

Abstract
Mitogen-activated protein kinase (MAPK) pathways couple intrinsic and extrinsic signals to hypertrophic growth of cardiomyocytes. The MAPK kinase MEK5 activates the MAPK ERK5. To investigate the potential involvement of MEK5–ERK5 in cardiac hypertrophy, we expressed constitutively active and dominant-negative forms of MEK5 in cardiomyocytes in vitro. MEK5 induced a form of hypertrophy in which cardiomyocytes acquired an elongated morphology and sarcomeres were assembled in a serial manner. The cytokine leukemia inhibitory factor (LIF), which stimulates MEK5 activity, evoked a similar response. Moreover, a dominant-negative MEK5 mutant specifically blocked LIF-induced elongation of cardiomyocytes and reduced expression of fetal cardiac genes without blocking other aspects of LIF-induced hypertrophy. Consistent with the ability of MEK5 to induce serial assembly of sarcomeres in vitro, cardiac-specific expression of activated MEK5 in transgenic mice resulted in eccentric cardiac hypertrophy that progressed to dilated cardiomyopathy and sudden death. These findings reveal a specific role for MEK5–ERK5 in the induction of eccentric cardiac hypertrophy and in transduction of cytokine signals that regulate serial sarcomere assembly.
Keywords: dilated cardiomyopathy/heart/leukemia inhibitory factor/mitogen-activated protein kinase
Introduction
Cardiac cells do not divide after birth, so both normal growth of the myocardium and stress-induced myocardial remodeling must take place through hypertrophic growth without cell division (MacLellan and Schneider, 2000). Cardiac hypertrophy can occur by an increase in width of myofibrils, resulting in a thickening of the myocardial wall or ‘concentric hypertrophy’, or by an increase in myofibril length, producing chamber dilation or ‘eccentric hypertrophy’. These contrasting forms of hypertrophy are coupled to parallel versus serial assembly of sarcomeres, respectively. In the case of normal physiological growth or exercise-induced hypertrophy, concentric and eccentric hypertrophy occur simultaneously and in a balanced manner, enabling the heart to increase pumping capacity in response to increased demand. Disease states that put stress on the heart can also induce hypertrophy; however, depending on the stimulus, either concentric or eccentric hypertrophy may predominate. Although hypertrophy may compensate initially for the additional demands placed on the heart by disease, continued stress almost inevitably results in decompensation and the development of hypertrophic or dilated cardiomyopathy. In order for any form of hypertrophic remodeling to occur, stress stimuli must activate signaling pathways that regulate protein synthesis, sarcomeric assembly and organization, and gene expression (Sugden and Clerk, 1998; Chien, 1999; Nicol et al., 2000).
Mitogen-activated protein kinase (MAPK) pathways provide an important connection between external stimuli that activate a wide variety of cell signaling systems and the nucleus. At the core of each MAPK cascade is a three-kinase module in which the most downstream member, the MAPK, is activated by a MAPK kinase (MAPKK or MEK), which is in turn activated by a MAPKK kinase (MAPKKK or MEKK) (English et al., 1999a). MAPKs can be divided into three major subfamilies based on sequence homology: the extracellularly responsive kinases (ERKs), the c-Jun N-terminal kinases (JNKs), also known as stress-activated protein kinases (SAPKs), and the p38-MAPKs. In the heart, all three classes of MAP kinases are activated by G-protein-coupled receptor (GPCR) agonists, stretch and certain types of stress, including ischemia (Sugden and Clerk, 1998; Abe et al., 2000; Ruwhof and van der Laarse, 2000). A critical role for MAPK pathways in the development of hypertrophy in vivo has been demonstrated by the finding that transgenic expression of a MAP kinase phosphatase in the mouse heart can attenuate hypertrophy induced by aortic banding and catecholamine infusion (Bueno et al., 2001). The role of individual MAPK pathways in various aspects of the hypertrophic response is more controversial (Sugden and Clerk, 1998).
ERK5, also known as big MAPK 1 (BMK1), has an N-terminal domain that is homologous to ERKs 1 and 2, but has unique C-terminal and loop-12 domains (Lee et al., 1995; Zhou et al., 1995). MEK5, the activating MAPKK for ERK5, is a highly specific ERK5 kinase and does not activate other MAPKs even when overexpressed in cultured cells (English et al., 1995; Zhou et al., 1995). MEK5–ERK5 signaling has been shown to be activated by growth stimuli including serum and ligands for tyrosine kinase and GPCRs (Kato et al., 1997; Kamakura et al., 1999; Fukuhara et al., 2000), as well as by oxidative and osmotic stress (Abe et al., 1996). Signaling by this MAPK module has not been studied in detail in cardiac cells; however, one report suggests that ERK5 may be regulated differently from ERK1/2 in these cells (Takeishi et al., 1999). Interestingly, the MEK1 inhibitors PD098059 and U0126 also inhibit activation of ERK5 (Kamakura et al., 1999), suggesting that functions previously attributed to ERK1/2 may also be mediated by ERK5.
Here we show that ERK5 is activated by hypertrophic stimuli and that constitutive activation of the MEK5 signaling pathway results in serial assembly of sarcomeres in cardiomyocytes in vitro, a process also induced by the interleukin-6 (IL-6) family cytokines, leukemia inhibitory factor (LIF) and cardiotrophin-1 (CT-1). Furthermore, adenoviral expression of dominant-negative MEK5 specifically blocks LIF-induced elongation of cardiomyocytes and reduces expression of a subset of fetal genes without blocking other aspects of LIF-induced hypertrophy such as parallel assembly of sarcomeres and increased cell size. We also show that activated MEK5 is sufficient to induce rapidly decompensating eccentric cardiac hypertrophy in transgenic mice, indicating a key role for MEK5 in the regulation of serial assembly of sarcomeres in vivo.
Results
Activation of ERK5 by hypertrophic agents and stress
Previous studies showed that ERK5 is strongly activated by ischemia in vivo, as well as oxidative and osmotic stress in cultured cells (Abe et al., 1996; Takeishi et al., 1999). This pathway has also been shown to be activated by signaling through the Gαq and G12/13 families of heterotrimeric G-proteins in fibroblasts (Fukuhara et al., 2000). To determine whether ERK5 might be a target of hypertrophic signaling pathways in cardiomyocytes, we treated primary neonatal rat cardiomyoctyes with the hypertrophic agonists phenylephrine (PE) and LIF, and the stress agents H2O2 and sorbitol (Figure 1). Following treatment, cardiomyocytes were harvested and ERK5 immunoprecipitation (IP) kinase assays were performed with a glutathione S-transferase (GST)–MEF2C substrate. Phosphorylation of GST–MEF2C indicated that ERK5 was activated 2- to 4-fold by hypertrophic agents, with activation peaking at ∼10 min and declining to basal levels or below by 60 min (Figure 1A and B). As shown previously (Abe et al., 1996), sorbitol gave a strong sustained activation of ERK5 (8-fold at 60 min), while H2O2 gave a lower but also sustained activation (5-fold at 60 min) (Figure 1C and D). Western blotting of immunoprecipitates with anti-ERK5 antibody showed that equivalent amounts of ERK5 protein were present in the kinase reactions (Figure 1). The endogenous ERK5 protein migrates at ∼100 kDa on SDS–PAGE, and phosphorylation of the protein has been shown to produce an upward shift in mobility. We did not observe this mobility shift in all of our experiments, probably because only a small percentage of the total protein was activated (Figure 1).

