzyme in the standard mixture. The reaction was initiated by adding [32P]NAD+ and 10 µg of the pre-incubated mitochondrial preparation.
If the influence of ADP-ribosylation on the activity of purified GDH was of interest, the assay was performed similarly, except that only unlabeled NAD+ (100 µM) was added and the amounts of protein were scaled up to suffice for several aliquots. The effect on endogenous GDH activity of the mitochondrial preparation was investigated using the supernatant after solubilization of the membranes (see below) for the assays.
Separation of the modified ∼53 kDa protein
The mitochondrial preparation was solubilized in the presence of 750 mM ε-aminocapronic acid, 50 mM Bis-Tris pH 7.0 and 2% Triton X-100. After centrifugation (100 000 g for 1 h at 4°C), the supernatant (∼4.5 mg of protein) was subjected to the ADP-ribosylation reaction for 60 min under standard conditions. This procedure led to the same labeling pattern as observed for the unsolubilized mitochondrial preparation. Proteins were then separated by blue native gel electrophoresis as described (Schägger and von Jagow, 1991). The wet gel was subjected to autoradiography and the protein band carrying the majority of incorporated label was excised carefully. The gel piece was then layered onto a 6–15% SDS–polyacrylamide gradient gel. After electrophoresis according to Laemmli (1970), the gel was stained with Coomassie Blue, dried and autoradiographed. Alternatively, the proteins were transferred onto a polyvinylidene difluoride (PVDF) membrane prior to the staining. In some experiments, after SDS–PAGE yielding the isolated labeled ∼53 kDa protein, the respective band was excised and subjected to ‘in-gel’ digestion with trypsin according to Eckerskorn and Lottspeich (1990). The resulting peptide mixture was then analyzed by mass spectrometry.
Protein sequencing
Protein sequencing was performed using an ABI 473A protein sequencer (Perkin Elmer/Applied Biosystems, Foster City, CA). N-terminal sequences of proteins were either obtained from a blot membrane or, in the case of the commercially available purified GDH, applied as liquid onto a glass fiber filter.
MALDI-TOF
Masses of tryptic peptides of the ∼53 kDa protein were determined as described before (Hinderlich et al., 1998) using a Reflex mass spectrometer (Bruker Daltonik, Bremen, Germany). The instrument was operated in linear or reflector mode. Several peptides were subjected to post-source decay analysis to establish their amino acid sequences (Chaurand et al., 1999). Data were analyzed using ExPasy tools provided by the Swiss Institute of Bioinformatics (http://www.expasy.ch) or the peptide-mass fingerprinting tool provided by the UCSF mass spectrometry facility (http://prospector.ucsf.edu/ucsfhtml3.2/msfit.htm).
ADP-ribosylation of GDH in vivo
Hep-G2 cells were grown in culture dishes at 37°C in 95% air, 5% CO2 in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal calf serum (FCS), 100 U/ml penicillin, 100 µg/ml streptomycin and 0.08 mU/ml insulin. For the labeling of endogenous proteins, the medium was changed to one deficient in FCS, hypoxanthine, antibiotics and a source of glutamine/glutamate. [8-14C]adenine (40 µCi/106 cells, 1 µmol/ml) and either 2 mM glutamate, 2 mM glutamine or 1 mM of each were added. The cells were harvested after 15 h, washed with phosphate-buffered saline and then lysed with 10 mM HEPES pH 7.8, 10 mM KCl, 0.2 mM EDTA, 1 mM DTT, 0.5 mM phenylmethylsulfonyl fluoride (PMSF) and 0.6% NP-40. Unbroken cells and nuclei were sedimented and the supernatant homogenized at 4°C in 0.2% Tween-20, 50 mM Tris–HCl pH 7.5, 150 mM NaCl and 5 mM EDTA using a Potter homogenizer. Following sedimentation of insoluble material by centrifugation at 15 000 g for 10 min, 2 µg of affinity-purified rabbit anti-GDH IgG were added and incubation conducted for 1 h at 4°C. Thereafter, 200 µl (bed volume) of protein A–Sepharose were added and incubation continued for 1 h. The mixture was then poured into a centrifuge column (0.6 ml) and the retained Sepharose washed three times with a solution consisting of 50 mM Tris–HCl pH 7.5, 150 mM NaCl and 0.2% Tween-20.
Bound proteins were eluted by adding SDS sample buffer and centrifugation. Proteins were separated in a 12% SDS–polyacrylamide gel and bound radioactivity revealed by autoradiography.
