

Abstract
Sec1p-like/Munc-18 (SM) proteins bind to t-SNAREs and inhibit ternary complex formation. Paradoxically, the absence of SM proteins does not result in constitutive membrane fusion. Here, we show that in yeast cells lacking the SM protein Vps45p, the t-SNARE Tlg2p is down-regulated, to undetectable levels, by rapid proteasomal degradation. In the absence of Vps45p, Tlg2p can be stabilized through abolition of proteasome activity. Surprisingly, the stabilized Tlg2p was targeted to the correct intracellular location. However, the stabilized Tlg2p is non-functional and unable to bind its cognate SNARE binding partners, Tlg1p and Vti1p, in the absence of Vps45p. A truncation mutant lacking the first 230 residues of Tlg2p no longer bound Vps45p but was able to form complexes with Tlg1p and Vti1p in the absence of the SM protein. These data provide us with two valuable insights into the function of SM proteins. First, SM proteins act as chaperone-like molecules for their cognate t-SNAREs. Secondly, SM proteins play an essential role in the activation process allowing their cognate t-SNARE to participate in ternary complex formation.
Keywords: endosome/membrane trafficking/Sec1p/SNARE/VPS
Introduction
The exchange of molecules between intracellular organelles of eukaryotic cells must be controlled in a highly regulated manner. The SNARE hypothesis was proposed to explain how transport vesicles, carrying cargo from one organelle, dock and fuse with their appropriate target membrane in a highly specific manner (Rothman, 1994). In this hypothesis, transport vesicles contain integral membrane proteins (v-SNAREs) that interact specifically with membrane proteins (t-SNAREs) found in the relevant target membrane. Formation of a stable, ternary complex between the correct set of SNARE proteins leads to membrane docking and fusion.
Central to the SNARE hypothesis is a high level of regulation of SNARE complex assembly. Two protein families have been implicated in this control: the small GTPase Rab family (Schimmoller et al., 1998) and the Sec1p-like protein family (Halachmi and Lev, 1996) or SM (Sec1/Munc18-related) proteins (Jahn and Sudhof, 1999). SM proteins are hydrophilic and bind to t-SNAREs with high affinity. Much of our understanding of the structure–function relationship between SM proteins and their cognate t-SNAREs has been obtained from studies in the mammalian synapse. Here, the SM protein Munc-18a/n-Sec1/RbSec1 binds to the t-SNARE syntaxin1a (Hata et al., 1993; Pevsner et al., 1994b). This interaction reduces the affinity of syntaxin1a for its v-SNARE, VAMP2, precluding SNARE complex formation (Pevsner et al., 1994a). Structural studies have revealed that syntaxin1a adopts two distinct conformations (Dulubova et al., 1999). Munc18a binds preferentially to the closed conformation in which three helical domains of syntaxin1a in the N-terminus (Habc) interact with the membrane-proximal helix in the C-terminus (H3). In the ternary complex, syntaxin adopts a more open conformation in which the Habc and H3 domains separate, allowing H3 to participate in the core complex.
From these data, a model has emerged in which Sec1p-like proteins act as negative regulators of SNARE complex assembly, possibly by holding syntaxin in the closed conformation. In order for membrane fusion to proceed, this complex must be disrupted, allowing syntaxin to undergo a conformational change so that it is able to form ternary complexes that mediate fusion. In support of this model, overproduction of SM proteins has been shown to inhibit specific membrane fusion events (Thurmond et al., 1998; Riento et al., 2000). Paradoxically, several studies indicate that loss of SM protein expression does not result in constitutive membrane transport as might have been predicted from this model. Rather, in the absence of SM proteins, there is a block in vesicle transport (Schekman, 1992; Verhage et al., 2000), indicating an additional role for SM proteins, possibly as positive regulators of membrane fusion.
We set out to investigate the molecular basis for the above paradox by focusing on the interaction between the t-SNARE Tlg2p, that regulates membrane traffic through the yeast endocytic system, and its SM protein Vps45p. Deletion of VPS45 resulted in a complete loss of Tlg2p through rapid proteasomal degradation. When Tlg2p expression was stabilized, the protein was targeted correctly but failed to interact with its cognate SNARE proteins, Tlg1p and Vti1p. Truncation of the N-terminal 230 residues from Tlg2p restored the ability to interact with Tlg1p and Vti1p in the absence of Vps45p. These data suggest that SM proteins act as chaperone-like molecules for their cognate t-SNAREs and also play an active role in the conformational switch from the closed to open state, thus activating the t-SNARE and facilitating membrane fusion.
