

Abstract
We have investigated the sites of N-terminally truncated cytochrome P4501A1 targeted to mitochondria (P450MT2) which interact with adrenodoxin (Adx), cytochrome P450 reductase (CPR) and bacterial flavodoxin (Fln). The binding site was mapped by a combination of in vitro mutagenesis, in vivo screening with a mammalian two-hybrid system, spectral analysis, reconstitution of enzyme activity and homology-based structural modeling. Our results show that part of an aqueous accessible helix (putative helix G, residues 264–279) interacts with all three electron donor proteins. Mutational studies revealed that Lys267 and Lys271 are crucial for binding to Adx, while Lys268 and Arg275 are important for binding to CPR and Fln. Additive effects of different electron donor proteins on enzyme activity and models on protein docking show that Adx and CPR bind in a non-overlapping manner to the same helical domain in P450MT2 at different angular orientations, while CPR and Fln compete for the same binding site. We demonstrate that evolutionarily divergent electron donor proteins interact with the same domain but subtly different contact points of P450MT2.
Keywords: adrenodoxin/cytochrome P450/electron transfer/P450 reductase/structural modeling
Introduction
Cytochrome P450 (P450) belongs to a multigene family of heme proteins that catalyze the NADPH-dependent oxidation of a wide variety of xenobiotic as well as physiologically important substrates (Okey et al., 1986; Gonzalez, 1990; Nebert, 1991). P450s are widely distributed in both prokaryotes and eukaryotes. In the metazoan organisms, large numbers of P450 forms involved in the metabolism of physiological substrates are localized in the endoplasmic reticulum (ER) and some also in mitochondria (Gonzalez, 1990; Okita and Masters, 1992). The P450s localized in the ER, routinely referred to as microsomal P450s, are organized in a transmembrane topology with almost all functional domains facing the cytosolic side (Tarr et al., 1983; Black, 1992). The microsomal P450s require a membrane-bound flavoprotein, NADPH cytochrome P450 reductase (CPR), for the transfer of electrons from NADPH (Okita and Masters, 1992). The constitutively expressed mitochondrial P450s involved in the physiological pathways of cholesterol and bile acid metabolism (P450scc, P45011β and P450c27), on the other hand, are extrinsic proteins, which require a soluble electron transport system consisting of an iron– sulfur protein, adrenodoxin (Adx), and a flavoprotein, adrenodoxin reductase (Adr), for enzymatic activity (Foster and Wilson, 1975; Mitani, 1979; Jefcoate, 1986). In support of an endosymbiotic origin of mitochondria, the mitochondrial P450s resemble the bacterial P450s, which also require soluble electron donor proteins, flavodoxin (Fln) and flavodoxin reductase (Flnr).
In contrast to this generality, recent studies in our laboratory on the mode of biogenesis of β-naphthoflavone (BNF)-inducible mitochondrial P450MT2 showed that the mitochondrially targeted P450 has a primary structure identical to that of the similarly induced P4501A1, except that it is N-terminally truncated to the +33 position (Addya et al., 1997; Ettickan et al., 2000). Our results showed that N-terminal cleavage by a cytosolic endoprotease activates the cryptic mitochondrial targeting signal, located between +33 and +44 of the protein. Most notably, P450MT2 (+33/1A1) showed high erythromycin N-demethylase (ERND) activity (Anandatheerthavarada et al., 1997, 1998), in contrast to low ERND activity of the intact, microsomal P4501A1, supported by CPR (Watkins et al., 1989; Anandatheerthavarada et al., 1997, 1998). The ERND activity and the previously reported arylhydrocarbon hydroxylase activity (AHH) of P450MT2 were supported by both Adx + Adr and CPR, though to different extents (Raza and Avadhani, 1988). Recent studies also showed that the steroid 17α hydroxylation and 7-ethoxyresorufin-O-deethylation activities of the microsomal P450c17 and P4501A2, respectively, were supported by Fln + Flnr and Adx + Adr systems, in addition to CPR (Jenkins and Waterman, 1994; Dong et al., 1996). Although the domains of Adx and CPR binding to different P450s have been mapped (Lambeth et al., 1979; Coghlan and Vickery, 1991; Shen and Kasper, 1995), the corresponding P450 regions involved in the functional association with electron transfer proteins remain unclear. More importantly, it is not known if these various electron donor proteins interact with the same or different domains of P450 protein.
In the present study, we have mapped the binding sites of P450MT2 using a combination of the mammalian two-hybrid system and reconstitution of activity with the wild-type and mutant enzymes. Results of biochemical and mutational experiments were fully supported by three-dimensional structural modeling and energy-minimized protein docking based on surface complementarity. Our results show that the two evolutionarily divergent electron transfer proteins, CPR and Adx, interact with P450MT2 through the same helical domain in a non-overlapping manner through different contact sites. The binding of Fln to P450MT2 overlaps both structurally and functionally with the binding of CPR.
Results
Mapping the Adx-binding domain of P450MT2 by the mammalian two-hybrid system
It is well established that the human and bovine Adx sequence region 74–86, rich in negatively charged amino acids, interacts with the conserved sequence domains of P450scc (376PLLKASIKETLRLH389) and other bona fide mitochondrial P450s through electrostatic interaction (Lambeth et al., 1979; Coghlan and Vickery, 1992; Wada and Waterman, 1992). A computer-based search revealed the presence of three motifs, termed MT2/I, MT2/II and MT2/III within P450MT2, which showed partial sequence similarity to the conserved Adx-binding domain of P450scc (Figure 1A). We mapped the Adx- and CPR-binding regions of P450MT2 using the mammalian matchmaker two-hybrid system. This system is based on the in vivo interaction between two proteins under test, one cloned in-frame with the Gal4 DNA-binding domain, and the other cloned in-frame with the VP16 transactivation domain, culminating in the activation of CAT gene expression. We cloned a series of N-terminal progressive deletions of P4501A1 (Figure 1A) in the pM vector containing the Gal4 DNA-binding domain. Bovine Adx and rat CPR cDNAs were cloned in the pVP16 vector containing the VP16 transactivation domain. Various 1A1 constructs in the pM plasmid were co-transfected with the Adx/VP16 plasmid or CPR/VP16 cDNA and the CAT reporter construct containing the Gal4 DNA-binding site. The level of CAT protein in transfected cells directly reflects the binding efficiency of the two proteins. As seen in Figure 1B, co-transfection of Adx and intact 1A1 cDNA constructs yielded a relative CAT protein level of 1, while the +5/1A1 and +33/1A1 cDNAs yielded 1.5- and 3.5-fold levels of intact 1A1, respectively. Co-transfection with CPR and intact 1A1 cDNAs, however, yielded a maximum CAT activity of 3.8, and progressively reduced activities of ∼3 and 2, respectively, with +5/1A1 and +33/1A1 cDNAs. In further support of our previous results with chemical cross-linking and in vitro reconstitution of activity (Anandatheerthavarada et al., 1998), these results demonstrate that CPR binds with highest efficiency to intact 1A1, while Adx binding is highest to N-terminally truncated P450MT2. The level of expression of CAT with both CPR and Adx cDNAs remained relatively constant with +100/1A1 and +200/1A1 cDNA constructs, while truncation to +300 and beyond drastically reduced the level of CAT protein with both CPR and Adx cDNAs. These results suggest that the P450MT2 domain binding to both Adx and CPR resides between amino acids 200 and 300.

