

Abstract
Yeast prions are inherited through proteins that exist in alternate, self-perpetuating conformational states. The mechanisms by which these states arise and are maintained are still poorly defined. Here we demonstrate for the first time that Sis1, a member of the Hsp40 chaperone family, plays a critical role in the maintenance of a prion. The prion [RNQ+] is formed by Rnq1, which is present in the same physical complex as Sis1, but only when Rnq1 is in the prion state. The G/F domain of Sis1 is dispensable for rapid growth on rich medium, but is required for [RNQ+] maintenance, distinguishing essential regions of Sis1 from those needed for prion interaction. A specific Sis1 deletion mutant altered the physical aggregation pattern of Rnq1 without curing the prion. This variant state propagated in a heritable fashion after wild-type Sis1 function was restored, indicating that multiple physical states are compatible with prion maintenance and that changes in chaperone activity can create prion variants. Using a prion chimera we demonstrate that the prion-determinant domain of Rnq1 is genetically sufficient for control by Sis1.
Keywords: chaperone/prion/Rnq1/Sis1/yeast
Introduction
The known prion proteins of Saccharomyces cerevisiae (Sup35, Ure2 and Rnq1) have the unique ability to exist in distinct and heritable alternative states. The stability of these states allows the faithful transmission of the prions [PSI+], [URE3] and [RNQ+], respectively, during mitotic and meiotic division (Wickner and Chernoff, 1999; Sondheimer and Lindquist, 2000). In the case of [URE3] and [PSI+], the shift to prion state causes an epigenetic phenotype because of the reduced function of the underlying prion protein. Although no phenotype has been linked to the [RNQ+] prion, we have previously shown that the prion determinant domain of Rnq1 can be fused to the C-terminal domain of Sup35 (Sup35C) to create a new prion, demonstrating that Rnq1 can epigenetically modify protein function in a heritable manner (Sondheimer and Lindquist, 2000).
After years of study, it is still not clear how a protein shifts between the prion and non-prion states in the cell. Wickner (1994), in his landmark paper which described the genetics of [URE3], hypothesized that the difference between the prion and non-prion states of Ure2 was conformational. Early support for this idea came from the discovery that the chaperone protein Hsp104 regulated the [PSI+] state (Chernoff et al., 1995). Chaperones assist the folding of other proteins by preventing the aggregation of unfolded or partially folded proteins, or by resolubilizing aggregated proteins (Bukau et al., 1999). The ability of chaperones to modulate protein structure and assembly may provide a mechanism for controlling the prion state.
Further studies have shown that expression of the chaperone Hsp104 is required for the propagation of all of the known yeast prions: [PSI+], [RNQ+] and [URE3] (Chernoff et al., 1995; Moriyama et al., 2000; Sondheimer and Lindquist, 2000). The interaction of [PSI+] with Hsp104 is complex because the chaperone must be present at physiological levels—neither deleted nor ectopically overexpressed—for the continued propagation of [PSI+] (Chernoff et al., 1995).
Hsp104 was first identified as a protein critical for induced thermotolerance (Sanchez and Lindquist, 1990). Hsp104 carries out this role by disaggregating protein complexes, a role that it carries out in vitro in combination with Ssa1, an Hsp70 family protein, and Ydj1, an Hsp40 family protein (Parsell et al., 1994; Glover and Lindquist, 1998). These connections are now being extended to studies of the maintenance and propagation of yeast prions in vivo. The capacity of Hsp104 overexpression to cure [PSI+] can be modified by chaperones in the Hsp70 family, specifically the Ssa and Ssb proteins (Chernoff et al., 1999; Newnam et al., 1999). Moreover, deletion of the SSA2 gene in a strain with a specific point mutation in SSA1 leads to an inability to propagate [PSI+] (Jung et al., 2000).
Other studies have begun to examine the role of chaperones in the Hsp40 family. Hsp40 proteins act as co-chaperones for Hsp70 proteins in a wide variety of cellular processes in many different organisms. The activities of these complexes include preventing protein aggregation, enhancing the folding of newly translated protein, and translocating proteins across organellar membranes (Craig et al., 1999). It has recently been shown that overexpression of the Hsp40 family member Ydj1 may slowly destabilize the [URE3] prion (Moriyama et al., 2000), and that overexpression of Ssa1, Ssb1 and Ydj1 may destabilize weak variants of [PSI+] (Kushnirov et al., 2000). A different Hsp40 protein, Sis1, co-immunoprecipitates from yeast cell lysates with Rnq1, the protein determinant of the [RNQ+] prion (Luke et al., 1991; M.Luke and K.T.Arndt, personal communication).
Sis1 is an essential protein, which is required for the initiation of translation (Zhong and Arndt, 1993). Like all other Hsp40 proteins, Sis1 contains a J domain, which allows the interaction of Hsp40 proteins with Hsp70 partners (Cheetham and Caplan, 1998). The J domain of Sis1 is followed by two glycine-rich regions, the glycine/phenylalanine (G/F) and glycine/methionine (G/M) regions, which are present in a subset of Hsp40 family proteins and have an unknown function. The C-terminus of the protein (CTD) binds to unfolded polypeptides in vitro (Lu and Cyr, 1998). Deletion and mutation analysis of Sis1 regions indicated that either the J domain together with the G/F domain, or the J domain with the G/M and CTD domains, is required for cell viability (Yan and Craig, 1999).