Results
Relationship between Tlg2p and Vps45p
SM proteins are peripheral membrane proteins thought to attach to membranes through their interaction with their cognate t-SNARE (Hata et al., 1993). The SM protein Vps45p has been shown to interact, both genetically and biochemically, with two different syntaxin homologues, Pep12p and Tlg2p (Burd et al., 1997; Webb et al., 1997; Nichols et al., 1998; Abeliovich et al., 1999). Consistent with a previous report (Nichols et al., 1998), we show that Tlg2p rather than Pep12p provides the main membrane binding site for Vps45p. In wild-type cells, 75% of Vps45p is membrane associated (Cowles et al., 1994), with the majority being found in the high speed, membrane pellet, P100. Vps45p has an identical subcellular distribution in cells lacking PEP12. In contrast, Vps45p was found in the cytosolic (S100) fraction of cells lacking Tlg2p. These data support a previous report (Nichols et al., 1998) that Tlg2p provides the major membrane binding site for Vps45p in vivo. To confirm that Vps45p and Tlg2p interact directly, we performed in vitro binding studies. Radiolabelled Vps45p, produced using an in vitro translation system, was incubated with bacterially produced GST or GST fusion proteins containing the cytosolic domains of the three t-SNAREs, Pep12p, Sed5p and Tlg2p. We observed a significant interaction between GST–Tlg2p and Vps45p in vitro (Figure 1B). In contrast, little if any binding of Vps45p to GST, GST–Pep12p or GST–Sed5p was observed (Figure 1B). These data are in agreement with previous studies showing that Vps45p co-precipitates with Tlg2p but not with Pep12p or Sed5p (Nichols et al., 1998).

Fig. 1. Vps45p interacts with Tlg2p. (A) Vps45p is membrane localized through Tlg2p. Wild-type (SF838-9D), pep12Δ (SGY39) and tlg2Δ (NOzY3) cells were fractionated by differential centrifugation to yield a low-speed pellet (P13), a high-speed pellet (P100) and a soluble fraction (S100). The amount of Vps45p, Pep12p, Tlg2p and Pgk1p in each fraction was assessed using immunoblot analysis. (B) Vps45p binds to the cytosolic domain of Tlg2p. Glutathione–Sepharose beads loaded with GST–Pep12p, GST–Sed5p, GST–Tlg2p, GST–Tlg2-Δ1–230, GST alone or empty beads (0) were incubated with [35S]methionine-labelled, in vitro-translated, Vps45p. GST fusions and associated proteins were eluted and analysed using SDS–PAGE and fluorography.
To substantiate the relationship between Vps45p and Tlg2p further, we examined the effect of loss of the SM protein on the t-SNARE. In agreement with a previous study (Nichols et al., 1998), we found that VPS45 is required for the stable expression of Tlg2p but not Pep12p. This is demonstrated in Figure 2, which also demonstrates that, like Pep12p, the cellular levels of other t-SNARE molecules, Vam3p, Sed5p and Tlg1p, are not affected by the loss of VPS45. Similarly, the levels of the two other SNAREs, Vti1p and Ufe1p, in vps45Δ cells were indistinguishable from those found in wild-type cells. Collectively, the data presented in Figures 1 and 2, together with previous reports (Nichols et al., 1998; Abeliovich et al., 1999), demonstrate that Tlg2p is the major t-SNARE that associates with Vps45p.
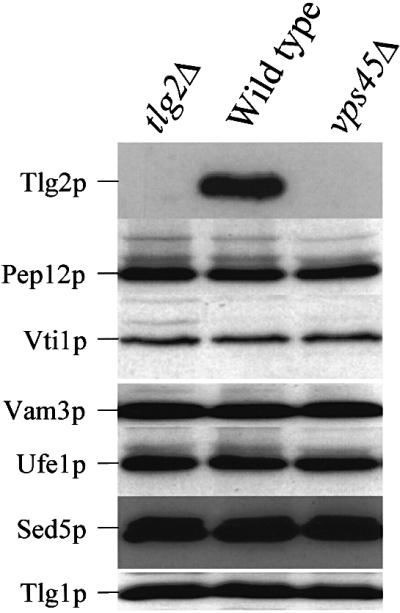
Fig. 2. Tlg2p is down-regulated upon loss of Vps45p. Proteins contained within whole-cell extracts prepared from wild-type (RPY10), vps45Δ (NOzY2) and tlg2Δ (NOzY4) cells were separated using SDS–PAGE before being transferred to nitrocellulose. The resulting filters were probed using antibodies that specifically recognize Tlg2p, Vti1p, Pep12p, Vam3p, Ufe1p, Sed5p or Tlg1p as indicated.