Fig. 1. Activation of endogenous ERK5 by hypertrophic and stress agents. Serum-deprived neonatal rat cardiomyoctyes were treated with (A) 100 µM PE, (B) 1000 U/ml LIF, (C) 200 µM H2O2 and (D) 0.3 M sorbitol for the indicated times, harvested and ERK5 kinase activity was measured. Top: ERK5 was immunoprecipitated from 200 µg of cellular lysate with an antibody specific for the C-terminal 20 amino acids. Kinase assays were performed with immunoprecipitated ERK5 using GST–MEF2C substrate in the presence of [γ-32P]ATP. GST–MEF2C phosphorylation was detected by autoradiography after SDS–PAGE. Middle: immunoblotting was performed on immuno precipitated material using rabbit anti-ERK5 antibody. Bottom: levels of 32P-phosphorylated GST–MEF2C were quantitated with a PhosphorImager. The averaged result ± SD of three independent experiments is shown.
Activated MEK5 induces serial assembly of sarcomeres in vitro
We next investigated the outcome of ERK5 activation in cardiomyoctyes using adenoviruses expressing three different forms of MEK5: wild-type MEK5 (AdMEK5WT), activated MEK5 with aspartate substitutions of Ser222 and Thr226 (AdMEK5DD) and dominant-negative MEK5 (AdMEK5KM) with a methionine substitution of ATP-binding Lys106 (English et al., 1999b). All three MEK5 derivatives were tagged with a hemagglutinin (HA) epitope.
Initially, the viruses were used to infect COS cells. Western blotting and IP kinase assays with anti-HA antibody and ERK5 kinase-dead substrate confirmed that the viruses expressed proteins of the correct size and that AdMEK5DD produced a constitutively active kinase (Figure 2A and B). Some kinase activity was also observed with the AdMEK5WT virus (Figure 2B). The viruses were then used to infect neonatal rat cardiomyocytes. Following infection, cells were serum starved for 72 h, fixed and stained with anti-sarcomeric α-actinin antibody (Figure 2C–H). Adenoviruses expressing β-galactosidase (Adβ-gal) and an activated form of MEK1 (AdMEK1CA) were used for comparison. Control cells infected with Adβ-gal lacked well developed sarcomeres (Figure 2C). Cells infected with AdMEK1CA or treated with PE had well assembled sarcomeric structures, an angular appearance and increased cell area (Figure 2D and F; Table I). Surprisingly, cardiomyocytes infected with AdMEK5DD were highly elongated (Figure 2E) when compared with either AdMEK1CA-infected cardiomyocytes or cardiomyocytes treated with PE for 48 h (Figure 2D and F). Although the average length of AdMEK5DD-infected cardiomyocytes was similar to the average length of AdMEK1CA-infected cardiomyocytes, this increase in length occurred without a corresponding increase in cell width, thus producing a 2-fold increase in average length to width ratio (Table I). In spite of their unusual elongated appearance, MEK5DD-expressing cardiomyocytes exhibited assembled sarcomeric structures comparable to those seen in PE-treated cells (Figure 2G and H).

Fig. 2. Activated MEK5 induces elongation of cultured neonatal rat cardiomyocytes. Adenoviruses expressing HA-tagged MEK5KM, MEK5WT and MEK5DD were used to infect COS cells at an m.o.i. of 100. (A) Lysates were prepared 48 h post-infection and 5 µg of protein were separated by SDS–PAGE and immunoblotted with anti-HA antibody. (B) Immunoprecipitations were performed on 100 µg of protein with anti-HA antibody. Kinase assays were performed with immunoprecipitated HA-MEK5 using GST–ERK5KMΔ substrate in the presence of [γ-32P]ATP. GST–ERK5KMΔ phosphorylation was detected after SDS–PAGE by autoradiography. Serum-deprived cardiomyocytes were infected at an m.o.i. of 100 with adenovirus expressing (C) β-galactosidase, (D) MEK1CA and (E and G) MEK5DD, or not infected and treated with (F and H) PE (100 µM). Cells were fixed 72 h post-infection and immunostained with anti-sarcomeric α-actinin antibody. Note that cells in (G) and (H) are shown at higher magnification than cells in (C–F). Bar, 20 µm.
Table I. Morphometric analysis of cardiomyocytes.
| Cell treatment | Area (µm2) | Major axis (µm) | Minor axis (µm) | Major/minor |
|---|---|---|---|---|
| Adβ-gal | 2167 ± 688 | 65 ± 11 | 42 ± 9.1 | 1.58 ± 0.39 |
| AdMEK1CA | 7146 ± 1560 | 134 ± 22* | 68 ± 12 | 2.38 ± 1.03** |
| AdMEK5DD | 3440 ± 1050 | 136 ± 37** | 34 ± 13** | 4.65 ± 2.28** |
| PE | 5745 ± 1560 | 110 ± 20 | 66 ± 12 | 1.71 ± 0.37 |
| PE + Adβ-gal | 6411 ± 2070 | 122 ± 23 | 66 ± 13 | 1.91 ± 0.44 |
| PE + AdMEK5KM | 4524 ± 1140* | 98 ± 15 | 59 ± 10 | 1.71 ± 0.35 |
| LIF | 3586 ± 1090 | 144 ± 36** | 33 ± 12** | 5.06 ± 2.35** |
| LIF + Adβ-gal | 3671 ± 874 | 148 ± 42** | 35 ± 14** | 5.37 ± 3.26** |
| LIF + AdMEK5KM | 4683 ± 889‡ | 101 ± 16‡ | 59 ± 10‡ | 1.78 ± 0.47‡ |
Neonatal rat cardiomyocytes were infected with virus and treated with PE (100 µM) or LIF (1000 U/ml) where indicated and as described in legends for Figures 2 and 3. Measurements were made using a computerized morphometric system. Thirty cells were examined for each adenovirus, and this was repeated for three independent experiments.
Values are means ± SD; *P <0.05 versus PE-treated (no virus); **P <0.001 versus PE-treated (no virus); ‡P <0.001 versus LIF-treated (no virus).
Dominant-negative MEK5 blocks LIF-induced hypertrophy
The elongated phenotype of MEK5DD-expressing cardiomyocytes was reminiscent of the phenotype induced by the cytokines CT-1 and LIF (Wollert et al., 1996). To determine whether MEK5 might participate in a LIF-activated signaling pathway that mediates serial assembly of sarcomeres, we infected cardiomyoctyes with β-gal- and MEK5KM-expressing adenoviruses, and analyzed the effects of LIF on these cells and on uninfected cells (Figure 3A–C; Table I). LIF induced dramatic elongation of uninfected and Adβ-gal-infected cardiomyocytes (Figure 3A and C; Table I). In contrast, cells infected with MEK5KM adenovirus failed to undergo LIF-induced elongation (Figure 3B; Table I). However, these cardiomyocytes still exhibited assembled sarcomeres and increased surface area (Figure 3B; Table I). Parallel sarcomeric assembly in response to PE occurred in uninfected as well as in Adβ-gal- and AdMEK5KM-infected cells (Figure 3D–F), although there was some reduction in total cell surface area in MEK5KM-infected cardiomyocytes (Table I). Therefore, it appears that MEK5 may be an essential component of a LIF-activated signaling pathway leading to cardiomyocyte elongation and serial assembly of sarcomeres. This result further demonstrates that parallel assembly of sarcomeres induced by LIF and PE does not require MEK5.