In this study we have evaluated the role of Sis1 in controlling the [RNQ+] state. As with Hsp104, wild-type Sis1 function is required for the maintenance of [RNQ+]. The interaction between Sis1 and Rnq1 is mediated by specific domains within the Sis1 protein, which are not required for the essential function of Sis1. The interaction between Sis1 and Rnq1 is mediated through a physical link, and alterations in Sis1 function have direct consequences upon the physical state of Rnq1 in the cell.
Results
The interaction of Rnq1 with Sis1 and Ssa1 depends on the presence of [RNQ+]
A previous study showed that Sis1 co-immunoprecipitated with Rnq1 from cell lysates (Luke et al., 1991). We have recently shown that the strain used in this study (W303) contains the [RNQ+] prion (Sondheimer and Lindquist, 2000). To determine whether the interaction of Sis1 and Rnq1 is dependent upon the prion state, we examined an isogenic pair of [RNQ+] and [rnq–] strains of the 33G-D373 background. Total Sis1 protein was immunoprecipitated and the associated proteins were analyzed by SDS–PAGE followed by blotting with anti-Rnq1 (α-Rnq1) antisera (Figure 1A). Sis1 associated with Rnq1 in the strain that was [RNQ+], but not in the isogenic [rnq–] strain. Similarly, a change in the Sis1 association with Rnq1 occurred in W303 strain when a [RNQ+] isolate was converted to the [rnq–] state by deletion of HSP104 (data not shown).
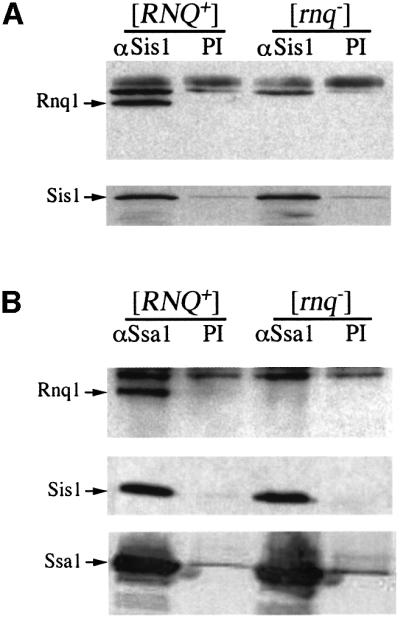
Fig. 1. Rnq1 associates with Sis1 and Ssa1 in a [RNQ+]-dependent fashion. Lysates from isogenic [RNQ+] or [rnq–] strains were immunoprecipitated with an α-Sis1 antibody or pre-immune (PI) serum (A). Proteins were resolved by SDS–PAGE and immunoblotted with α-Rnq1 (top panel) or α-Sis1 (bottom panel). The experiment was repeated with α-Ssa1 immunoprecipitation (B). The first panel shows western blot detection of Rnq1, the second of Sis1, and the bottom panel of Ssa1. The association between Sis1 and Ssa1 is maintained independently of the presence of [RNQ+].
Sis1 is a co-chaperone for the Hsp70 family member Ssa1 in vitro (Lu and Cyr, 1998). Therefore, we also characterized the interaction of Ssa1 with Rnq1 and Sis1. Ssa1 antibody precipitated Sis1 from cell lysates, an interaction that was not affected by the presence or absence of the prion. However, Rnq1 co-immunoprecipitated with Ssa1 only in strains containing the [RNQ+] prion (Figure 1B). Thus, the recovery of detectable levels of Rnq1 in association with Sis1 or Ssa1 is dependent upon the prion state of Rnq1.
The G/F region of Sis1 is required for the maintenance of [RNQ+]
The prion-dependent interaction of Rnq1 with the chaperones Ssa1 and Sis1 suggests two possibilities. The chaperones may have a productive biological interaction with the [RNQ+] prion, assisting in its maintenance. Alternatively, the chaperones might bind to the prion state simply because they recognize it as a misfolded protein. If Sis1 plays an active role in protecting or maintaining the [RNQ+] prion, then mutations that interfere with Sis1 function might lead to a loss of the prion. However, if Sis1 binds Rnq1 merely as a consequence of its misfolded state, we would expect such mutations to have no effect upon [RNQ+] propagation.
Because the deletion of SIS1 is lethal, we tested these possibilities by replacing wild-type SIS1 with deletion mutants that support cell viability, using a [RNQ+] W303 background. The strains expressed either wild-type SIS1 gene or one of three different mutations: sis1(ΔG/M ΔCTD), sis1(ΔCTD) or sis1(ΔG/F) (Figure 2A). We also tested the effect of overexpressing the chaperones Hsp104 or Sis1 in the wild-type [RNQ+] background. All of these strains exhibited similar growth rates on rich media (data not shown).

Fig. 2. Chaperone effects on Rnq1 state: centrifugation analysis. Sis1 deletion mutants were created by a plasmid shuffle strategy. A [RNQ+] strain with a deletion of the genomic copy of SIS1 rescued by a URA3 plasmid expressing the full-length protein was transformed with various SIS1 constructs on TRP1 plasmids (A). By selecting these strains on 5-FOA, colonies were identified that expressed only the deletion construct. Lysates were prepared from these strains and were used as total (T) fractions or separated into supernatant (S) and pellet (P) fractions by centrifugation (B). The strain expressing sis1(ΔG/F) lost the [RNQ+] centrifugation pattern: Rnq1 moved from the pellet to the soluble fraction. The polyclonal α-Rnq1 antiserum recognizes two cross-reacting bands (below and above the Rnq1 band) in yeast lysates. These cross-reacting bands are also present in lysates of strains deleted for the RNQ1 gene (Sondheimer, 2000). (C) Overexpression of chaperones does not cause a change in prion state. Wild-type [RNQ+] W303 were transformed with plasmids expressing either HSP104 or SIS1 from the GPD promoter, and strains were analyzed as described above. The expression of chaperones was verified by western blotting (data not shown).