Fig. 1. Mapping of P450MT2 domains needed for binding to Adx and CPR by using the mammalian two-hybrid system. (A) The N-terminal deletions of P4501A1 and the location of sequences showing partial identity to the conserved Adx-binding domain of P450c27. (B) The level of CAT protein indicating the extent of interaction between the various 1A1 deletion proteins and Adx or CPR proteins expressedin COS cells. The details of mammalian two-hybrid screening, transfection and measurement of CAT protein by ELISA are given in Materials and methods.
Detailed characterization of binding sites by chemical cross-linking and reconstitution of enzyme activity
To gain further insight into the precise sequence region of P450MT2 interacting with Adx, CPR and Fln, we carried out chemical cross-linking experiments. As shown in Figure 2A, 35S-labeled Adx was cross-linked to unlabeled P450MT2, and the cross-linked products immunoprecipitated with P4501A1 antibody. Results show that MT2/I peptide inhibited cross-linking in a concentration-dependent manner, causing a nearly complete inhibition at 70-fold molar concentration of the peptide. MT2/II and MT2/III peptides, on the other hand, failed to compete for cross-linking even at 70-fold molar excess. These results suggest that sequence region 264–278 representing the MT2/I peptide is involved in interaction with Adx. We also tested cross-linking to Fln since this flavoprotein has been shown to interact functionally with bacterially expressed purified P4501A2, a close relative of P4501A1 (Dong et al., 1996). Results of cross-linking and competition with various peptides in Figure 2B show that the same MT2/I region is involved in interaction with Fln. Surprisingly, the extent of cross-linking to labeled CPR was also inhibited by MT2/I peptide (Figure 2C), indicating that the same sequence region is involved in binding to all the three electron transfer proteins.

Fig. 2. Inhibition of chemical cross-linking of Adx, Fln and CPR to P450MT2 by sequence-specific peptides. Cross-linking was carried out with unlabeled P450MT2 and 35S-labeled Adx (A), Fln (B) or CPR (C) as described in Materials and methods, and the products were immunoprecipitated with P4501A1 antibody. The immunoprecipitates were resolved on 12% SDS–polyacrylamide gels and subjected to fluorography. The indicated amounts of MT2/I, MT2/II and MT2/III peptides were added before the addition of cross-linking agent, EDC. CLP = cross-linked product.
The precise mode of interaction of P450MT2 with three different electron transfer systems was investigated further by reconstitution of ERND activity in Adx + Adr, Fln + Flnr and CPR systems. The ERND activity supported by these systems and also the extent of inhibition of activity with wild-type and variously mutated MT2/I peptides was studied. As shown at the top of Figure 3A, KK Mut peptide contains K267N and K271N substitutions, and KK&KR Mut peptide contains K267N, K268N, K271N and R275N substitutions in the MT2/I peptide sequence. P450MT2 yielded 2.5- to 3-fold higher ERND activity in Adx + Adr and Fln + Flnr systems than with the CPR system (Figure 3A). A 70-fold molar excess of MT2/I peptide drastically inhibited ERND activity supported by all three of the electron donor systems. MT2/II and MT2/III peptides, on the other hand, did not significantly inhibit the activity with any of the three electron donor systems. Furthermore, AdxC peptide from the C-terminal acidic domain of Adx inhibited reactions only with the Adx + Adr system, with no significant effect on the CPR and Fln + Flnr systems, demonstrating the specificity of competition with added peptides. A 70-fold molar excess of KK Mut peptide failed to inhibit the activity supported by Adx + Adr, further indicating that K267 and K271 of P450MT2 are involved in interaction with Adx. Surprisingly, however, KK Mut peptide effectively inhibited both the CPR-supported and Fln + Flnr-supported activities, suggesting that residues of MT2/I other than K267 and K271 are involved in interaction with these two electron transfer proteins. KK&KR Mut peptide, with all four positive residues mutated, did not inhibit activity with any of the three electron donor protein systems, indirectly indicating that K268 and R275 are more important for interaction with Fln and CPR. These results strongly suggest that the three proteins under study interact with the same domain of P450MT2, but the contact sites for the different electron donors vary.

Fig. 3. Mode of interaction of different electron transfer proteins with P450MT2. (A) Effects of different peptides on the ERND activity of P450MT2 supported by different electron transfer systems. The amino acid sequences of the human Adx C-terminal acidic domain, wild-type and mutated MT2/I, and wild-type MT2/II and MT2/III peptides are shown at the top. ERND activity was reconstituted with either Adx/Adr, Fln/Flnr or CPR as described in Materials and methods, in the presence (70-fold molar excess over P450) or absence of the indicated peptides. (B) Additive or inhibitory effects of different electron transfer proteins on each other. Reconstitution was carried out with saturating levels of CPR (0. 75 µM) or Fln (1.75 µM)/Flnr (0.175 µM) and the effects of added Adx alone (0.5 µM), Fln alone (1 µM) or Adx (0.5 µM)/Adr (0.05 µM) on the activity were tested. Other conditions of reconstitution were as described in Materials and methods.
To gain further insight into the mode of interaction with P450MT2, we examined whether the effects of individual electron transfer proteins are exclusive of each other or additive. Titration of the ERND activity of P450MT2 with different concentrations of electron transfer proteins (not shown) yielded a maximum activity of 1 nmol of H[14C]HO/min/nmol P450 with 0.75 µM CPR, ∼2.5 nmol H[14C]HO/min/nmol P450 with 1.75 µM Fln (and 0.175 µM Flnr) and 2.8 nmol H[14C]HO/min/nmol P450 with 0.75 µM Adx (and 0.075 µM Adr). As seen in Figure 3B, 0.5 µM Adx alone added to the reaction mixture, in the presence of saturating levels of CPR (0.75 µM), did not have any significant effect on the ERND activity, while addition of 1 µM Fln alone inhibited the activity by ∼75%. Addition of 0.5 µM Adx + 0.05 µM Adr to the reaction containing saturating levels of CPR increased the activity to 3.2 nmol H[14C]HO/min/nmol P450. Results also show that addition of 0.5 µM Adx alone to a reaction mixture containing saturating levels of Fln and Flnr did not have any effect on the ERND activity of P450MT2, while a combination of Adx (0.5 µM) and Adr (0.05 µM) yielded increased activity of 4.2 nmol H[14C]HO/min/nmol P450. These results suggest that CPR and Fln bind to P450 MT2 in an overlapping or competing manner, while Adx binding does not overlap with the former two proteins.