Fig. 3. Dominant-negative MEK5 blocks LIF-induced elongation of neonatal rat cardiomyocytes. Cardiomyocytes were either not infected or infected with adenovirus at an m.o.i. of 100, serum deprived, and at 24 h post-infection treated either with LIF (1000 U/ml) or PE (100 µM) for an additional 48 h prior to fixation and immunostaining with anti-sarcomeric α-actinin. (A) Uninfected cells treated with LIF. (B) AdMEK5KM-infected cells treated with LIF. (C) Adβ-gal-infected cells treated with LIF. (D) Uninfected cells treated with PE. (E) AdMEK5KM-infected cells treated with PE. (F) Adβ-gal-infected cells treated with PE. Bar, 20 µm.
MEK5 synergizes with hypertrophic signaling pathways to induce fetal gene expression
To determine whether elevated MEK5 could also transduce signals that regulate fetal gene expression, we treated cardiomyocytes infected with AdMEK5WT, AdMEK5KM or Adβ-gal with LIF or PE and analyzed expression of atrial natriuretic factor (ANF), brain natriuretic peptide (BNP) and skeletal α-actin (Figure 4A). Skeletal α-actin expression was induced 4- to 5-fold in Adβ-gal-infected cells treated with LIF or PE, and this induction was slightly higher in cells infected with AdMEK5WT (Figure 4A). Likewise, ANF and BNP expression were induced 3- to 4-fold by both agonists and this induction was even higher in cells infected with AdMEK5WT (Figure 4A). Agonist induction of all three of these fetal genes was partially or completely blocked by Ad-MEK5KM (Figure 4A). In the absence of PE or LIF treatment, Ad-MEK5DD had only a slight effect on ANF or BNP expression; however, the constitutively active kinase strongly induced skeletal α-actin expression (Figure 4B). The control Adβ-gal did not significantly affect fetal gene expression in the presence or absence of agonist.

Fig. 4. MEK5 signaling contributes to the regulation of cardiomyocyte fetal gene expression by PE and LIF. (A) Cardiomyocytes were either not infected or infected with MEK5WT, MEK5KM or β-gal adenoviruses at an m.o.i. of 20 and serum deprived. At 36 h post-infection, cells were either not treated or treated with 50 µM PE (black bar) or 1000 U/ml LIF (white bar) for an additional 24 h. RNA was prepared and used for dot-blots with oligonucleotide probes specific for skeletal α-actin, ANF or BNP. Signal intensity was quantitated using a PhosphorImager. The average fold induction ± SD of three independent experiments is shown. Fold induction is relative to uninfected cells without PE or LIF treatment. (B) Cardiomyocytes were either not infected (–) or infected with MEK5DD or β-gal adenoviruses at an m.o.i. of 20 and serum deprived. At 48 h post-infection, the cells were harvested and RNA was prepared. Transcript levels for α-skeletal actin, ANF or BNP were determined as described in (A). Fold induction is relative to uninfected cells.
Activated MEK5 induces dilated cardiomyopathy in mice
To determine whether activated MEK5 is also capable of inducing cardiomyocyte hypertrophy in vivo, we created transgenic mice that overexpressed MEK5DD in the heart under the control of the α-MHC promoter. The level of transgene expression in five different lines of transgenic mice was determined by anti-HA western blotting (Figure 5A). Although all of the transgenic lines showed a similar phenotype, we will only describe data from line 367, which was one of the more severely affected lines.

Fig. 5. Expression of MEK5 and ERK5 in wild-type and MEK5DD transgenic mice. Lysates were prepared from wild-type (WT) and transgenic (TG) hearts, and 20 µg of protein were separated by SDS–PAGE. (A) Expression of HA-tagged MEK5DD was analyzed in different lines of transgenic mice by immunoblotting with anti-HA antibody. Lines of MEK5DD transgenic mice are indicated by identifying numbers. For each line, lysate was prepared from two hearts and loaded in adjacent lanes. Expression of (B) MEK5 and (C) ERK5 was analyzed in wild-type and line 367 MEK5DD transgenic mice by immunoblotting with L610 rabbit anti-MEK5 antiserum and rabbit anti-ERK5. Bands that are either non-specific (asterisk) or degradation products (arrowhead) are indicated. Note the reduced mobility of ERK5 in transgenic animals relative to wild type.
We compared levels of HA-MEK5DD with endogenous MEK5 in wild-type and transgenic mice by anti-MEK5 western blotting (Figure 5B). There are two isoforms of the MEK5 protein, MEK5α and MEK5β, produced by alternative splicing. These proteins are identical except that MEK5α has an 89 amino acid extension at its N-terminus. Because we used MEK5β and mutants thereof for our overexpression studies in cultured cardiomyocytes and transgenic mice, MEK5DD co-migrates with endogenous MEK5β. We found an 8-fold increase in levels of total MEK5β (HA-MEK5DD + endogenous MEK5β) in transgenic animals relative to wild type, and this did not change with age. Endogenous MEK5α expression was unchanged in MEK5DD transgenic animals (data not shown). Levels of endogenous ERK5 expression were not altered in transgenic hearts relative to wild type; however, a more slowly migrating band was observed in extracts from transgenic hearts relative to wild type, particularly in 1-week-old animals. Presumably the more slowly migrating band represents phospho-ERK5 (Figure 5C). This suggests that expression of activated MEK5 in transgenic hearts induces phosphorylation of endogenous ERK5.
Transgenic mice appeared normal at birth and thrived but, by 4–5 weeks of age, they began to die, apparently from heart failure (Figure 6). By ∼8 weeks of age, approximately half of the transgenic mice had died. Past 8 weeks of age, transgenic mice continued to die prematurely, although some lived as long as 18 weeks.

Fig. 6. Survival curve for wild-type and MEK5DD transgenic mice. F1 hemizygous transgenic mice were generated by backcrossing the transgenic founder mouse with C57B6 mice. The open circles represent the percentage survival of wild-type (WT) F1 mice (n = 24); the closed circles represent percentage survival of transgenic (TG) F1 mice (n = 24).
At 3 weeks of age, transgenic hearts appeared normal, but by 6 weeks of age many of the transgenic hearts were enlarged (Figure 7A). Mice that survived until 12 weeks of age showed even more pronounced ventricular dilation. In addition, atrial enlargement and formation of large atrial thrombi frequently were observed. Sectioning of lungs and liver in 8- and 12-week-old mice revealed the presence of congestion (data not shown), suggesting diminished cardiac performance characteristic of congestive heart failure.

Fig. 7. MEK5DD transgenic hearts show progressive dilation and thinning of ventricular walls with age. (A) Hearts were removed from wild-type and MEK5DD transgenic mice at 3, 6 and 12 weeks of age. Hearts were fixed in 10% PBS-buffered formalin and photographed. (B) Hearts from 12-week-old MEK5DD-transgenic and wild-type mice were fixed and sectioned longitudinally or at the midsagittal level parallel to the base and stained with hematoxylin–eosin. ra, right atrium; la, left atrium; rv, right ventricle; lv, left ventricle.
Sectioning of transgenic hearts revealed that the walls of both the right and left ventricular chambers were extremely thin relative to wild type (Figure 7B). Closer examination of sections revealed a decrease in cross-sectional area of myocytes in the hearts of 8-week-old MEK5DD transgenic mice (Figure 8A, B and D). The decreased myocyte cross-sectional area observed in MEK5DD transgenic hearts contrasts sharply with the dramatic increase in myocyte cross-sectional area in the hearts of mice that overexpress an activated form of the calcium-regulated phosphatase calcineurin in the heart (Figure 8A and C) (Molkentin et al., 1998).