The modified strains were assayed for the presence of [RNQ+] by high-speed centrifugation of cell lysates. Rnq1 protein is soluble in lysates of [rnq–] strains, but pellets from [RNQ+] lysates after centrifugation (Sondheimer and Lindquist, 2000). Overexpression of Sis1 or Hsp104 did not modify the sedimentation properties of Rnq1 (Figure 2C). Deletion of the CTD domain of Sis1, or the G/M and CTD domains together, also had no effect in the W303 strain. However, a deletion of the G/F region of Sis1 abolished sedimentation of Rnq1 (Figure 2B).
Mutations in Sis1 alter the appearance of Rnq1–GFP aggregation
To analyze further the changes in the physical state of Rnq1 that are associated with the Sis1 mutations, we monitored the distribution of Rnq1 in vivo by transient expression of an Rnq1–green fluorescent protein fusion (Rnq1–GFP). As described previously, the Rnq1–GFP fusion provides a marker for the distribution of the endogenous protein; in [RNQ+] strains, the fusion protein appears as an aggregate, usually one major focus per cell, against a darker background, whereas in [rnq–] strains the fusion protein is diffusely distributed (Sondheimer and Lindquist, 2000). In a W303 strain carrying a full-length Sis1 expression plasmid to cover the genomic SIS1 deletion, the [RNQ+] pattern of Rnq1–GFP expression was recapitulated (Figure 3A). In contrast, the Rnq1–GFP pattern in the sis1ΔG/F strain was diffuse, consistent with the apparent solubility of Rnq1 in these strains in the centrifugation assay (Figure 3B). Unexpectedly, the sis1(ΔG/MΔCTD) replacement, which had a centrifugation pattern like that of the [RNQ+] strain carrying the wild-type gene for Sis1, showed a third, distinct pattern of Rnq1–GFP distribution. Less than 2% of the sis1(ΔG/M ΔCTD)-expressing cells contained a single large focus of Rnq1–GFP. In the remaining cells, the fusion protein was dispersed into numerous tiny aggregates of protein that moved rapidly throughout the cytoplasm (Figure 3C and D).

Fig. 3. Sis1 deletion mutants alter the localization of Rnq1–GFP. W303 strains deleted for SIS1 and containing the Sis1 replacement plasmids were transformed with a plasmid encoding Rnq–GFP from a CUP1 promoter. The fusion protein was expressed for 4 h by the addition of 50 µM CuSO4 and cells were viewed unfixed using a confocal microscope. Images are from a single Z-section unless noted otherwise. (A) Fluorescence in the strain containing the wild-type SIS1 (TRP1+) plasmid. (B) The strain containing the sis1(ΔG/F) (TRP1+) plasmid; the Rnq1–GFP fusion protein is diffusely present within the cell. (C) The strain containing the sis1(ΔG/MΔCTD) (TRP1+) plasmid. The appearance of this strain was unique. Rnq1–GFP was found in tiny particles that moved rapidly within the cell. (D) Three-dimensional rendering of the same cell reconstructed from serial confocal slices. The reconstructed image has been false colored by depth of field to show the three-dimensional filling of the cell by small Rnq1–GFP aggregates. To demonstrate the propagation of the prion state, the Sis1 mutants were used to cytoduce recipient strains of the 10B-H49 background. The recipient strains were [rnq–] prior to cytoduction and carried the wild-type SIS1 gene. Cytoductant progeny from the cross of the sis1(ΔG/F) strain to the 10B-H49 recipient produced the same pattern as their sis1(ΔG/F)parent (E). Cytoductant progeny from the cross of the sis1(ΔG/MΔCTD) strain recapitulated the appearance of the sis1(ΔG/MΔCTD) parent 20–70% of the time in four separate experiments using isolates from two different cytoduction events (F).
The Sis1ΔG/F mutant is unable to propagate the prion
The definitive genetic test for the maintenance or loss of the [RNQ+] prion is the ability to transmit the [RNQ+] prion to [rnq–] partners through cytoplasmic contact. This property is most easily tested by cytoduction (Figure 4A). In cytoduction, strains with karyogamy deficiencies (e.g. kar1-1 strains) fuse to form a heterokaryon with a mixed cytoplasm and two unfused nuclei (Conde and Fink, 1976). This allows the transmission of prion state, but not the exchange of genetic information between the nuclei. Upon division, the heterokaryon creates daughter cells with only one of the two parental nuclei. These haploids can be selected using genetic markers from the initial [rnq–] parent, propagated for many generations to dilute out the initial protein content, and later examined for the maintenance of the [RNQ+] prion in the newly synthesized Rnq1 protein.