Mutations in P450MT2 that differently affect interaction with different electron donor proteins
In a previous study, we showed that Adx, CPR and erythromycin induce the high spin state in bacterially expressed purified P450MT2 (Anandatheerthavarada et al., 1998). In the present study, we examined whether K267N/K271N and K268N/R275N mutations differently affect the ability of different electron donor proteins to induce the spin state change in P450MT2 or globally affect its structure. Figure 4, left panel, shows the electrophoretic patterns of the bacterially expressed, purified wild-type and mutant forms of P450MT2. The gel patterns show >80% purity for all three proteins. The immunoblot presented in the right hand panel of Figure 4 shows that both the wild-type and mutant proteins cross-react with antibody to P4501A1. Results of spectral analysis in Figure 5A show that erythromycin induced the high-spin state in wild-type P450MT2, KK Mut (K267N/K271N) and KR Mut (K268N/R275N) proteins, and the extent of change was similar, as seen by the ΔOD at 420 and 390 nm. These results along with the CD spectral analysis (not shown) suggest no significant structural aberrations in the mutant proteins.

Fig. 4. Bacterial expression and purification of wild-type and mutant P450MT2. (A) Wild-type, KK Mut (K267N, K271/N) and KR Mut (K268N, R275N) proteins were expressed in E.coli DH5α cells and purified by nickel chelate column chromatography as described in Materials and methods. A 2.5 µg aliquot of protein in each case was subjected to electrophoresis on 14% polyacrylamide gels and visualized by staining with Coomassie blue. (B) A duplicate gel as in (A) was subjected to immunoblot analysis using P4501A1-specific polyclonal antibody (1:3000 dilution). The amino acid residues substituted in KK Mut and KR Mut are indicated at the bottom of the gel.

Fig. 5. Extent of binding of wild-type and mutated P450MT2 to various electron transfer proteins by spectral shift measurements. Shifts in the spin state of bacterially expressed and purified wild-type (WT), KK Mut and KR Mut P450MT2 by added substrate or various electron transfer proteins were measured spectrophotometrically as described in Materials and methods. Effects of (A) erythromycin (ERM, 0.5 mM), (B) Adx (6 µM), (C) CPR (4 µM) and (D) Fln (6 µM). In (B–D), the effect of a 70-fold molar excess of P450MT2/I peptide was used as a positive control. (E) The effects of increasing amounts (0.2–0.6 µM) of Fln. The table in (F) shows the Kd for P450MT2 binding to various electron transfer proteins. The Kd value for Fln was calculated from ΔOD in (E), and the values for Adx and CPR were as reported before (Anandatheerthavarada et al., 1998).
We also compared the Adx-, Fln- and CPR-induced spin state changes in the wild-type and the two mutant proteins (KK Mut and KR Mut) to confirm that the spin state changes result from interaction through the same sequence domains. Results in Figure 5B and C show that Adx and CPR induce the high-spin state in wild-type P450MT2, though the former seems to induce a greater change as judged by a higher ΔOD. Furthermore, CPR induced the high-spin state in KK Mut protein to an extent similar to that of wild-type protein, though it failed to induce spin state changes in the KR Mut protein (Figure 5C and F). In contrast, Adx failed to induce any significant change in spin state with KK Mut protein but efficiently induced a change in the spin state with KR Mut protein (Figure 5B and F). These results support the notion that P450MT2 interacts with these two electron transfer proteins through different contact points. Results with Fln were similar to those obtained with CPR (Figure 5D and F), except that the latter induced a greater change in spin state, similar to Adx. As shown in Figure 5E and F, Fln interacted with P450MT2 with a Kd of 0.6 µM, which is similar to the previously reported affinity of Adx binding to this P450 (Anandatheerthavarada et al., 1998), but significantly higher than that shown for P450c17 (Jenkins and Waterman, 1994).
A possible difference in the mode of interaction of P450MT2 with Adx and Fln/CPR systems was ascertained further by using three additional approaches. First, as shown in Figure 6A, chemical cross-linking experiments showed that Adx, CPR and Fln cross-linked efficiently to bacterially expressed P450MT2 (+33/1A1) as well as to that purified from rat liver mitochondria. Results also show that KR Mut protein cross-linked to Adx at a level similar to wild-type P450MT2, while the KK Mut protein showed vastly reduced cross-linking. The efficiency of cross-linking to Fln and CPR was quite distinct in that KK Mut protein cross-linked at a level similar to the wild-type P450MT2, while the KR Mut protein showed vastly reduced cross-linking to both of these proteins. In Figure 6B, in vivo interaction of wild-type or mutated 1A1 with Adx or CPR electron transfer proteins was studied by the mammalian matchmaker system. In this system, the level of reporter protein, CAT, is directly proportional to the extent of interaction between the two test proteins. Results show that 1A1 protein interacts with CPR with ∼4-fold higher efficiency than with Adx protein, while the N-terminal truncated P450MT2 interacted with Adx with a 2-fold higher efficiency than with CPR. These results are in keeping with our previous results on cross-linking and reconstitution of ERND activity (Anandatheerthavarada et al., 1998). Furthermore, KK Mut protein interacted with Adx with a vastly reduced efficiency, while its interaction with CPR protein was nearly the same as that with wild-type P450MT2. In contrast, KR Mut protein interacted with Adx with an efficiency similar to the wild-type protein, while the extent of interaction with CPR was drastically reduced. Finally, reconstitution of ERND activity (Figure 6C) shows that KK Mut protein is marginally active with the Adx + Adr system, but shows activity similar to wild-type P450MT2 with both CPR and Fln + Flnr. In contrast, KR Mut protein shows ERND activity similar to wild-type P450MT2 in an Adx + Adr-supported system, but a vastly reduced activity with both CPR and Fln + Flnr. These results confirm that K267 and K271 are required for interaction with Adx, while K268 and R275 are required for interaction with CPR and Fln proteins.
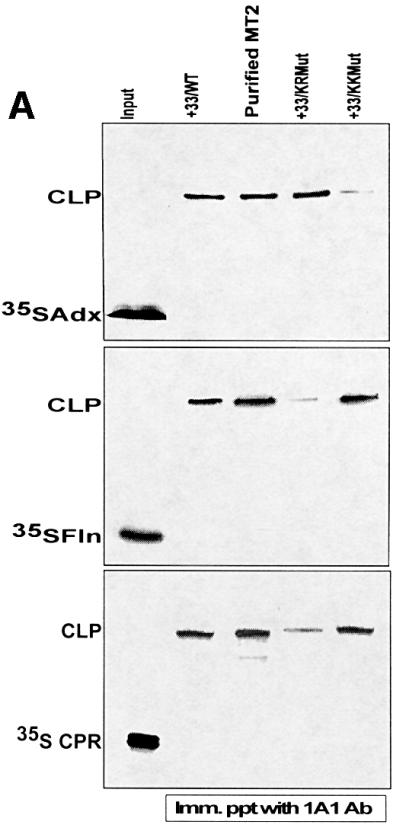

Fig. 6. Adx and CPR/Fln interact with P450MT2 through different sites of the P450MT2/I domain. Wild-type bacterially expressed +33/1A1, KK Mut and KR Mut proteins, and P450MT2 purified from rat liver mitochondria, were tested for their ability to interact with different electron transfer proteins by different approaches. (A) Chemical cross-linking of unlabeled P450MT2, KK Mut and KR Mut proteins to 35S-labeled Adx, Fln and CPR as indicated. Cross-linked products (CLP) were immunoprecipitated with antibody to P4501A1 and analyzed by electrophoresis on 12% polyacrylamide gels as described in Figure 2. (B) Extent of interaction of wild-type, KK Mut and KR Mut P450MT2 with Adx and CPR tested by mammalian two-hybrid screening as described in Figure 1. (C) ERND activity of the wild-type and mutant proteins, reconstituted with either Adx/Adr, Fln/Flnr or CPR as described in Figure 3 and Materials and methods.