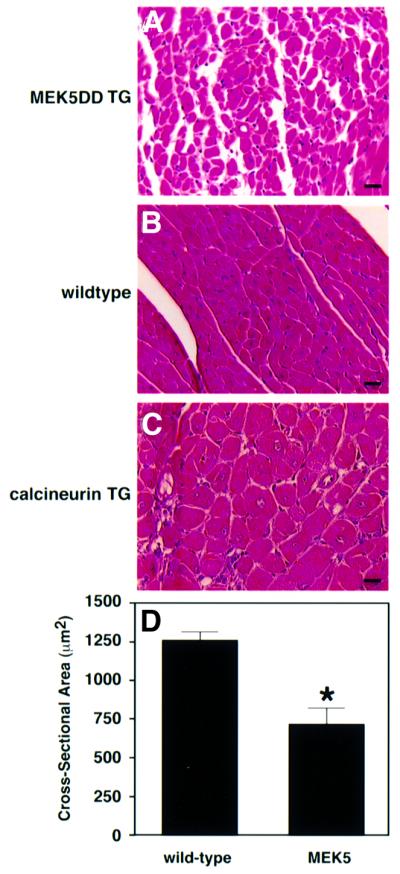
Fig. 8. MEK5DD transgenic hearts show reduced myofiber cross-sectional area relative to wild type. Hearts were removed from 8-week-old wild-type and from MEK5DD and calcineurin transgenic mice, fixed, sectioned and stained with hematoxylin–eosin. Dramatic differences in myocyte cross-sectional area are apparent in hematoxylin–eosin-stained sections from (A) MEK5DD transgenic hearts, (B) wild-type hearts and (C) calcineurin transgenic hearts. Bar, 20 µm. (D) The cross-sectional area of myocytes from 8-week-old wild-type and MEK5DD transgenic mice was quantitated using a computerized morphometric system. Measurements were made on equivalent sections from five wild-type and five transgenic hearts, and, within each section, measurements were taken from left and right ventricle, septum and papillary muscle (10 measurements each). The average result ± SD is shown; *P <0.001.
Average cross-sectional areas of cardiac myocytes from MEK5DD transgenic and wild-type mice were determined at 1, 2 and 3 weeks of age. At 1 and 2 weeks of age, myocyte cross-sectional area was not significantly different between transgenic and control; however, by 3 weeks of age, average myocyte cross-sectional area was decreased by 12% in MEK5DD transgenic hearts relative to wild type (data not shown). The cross-sectional area of cardiac myocytes in wild-type hearts increased as mice aged, whereas that of cardiac myocytes from transgenic mice increased very little with age, with the result that the cross-sectional area of cardiac myocytes in 8-week-old transgenic hearts was 44% less than control (Figure 8D). Ventricle weight/tibia length ratios of MEK5DD transgenic mice were not significantly different from those of wild-type animals (data not shown). The fact that MEK5DD transgenic and wild-type ventricles were similar in mass, despite significantly decreased myocyte cross-sectional area, suggests that myocytes in MEK5DD transgenic hearts undergo growth by eccentric hypertrophy. Apart from the abnormal hypertrophy of cardiomyocytes, MEK5DD transgenic hearts seemed remarkably healthy. Trichrome staining did not reveal any evidence of fibrosis, and TUNEL assays did not indicate that levels of apoptosis were elevated even in severely dilated MEK5DD hearts compared with wild-type hearts.
Functional analysis of transgenic mice
We hypothesized that the eccentric hypertrophy of MEK5DD transgenic hearts resulted in decreased cardiac performance and, eventually, congestive heart failure. Indeed, transthoracic echocardiography showed significant left ventricular dilation, as assessed by the end diastolic diameter (LVED), at 6 and 10 weeks of age compared with wild-type controls (Table II). In addition, septal and posterior wall thickness was reduced compared with wild type (Table II). These measurements indicate a phenotype of primary dilated cardiomyopathy without ventricular hypertrophy. Furthermore, at 6 weeks of age, MEK5DD transgenic hearts displayed a highly significant reduction of fractional shortening (Table II). This reduction was even more pronounced at 10 weeks of age, indicating a progressive worsening of ventricular function. These data suggest that cardiac failure due to dilated cardiomyopathy may be the cause of death in MEK5DD transgenic animals. Alternatively, severe arrhythmias could have resulted in the sudden death of transgenic animals.
Table II. Echocardiography of MEK5DD transgenic mice demonstrates eccentric hypertrophy and decreased fractional shortening.
| Non-transgenic | MEK5DD-TG | % change | |
|---|---|---|---|
| 6 weeks | |||
| IVSD | 0.67 ± 0.01 | 0.66 ± 0.004 | –1 |
| IVSS | 1.31 ± 0.01 | 1.04 ± 0.01 | –21** |
| LVPWD | 0.65 ± 0.01 | 0.60 ± 0.01 | –5 |
| LVPWS | 1.08 ± 0.02 | 0.90 ± 0.01 | –17 |
| LVED | 3.57 ± 0.03 | 4.47 ± 0.06 | +28* |
| LVES | 1.92 ± 0.02 | 3.39 ± 0.07 | +76** |
| FS | 0.46 ± 0.04 | 0.25 ± 0.06 | –47** |
| 10 weeks | |||
| IVSD | 0.61 ± 0.004 | 0.57 ± 0.003 | –4 |
| IVSS | 1.15 ± 0.01 | 0.91 ± 0.01 | –21* |
| LVPWD | 0.59 ± 0.01 | 0.54 ± 0.01 | –5 |
| LVPWS | 1.08 ± 0.01 | 0.77 ± 0.01 | –29** |
| LVED | 3.85 ± 0.04 | 5.58 ± 0.08 | +45** |
| LVES | 2.31 ± 0.05 | 4.65 ± 0.09 | +101** |
| FS | 0.41 ± 0.06 | 0.17 ± 0.06 | –58** |
Measurements are given in millimeters, except fractional shortening (FS), which is unitless. Values were determined from three separate M-mode measurements and averaged between six mice in the non-transgenic group and 10 mice in the transgenic group.
Values are mean ± SEM; *P <0.05; **P <0.001.
IVSD, interventricular septum diastole; IVSS, interventricular septum systole; LVPWD, left ventricular posterior wall diastole; LVPWS, left ventricular posterior wall systole; LVED, left ventricular end diastolic dimension; LVES, left ventricular end systolic dimension.
Induction of fetal gene expression in MEK5DD transgenic hearts
To determine whether MEK5DD transgenic hearts showed stress-associated patterns of gene expression, we performed dot-blot analysis on RNA extracted from hearts of 8-week-old wild-type and transgenic animals (Figure 9A). α-MHC, sarcoplasmic reticulum calcium ATPase-2a (SERCA2a) and phospholamban (PLB), which are typically down-regulated in failing hearts, were all down-regulated in hearts from MEK5DD transgenics (Figure 9B). Conversely, β-MHC, skeletal α-actin and ANF, which are typically induced during heart failure, were up-regulated by 12-, 5- and 30-fold, respectively, in MEK5DD transgenic hearts (Figure 9B).