Chemical stability of the protein–ADP-ribose linkages
[32P]ADP-ribosylated mitochondrial proteins or GDH were separated by 12% SDS–PAGE and then transferred onto a PVDF membrane. The bands corresponding to GDH or the mitochondrial ∼53 kDa band were excised and then treated individually with the reagents indicated in the figure legends. Release of radioactivity was quantitated by Cerenkov counting.
Assay of GDH activity
l-glutamate formation was followed spectrophotometrically at 340 nm in a medium consisting of 50 mM NH4Cl, 200 µM NADH, 2.5 mM α-ketoglutarate and 100 mM Tris–HCl pH 7.5 (Mazon, 1978). Measure ments of endogenous GDH present in the mitochondrial preparation were carried out using mitochondrial preparations solubilized in the presence of 0.3% lauryl dimethylamine N-oxide. Unsolubilized material was removed by centrifugation. Following the solubilization, there was no measurable consumption of NADH in the absence of α-ketoglutarate.
The influence of ADP-ribosylation on GDH activity was monitored using samples withdrawn from the ADP-ribosylation assays. For analysis of the purified GDH, the added mitochondrial preparation was removed by centrifugation.
Quantitation of ADP-ribose incorporation into GDH
Purified GDH (1 mg/ml) was ADP-ribosylated in the presence of 1 mg/ml mitochondrial preparation and 100 µM [32P]NAD+ (10 µCi/ml) under the conditions described above. After several time intervals (0–60 min), aliquots (30 µl) were withdrawn. Thereafter, 5 mM unlabeled NAD+ (final concentration) was added and the mitochondria sedimented. The supernatant (20 µl) was immediately subjected to gel filtration by HPLC. The column (Bio-Select SEC 250-5; 300 × 7.8 mm; Bio-Rad) was operated with a buffer containing 20 mM Tris–HCl pH 8.0 and 200 mM NaCl. Fractions containing GDH (as established by SDS–PAGE) were pooled and both their radioactivity and protein content determined. The results were corrected for the data obtained from control samples lacking either GDH or mitochondria.
Protein determination
Protein was determined by a biuret procedure or the BCA assay from Pierce using bovine serum albumin as standard. All data presented were obtained using at least two different mitochondrial preparations.
Acknowledgments
Acknowledgements
We thank Dr Koch-Nolte (Hamburg) for the generous gift of FLAG-tagged RT6.2. This study was supported by the Fonds der Chemischen Industrie.
References
- Aktories K. (1997) Rho proteins: targets for bacterial toxins. Trends Microbiol., 5, 282–288. [DOI] [PubMed] [Google Scholar]
- Boyer P.D. (1997) The ATP synthase—a splendid molecular machine. Annu. Rev. Biochem., 66, 717–749. [DOI] [PubMed] [Google Scholar]
- Cervantes-Laurean D., Minter,D.E., Jacobson,E.L. and Jacobson,M.K. (1993) Protein glycation by ADP-ribose: studies of model conjugates. Biochemistry, 32, 1528–1534. [DOI] [PubMed] [Google Scholar]
- Cervantes-Laurean D., Loflin,P.T., Minter,D.E., Jacobson,E.L. and Jacobson,M.K. (1995) Protein modification by ADP-ribose via acid-labile linkages. J. Biol. Chem., 270, 7929–7936. [DOI] [PubMed] [Google Scholar]
- Chaurand P., Luetzenkirchen,F. and Spengler,B. (1999) Peptide and protein identification by matrix-assisted laser desorption ionization (MALDI) and MALDI-post-source decay time-of-flight mass spectrometry. J. Am. Soc. Mass Spectrom., 10, 91–103. [DOI] [PubMed] [Google Scholar]
- Cho S.W., Cho,E.H., Hwang,S.H. and Choi,S.Y. (1999) Alteration of the NAD+/NADH ratio in CHO cells by stable transfection with human cytosolic glycerol-3-phosphate dehydrogenase: resistance to oxidative stress. Mol. Cells, 28, 91–98. [PubMed] [Google Scholar]
- Eckerskorn C. and Lottspeich,F. (1990) Combination of two-dimensional gel electrophoresis with microsequencing and amino acid composition analysis: improvement of speed and sensitivity in protein characterization. Electrophoresis, 11, 554–561. [DOI] [PubMed] [Google Scholar]
- Fisher H.F. (1985) l-glutamate dehydrogenase from bovine liver. Methods Enzymol., 113, 16–27. [DOI] [PubMed] [Google Scholar]