Structural bases of interaction of P450MT2 with different electron transfer proteins
A ribbon diagram of the structure of P450MT2 based on the sequence homology with P450BM3, P450CAM and P4502C5 is presented in Figure 7A. The modeling was based mainly on the coordinates of the BM3 crystal structure reported before (Ravichandran et al., 1993). The model resembles the recently described crystal structure of P4502C5 (Williams et al., 2000) in many respects. The putative extrinsic membrane association domain, immediately past the N-terminal transmembrane domain, predicted on the basis of P4502C5 structure, is indicated by an arrow. The sequence of the P450MT2/I region implicated in binding to different electron transfer proteins is part of a long helix G, immediately facing helix F, which is suggested to lie close to the heme group in P4502C5 (Williams et al., 2000). Furthermore, the four positively charged residues (K267, K268, K271 and R275) are fully exposed to the surface, though at different angles. Residues K267 and K271 project out at an angle from the central core of helix G 90° to that of K268 and R275, though both pairs face the aqueous exterior (Figure 7G).

Fig. 7. Molecular modeling of P450MT2 and energy-minimized docking of various electron transfer proteins on helix G. Details of molecular modeling and protein docking were as described in Materials and methods. (A) A ribbon molecular model of P450MT2. (B) Docking of Adx on P450MT2 through helix G (yellow). (C) Docking of Fln on P450MT2 through helix G. (D) Docking of CPR on P450MT2 through helix G. (E) Formation of a ternary complex of P450MT2, Fln and Adx. The latter two are docked on P450MT2 through the same domain (helix G). (F) Ternary complex formation between P450MT2, CPR and Adx. Note that both Adx and Fln are docked on P450MT2 through the same helical domain (helix G). (G) Different angular orientations of basic residues K267/K271 and K268/R275. Wiggly, unmarked arrows on violet (B, E and F), lavender (C and E) and dark blue (D and F) point to acidic residues of electron donor proteins in contact with P450MT2.
Figure 7B–D shows the docking of Adx, Fln and CPR to the P450MT2 protein under energy-minimized conditions. All three proteins are docked on P450MT2 through the long helix G (shown in yellow). Additionally, Adx interacts through K267 and K271 (labeled in green, Figure 7B), while both Fln and CPR appear to interact through K268 and R275 (labeled in green, Figure 7C and D). These models further support the notion that all three electron transfer proteins interact with P450MT2 through the same helical domain but through different positively charged amino acid residues. This possibility was supported further by the results of double docking, which show that Adx and Fln or Adx and CPR could be docked on to helix G simultaneously to form energetically stable ternary complexes without any apparent steric hindrance between the interacting proteins. In support of the biochemical and mutational data, our attempts to dock Fln and CPR together were unsuccessful and did not yield energetically favorable complexes. The energy values (ΔG′) for surface interaction of Adx, Fln and CPR with P450MT2 are summarized in Table I.
Table I. Energy considerations for electron transport proteins binding to P450MT2.
| Proteins docked | Bonds | Angles | Torsion | Improper | Non-bonded | Electrostatic | Total energy |
|---|---|---|---|---|---|---|---|
| MT2 + Adx | 842.720 | 89.847 | 3217.963 | 1086.382 | –18502.55 | –17096.71 | –26962.34 |
| MT2 + Fln | 884.354 | 3657.025 | 3281.348 | 1117.374 | –21187.97 | –19160.89 | –31407.97 |
| MT2 + CPR | 681.173 | 5019.289 | 4543.929 | 1446.677 | –31107.72 | –27683.97 | –47100.01 |
| MT2 + CPR + Adx | 911.503 | 6563.837 | 6101.497 | 1998.476 | –41748.90 | –37891.63 | –64065.217 |
| MT2 + Fln + Adx | 958.718 | 4104.564 | 3824.579 | 1305.631 | –24699.77 | –21380.53 | –35886.808 |
| MT2 | 780.130 | 3157.161 | 2652.394 | 1005.427 | –14345.08 | –14843.26 | –21593.228 |
Energy change is given as kJ/mol.
Energy computations were performed with the GROMOS96 complementation with the Swiss-PDB viewer.
Discussion
The endosymbiotic theory on the origin of mitochondria implies that evolution of mitochondria and the ER diverged very early during eukaryotic evolution. In view of this, recent observations in our laboratory on the mitochondrial localization of N-terminal truncated microsomal P4501A1 (referred to as P450MT2) was indeed surprising (Raza and Avadhani, 1988; Addya et al., 1997; Anandatheerthavarada et al., 1999b). Even more surprising was the observation that the mitochondrially imported P450 readily accepted electrons from two evolutionarily divergent electron donor proteins, Adx/Adr and CPR, for catalyzing the ERND activity (Anandatheerthavarada et al., 1998, 1999b; Ettickan et al., 2000). Our results showing a marked difference in the substrate specificity of P450MT2 and P4501A1, and their preference for two different electron donor proteins also suggested a conformational change in the mitochondrial form possibly due to loss of the N-terminal transmembrane domain. Previously, we (Niranjan and Avadhani, 1980; Niranjan et al., 1984; Raza and Avadhani 1988; Anandatheerthavarada et al., 1997) observed that the release of BNF-, phenobarbital- and dexamethazone-induced P450s from mitochondrial inner membrane preparations required extraction with either high salt buffer or alkaline Na2CO3. These results suggest that both mitochondrial P450MT2, lacking the N-terminal trans membrane domain, and P450MT4 (phosphorylated form of P4502B1), containing an intact N-terminal transmembrane domain, are associated with the mitochondrial inner membrane in a membrane-extrinsic orientation (Shayiq et al., 1991; Anandatheerthavarada et al., 1997, 1999b). Crystal structure analysis of N-terminally truncated P4502C5 (Williams et al., 2000) indeed lends support to this possibility. Thus, inside the mitochondrial inner membrane environment, P450MT2 (Anadatheerthavarada et al., 1998), P4502B1, 2E1 and 3A1/2 (Anandatheerthavarada et al., 1999a; Robin et al., 2001) preferentially interact with the same C-terminal acidic domain of Adx, which was shown to be involved in interaction with P450scc. The difference in the membrane topologies of the microsomal-associated versus the mitochondrial-imported P450s may underlie the biochemical and evolutionary distinction between P450s belonging to the two membrane compartments. Here we address the question of how the electrons from NADPH may be channeled through structurally and evolutionarily divergent electron donor proteins into P450MT2.