Fig. 4. Cytoduction of [RNQ+] from strains with Sis1 replacements. W303 strains (initially [RNQ+]) with different SIS1 expression plasmids were created as described in Figure 2. These strains were used to cytoduce the [rnq–] strain 10B-H49 (A). In cytoduction, a strain containing the desired phenotypic element (the donor) is co-incubated with a strain lacking the element (the recipient). The two cell types fuse cytoplasmic content, but not their nuclei, preventing gene exchange. Haploid buds form from the heterokaryon. Desired offspring buds can be selected using nuclear markers from the recipient and propagated into colonies. The resulting 10B-H49 cytoductants were assayed for the presence of [RNQ+] by centrifugation. The (sis1 ΔG/F) replacement strain was unable to cytoduce [RNQ+] to the 10B-H49 recipient (B). The TRP1 plasmid containing the different SIS1 constructs was not transmitted during the cytoduction events, as demonstrated by the failure of the recipient strains to grow on defined media lacking tryptophan (data not shown).
The W303 Sis1 strains described above were tested for prion cytoduction using a [rnq–] strain from the 10B-H49 background as a recipient. The cytoductant progeny were assayed for the presence of [RNQ+] by centrifugation (Figure 4B). The strain expressing sis1(ΔG/F) was unable to pass the prion to the 10B-H49 recipient via cytoduction. All other strains successfully cytoduced the prion conformation to their partner. We confirmed findings from the centrifugation assay with Rnq1–GFP localization: the fusion protein was coalescent in cytoductant progeny from SIS1 or sis1(ΔCTD) strains, but not in the progeny from sis1(ΔG/F) cytoduction [Figure 3E and data not shown; see below for experiments with the sis1(ΔG/MΔCTD) strain]. These results indicate that the isogenic recipient strains propagated the differences in Rnq1 conformational state in a stable, heritable manner even though they did not differ at the SIS1 locus. Thus, the deletion of the Sis1 G/F domain not only alters the solubility and localization of Rnq1, but also eliminates the transmissible [RNQ+] state.
Propagation of a distinct aggregated Rnq1 state
Studies of the [PSI+] prion have found that Sup35 can exist in variant states that share the epigenetic characteristics of [PSI+], but have distinct phenotypic and biochemical characteristics (Derkatch et al., 1996). Variants exhibit a milder version of the nonsense-suppression phenotype associated with [PSI+] and have decreased mitotic stability (Zhou et al., 1999). The variants are likely to represent the protein’s capacity to fold into multiple prion structures that have different stability and different inhibition of functional activity. The prion strain phenomena of yeast may be related to the pathogenic strain variants of the mammalian prion diseases.
We investigated the possibility that the ‘small aggregate’ state of [RNQ+] isolated in the W303 sis1(ΔG/M ΔCTD) strain might also be a distinct prion strain of [RNQ+]. The most definitive way to test this possibility is by cytoduction of the ‘small aggregate’ [RNQ+] variant to a [rnq–] cell that has wild-type SIS1, and asking whether the ‘small aggregate’ phenotype is propagated in the new background. The 10B-H49 [rnq–] cytoductants that were transiently exposed to the cytoplasm from the W303 sis1(ΔG/MΔCTD) strain during cytoduction continued to propagate the variant state of [RNQ+]. Approximately half of the cytoductant progeny derived from a cross between 10B-H49 [rnq–] and W303 sis1(ΔG/MΔCTD) [RNQ+] exhibited a fluorescence pattern identical to the original sis1(ΔG/MΔCTD) parent: small clusters of protein rather than the large aggregate pattern observed in the original [RNQ+] strain (Figure 3F). The incomplete transmission of the ‘small aggregate’ distribution may be due to a modestly reduced stability of this phenotype over the many generations of growth needed to complete the experiment. However, if the ‘small aggregate’ distribution pattern of Rnq1 had been merely due to the inability of sis1(ΔG/MΔCTD) strains to package Rnq1 into [RNQ+] aggregates properly, these forms would have been eliminated immediately in the SIS1 cytoductant-recipient background. The maintenance of the ‘small aggregate’ state suggests that Rnq1 can exist in more than one type of heritable conformation in wild-type strains.
Sis1 deletion mutants can not propagate the hybrid prion [RPS+]
Previous work has demonstrated that the prion domain (PrD) of Rnq1 can be transferred to another open reading frame to cause a prion-like modification of its function (Sondheimer and Lindquist, 2000). The [RPS+] state is caused by the prion-like behavior of a protein consisting of the PrD (residues 153–405) of Rnq1 and the highly charged middle domain (M) and functional C-terminal domain (C) of Sup35 (Rmc). The availability of this system allowed us to determine whether the Sis1 sensitivity of the [RNQ+] prion could be transferred to [RPS+] by the Rnq1 PrD.
Sis1 deletion mutants were created as described above in a 74D-694 strain carrying the hybrid RMC gene in place of SUP35; this strain was both [RNQ+] and [RPS+] initially. This particular background was used because it contains a premature stop codon in the ADE1 gene, which can be suppressed by [PSI+] and [RPS+]. That is, [PSI+] and [RPS+] cells will grow on media lacking adenine, whereas [psi–] and [rps–] cells will not.
74D-694 cells expressing the mutant sis1(ΔG/F) gene had a wild-type growth rate on rich media, but failed to grow on SD-ade, indicating that [RPS+] had been lost (Figure 5A). By contrast, the strains carrying the sis1(ΔCTD) or sis1(ΔG/MΔCTD) mutants showed a reduced growth rate on YPD, an effect that was not observed in the W303 background. Furthermore, both of these mutations affected the stability of [RPS+]: neither strain showed initial growth on SD-ade, although a very small number of Ade+ colonies did appear after prolonged incubation (data not shown). Neither the loss of the [RPS+] prion nor the defect in normal growth was due to changes in the availability of the Rmc fusion protein; expression of the fusion was similar in 74D-694 strains regardless of the SIS1 allele being studied (S.Uptain and S.Lindquist, unpublished).