Fig. 9. Induction of fetal gene expression in MEK5DD transgenic hearts. RNA was prepared from wild-type and transgenic hearts. (A) RNA dot-blots were prepared with 1 µg of RNA/dot and probed with oligonucleotide probes specific for the indicated gene. (B) The average fold induction or repression of gene expression ± SD for MEK5DD transgenic animals relative to wild type is shown. Signal intensity was quantitated using a PhosphorImager.
Discussion
In this study, we investigated the role of the MEK5–ERK5 signaling module in hypertrophic signaling in cardiac myocytes in vitro and in vivo. Our results demonstrate that ERK5 is activated by PE and LIF, as well as by the stress stimuli H2O2 and sorbitol. Adenoviral-mediated expression of constitutively activated MEK5 induced cardiomyocytes to assume a highly elongated morphology, reminiscent of the phenotype induced by LIF and the related cytokine CT-1. Consistent with the potential involvement of MEK5 in the LIF signaling pathway, a dominant-negative MEK5 mutant blocked LIF-induced elongation of cardiomyocytes. In contrast, dominant-negative MEK5 had no effect on LIF-induced assembly of sarcomeres and actually increased cell area relative to LIF treatment alone. The outcome of MEK5 activation in vitro is highly unique: no other signaling molecule has been shown to be sufficient to induce the elongated phenotype typical of CT-1/LIF-activated signaling in cardiomyocytes. These effects of MEK5 may reflect a similar function in vivo because transgenic mice that overexpress activated MEK5 under control of the α-MHC promoter develop severe dilated cardiomyopathy characterized by thinning of the ventricular walls and decreased cross-sectional area of individual myocytes.
LIF and CT-1 signaling in the heart
LIF and CT-1 belong to the IL-6 family of cytokines and bind to a heterodimer of gp130 and the LIF receptor (Wollert and Chien, 1997). A plethora of hormones and peptide growth factors can stimulate a hypertrophic phenotype in cultured cardiomyocytes; however, LIF and CT-1 are unique in their ability to induce primarily serial assembly of sarcomeres and an elongated morphology (Wollert et al., 1996). The mechanism by which LIF and CT-1 induce serial assembly of sarcomeres is not known. Several signaling pathways have been implicated in the induction of the hypertrophic phenotype by LIF and CT-1 (Kunisada et al., 1996; Oh et al., 1998; Kato et al., 2000). However, with the exception of the original characterization of CT-1-induced hypertrophy (Wollert et al., 1996), all of these studies have defined morphological hypertrophy as an overall increase in cell area without describing contributions of length and width. One study implicated Janus kinase (JAK)/signal transducer and activator of transcription (STAT) but not ERK1/2 or phosphatidylinositide 3-kinase (PI3-K) in CT-1-induced organization of sarcomeres (Kodama et al., 2000), but the distinction between serial versus parallel assembly of sarcomeres was not addressed.
Regulation of myofibril assembly and cardiomyocyte morphology
Hypertrophy induced by GPCR agonists as well as normal physiological growth in vivo involves a balance between parallel and serial assembly of sarcomeres. In the case of LIF treatment or overexpression of activated MEK5, the balance is shifted so that serial assembly of sarcomeres predominates. However, in spite of the fact that AdMEK5DD-infected cells appear highly elongated, morphological measurement shows that AdMEK5DD- and AdMEK1CA-infected cells are comparable in length (Table I), and that the length to width ratio is dramatically altered in AdMEK5DD-infected cells. This implies that although serial assembly of sarcomeres occurs normally in these cells, parallel assembly is almost entirely absent. This distinction is illustrated further by results with AdMEK5KM. LIF-treated cells infected with the dominant-negative MEK5 adenovirus are similar to PE-treated or AdMEK1CA-infected cardiomyocytes in appearance, suggesting that a balance between parallel and serial assembly of sarcomeres has been restored. This result can be explained by a model in which LIF and MEK5DD induce myofibril formation and at the same time interfere specifically with assembly of sarcomeres in parallel (Figure 10). Relief of this interference by dominant-negative MEK5 would then allow parallel assembly of sarcomeres in response to LIF. Results with activated and dominant-negative MEK5 further imply that although activated MEK5 is sufficient to induce a specific pattern of sarcomere assembly, MEK5 signaling is not essential for sarcomere formation per se. The ability of LIF and MEK5 to regulate parallel and serial assembly of sarcomeres differentially implies that there is something inherently different about these two processes. Further investigation of the mechanism by which MEK5 directs sarcomere assembly should provide novel insight into regulation of myofibril formation.

Fig. 10. A model for LIF-induced cardiomyocyte elongation mediated by MEK5 inhibition of parallel assembly of sarcomeres. MEK5 induces cardiomyocyte elongation by interfering with parallel assembly of sarcomeres. Other signaling molecules implicated downstream of LIF include MEK1, JAK/STAT, PI3-K, CaMK and calcineurin.
Regulation of cardiomyocyte gene expression
Treatment of cardiomyocytes with LIF and CT-1 induces expression of fetal and immediate early genes (Wollert et al., 1996). Inhibitor studies have shown that activation of ERK1/2, Ca2+/calmodulin-dependent protein kinase (CaMK)II and IV, and calcineurin signaling all contribute to full induction of ANF, BNP and α-skeletal actin by IL-6 family cytokines (Wollert et al., 1996; Kato et al., 2000; Kodama et al., 2000). Additional signaling pathways may play more limited roles (Kodama et al., 2000). Our results demonstrate that dominant-negative MEK5 partially blocks and wild-type MEK5 increases induction of ANF, BNP and α-skeletal actin by PE and LIF. Activated MEK5 also strongly induced α-skeletal actin, but only weakly induced ANF and BNP. Therefore, it is likely that MEK5 synergizes with MEK1/2 or calcium-regulated signaling pathways to induce the fetal gene expression program fully.
Interaction of MEK5 with other signaling pathways
It seems paradoxical that ERK5 kinase activity is increased in response to stimuli that have dramatically different effects on cardiomyocyte phenotype. For example, LIF, PE and stress agents all activate ERK5, but in each case the outcome is different: LIF induces cellular elongation; PE induces overall hypertrophy; and H2O2 and sorbitol induce rapid apoptosis. Combinatorial activation of signaling pathways may be a key factor in determining the outcome of MAPK activation in cardiomyocytes. Expression of activated MKK6, a p38-specific MAPKK, in cardiomyocytes is sufficient to induce all the characteristic features of hypertrophy, whereas activated MKK3, another p38-specific MAPKK, can induce either hypertrophy or apoptosis, depending on which isoform of p38 is co-expressed (Wang et al., 1998a,b). The level and temporal pattern of activation may also be key factors in determining cardiomyocyte response. For example, low levels of Gαq signaling have been associated with hypertrophy, whereas higher levels can induce apoptosis (Adams et al., 1998). ERK1/2 signaling has been shown to protect cardiomyocytes from stress-induced apoptosis and ERK5 may have a similar function (Bueno et al., 2000). It is also important to note that increased catalytic activity of ERK5 may not be the only relevant outcome of MEK5 signaling. MEK5 may have other uncharacterized targets and ERK5 may have functions that are independent of kinase activity. It is of note that a recent study showed that ERK5 activation of the MEF2 transcription factor did not require the catalytic domain of the kinase (Kasler et al., 2000).