The amino acid residues of P450MT2 that participate in the interaction with Adx, CPR and Fln were fine mapped by studying the interaction of mutant P450MT2 containing K267N/K271N or K278N/R275N substitutions with different electron donor proteins by three approaches: (i) spectral analysis to measure the spin state changes in purified proteins; (ii) reconstitution of ERND activity with purified proteins; and (iii) in vivo interaction of mutant proteins with Adx or CPR in the mammalian two-hybrid system. Both K267N mutant (results not shown) and KK mutant (K267N and K271N) proteins lost the ability to bind to Adx but not to CPR in chemical cross-linking, reconstitution of ERND activity and in vivo interaction in the two-hybrid screening systems. Mutations K268N and R275N, on the other hand, selectively abolished interaction with CPR without significantly affecting interaction with Adx. These mutant proteins still bound to the substrate erythromycin as efficiently as the parent wild-type P450MT2, as tested by the spin state changes. Furthermore, they exhibited normal CO reduced spectra, suggesting that mutations at the positively charged residues did not affect the overall structure of the P450. Furthermore, enzyme reconstitution studies showing the ability of the mutant proteins to catalyze ERND activity either with Adx/Adr or with CPR ensured the structural integrity of the mutant proteins. The mutational studies implicate a pivotal role for K267 and K271 for interaction with Adx, as opposed to K268 and R275, which are critical for interaction with CPR and Fln. These results confirm and extend previous studies on the biochemical and mutational analyses, as well as homology-based molecular modeling, which suggested that interaction of CPR and Adx with various P450s involves electrostatic interaction (Coghlan and Vickery, 1991, 1992; Shen and Kasper, 1995; Shen and Strobel, 1995; Graham and Peterson, 1999; Pikuleva et al., 1999; Davydov et al., 2000).
The interacting residues K267, K268, K271 and R275 are localized in a relatively small area of P450MT2. Although not rigorously proven, some biochemical studies imply extensive surface interaction between CPR and P450s (Voznesensky and Shenkman, 1992). In view of this, simultaneous/differential binding of Adx and CPR/Fln raises a question as to how these interactions are mediated without any steric effects. To address this question, we generated a homology-based model for P450MT2 and other P450s targeted to both the microsomes and mitochondria (latter results not shown). According to the model, the MT2/I sequence region shown to be involved in interaction with all three electron donor proteins is organized as helix G (see Figure7A) with the positive charges exposed on the surface. Interestingly, K267/K271 and K268/R275 pairs that are implicated in selective binding to Adx and Fln/CPR proteins show different angular orientation (Figure 7G). Many of the structural features, including the putative extrinsic membrane association domain, the heme-binding domain, etc., revealed by the crystal structure of P4502C5 (Cosme and Johnson, 2000; Williams et al., 2000) are also apparent in the present structural model for P450MT2. Energy-minimized docking of Adx, Fln and CPR proteins confirms that helix G is both the structurally and the energetically most favored site of interaction with all three electron transfer proteins. Furthermore, our molecular modeling experiments for the first time indicate the formation of ternary complexes of P450MT2 with Adx and CPR or Adx and Fln with no apparent steric interference or energy loss. It should be noted that the structural details of CPR (Wang et al., 1997) are based on the analysis of an N-terminally truncated form, which recently was shown to be functional in transferring electrons to P450 from an electrochemically reduced dye mediator (Estabrook et al., 1996). Our modeling data not only support the interaction of truncated CPR with P450, but also confirm that both the docking angle and the contact points for interaction of CPR/Fln with P452MT2 are different from those of Adx. Calculations based on the surface-exposed residues of P450 protein with bound electron transport proteins indicate that interaction of P450MT2 with Adx involves <15 residues and that with CPR involves ∼50 residues (results not shown). The additive effects of Adx/Adr on the activity of P450MT2 reconstituted with saturating levels of CPR or Fln/Flnr provide direct support for the simultaneous binding of Adx and CPR/Fln without apparent interference. These results provide new insights into the mode of P450MT2 interaction with three different electron transport proteins.
Using in vitro mutagenesis, Shen and Kasper (1995) showed that substitutions at two clusters of acidic residues of CPR, namely 207DDD209 and 213EED215, selectively affected the ability of the electron transfer protein to interact with cytochrome c and phenobarbital-induced P4502B1, respectively. Similarly, Jenkins et al. (1997) showed that D144 and E145 of Anabaena Fln are critical for transferring electrons to P450c17 for substrate oxidation. Although the major focus of the present study was to map the domain(s) of P450MT2 that may be involved in receiving electrons from different electron transfer proteins, our results on competition with synthetic peptides and site-specific mutations are consistent with the biochemical and mutational data on the domains of CPR, Fln and Adx binding to various P450s (Jenkins 1994; Shen and Kasper, 1995; Shen and Strobel, 1995; Pikuleva et al., 1999). Although not shown, the structural analysis indicates that the 207–215 region of CPR and the 126–145 sequence of Anabaena Fln, housing clusters of negatively charged amino acids, are involved in interaction with the K268 and/or R275 residues of P450MT2. Furthermore, in direct agreement with the predictions of Jenkins and Waterman (1998), the present biochemical and structural modeling results show that the mode of interaction of Fln is very similar to that of CPR.
In a series of studies, Strobel and co-workers (Shen and Strobel, 1995; Cvrk et al., 1996) used peptide-specific antibodies to map the putative CPR-interacting domain of P4501A1 to a helical region spanning residues 269–281. Our results on the detailed mapping of binding sites are consistent with the general predictions of these studies. Results on modeling (not presented) show that the putative helix G is conserved in a number of P450s, including P4502B1, 2E1, 2C12 and 2D6 that are targeted to both the microsomal and mitochondrial compartments, and in vitro reconstitution of mitochondrial counterparts of these P450s is inhibited by MT2/I peptide. These results are also consistent with recent homology-based modeling (Graham and Peterson, 1999) showing conserved cationic patches on the proximal face of P450s, the putative ‘evolutionary trace’, possibly representing the CPR-binding site. At the present time, the precise proximity of helix G to the heme group or how the electrons from helix G are transferred to the heme group remain unclear. It is also not clear if helix G, which is conserved in all the P450s exhibiting dual targeting to both the microsomal and mitochondrial compartments, provides sites for binding to electron transfer proteins or if other sites, such as helix F, provide sites in some cases. It is possible that binding of Adx or CPR/Fln can induce conformational changes in P450MT2, which may facilitate the positioning of helix G in close proximity to the heme, allowing electron flow. This possibility can be verified only through high resolution crystallographic studies of P450MT2– electron transport protein complexes. Nevertheless, in addition to providing insights into the specific sites of interaction with electron transfer proteins, results of this study show for the first time that P450MT2 interacts with three evolutionarily divergent electron transfer proteins through the same domain, but through different contact points.
Materials and methods
Purification of P450 reductase, Adx, Adr and P450MT2
Mitochondrial P450MT2 from BNF-treated rat livers was purified using a combination of PEG fractionation and chromatography on ω-octylaminoagarose, DEAE–Sephacel and hydroxylapatite columns essentially as described before (Raza and Avadhani, 1988). Adx and Adr were purified from the bovine adrenal mitochondria by the method of Foster and Wilson (1975). NADPH cytochrome P450 reductase was purified from phenobarbital-treated rat liver microsomes by AMP–Sepharose chromatography according to Yasukochi and Masters (1976). Purified proteins were >85–90% homogeneous as tested by SDS–PAGE. The specific activities of purified P450s were in the range of 12–14 nmol/mg of protein and that of NADPH P450 reductase was 45 µmol of cytochrome c reduced/min/mg of protein.