Fig. 5. Deletion of Sis1 G/F or CTD regions eliminates the hybrid [RPS+] prion and [RNQ+] in 74D-694 cells. The presence of [RPS+] allows survival on media lacking adenine and growth into white colonies on YPD because of the epigenetic inactivation of the Sup35 C-terminal region. Sis1-replacement derivatives from a 74D-694 [RPS+] background were created as described above. These were plated by 5-fold serial dilution to YPD and SD-ade (A). Yeast deleted for the CTD or both the G/M region and the CTD of Sis1 had a notable slow-growth phenotype at 30°C. These colonies were red on YPD and showed no apparent growth on SD-ade within a week. Strains deleted for G/F had a wild-type growth rate on YPD, but also lost the ability to grow on SD-ade. To investigate the possibility that Sis1 plays a strain-specific role in prion maintenance in 74D-694, we examined the effect of the Sis1 deletion proteins on [RNQ+] maintenance in these strains by centrifugation assay (B). All tested SIS1 deletions led to a loss of [RNQ+].
The different effects of sis1(ΔCTD) and sis1(ΔG/M ΔCTD) on [RNQ+] propagation in W303 and [RPS+] propagation in 74D-694 could be due to an intrinsic difference between the two prions in their requirement for Sis1 function. Alternatively, the difference might be due to the distinct interaction of Sis1 with the two yeast backgrounds. To distinguish these alternatives we evaluated the state of Rnq1 in the presence of the different Sis1 chaperone mutants in 74D-694. Using Rnq1 centrifugation we observed that [RNQ+], like [RPS+], was eliminated by all three tested modifications of SIS1 (ΔG/F, ΔG/MΔCTD or ΔCTD), but not by the replacement of SIS1 with another wild-type copy (Figure 5B). These results indicate that Sis1 effects are dependent upon the genetic background studied. More importantly, we find that the sensitivity of the Rnq1 prion domain to Sis1 mutation is similar whether the domain is regulating the [RNQ+] prion or the synthetic [RPS+] state.
Discussion
We have demonstrated that the Sis1 chaperone is required for the propagation and inheritance of the [RNQ+] prion. Sis1 is physically associated with the Rnq1 protein, but strikingly, this association is only present when Rnq1 is in the prion state. By two physical assays, differential centrifugation and localization of Rnq1–GFP, we show that strains with the sis1(ΔG/F) mutation lose the physical characteristics of the Rnq1 prion state. Furthermore, using cytoduction, we demonstrate that these strains fail to propagate the prion state to wild-type (SIS1) recipients. Thus, the physical structure of [RNQ+] is linked to a chaperone that is required for its own propagation, a striking confirmation of the relationship between prion propagation and the folded state of Rnq1. This provides the strongest evidence to date that a chaperone’s effects on prion propagation are direct.
When SIS1 was replaced with sis1(ΔG/MΔCTD) in W303 [RNQ+] cells, an unexpected pattern of Rnq1–GFP fluorescence was found: instead of one or two large foci within each cell, there were many tiny foci of protein. The ‘small aggregate’ state might have simply represented a failure of the modified Sis1 protein to provide a proper folding environment for the mature prion. However, the transmission of this variant state by cytoduction into wild-type [rnq–] SIS1 recipients and the propagation of this state in the new background indicate that the ‘small aggregate’ variant is distinct, heritable and stable. This finding is not without precedent: variants of the [PSI+] prion, which are phenotypically distinct, have also been discovered (Derkatch et al., 1996; Zhou et al., 1999). What is perhaps most remarkable about the Rnq1 ‘small aggregate’ state is that it is initiated by exposure to the mutant SIS1(ΔG/M ΔCTD) protein, but does not require the continued presence of SIS1(ΔG/MΔCTD) to propagate. The discovery that a chaperone protein is involved in the creation of a yeast prion variant may provide new avenues of research into similar phenomena in other prions, most notably the described strain variants of the mammalian transmissible spongiform encephalopathies.
The relative importance of the four different domains of Sis1 to the overall function of the chaperone is a matter of ongoing research. Here we have demonstrated a specific role for one of these domains in vivo: the G/F domain of Sis1 is necessary for propagation of the [RNQ+] prion. The G/F domain is, however, completely dispensable for rapid growth.
The loss of the G/M and/or CTD regions of Sis1 has different consequences depending on the background studied. The 74D-694 cells bearing the sis1(ΔCTD) or sis1(ΔG/MΔCTD) mutations lose the [RNQ+] prion, whereas W303 strains with the same mutations do not. Additionally, these same 74D-694 strains are unable to grow at wild-type rates on rich media. The loss of the prion in 74D-694 sis1(ΔCTD) and sis1(ΔG/MΔCTD) strains may be due to either a failure of the truncated Sis1 proteins to interact with Rnq1 in 74D-694 or to the general growth defect in 74D-694 cells caused by the partial loss of Sis1 activity. It has been shown that the role of the C-terminal regions of Sis1 in cell growth and viability is influenced by the activity of other chaperones within the cell; W303 strains that lack regions of the Hsp40 homolog Ydj1 are inviable when SIS1 is replaced by sis1(ΔG/MΔCTD) (Johnson and Craig, 2001). The observed loss of wild-type growth rate in 74D-694 strains carrying the sis1(ΔCTD) and sis1(ΔG/MΔCTD) mutations is not related to any prion phenomena. Therefore, the loss of [RNQ+] in 74D-694 sis1(ΔCTD) and sis1(ΔG/MΔCTD) cells may be incidental to the growth defect caused by the loss of Sis1 activity.