- Fonnum F. (1984) Glutamate: a neurotransmitter in mammalian brain. J. Neurochem., 42, 1–11. [DOI] [PubMed] [Google Scholar]
- Frei B. and Richter,C. (1988) Mono(ADP-ribosylation) in rat liver mitochondria. Biochemistry, 27, 529–535. [DOI] [PubMed] [Google Scholar]
- Galione A., Cui,Y., Empson,R., Iino,S., Wilson,H. and Terrar,D. (1998) Cyclic ADP-ribose and the regulation of calcium-induced calcium release in eggs and cardiac myocytes. Cell. Biochem. Biophys., 28, 19–30. [DOI] [PubMed] [Google Scholar]
- Hilz H., Koch,R., Fanick,W., Klapproth,K. and Adamietz,P. (1984) Nonenzymic ADP-ribosylation of specific mitochondrial polypeptides. Proc. Natl Acad. Sci. USA, 81, 3929–3933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinderlich S., Nöhring,S., Weise,C., Franke,P., Stäsche,R. and Reutter,W. (1998) Purification and characterization of N-acetylglucos amine kinase from rat liver—comparison with UDP-N-acetyl glucosamine 2-epimerase/N-acetylmannosamine kinase. Eur. J. Biochem., 252, 133–139. [DOI] [PubMed] [Google Scholar]
- Hopp T.P., Prickett,K.S., Price,V., Libby,R.T., March,C.J., Cerreti,P., Urdal,D.L. and Conlon,P.J. (1988) A short polypeptide marker sequence useful for recombinant protein identification and purification. Biotechnology, 6, 1205–1210. [Google Scholar]
- Jacobson E.L., Cervantes-Laurean,D. and Jacobson,M.K. (1994) Glycation of proteins by ADP-ribose. Mol. Cell. Biochem., 138, 207–212. [DOI] [PubMed] [Google Scholar]
- Jacobson M.K., Loflin,P.T., Aboul-Ela,N., Mingmuang,M., Moss,J. and Jacobson,E.L. (1990) Modification of plasma membrane protein cysteine residues by ADP-ribose in vivo. J. Biol. Chem., 265, 10825–10828. [PubMed] [Google Scholar]
- Jorcke D., Ziegler,M., Herrero-Yraola,A. and Schweiger,M. (1998) Enzymic, cysteine-specific ADP-ribosylation in bovine liver mitochondria. Biochem. J., 332, 189–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Julliard J.H. and Smith,E.L. (1979) Partial amino acid sequence of the glutamate dehydrogenase of human liver and a revision of the sequence of the bovine enzyme. J. Biol. Chem., 254, 3427–3438. [PubMed] [Google Scholar]
- Koch-Nolte F., Petersen,D., Balasubramanian,S., Haag,F., Kahlke,D., Willer,T., Kastelein,R., Bazan,F. and Thiele,H.-G. (1996) Mouse T cell membrane proteins Rt6-1 and Rt6-2 are arginine/protein mono(ADP-ribosyl)transferases and share secondary structure motifs with ADP-ribosylating bacterial toxins. J. Biol. Chem., 271, 7686–7693. [DOI] [PubMed] [Google Scholar]
- Kun E., Zimber,P.H., Chang,A.C.Y., Puschendorf,B. and Grunicke,H. (1975) Macromolecular enzymatic product of NAD+ in liver mitochondria. Proc. Natl Acad. Sci. USA, 72, 1436–1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laemmli U.K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature, 227, 680–685. [DOI] [PubMed] [Google Scholar]
- Lee H. and Iglewski,W.J. (1984) Cellular ADP-ribosyltransferase with the same mechanism of action as diphtheria toxin and Pseudomonas toxin A. Proc. Natl Acad. Sci. USA, 81, 2703–2707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H.C. (1997) Mechanisms of calcium signaling by cyclic ADP-ribose and NAADP. Physiol. Rev., 77, 1133–1164. [DOI] [PubMed] [Google Scholar]
- Ludden P.W. (1994) Reversible ADP-ribosylation as a mechanism of enzyme regulation in procaryotes. Mol. Cell. Biochem., 138, 123–129. [DOI] [PubMed] [Google Scholar]
- Lupi R, Corda,D. and Di Girolamo,M. (2000) Endogenous ADP-ribosylation of the G protein β subunit prevents the inhibition of type 1 adenylyl cyclase. J. Biol. Chem., 275, 9418–9424. [DOI] [PubMed] [Google Scholar]
- Masmoudi A. and Mandel,P. (1987) ADP-ribosyl transferase and NAD glycohydrolase activities in rat liver mitochondria. Biochemistry, 26, 1965–1969. [DOI] [PubMed] [Google Scholar]