Mammalian matchmaker two-hybrid system
The mammalian two-hybrid system was purchased from Clontech. The interaction between Adx and P450 MT2b (+33/1A1), hereafter referred to as P450MT2, or between CPR and P450MT2 was studied according to the manufacturer’s protocol. Briefly, Adx cDNA was cloned in the pVP16 vector between EcoRI and XbaI sites, while P450 reductase cDNA was cloned in the EcoRI site. This results in the expression of the in-frame fusion between the VP16 transcription activation domain and the Adx or CPR proteins. The progressive N-terminal deletions of P4501A1 and point mutations in the MT2/I sequence region were cloned in the HindIII and XbaI restriction sites of the pM vector. This cloning results in the in-frame fusion of the Gal4 DNA-binding domain with the P450 proteins to be tested. The transient transfections of VP16–Adx and Gal4-DBD–P450 cDNA constructs and the CAT reporter containing the basal promoter and upstream Gal4 DNA-binding motifs were carried out in COS cells using Fugene 6 reagent. After 62 h, cells were harvested, lysed and the level of CAT expression was measured by using the CAT-ELISA assay kit purchased from Boehringer-Amersham (Roche Molecular Biochemicals).
Spectral measurements
Spectroscopic changes in the spin state due to Adx, CPR and Fln binding to wild-type and mutant P450MT2 were measured (Kido and Kimura, 1979; Anandatheerthavarada et al., 1998) in a Cary 1E spectrophotometer using 2 µM P450 in 35 mM phosphate buffer (pH 7.4), 0.1% Emulgen 911, 0.1 mM EDTA, 0.1 mM dithiothreitol (DTT) and 20% glycerol. Each sample was analyzed for the CO reduced spectrum (Matsubara et al., 1976) in order to confirm that the changes in the absorbency were not due to denaturation of the P450 proteins during spectral analysis.
In vitro reconstitution of monooxygenase activity
Reconstitution of monooxygenase activity for ERND was carried out essentially as described (Brian et al., 1990; Anandatheerthavarada et al., 1998) in a buffer containing 50 mM Tris–HCl, pH 7.4, 20 mM MgCl2, 0.4 mM [N-methyl-14C]erythromycin (1 µCi/mmol), dilauroylphosphatidylcholine (20 µg/ml mixed vesicles), 60 pmol of P450 and, unless otherwise stated, either 0.2 nmol of Adx, 0.02 nmol of Adr or 0.1 nmol of P450 reductase, or 0.3 nmol of Fln or 0.04 nmol Flnr in 200 µl reaction volumes. The final reaction product, H[14C]HO, was measured according to Yang et al. (1991). All the significant values reported here represented more than five times the background values.
Protein cross-linking studies
Transcription-linked translation for generating 35S-labeled wild-type Adx, Fln and CPR was carried out as described (Addya et al., 1997). Cross-linking reactions (100 µl volume) were carried out in 50 mM phosphate buffer (pH 7.4) containing 10% (v/v) glycerol, 0.05% Emulgen 911, 0.1 mM DTT, 0.1 mM EDTA, 0.5 µM unlabeled P450 proteins and 40 000 c.p.m. of 35S-labeled Adx, Fln or CPR (Tamburini and Schenkman, 1987; Anandatheerthavarada et al., 1998). Cross-linking was initiated by adding 9 mM 1-ethyl-3-(3-dimethylamino-propyl) carbodiimide (EDC) (Mauk and Mauk, 1989). The reaction was carried out for 60 min at room temperature, and was terminated by adding 10 vols of phosphate-buffered saline. The cross-linked complexes were subjected to immunoprecipitation with antibodies to P450MT2 and Adx (Bhat and Avadhani, 1985; Anandatheerthavarada et al., 1998). The immunoprecipitates were analyzed on 12% SDS–polyacrylamide gels (Laemmli, 1970) and detected by fluorography or scanning through a BioRad GS525 molecular imager.
Bacterial expression and purification of P450 and electron transfer proteins
cDNAs encoding C-terminal histidine-tagged P450MT2 and the mutant forms of the protein were generated as described before (Anandatheerthavarada et al., 1998). The cDNA constructs were cloned in the bacterial expression vector PCW and expressed in Escherichia coli strain DH5α as described (Clark and Waterman, 1991; Parikh et al., 1997). The proteins were purified to ∼65–80% purity (9–12 nmol of P450/mg of protein) by using a combination of Ni2+–agarose, DEAE–cellulose and hydroxylapatite column chromatography. Expression and purification of Fln and Flnr were as described before (Jenkins and Waterman, 1998).
Synthetic peptides
Peptides were synthesized using an ABI Model 431A synthesizer and purified by HPLC on a reverse phase column. A list of the various synthetic peptides used in this study is presented in Figures 1 and 3.
Molecular modeling
A three-dimensional structural model for P450MT2 was developed using the Swiss-Model software (Guex and Peitsch, 1997). The molecular model for P450MT2 was built based on the sequence homology to P4502c17 and BM3 (1BVY; Ravichandran et al., 1993). Models for Fln (Lostao et al., 2000), Adx (Muller et al., 1998; Pikuleva et al., 2000) and CPR (Wang et al., 1997) were built based on the X-ray structure coordinates reported before, and listed in the PDB data bank, using the Swiss-Model software. The accuracy of the models was verified by energy calculations using Gramos 96 software, provided by the Swiss PDB viewer. Models for heteromeric complexes of P450MT2 and various electron transfer proteins were generated by manual docking of individual structures using INSIGHT (MSI Inc., San Diego, CA). The solvent-accessible surfaces were generated for P450MT2, Adx, Fln and CPR. Using surface complementarity and biochemical data, the structures of Adx, Fln and CPR were docked on helix G of P450MT2.