The [RNQ+] prion was not cured by the overexpression of Sis1 or Hsp104, chaperones that the prion depends upon for maintenance. This is in contrast to both [PSI+] and [RPS+], which can be cured by overexpression of Hsp104 (Chernoff et al., 1995; Sondheimer and Lindquist, 2000). The difference in Hsp104-mediated curing of [RPS+] and [RNQ+] prions is intriguing because in both cases the same sequence, Rnq1(153–402), is the prion-determining domain. One possible explanation for this difference could be a distinct role of Hsp104 in the 74D-694 and W303 backgrounds, allowing the curing of protein elements containing the Rnq1 PrD only in the 74D-694 background. Another possibility is that a region of Sup35 outside of the prion-determining domain may be required for [PSI+] curing by high-dosage Hsp104. It is noteworthy that [URE3], like [RNQ+], is cured by the deletion, but not the overexpression, of Hsp104. It is possible that Hsp104 may be required for yeast prion maintenance generally and that the elimination of [PSI+] by Hsp104 overexpression may be due to a specific interaction with Sup35.
We have also found that the Hsp70 family protein Ssa1 is in the same protein complex as Rnq1 when Rnq1 is in the prion state. Directly assessing the effects of Ssa1 deficiencies in vivo proved difficult because of the functional equivalence and coordinated regulation of the Ssa1–4 proteins (Craig et al., 1999). We did find that deleting both SSA1 and SSA2 in the W303 background did not affect [RNQ+] (data not shown). Other investigators have found that alterations in Ssa activity cure [PSI+] (Jung et al., 2000), and further study may uncover a requirement for Ssa activity in [RNQ+] maintenance.
The role of chaperones in the maintenance of prions is slowly becoming apparent. The response of chaperones to prions is clearly distinct from that to aggregates of denatured proteins. Aggregates of denatured proteins induce the expression of chaperones and are generally a substrate for chaperone-mediated elimination (Parsell et al., 1994; Chai et al., 1999). By contrast, none of the yeast prions induces a heat shock response that causes its own elimination; even the curing of [PSI+] by Hsp104 only occurs when the chaperone is overexpressed ectopically (Singh et al., 1979; Chernoff et al., 1995).
Recent evidence has suggested that [PSI+] is an adaptive state that generates useful phenotypic diversity (True and Lindquist, 2000). Thus, it may be that yeast prions are a normal, rather than pathological, state of the cell, and that chaperones function to maintain prions rather than to disperse them. Prions are a remarkable epigenetic mechanism for the inheritance of protein conformation and phenotype. A more complete analysis of the role of chaperones in all known prion systems may suggest common pathways for the coordination and control of prions.
Materials and methods
Cloning, strains and media
A complete list of all strains and plasmids used is displayed in Tables I and II, respectively. Isogenic [RNQ+] and [rnq–] strains were created in the strain 10B-H49 (MATα, ade2-1, SUQ5, lys1-1, his3-11,15, leu2-3, kar1-1, ura3::KANMX4) by cytoduction. Cytoduction donors and recipients were plated together on rich media and cytoductants were selected on SGlycerol-trp media. Sis1 replacement experiments used the W303 strain (MATa, can1-100, his3-11,15, leu2-3,112, trp1-1, ura3-1, ade2-1) or 74D-694 [MATa, ade1-14(UGA), trp1-289(UAG), his3Δ-200, ura3-52, leu2-3,112, sup35::RMC].
Table I. Strains.
| Strain name | Genotype | Source |
|---|---|---|
| 33G-D373 [RNQ+] | MATa, ade, his, leu, lys, phe, trp, ura, kar1-15 | this study |
| 33G-D373 [rnq–] | MATa, ade, his, leu, lys, phe, trp, ura, kar1-15 | this study |
| W303 | MATa, his, leu, trp, ura, ade, [RNQ+] | R.Rothstein |
| W303 Δsis1 pURA-SIS1 | MATa, his, leu, trp, ade, sis1::KANR, pURA-SIS1 | this study |
| 10B-H49 | MATα, ade, lys, his, leu, kar, ura3::KANR, [rnq–] | this study |
| 74D-694 | MATa, ade1-14, trp, his, ura, leu, [RNQ+] | Chernoff et al. (1993) |
| 74D-694 [RPS+] | MATa, ade1-14, trp, his, ura, leu, sup35::RMC | Sondheimer (2000) |
| W303 ssa1 ssa2 | MATa, trp, ura, lys, ade, ssa1::HIS3, ssa2::LEU2 | Sanchez et al. (1993) |
Disruptions of Sis1 were created in strains containing a URA3+ marked Sis1 expression plasmid to provide a continuous source of wild-type Sis1. Disruption was performed using the long flanking homology method with a kanamycin resistance marker (Wach, 1996). First round primers were: 5′-CTTCTATAACATGATCAGTAATAGC-3′; 5′-GGGGATCCGTCGACCTGCAGCGTACGCATTATTAGTTCTGTATACTATAC-3′; 5′-GTTTAAACGAGCTCGAATTCATCGATCTAAACGACGCTCAAAAACGTGC-3′; and 5′-CTCGTGTTATGGAGCAATTGC-3′. Disruption of chromosomal SIS1 was verified by failure to grow on 5-fluoroorotic acid (5-FOA). Strains were then transformed with TRP1-containing plasmids encoding the appropriate Sis1 replacement driven by the endogenous promoter. The URA3 plasmid was counterselected on 5-FOA, and Sis1 replacement was verified by western blotting. Studies of the SIS1 mutant strains were based upon two isolated colonies each from three separate transformation and plasmid shuffle events.