- Mazon M.J. (1978) Effect of glucose starvation on the nicotinamide adenine dinucleotide phosphate-dependent glutamate dehydrogenase of yeast. J. Bacteriol., 133, 780–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy A.D., Walker,J.M. and Tipton,K.F. (1980) Purification of glutamate dehydrogenase from ox brain and liver. Evidence that commercially available preparations of the enzyme from ox liver have suffered proteolytic cleavage. Biochem. J., 191, 605–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald L.J. and Moss,J. (1993a) Nitric oxide-independent, thiol-associated ADP-ribosylation inactivates aldehyde dehydrogenase. J. Biol. Chem., 268, 17878–17882. [PubMed] [Google Scholar]
- McDonald L.J. and Moss,J. (1993b) Stimulation by nitric oxide of an NAD linkage to glyceraldehyde-3-phosphate dehydrogenase. Proc. Natl Acad. Sci. USA, 90, 6238–6241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald L.J., Wainschel,L.A., Oppenheimer,N.J. and Moss,J. (1992) Amino acid-specific ADP-ribosylation: structural characterization and chemical differentiation of ADP-ribose–cysteine adducts formed nonenzymatically and in a pertussis toxin-catalyzed reaction. Biochemistry, 31, 11881–11887. [DOI] [PubMed] [Google Scholar]
- McGeer E.G. and McGeer,P.L. (1976) Duplication of biochemical changes of Huntington’s chorea by intrastriatal injections of glutamic and kainic acids. Nature, 263, 517–519. [DOI] [PubMed] [Google Scholar]
- Moss J. and Vaughan,M. (1988) ADP-ribosylation of guanyl nucleotide-binding regulatory proteins by bacterial toxins. Adv. Enzymol., 61, 303–379. [DOI] [PubMed] [Google Scholar]
- Moss J., Stanley,S.J. and Watkins,P.A. (1980) Isolation and properties of an NAD- and guanidine-dependent ADP-ribosyltransferase from turkey erythrocytes. J. Biol. Chem., 255, 5838–5840. [PubMed] [Google Scholar]
- Oei S.L., Griesenbeck,J. and Schweiger,M. (1997) The role of poly(ADP-ribosyl)ation. Rev. Physiol. Biochem. Pharmacol., 131, 127–173. [DOI] [PubMed] [Google Scholar]
- Okazaki I.J. and Moss,J. (1996) Structure and function of eukaryotic mono-ADP-ribosyltransferases. Rev. Physiol. Biochem. Pharmacol., 129, 51–104. [DOI] [PubMed] [Google Scholar]
- Richter C. and Kass,G.E.N. (1991) Oxidative stress in mitochondria: its relationship to cellular Ca2+ homeostasis, cell death, proliferation, and differentiation. Chem. Biol. Interact., 77, 1–23. [DOI] [PubMed] [Google Scholar]
- Richter C., Winterhalter,K.H., Baumhüter,S., Lötscher,H.-R. and Moser,B. (1983) ADP-ribosylation in inner membrane of rat liver mitochondria. Proc. Natl Acad. Sci. USA, 80, 3188–3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxty B.A. and van Heyningen,S. (1995) The purification of a cysteine-dependent NAD+ glycohydrolase activity from bovine erythrocytes and evidence that it exhibits a novel ADP-ribosyltransferase activity. Biochem. J., 310, 931–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schägger H. and von Jagow,G. (1991) Blue native electrophoresis for isolation of membrane protein complexes in enzymatically active form. Anal. Biochem., 199, 223–231. [DOI] [PubMed] [Google Scholar]
- Tanuma S., Kawashima,K. and Endo,H. (1987) An NAD:cysteine ADP-ribosyltransferase is present in human erythrocytes. J. Biochem., 101, 821–824. [DOI] [PubMed] [Google Scholar]
- Ziegler M. (2000) New functions of a long known molecule. Emerging roles of NAD in cellular signaling. Eur. J. Biochem., 267, 1550–1564. [DOI] [PubMed] [Google Scholar]
- Ziegler M., Jorcke,D., Zhang,J., Schneider,R., Klocker,H., Auer,B. and Schweiger,M. (1996) Characterization of detergent-solubilized beef liver mitochondrial NAD+ glycohydrolase and its truncated hydrosoluble form. Biochemistry, 35, 5207–5212. [DOI] [PubMed] [Google Scholar]
- Ziegler M., Jorcke,D. and Schweiger,M. (1997) Metabolism of cyclic ADP-ribose: a new role for NAD+ glycohydrolases. Rev. Physiol. Biochem. Pharmacol., 131, 89–126. [DOI] [PubMed] [Google Scholar]
- Zolkiewska A., Nightingale,M.S. and Moss,J. (1992) Molecular characterization of NAD:arginine ADP-ribosyltransferase from rabbit skeletal muscle. Proc. Natl Acad. Sci. USA, 89, 11352–11356. [DOI] [PMC free article] [PubMed] [Google Scholar]