References
- Addya S., Anandatheerthavarada,H.K., Biswas,G., Bhagwat,S.V., Mullick,J. and Avadhani,N.G. (1997) Targeting of NH-terminal-processed microsomal protein to mitochondria: a novel pathway for the biogenesis of hepatic mitochondrial P450MT2. J. Cell Biol., 139, 589–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anandatheerthavarada H.K., Addya,S., Dwivedi,R.S., Biswas,G., Mullick,J. and Avadhani,N.G. (1997) Localization of multiple forms of inducible cytochromes P450 in rat liver mitochondria: immunological characteristics and patterns of xenobiotic substrate metabolism. Arch. Biochem. Biophys., 339, 136–150. [DOI] [PubMed] [Google Scholar]
- Anandatheerthavarada H.K., Addya,S., Mullick,J. and Avadhani,N.G. (1998) Interaction of adrenodoxin with P450 1A1 and its truncated form P450MT2 through different domains: differential modulation of enzyme activities:. Biochemistry, 37, 1150–1160. [DOI] [PubMed] [Google Scholar]
- Anandatheerthavarada H.K., Biswas,G., Mullick,J., Babu,S.V.N., Laszlo,O., Pain,D. and Avadhani,N.G. (1999a) Dual targeting of cytochrome P4502B1 to mitochondria and microsomes involves a novel signal activation by phosphorylation at Ser128. EMBO J., 18, 5494–5504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anandatheerthavarada H.K., Vijayasarathy,C., Bhagwat,S.V., Biswas, G., Mullick,J. and Avadhani,N.G. (1999b) Physiological role of the N-terminal processed P4501A1 targeted to mitochondria in erythromycin metabolism and reversal of erythromycin-mediated inhibition of mitochondrial protein synthesis. J. Biol. Chem., 274, 6617–6625. [DOI] [PubMed] [Google Scholar]
- Bhat N.K. and Avadhani,N.G. (1985) Transport of proteins into hepatic and nonhepatic mitochondria: specificity of uptake and processing of precursor forms of carbamoyl-phosphate synthetase I. Biochemistry, 24, 8107–8113. [DOI] [PubMed] [Google Scholar]
- Black S.D. (1992) Membrane topology of the mammalian P450 cytochromes. FASEB. J., 6, 680–685. [DOI] [PubMed] [Google Scholar]
- Brian W.R., Sari,M., Iwasaki,M., Shimada,T., Kaminisky,L.S. and Guengerich,F.P. (1990) Catalytic activities of human liver cytochrome P-450 IIIA4 expressed in Saccharomyces cerevisiae. Biochemistry, 29, 11280–11292. [DOI] [PubMed] [Google Scholar]
- Clark B.J. and Waterman,M.R. (1991) The hydrophobic amino-terminal sequence of bovine 17α-hydroxylase is required for the expression of functional hemoprotein in COS 1 cells. J. Biol. Chem., 266, 5898–5904. [PubMed] [Google Scholar]
- Coghlan V.M. and Vickery,L.E. (1991) Site-specific mutations in human ferredoxin that affect binding to ferredoxin reductase and cytochrome P450scc. J. Biol. Chem., 266, 18606–18612. [PubMed] [Google Scholar]
- Coghlan V.M. and Vickery,L.E. (1992) Electrostatic interactions stabilizing ferredoxin electron transfer complexes. Disruption by ‘conservative’ mutations. J. Biol. Chem., 267, 8932–8935. [PubMed] [Google Scholar]
- Cosme J. and Johnson,E.F. (2000) Engineering microsomal cytochrome P450 2C5 to be a soluble, monomeric enzyme. Mutations that alter aggregation, phospholipid dependence of catalysis and membrane binding. J. Biol. Chem., 275, 2545–2553. [DOI] [PubMed] [Google Scholar]
- Cvrk T., Hodek,P. and Strobel,H.W. (1996) Identification and characterization of cytochrome P4501A1 amino acid residues interacting with a radiolabeled photoaffinity diazido-benzphetamine analogue. Arch. Biochem. Biophys., 330, 142–152. [DOI] [PubMed] [Google Scholar]
- Davydov D.R., Kariakin,A.A., Petushkova,N.A. and Peterson,J.A. (2000) Association of cytochromes P450 with their reductases: opposite sign of the electrostatic interactions in P450BM-3 as compared with the microsomal 2B4 system. Biochemistry, 39, 6489–6497. [DOI] [PubMed] [Google Scholar]
- Dong M.S., Yamazaki,H., Guo,Z. and Guengerich,F.P. (1996) Recombinant human cytochrome P450 1A2 and an N-terminal-truncated form: construction, purification, aggregation properties and interactions with flavodoxin, ferredoxin and NADPH-cytochrome P450 reductase. Arch. Biochem. Biophys., 327, 11–19. [DOI] [PubMed] [Google Scholar]
- Estabrook R.W., Shet,M.S., Fisher,C.W., Jenkins,C.M. and Waterman, M.R. (1996) The interaction of NADPH-P450 reductase with P450: an electrochemical study of the role of the flavin mononucleotide-binding domain. Arch. Biochem. Biophys., 333, 308–315. [DOI] [PubMed] [Google Scholar]
- Ettickan B., Anadatheerthavarada,H.K., Bhagwat,S.V., Biswas,G., Fang, J.-K. and Avadhani,N.G. (2000) Accumulation of mitochondrial P450MT2, N-terminal truncated P4501A1 in rat brain during chronic treatment with β-naphthoflavone: a role in the metabolism of neuroactive drugs. J. Biol. Chem., 275, 34415–34423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster R.P. and Wilson,L.D. (1975) Purification and characterization of adrenodoxin reductase from bovine adrenal cortex. Biochemistry, 14, 1477–1484. [DOI] [PubMed] [Google Scholar]
- Gonzalez F.J. (1990) Molecular genetics of P450 superfamily. Pharmacol. Ther., 45, 1–38. [DOI] [PubMed] [Google Scholar]
- Graham S.E. and Peterson,J.A. (1999) How similar are P450s and what can their differences teach us? Arch. Biochem. Biophys., 369, 24–29. [DOI] [PubMed] [Google Scholar]
- Guex N. and Peitsch,M.C. (1997) SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modelling. Electrophoresis, 18, 2714–2723. [DOI] [PubMed] [Google Scholar]
- Jefcoate C.R. (1986) Cytochrome P450 enzymes in sterol biosysnthesis and metabolism. In Ortiz de Montellano,P.R. (ed.), Cytochrome P-450: Structure, Mechanism and Biochemistry. Plenum Publishing, New York, pp. 387–428.
- Jenkins C.M. and Waterman,M.R. (1994) Flavodoxin and NADPH-flavodoxin reductase from Escherichia coli support bovine cytochrome P450c17 hydroxylase activities. J. Biol. Chem., 269, 27401–27408. [PubMed] [Google Scholar]
- Jenkins C.M. and Waterman,M.R. (1998) NADPH-flavodoxin reductase and flavodoxin from Escherichia coli characteristics: as a soluble microsomal P450 reductase. Biochemistry, 37, 6106–6113. [DOI] [PubMed] [Google Scholar]
- Jenkins C.M., Genzor,C.G., Fillat,M.F., Waterman,M.R. and Gomez-Moreno,C. (1997) Negatively charged Anabaena flavodoxin residues (Asp144 and Glu145) are important for reconstitution of cytochrome P450 17α-hydroxylase activity. J. Biol. Chem., 272, 22509–22513. [DOI] [PubMed] [Google Scholar]
- Kido T. and Kimura,T. (1979) The formation of binary and ternary complexes of cytochrome P-450scc with adrenodoxin and adrenodoxin reductase, adrenodoxin complex. The implication in ATCH function. J. Biol. Chem., 254, 11806–11815. [PubMed] [Google Scholar]
- Laemmli U.K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature, 227, 6800–6805. [DOI] [PubMed] [Google Scholar]
- Lambeth D.J., Seybert,D.W. and Kamin,H. (1979) Ionic effects on adrenal steroidogenic electron transport. The role of adrenodoxin as an electron shuttle. J. Biol. Chem., 254, 7255–7264. [PubMed] [Google Scholar]
- Lostao A., El Harrous,M., Daoudi,F., Romero,A., Parody-Morreale,A. and Sancho,J.J. (2000) Dissecting the energetics of the apoflavodoxin–FMN complex. J. Biol. Chem., 275, 9518–9526. [DOI] [PubMed] [Google Scholar]
- Matsubara T., Koike,M., Touchi,A., Tochino,Y. and Sugeno,K. (1976) Quantitative determination of cytochrome P450 in rat liver homogenate. Anal. Biochem., 76, 596–603. [DOI] [PubMed] [Google Scholar]
- Mauk M.R. and Mauk,A.G. (1989) Crosslinking of cytochrome c and cytochrome b5 with a water-soluble carbodiimide. Eur. J. Biochem., 186, 473–486. [DOI] [PubMed] [Google Scholar]
- Mitani F. (1979) Cytochrome P450 in adrenocortical mitochondria. Mol. Cell. Biochem., 24, 21–43. [DOI] [PubMed] [Google Scholar]
- Muller A., Muller,J.J., Muller,Y.A., Uhlmann,H., Bernhardt,R. and Heinemann,U. (1998) New aspects of electron transfer revealed by the crystal structure of a truncated bovine adrenodoxin, Adx(4–108). Structure, 6, 269–280. [DOI] [PubMed] [Google Scholar]
- Nebert D.W. (1991) Proposed role of drug-metabolizing enzymes: regulation of steady state levels of the ligands that effect growth, homeostasis, differentiation and neuroendocrine functions. Mol. Endocrinol., 9, 1203–1214. [DOI] [PubMed] [Google Scholar]
- Niranjan B.G. and Avadhani,N.G. (1980) Activation of aflatoxin B1 by a mono-oxygenase system localized in rat liver mitochondria. J. Biol. Chem., 255, 6575–6578. [PubMed] [Google Scholar]
- Niranjan B.G., Wilson,N.M., Jefcoate,C.R. and Avadhani,N.G. (1984) Hepatic mitochondrial cytochrome P450 system. J. Biol. Chem., 259, 12495–12501. [PubMed] [Google Scholar]
- Okey A.B., Roberts,E.A., Harper,P.A. and Denison,M.S. (1986) Induction of drug-metabolizing enzymes: mechanisms and consequences. Clin. Biochem., 19, 132–141. [DOI] [PubMed] [Google Scholar]
- Okita R.T. and Masters,B.S.S. (1992) Biotransformations: the cytochromes P-450. In Devlin,T.M. (ed.), Text Book of Biochemistry with Clinical Correlations, 3rd edn. Wiley-Liss, New York, pp. 981–1000.