Role of apoptosis in dilated cardiomyopathy
In many cases of concentric hypertrophy, a transition to dilated cardiomyopathy occurs during the end stages of heart failure. Although there is evidence to suggest that apoptosis of cardiomyocytes may be responsible for this transition (MacLellan and Schneider, 1997), Gerdes and co-workers have found that myocyte lengthening alone can account for chamber dilation in the progression to heart failure of the spontaneously hypertensive rat (Tamura et al., 1998). We examined dilated MEK5DD transgenic hearts, but found no evidence for increased levels of apoptosis or necrosis.
Comparison of MEK5DD transgenic with other mouse models of cardiomyopathy
A large number of mouse models of heart failure have been described, many of them produced by overexpression of constitutively active signaling molecules (Ikeda et al., 2000). We have been particularly interested in the role that calcium-dependent signaling pathways play in the development of hypertrophic cardiomyopathy (Frey et al., 2000). We have shown previously that cardiac-specific expression of constitutively active forms of two calcium-dependent signaling molecules, calcineurin and CaMKIV, is sufficient to induce compensated concentric hypertrophy in mice (Molkentin et al., 1998; Passier et al., 2000). In both cases, the concentric hypertrophy eventually decompensates and the hearts undergo some degree of dilation. We have not yet determined whether MEK5– ERK5 signaling operates downstream of calcium-dependent signaling in these models, although ERK5 activation by H2O2 has been shown previously to be calcium dependent (Abe et al., 1996).
Although experiments in cultured cardiomyocytes have provided substantial evidence supporting a role for each of the three major MAPK pathways in hypertrophy (Sugden and Clerk, 1998), until recently the sufficiency of these molecules to induce a hypertrophic phenotype in vivo had not been examined. Surprisingly, despite similar effects in vitro, overexpression of these signaling molecules in vivo in the mouse heart produces very distinct phenotypes. While expression of constitutively active MEK1 in the mouse heart was sufficient to drive a compensated concentric hypertrophy, expression of transforming growth factor-β-activated kinase (TAK1), a MAPKKK for the p38 pathway, produced cardiac hypertrophy with interstitial fibrosis, apoptosis and severe myocardial dysfunction (Bueno et al., 2000; Zhang et al., 2000). MEK5 and ERK5 are most closely related to components of the ERK1/2 signaling cascade, but appear to have distinct functions in regulating sarcomere assembly in cultured cardiomyocytes. Activation of MEK5 in the mouse heart produces a decompensated eccentric hypertrophy: a phenotype in stark contrast to the compensated concentric hypertrophy observed in MEK1 transgenic mice.
There is a tendency generally to categorize different mouse models of cardiomyopathy as ‘dilated’ or ‘hypertrophic’ without more detailed consideration of the phenotypes. However, closer examination reveals that even among mouse models of dilated cardiomyopathy, disease phenotypes vary drastically. Hearts from desmin null mice have been shown to progress through a concentric hypertrophy phase prior to dilation, and dilated hearts from mice overexpressing a dominant-negative mutant of the transcription factor CREB exhibit a mixture of hypertrophied and atrophied cardiomyocytes (Fentzke et al., 1998; Milner et al., 1999). In contrast, transgenic expression of activated MEK5 produces a homogenous decrease in myocyte diameter without significant increases in apoptosis, necrosis or fibrosis. In vivo, it is unlikely that a signaling pathway will be activated in isolation, so that the effects of MEK5 signaling may depend on the simultaneous activation of other signaling pathways, including other MAPK signaling pathways, Ca2+-regulated signaling molecules and rho-family small GTP-binding proteins. However, the unique nature of the MEK5DD-induced phenotype in vitro and in vivo suggests that further examination of the mechanism of MEK5-induced eccentric hypertrophy may provide novel insight into the fundamental mechanisms underlying regulation of sarcomere assembly and the role that this process plays in development of dilated cardiomyopathy.
Materials and methods
Immunoprecipitation, kinase assays and immunoblotting
Immunoprecipitations (IPs) and kinase assays were performed as described (English et al., 1999b). Substrates used were GST fused to MEF2C amino acids 204–321 or GST–ERK5KMΔ, a truncated catalytically inactive form of ERK5 with Lys83 mutated to methionine. Following incubation for 30 min at 30°C, samples were separated by SDS–PAGE and transferred to a polyvinylidene difluoride (PVDF) membrane. The level of 32P-labeled GST fusion protein on the membrane was analyzed using a PhosphorImager (Molecular Dynamics). The membrane was then immunoblotted with anti-ERK5 rabbit polyclonal antibody (StressGen) at a final concentration of 150 ng/ml and proteins were visualized using a chemiluminescence system (Santa Cruz). Other antibodies were used at the following concentrations or dilutions for immunoblotting: anti-HA high affinity rat monoclonal antibody (Roche), 50 ng/ml; anti-MEK5 mouse monoclonal M72220 (Transduction Laboratories), 250 ng/ml; anti-MEK5 rabbit polyclonal L610 (English et al., 1995), 1:500.
Construction of adenoviruses and other DNA constructs
All constructs were generated from cDNAs encoding the MEK5β splice isoform (English et al., 1995). MEK5 mutants have been described previously (English et al., 1999b). MEK5 cDNAs were HA tagged by PCR and cloned into the vector pAC-CMV. Recombinant adenoviruses were generated by co-transfection of pAC-CMV constructs along with pJM17 into 293 cells using the calcium phosphate precipitation method (Becker et al., 1994). Primary lysates were amplified by reinfection of 293 cells. Titering of viruses was performed on 293 monolayers using the agar overlay method. Titers were in the range of 0.5–1 × 109 plaque-forming units (p.f.u.)/ml. The Ad-MEK1CA and β-galactosidase adenoviruses were a gift from L.J.Klesse and L.F.Parada (Klesse et al., 1999).
Cardiomyocyte culture
Cardiomyocyte cultures were prepared by dissociation of 1-day-old neonatal rat hearts and were plated differentially to remove fibroblasts. Cells were plated in 4:1 Dulbecco’s modified Eagle’s medium (DMEM):199 medium with 10% horse serum and 5% fetal calf serum at a density of 5 × 104 cells/cm2 for immunofluorescence experiments and at a density of 2 × 104 cells/cm2 for RNA and kinase experiments. Eighteen hours after plating, cells were changed into serum-free media and either incubated for an additional 48 h prior to treatment with hypertrophic agents or infected with adenovirus at a multiplicity of infection (m.o.i.) of 100. In cases where adenovirus-infected cells were treated with LIF or PE, these agents were added 24 h after the initial adenovirus infection. Treatment with hypertrophic agents was continued for 24 h in cases where RNA was harvested and for 48 h in cases where cells were fixed for immunostaining.
Immunofluorescence
For immunofluorescence, cells were grown on glass coverslips coated with 4 µg/cm2 laminin (Gibco-BRL). Cells were fixed in 3.7% formaldehyde on ice for 30 min, permeabilized with 0.1% Triton X-100 in phosphate-buffered saline (PBS) and blocked with 5% serum in PBS for 1 h at room temperature. Cells were incubated with monoclonal anti-α-actinin (sarcomeric) clone EA-53 (Sigma) at a dilution of 1:40 in blocking buffer for 1 h at 37°C, washed and incubated with fluorescein-conjugated horse anti-mouse IgG antibody (Vector) at a dilution of 1:200 in blocking buffer for 1 h at 37°C. Following secondary antibody incubation, cells were washed with PBS and rinsed briefly with 2 µg/ml Hoechst in H2O.