Proteasomal degradation of Tlg2p
There are two sites of protein degradation in yeast cells; the vacuole and the proteasome. Strains harbouring the pep4-3 mutation lack vacuolar protease activity (Woolford et al., 1986), while proteasome activity can be abolished through mutations in either catalytic (Pre1p, Pre2p) (Heinemeyer et al., 1991, 1993) or regulatory (Cim3p) subunits (Ghislain et al., 1993). Figure 3A demonstrates that loss of proteasome activity in cells lacking VPS45 restored the level of Tlg2p to that found in wild-type cells. In contrast, loss of vacuolar protease activity had no affect on the amount of Tlg2p found in vps45Δ cells. These data suggest that in the absence of Vps45p, Tlg2p is synthesized, but subsequently is degraded by the proteasome.

Fig. 3. Loss of VPS45 results in degradation of Tlg2p by the proteasome. (A) Proteins contained within whole-cell extracts prepared from wild-type (RPY10), vps45Δ (NOzY2), pep4-3 (SF838–9D), pep4-3 vps45Δ (NOzY1), pre1-1 pre2-2 (LHY58), pre1-1 pre2-2 vps45Δ (NOzY13), cim3-1 (LHY913) and cim3-1 vps45Δ (NOzY8) were separated using SDS–PAGE before being transferred to nitrocellulose. The resulting filter was probed using antibodies that specifically recognize Tlg2p. (B) Proteins synthesized by wild-type (RPY10), vps45Δ (NOzY2), vps45Δ pep4-3 (NOzY1) and vps45Δ pre1-1 pre2-2 (NOzY13) cells were labelled using [35S]cysteine/methionine for 10 min prior to the addition of excess unlabelled cysteine and methionine. At the times indicated, Tlg2p was immunoprecipitated and samples were analysed using SDS–PAGE and fluorography.
In order to look at the kinetics of degradation of Tlg2p in cells lacking Vps45p, the half-life of the t-SNARE was examined using pulse–chase analysis. In wild-type cells, Tlg2p has a half-life of >2 h (Figure 3B), whereas in vps45Δ cells, although initially synthesized to levels comparable with wild-type cells, Tlg2p was degraded with a half-life of ∼20 min. As expected from the data presented in Figure 3A, the rapid turnover of Tlg2p in the absence of Vps45p occurs in both the presence and absence of active vacuolar proteases. In contrast, abolition of proteasome activity, through mutations in PRE1 and PRE2, stabilizes Tlg2p in the absence of VPS45. The data presented in Figure 3 show that, in the absence of Vps45p, the t-SNARE Tlg2p is degraded rapidly by the proteasome.
Tlg2p is not functional in the absence of VPS45
Tlg2p has been found in a SEC18-dependent complex with the t-SNARE Tlg1p and the v-SNARE Vti1p (Coe et al., 1999). Figure 4A shows that both Tlg1p and Vti1p co-precipitate with Tlg2p in wild-type or proteasome- deficient cells. In contrast, neither Tlg1p nor Vti1p could be co-precipitated with Tlg2p in the proteasome-deficient cells lacking VPS45. These data indicate that, although abolition of proteasome activity prevents the degradation of Tlg2p in the absence of VPS45, the stabilized protein is unable to form complexes with its usual binding partners.

Fig. 4. Vps45p is required for Tlg2p to form ternary complexes with Tlg1p and Vti1p. (A) Stabilized Tlg2p does not co-precipitate Tlg1p and Vti1p. Tlg2p-containing complexes were immunoprecipitated from the following strains: wild-type (LHY55), tlg2Δ (NozY4), vps45Δ (NozY11), pre1-1 pre2-2 (LHY58) and vps45Δ pre1-1 pre2-2 (NOzY13). Immunoblot analysis was used to detect the presence of Tlg1p and Vti1p in the immuno precipitated complexes. As a control, proteins precipitated using pre-immune serum from wild-type and tlg2Δ cells (PI) were also analysed. (B) Tlg2p is no longer found in SNARE complexes following rapid inactivation of Vps45p. Tlg2p-containing complexes were immunoprecipitated from wild-type (SF838-9D) and vps45-ts (RPY15) cells. Cells were grown to early log phase at 25°C before being shifted to 37°C for the indicated times. Immunoblot analysis was used to detect the presence of Tlg1p and Vti1p in the immunoprecipitated complexes.