[RNQ+] and [rnq–] versions of the 33G-D73 strain were created using a cytoduction protocol. First, the KAR1 locus of the strains was replaced with a kar1-15 allele using pop-in/pop-out replacement (the plasmid was a gift of M.Rose). The resulting strains were then cytoduced by either W303 wild-type ([RNQ+]) or W303 Δrnq1 ([rnq–]) strains. This created an isogenic strain pair, one [RNQ+] and the other [rnq–], as verified by centrifugation assay (data not shown).
Prion analyses
Density-based centrifugation of cell extracts was performed as described previously (Sondheimer and Lindquist, 2000). To analyze the localization of Rnq1 protein, strains were transformed with a plasmid expressing Rnq1–GFP from a CUP1 promoter. The strains were induced by growth in 50 µM CuSO4 for 4 h. Cells were viewed unfixed using a confocal microscope (Axiovert) with Zeiss LSM510 software (University of Chicago CDIF). To improve resolution, the SIS1(ΔG/MΔCTD) strain was photographed on a stage cooled to 14°C.
Immunoprecipitation
Cells were broken by vortexing for 2 min at 4°C in lysis buffer (150 mM KCl, 20 mM Tris pH 8, 1 mM EDTA, 1 mM dithiothreitrol, 1 mM phenylmethylsulfonyl fluoride, 200 µM TPCK, 20 µM leupeptin, 1 µM pepstatin A, 0.2% Triton X-100, 0.2% SDS) with glass beads. Crude lysates were cleaned by a 14 000 r.p.m. spin for 5 min in an Eppendorf benchtop centrifuge. Lysates were incubated for 1 h with 1 µl of pre-immune serum and 20 µl of protein A beads. Pre-cleared lysates were centrifuged again (14 000 r.p.m. for 10 s) and supernatants were mixed with 40 µl of α-Sis1 cross-linked protein A beads and re-incubated. Precipitates were washed with lysis buffer three times before resuspension in gel loading buffer for SDS–PAGE and western analysis.
Table II. Plasmids.
| Plasmid name | Markers | Description | Source |
|---|---|---|---|
| pURA-SIS1 | CEN, URA3 | full-length copy of SIS1 with native promoter | Yan and Craig (1999) |
| pTRP-SIS1 | CEN, TRP1 | full-length copy of SIS1 with native promoter | Yan and Craig (1999) |
| pTRP-SIS1ΔG/MΔCTD | CEN, TRP1 | truncation of SIS1 (1–121) with native promoter | Yan and Craig (1999) |
| pTRP-SIS1ΔCTD | CEN, TRP1 | truncation of SIS1 (1–172) with native promoter | Yan and Craig (1999) |
| pTRP-SIS1ΔG/F | CEN, TRP1 | truncation of SIS1 (Δ71–121) with native promoter | Yan and Craig (1999) |
| pHIS-GPD-104 | 2Δ, HIS | Hsp104 overexpression plasmid | Sanchez and Lindquist (1990) |
| pTRP-GPD-SIS1 | CEN, TRP1 | Sis1 overexpression plasmid | W.Yan, unpublished |
| pURA-RNQ1–GFP | CEN, URA3 | expresses Rnq1–GFP fusion from CUP1 promoter | Sondheimer and Lindquist (2000) |
Acknowledgments
Acknowledgements
We would like to thank the members of the Lindquist and Craig laboratories for helpful discussions of these data. N.S. was supported by NIH grants 5 T32 GM07183-24 and 5 T32 GM07281. N.L. was supported by NIH fellowship 5 F31 GM18507-05. This work was supported by the Howard Hughes Medical Institute and NIH grants GM25874 and GM27870.
References
- Bukau B., Schmid,F. and Buchner,J. (1999) Assisted protein folding. In Bukau,B. (ed.), Molecular Chaperones and Folding Catalysts. Harwood Academic, Amsterdam, The Netherlands.
- Chai Y., Koppenhafer,S.L., Bonini,N.M. and Paulson,H.L. (1999) Analysis of the role of heat shock protein (Hsp) molecular chaperones in polyglutamine disease. J. Neurosci., 19, 10338–10347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheetham M. and Caplan,A. (1998) Structure, function and evolution of DnaJ: conservation and adaptation of chaperone function. Cell Stress Chaperones, 3, 28–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chernoff Y.O., Derkatch,I.L. and Inge-Vechtomov,S.G. (1993) Multicopy sup35 gene induces de-novo appearance of psi-like factors in the yeast Saccharomyces cerevisiae. Curr. Genet., 24, 268–270. [DOI] [PubMed] [Google Scholar]
- Chernoff Y.O., Lindquist,S.L., Ono,B., Inge-Vechtomov,S.G. and Liebman,S.W. (1995) Role of the chaperone protein Hsp104 in propagation of the yeast prion-like factor [psi+]. Science, 268, 880–884. [DOI] [PubMed] [Google Scholar]
- Chernoff Y.O., Newnam,G.P., Kumar,J., Allen,K. and Zink,A.D. (1999) Evidence for a protein mutator in yeast: role of the Hsp70-related chaperone Ssb in formation, stability and toxicity of the [PSI] prion. Mol. Cell. Biol., 19, 8103–8112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conde J. and Fink,G.R. (1976) A mutant of Saccharomyces cerevisiae defective for nuclear fusion. Proc. Natl Acad. Sci. USA, 73, 3651–3655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig E., Yan,W. and James,P. (1999) Genetic dissection of the Hsp70 chaperone system of yeast. In Bukau,B. (ed.), Molecular Chaperones and Folding Catalysts. Harwood Academic, Amsterdam, The Netherlands, pp. 139–162.