Transgenic mice and genotyping
An expression plasmid encoding a constitutively active form of MEK5β (MEK5DD) was subcloned into pBluescript between the α-myosin heavy chain promoter and the human growth hormone poly(A) tail (Gulick et al., 1991). The MEK5DD sequence was preceded by an HA epitope tag. Plasmid DNA was removed by NotI digestion, and the linearized MEK5DD construct was gel purified.
(C3HB6)F1 mice were superovulated by standard procedures and fertilized eggs were injected with linearized DNA (2 ng/µl). Injected eggs were transferred to the oviducts of pseudopregnant ICR mice. Offspring were analyzed for the presence of the transgene by Southern analysis of genomic DNA using a 32P-labeled human growth hormone cDNA fragment as a probe.
Transthoracic echocardiography
Cardiac function of wild-type and MEK5 transgenic mice was evaluated non-invasively by transthoracic echocardiography. Mice at the age of 6–10 weeks were anesthetized with 2.5% avertin (15 µl/g body weight). The ventral chest was shaved and the animal placed on a thermally controlled table in a slight left lateral decubitus position. Echocardio graphy was performed using a Hewlett Packard (Andover, MA) Sonos 5500 Ultrasound system with a 12 MHz transducer. Heart rate was determined by electrocardiogram analysis. At least three independent M-mode measurements per animal were obtained by an examiner blinded to the genotype of the animal. End systolic and end diastolic chamber diameter, interventricular septum and posterior wall thickness, as well as left ventricular fractional shortening {FS% = [(LVEDD – LVESD)/LVEDD] × 100}, were determined in a short axis view at the level of the papillary muscles.
Histology and morphometric analysis
Hearts from wild-type and transgenic mice were collected, fixed in 10% formalin buffered with PBS, dehydrated in ethanol, transferred to xylene, and then to paraffin. Paraffin-embedded hearts were sectioned at 4 µm and subsequently stained with hematoxylin and eosin or with Masson trichrome. Myocyte cross-sectional areas were measured from wild-type and MEK5DD transgenic heart sections (n = 10) using a computerized morphometric system (Scion Image, National Institutes of Health). Sections from different regions of the heart (left and right ventricle, septum and papillary muscle) were measured at a 40× magnification. Myocyte cross-sectional area was measured per nucleus and only myocytes that were cut in the same direction were included in the measurements. As criteria, the position and shape of the nucleus within the myocyte were used. The same software was used to quantitate area, perimeter, and major and minor axes of cardiomyocytes grown on coverslips.
RNA isolation and dot-blot analysis
Total RNA was purified with Trizol reagent (Gibco-BRL) as recommended. RNA from wild-type and transgenic hearts, as well as from cultured cardiomyocytes, was subjected to dot-blot hybridization against a panel of oligonucleotide probes as described (Jones et al., 1996).
Acknowledgments
Acknowledgements
We thank L.J.Klesse and L.F.Parada for reagents; B.Mercer for generating transgenic mice; W.Simpson and J.Page for editorial assistance; and A.Tizenor for graphics. This work was supported by grants from the National Institutes of Health and the Texas Advanced Technology Program to E.N.O. and by National Institutes of Health (DK34128) to M.C. R.L.N. was supported by an NIH postdoctoral fellowship, N.F. was supported by the Deutsche Forschungsgemeinschaft, and G.P. was supported by Pharmacological Sciences Training Grant G1907062-25.
References
- Abe J., Kusuhara,M., Ulevitch,R.J., Berk,B.C. and Lee,J.D. (1996) Big mitogen-activated protein kinase 1 (BMK1) is a redox-sensitive kinase. J. Biol. Chem., 271, 16586–16590. [DOI] [PubMed] [Google Scholar]
- Abe J., Baines,C.P. and Berk,B.C. (2000) Role of mitogen-activated protein kinases in ischemia and reperfusion injury: the good and the bad. Circ. Res., 86, 607–609. [DOI] [PubMed] [Google Scholar]
- Adams J.W., Sakata,Y., Davis,M.G., Sah,V.P., Wang,Y., Liggett,S.B., Chien,K.R., Brown,J.H. and Dorn,G.W. (1998) Enhanced Gαq signaling: a common pathway mediates cardiac hypertrophy and apoptotic heart failure. Proc. Natl Acad. Sci. USA, 95, 10140–10145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker T.C., Noel,R.J., Coats,W.S., Gomez-Foix,A.M., Alam,T., Gerard,R.D. and Newgard,C.B. (1994) Use of recombinant adenovirus for metabolic engineering of mammalian cells. Methods Cell Biol., 43, 161–189. [DOI] [PubMed] [Google Scholar]
- Bueno O.F. et al. (2000) The MEK1–ERK1/2 signaling pathway promotes compensated cardiac hypertrophy in transgenic mice. EMBO J., 19, 6341–6350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bueno O.F., De Windt,L.J., Lim,H.W., Tymitz,K.M., Witt,S.A., Kimball,T.R. and Molkentin,J.D. (2001) The dual-specificity phosphatase MKP-1 limits the cardiac hypertrophic response in vitro and in vivo. Circ. Res., 88, 88–96. [DOI] [PubMed] [Google Scholar]
- Chien K.R. (1999) Stress pathways and heart failure. Cell, 98, 555–558. [DOI] [PubMed] [Google Scholar]
- English J.M., Vanderbilt,C.A., Xu,S., Marcus,S. and Cobb,M.H. (1995) Isolation of MEK5 and differential expression of alternatively spliced forms. J. Biol. Chem., 270, 28897–28902. [DOI] [PubMed] [Google Scholar]
- English J., Pearson,G., Wilsbacher,J., Swantek,J., Karandikar,M., Xu,S. and Cobb,M.H. (1999a) New insights into the control of MAP kinase pathways. Exp. Cell Res., 253, 255–270. [DOI] [PubMed] [Google Scholar]
- English J.M., Pearson,G., Hockenberry,T., Shivakumar,L., White,M.A. and Cobb,M.H. (1999b) Contribution of the ERK5/MEK5 pathway to Ras/Raf signaling and growth control. J. Biol. Chem., 274, 31588–31592. [DOI] [PubMed] [Google Scholar]
- Fentzke R.C., Korcarz,C.E., Lang,R.M., Lin,H. and Leiden,J.M. (1998) Dilated cardiomyopathy in transgenic mice expressing a dominant-negative CREB transcription factor in the heart. J. Clin. Invest., 101, 2415–2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey N., McKinsey,T.A. and Olson,E.N. (2000) Decoding calcium signals involved in cardiac growth and function. Nature Med., 6, 1221–1227. [DOI] [PubMed] [Google Scholar]
- Fukuhara S., Marinissen,M.J., Chiariello,M. and Gutkind,J.S. (2000) Signaling from G protein-coupled receptors to ERK5/Big MAPK 1 involves Gαq and Gα12/13 families of heterotrimeric G proteins. Evidence for the existence of a novel Ras and Rho-independent pathway. J. Biol. Chem., 275, 21730–21736. [DOI] [PubMed] [Google Scholar]