- Parikh A., Gillam,E.M.J. and Guengerich,F.P. (1997) Drug metabolism by Escherichia coli expressing human cytochromes P450. Nature Biotechnol., 15, 784–788. [DOI] [PubMed] [Google Scholar]
- Pikuleva I.A., Cao,C. and Waterman,M.R. (1999) An additional electrostatic interaction between adrenodoxin and P450c27 (CYP27A1) results in tighter binding than between adrenodoxin and P450scc (CYP11A1). J. Biol. Chem., 274, 2045–2052. [DOI] [PubMed] [Google Scholar]
- Pikuleva I.A., Tesh,K., Waterman,M.R. and Kim,Y. (2000) The tertiary structure of full-length bovine adrenodoxin suggests functional dimers. Arch. Biochem. Biophys., 373, 44–55. [DOI] [PubMed] [Google Scholar]
- Ravichandran K.G., Boddupalli,S.S., Hasermann,C.A., Peterson,J.A. and Deisenhofer,J. (1993) Crystal structure of hemoprotein domain of P450BM-3, a prototype for microsomal P450s. Science, 261, 731–736. [DOI] [PubMed] [Google Scholar]
- Raza H. and Avadhani,N.G. (1988) Hepatic mitochondrial cytochrome P-450 system. Purification and characterization of two distinct forms of mitochondrial cytochrome P-450 from β-naphthoflavone-induced rat liver. J. Biol. Chem., 263, 9533–9541. [PubMed] [Google Scholar]
- Robin M.-A., Anandatheerthavarada,H., Fang,J.-K., Cudic,M., Otvos,L. and Avadhani,N.G. (2001) Mitochondrial targeted cytochrome P4502E1 (P450MT5) contains an intact N-terminus and requires mitochondrial specific electron transfer proteins for activity. J. Biol. Chem., in press. [DOI] [PubMed] [Google Scholar]
- Shayiq R.M., Addya,S. and Avadhani,N.G. (1991) Constitutive and inducible forms of cytochrome P450 from hepatic mitochondria. Methods Enzymol., 206, 587–594. [DOI] [PubMed] [Google Scholar]
- Shen A.L. and Kasper,C.B. (1995) Role of acidic residues in the interaction of NADPH-cytochrome P450 oxidoreductase with cytochrome P450 and cytochrome c.J. Biol. Chem., 270, 27475–27480. [DOI] [PubMed] [Google Scholar]
- Shen S. and Strobel,H.W. (1995) Functional assessment of specific amino acid residues of cytochrome P4501A1 using anti-peptide antibodies. Arch. Biochem. Biophys., 320, 162–169. [DOI] [PubMed] [Google Scholar]
- Tamburini P.P. and Schenkman,J.B. (1987) Purification to homogeneity and enzymological characterization of a functional covalent complex composed of cytochromes P-450 isozyme 2 and b5 from rabbit liver. Proc. Natl Acad. Sci. USA, 84, 11–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarr G.E., Black,S.D., Fujita,V.S. and Coon,M.J. (1983) Complete amino acid sequence and predicted membrane topology of phenobarbital-induced cytochrome P450 (isozyme 2) from rabbit liver microsomes. Proc. Natl Acad. Sci. USA, 80, 6552–6556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voznesensky A.I. and Schenkman,J.B. (1992) The cytochrome P450 2B4–NADPH cytochrome P450 reductase electron transfer complex is not formed by charge-pairing. J. Biol. Chem., 267, 14669–14676. [PubMed] [Google Scholar]
- Wada A. and Waterman,M.R. (1992) Identification by site-directed mutagenesis of two lysine residues in cholesterol side chain cleavage cytochrome P450 that are essential for adrenodoxin binding. J. Biol. Chem., 267, 22877–22882. [PubMed] [Google Scholar]
- Wang M., Roberts,D.L., Paschke,R., Shea,T.M., Masters,B.S.S. and Kim,J.J.J. (1997) Three-dimensional structure of NADPH-cytochrome P450 reductase: prototype for FMN- and FAD-containing enzymes. Proc. Natl Acad. Sci. USA, 94, 8411–8416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins P.B., Murray,S.A., Winkelman,L.G., Heuman,D.M., Wrighton, S.A. and Guzelian,P.S. (1989) Erythromycin breath test as an assay of glucocorticoid-inducible liver cytochromes P-450. Studies in rats and patients. J. Clin. Invest., 83, 688–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams P.A., Cosme,J., Sridhar,V., Johnson,E.F. and McRee,D.E. (2000) Mammalian microsomal cytochrome P450 monooxygenase: structural adaptations for membrane binding and functional diversity. Mol. Cell, 5, 121–131. [DOI] [PubMed] [Google Scholar]
- Yang C.S., Patteu,C.J., Ishizaki,H. and Yoo,J.H. (1991) Induction, purification and characterization of cytochrome P450IIE. Methods Enzymol., 206, 595–603. [DOI] [PubMed] [Google Scholar]
- Yasukochi Y. and Masters,B.S.S. (1976) Some properties of a detergent-solubilized NADPH-cytochrome c(cytochrome P-450) reductase purified by biospecific affinity chromatography. J. Biol. Chem., 251, 5337–5344. [PubMed] [Google Scholar]