- Derkatch I.L., Chernoff,Y.O., Kushnirov,V.V., Inge-Vechtomov,S.G. and Liebman,S.W. (1996) Genesis and variability of [PSI] prion factors in Saccharomyces cerevisiae. Genetics, 144, 1375–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glover J.R. and Lindquist,S. (1998) Hsp104, Hsp70 and Hsp40: a novel chaperone system that rescues previously aggregated proteins. Cell, 94, 73–82. [DOI] [PubMed] [Google Scholar]
- Johnson J.L. and Craig,E.A. (2001) An essential role for the substrate-binding region of Hsp40s in Saccharomyces cerevisiae. J. Cell Biol., 152, 851–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung G., Jones,G., Wegrzyn,R. and Masison,D. (2000) A role for cytosolic Hsp70 in yeast [PSI+] prion propagation and [PSI+] as a cellular stress. Genetics, 156, 559–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kushnirov V.V., Kryndushkin,D.S., Boguta,M., Smirnov,V.N. and Ter-Avanesyan,M.D. (2000) Chaperones that cure yeast artificial [Psi+] and their prion-specific effects. Curr. Biol., 10, 1443–1446. [DOI] [PubMed] [Google Scholar]
- Lu Z. and Cyr,D.M. (1998) Protein folding activity of Hsp70 is modified differentially by the Hsp40 co-chaperones Sis1 and Ydj1. J. Biol. Chem., 273, 27824–27830. [DOI] [PubMed] [Google Scholar]
- Luke M.M., Sutton,A. and Arndt,K.T. (1991) Characterization of SIS1, a Saccharomyces cerevisiae homologue of bacterial DnaJ proteins. J. Cell Biol., 114, 623–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriyama H., Edskes,H.K. and Wickner,R.B. (2000) [URE3] prion propagation in Saccharomyces cerevisiae: requirement for chaperone Hsp104 and curing by overexpressed chaperone Ydj1. Mol. Cell. Biol., 20, 8916–8922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newnam G.P., Wegrzyn,R.D., Lindquist,S.L. and Chernoff,Y.O. (1999) Antagonistic interactions between yeast chaperones Hsp104 and Hsp70 in prion curing. Mol. Cell. Biol., 19, 1325–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsell D.A., Kowal,A.S., Singer,M.A. and Lindquist,S. (1994) Protein disaggregation mediated by heat-shock protein Hsp104. Nature, 372, 475–478. [DOI] [PubMed] [Google Scholar]
- Sanchez Y. and Lindquist,S.L. (1990) Hsp104 required for induced thermotolerance. Science, 248, 1112–1115. [DOI] [PubMed] [Google Scholar]
- Sanchez Y., Parsell,D.A., Taulien,J., Vogel,J.L., Craig,E.A. and Lindquist,S. (1993) Genetic evidence for a functional relationship between Hsp104 and Hsp70. J. Bacteriol., 175, 6484–6491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A., Helms,C. and Sherman,F. (1979) Mutation of the non-Mendelian suppressor [PSI+] in yeast by hypertonic media. Proc. Natl Acad. Sci. USA, 76, 1952–1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sondheimer N. (2000) The identification of novel prion elements in the yeast Saccharomyces cerevisiae. Doctoral dissertation, University of Chicago, Chicago, IL, pp. 1–161.
- Sondheimer N. and Lindquist,S. (2000) Rnq1: an epigenetic modifier of protein function in yeast. Mol. Cell, 5, 163–172. [DOI] [PubMed] [Google Scholar]
- True H. and Lindquist,S. (2000) A yeast prion provides a mechanism for genetic variation and phenotypic diversity. Nature, 407, 477–483. [DOI] [PubMed] [Google Scholar]
- Wach A. (1996) PCR-synthesis of marker cassettes with long flanking homology regions for gene disruptions in S. cerevisiae. Yeast, 12, 259–265. [DOI] [PubMed] [Google Scholar]
- Wickner R.B. (1994) [URE3] as an altered URE2 protein: evidence for a prion analog in Saccharomyces cerevisiae. Science, 264, 566–569. [DOI] [PubMed] [Google Scholar]
- Wickner R. and Chernoff,Y. (1999) Prions of fungi: [URE3], [PSI] and [Het-s] discovered as heritable traits. In Prusiner,S. (ed.), Prion Biology and Diseases. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 229–272.
- Yan W. and Craig,E.A. (1999) The glycine–phenylalanine-rich region determines the specificity of the yeast Hsp40 Sis1. Mol. Cell. Biol., 19, 7751–7758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong T. and Arndt,K.T. (1993) The yeast Sis1 protein, a Dnaj homologue, is required for the initiation of translation. Cell, 73, 1175–1186. [DOI] [PubMed] [Google Scholar]
- Zhou P., Derkatch,I.L., Uptain,S.M., Patino,M.M., Lindquist,S. and Liebman,S.W. (1999) The yeast non-Mendelian factor [ETA+] is a variant of [PSI+], a prion-like form of release factor eRF3. EMBO J., 18, 1182–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]