- Gulick J., Subramaniam,A., Neumann,J. and Robbins,J. (1991) Isolation and characterization of the mouse cardiac myosin heavy chain genes. J. Biol. Chem., 266, 9180–9185. [PubMed] [Google Scholar]
- Ikeda Y. and Ross,J.,Jr (2000) Models of dilated cardiomyopathy in the mouse and the hamster. Curr. Opin. Cardiol., 15, 197–201. [DOI] [PubMed] [Google Scholar]
- Jones W.K. et al. (1996) Ablation of the murine α myosin heavy chain gene leads to dosage effects and functional deficits in the heart. J. Clin. Invest., 98, 1906–1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamakura S., Moriguchi,T. and Nishida,E. (1999) Activation of the protein kinase ERK5/BMK1 by receptor tyrosine kinases. Identification and characterization of a signaling pathway to the nucleus. J. Biol. Chem., 274, 26563–26571. [DOI] [PubMed] [Google Scholar]
- Kasler H.G., Victoria,J., Duramad,O. and Winoto,A. (2000) ERK5 is a novel type of mitogen-activated protein kinase containing a transcriptional activation domain. Mol. Cell. Biol., 20, 8382–8389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato T., Sano,M., Miyoshi,S., Sato,T., Hakuno,D., Ishida,H., Kinoshita-Nakazawa,H., Fukuda,K. and Ogawa,S. (2000) Calmodulin kinases II and IV and calcineurin are involved in leukemia inhibitory factor-induced cardiac hypertrophy in rats. Circ. Res., 87, 937–945. [DOI] [PubMed] [Google Scholar]
- Kato Y., Kravchenko,V.V., Tapping,R.I., Han,J., Ulevitch,R.J. and Lee,J.D. (1997) BMK1/ERK5 regulates serum-induced early gene expression through transcription factor MEF2C. EMBO J., 16, 7054–7066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klesse L.J., Meyers,K.A., Marshall,C.J. and Parada,L.F. (1999) Nerve growth factor induces survival and differentiation through two distinct signaling cascades in PC12 cells. Oncogene, 18, 2055–2068. [DOI] [PubMed] [Google Scholar]
- Kodama H. et al. (2000) Significance of ERK cascade compared with JAK/STAT and PI3-K pathway in gp130-mediated cardiac hypertrophy. Am. J. Physiol. Heart Circ. Physiol., 279, H1635–H1644. [DOI] [PubMed] [Google Scholar]
- Kunisada K., Hirota,H., Fujio,Y., Matsui,H., Tani,Y., Yamauchi-Takihara,K. and Kishimoto,T. (1996) Activation of JAK–STAT and MAP kinases by leukemia inhibitory factor through gp130 in cardiac myocytes. Circulation, 94, 2626–2632. [DOI] [PubMed] [Google Scholar]
- Lee J.D., Ulevitch,R.J. and Han,J. (1995) Primary structure of BMK1: a new mammalian map kinase. Biochem. Biophys. Res. Commun., 213, 715–724. [DOI] [PubMed] [Google Scholar]
- MacLellan W.R. and Schneider,M.D. (1997) Death by design. Programmed cell death in cardiovascular biology and disease. Circ. Res., 81, 137–144. [DOI] [PubMed] [Google Scholar]
- MacLellan W.R. and Schneider,M.D. (2000) Genetic dissection of cardiac growth control pathways. Annu. Rev. Physiol., 62, 289–319. [DOI] [PubMed] [Google Scholar]
- Milner D.J., Taffet,G.E., Wang,X., Pham,T., Tamura,T., Hartley,C., Gerdes,A.M. and Capetanaki,Y. (1999) The absence of desmin leads to cardiomyocyte hypertrophy and cardiac dilation with compromised systolic function. J. Mol. Cell. Cardiol., 31, 2063–2076. [DOI] [PubMed] [Google Scholar]
- Molkentin J.D., Lu,J.R., Antos,C.L., Markham,B., Richardson,J., Robbins,J., Grant,S.R. and Olson,E.N. (1998) A calcineurin-dependent transcriptional pathway for cardiac hypertrophy. Cell, 93, 215–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicol R.L., Frey,N. and Olson,E.N. (2000) From the sarcomere to the nucleus: role of genetics and signaling in structural heart disease. Annu. Rev. Gen. Genet., 1, 179–223. [DOI] [PubMed] [Google Scholar]
- Oh H., Fujio,Y., Kunisada,K., Hirota,H., Matsui,H., Kishimoto,T. and Yamauchi-Takihara,K. (1998) Activation of phosphatidylinositol 3-kinase through glycoprotein 130 induces protein kinase B and p70 S6 kinase phosphorylation in cardiac myocytes. J. Biol. Chem., 273, 9703–9710. [DOI] [PubMed] [Google Scholar]
- Passier R. et al. (2000) CaM kinase signaling induces cardiac hypertrophy and activates the MEF2 transcription factor in vivo. J. Clin. Invest., 105, 1395–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruwhof C. and van der Laarse,A. (2000) Mechanical stress-induced cardiac hypertrophy: mechanisms and signal transduction pathways. Cardiovasc. Res., 47, 23–37. [DOI] [PubMed] [Google Scholar]
- Sugden P.H. and Clerk,A. (1998) Cellular mechanisms of cardiac hypertrophy. J. Mol. Med., 76, 725–746. [DOI] [PubMed] [Google Scholar]
- Takeishi Y., Abe,J., Lee,J.D., Kawakatsu,H., Walsh,R.A. and Berk,B.C. (1999) Differential regulation of p90 ribosomal S6 kinase and big mitogen-activated protein kinase 1 by ischemia/reperfusion and oxidative stress in perfused guinea pig hearts. Circ. Res., 85, 1164–1172. [DOI] [PubMed] [Google Scholar]
- Tamura T., Onodera,T., Said,S. and Gerdes,A.M. (1998) Correlation of myocyte lengthening to chamber dilation in the spontaneously hypertensive heart failure (SHHF) rat. J. Mol. Cell. Cardiol., 30, 2175–2181. [DOI] [PubMed] [Google Scholar]
- Wang Y., Huang,S., Sah,V.P., Ross,J.,Jr, Brown,J.H., Han,J. and Chien,K.R. (1998a) Cardiac muscle cell hypertrophy and apoptosis induced by distinct members of the p38 mitogen-activated protein kinase family. J. Biol. Chem., 273, 2161–2168. [DOI] [PubMed] [Google Scholar]
- Wang Y., Su,B., Sah,V.P., Brown,J.H., Han,J. and Chien,K.R. (1998b) Cardiac hypertrophy induced by mitogen-activated protein kinase kinase 7, a specific activator for c-Jun NH2-terminal kinase in ventricular muscle cells. J. Biol. Chem., 273, 5423–5426. [DOI] [PubMed] [Google Scholar]
- Wollert K.C. and Chien,K.R. (1997) Cardiotrophin-1 and the role of gp130-dependent signaling pathways in cardiac growth and development. J. Mol. Med., 75, 492–501. [DOI] [PubMed] [Google Scholar]
- Wollert K.C. et al. (1996) Cardiotrophin-1 activates a distinct form of cardiac muscle cell hypertrophy. Assembly of sarcomeric units in series via gp130/leukemia inhibitory factor receptor-dependent pathways. J. Biol. Chem., 271, 9535–9545. [DOI] [PubMed] [Google Scholar]
- Zhang D., Gaussin,V., Taffect,G.E., Belaguli,N.S., Yamada,M., Schwartz,R.J., Michael,L.H., Overbeek,P.A. and Schneider,M.D. (2000) TAK1 is activated in the myocardium after pressure overload and is sufficient to provoke heart failure in transgenic mice. Nature Med., 6, 556–563. [DOI] [PubMed] [Google Scholar]
- Zhou G., Bao,Z.Q. and Dixon,J.E. (1995) Components of a new human protein kinase signal transduction pathway. J. Biol. Chem., 270, 12665–12669. [DOI] [PubMed] [Google Scholar]