One explanation for the inability of the stabilized Tlg2p, produced by proteasome-deficient vps45Δ mutant cells, to bind to Tlg1p and Vti1p is that the t-SNARE is mistargeted. To investigate this, we performed equilibrium density centrifugation. Figure 5 demonstrates that in wild-type cells Tlg2p was enriched in one single membrane peak (fractions 5–8) that was well separated from peaks containing markers of the vacuolar membrane (Vph1p, fractions 1 and 2) and the endoplasmic reticulum (ER; Dpm1p, fractions 1–3). In cells lacking both VPS45 and proteasome activity (vps45Δ pre1-1 pre2-2), the stabilized Tlg2p fractionated to an identical position in the gradient (fractions 5–8) to that observed in wild-type cells. To confirm that Tlg2p was localized correctly in vps45Δ pre1-1 pre2-2 cells, we studied the localization of a Tlg2p–green fluorescent protein (GFP) fusion protein. In wild-type cells, the Tlg2p–GFP fusion protein gave a punctate labelling pattern characteristic of Golgi/endosomal marker proteins (Seron et al., 1998). An indistinguishable labelling pattern was observed for the same Tlg2p–GFP fusion protein in vps45Δ pre1-1 pre2-2 mutant cells (data not shown).

Fig. 5. Tlg2p is targeted independently of Vps45p. Total membranes from wild-type (LHY55) and vps45Δ pre1-1 pre2-2 (NOzY13) cells were fractionated to equilibrium on a 40–65% self-forming sorbitol gradient. The amount of Tlg2p, Vph1p, Dpm1p and Pep12p in each fraction was assessed through immunoblot analysis.
To demonstrate further the inability of Tlg2p to form SNARE complexes in the absence of Vps45p function, we performed similar experiments to those described in Figure 4A in cells harbouring a temperature-sensitive allele of VPS45. At 25°C, these cells faithfully deliver carboxypeptidase Y (CPY) to the vacuole, whereas at the non-permissive temperature (37°C) VPS45 is no longer functional and these cells missort CPY to the cell surface (Piper et al., 1994). As shown in Figure 4B, at 25°C Tlg2p forms a complex with both Tlg1p and Vti1p as in wild-type cells. However, after a brief period at the non-permissive temperature (37°C), although Tlg2p can still be immunoprecipitated efficiently from these cells, it no longer binds to either Tlg1p or Vti1p. Interestingly, we observed a reduction in Tlg2p protein levels after 20 min at 37°C and, after 60 min at the non-permissive temperature, no Tlg2p was detectable in these cells (Figure 4B). These data indicate that loss of Vps45p function is closely linked both to the stability of Tlg2p, and also to its ability to form SNARE complexes.
Tlg2p-Δ1–230 forms SNARE complexes independently of VPS45
The observation that Tlg2p does not bind to Tlg1p or Vti1p in the absence of Vps45p suggests a positive requirement for the SM protein in SNARE complex assembly. To explore the role of Vps45p in SNARE complex assembly further, we set out to construct a version of Tlg2p with reduced affinity for Vps45p and examine its biochemical and functional properties. It has been proposed that members of the syntaxin/t-SNARE family are characterized by the presence of a C-terminal core complex-forming helix (H3) and an N-terminal three-helix bundle (Habc) (Dulubova et al., 2001). Interactions between the H3 and Habc domains have been shown to give rise to a closed syntaxin conformation which binds with high affinity to its cognate SM protein (Dulubova et al., 1999). In the open conformation, the H3 domain is thought to be exposed, allowing syntaxin to participate in SNARE complex formation (Dulubova et al., 1999). It has been shown previously that deletion of the N-terminal Habc domain of either syntaxin1a or Sso1p results in proteins that form SNARE complexes and/or catalyse membrane fusion considerably faster than the full-length proteins (Nicholson et al., 1998; Parlati et al., 1999). These mutants presumably do not adopt the closed conformation due to the absence of the Habc helices, and thus the H3 helix is exposed to participate in SNARE complexes. Although detailed structural studies have not yet been performed on Tlg2p, preliminary NMR studies on Sed5p, Tlg2p and Pep12p suggest that all syntaxins may contain an N-terminal three-helix domain like that found in syntaxin1a, Vam3p and Sso1p (Dulubova et al., 2001). We constructed a truncated version of Tlg2p lacking the N-terminal 230 residues of the protein (Tlg2p-Δ1–230). The gene encoding this truncation mutant was placed under the control of the inducible GAL1/10 promoter (GAL-TLG2-Δ1–230). Initially, we examined the ability of this truncated t-SNARE to interact with Vps45p both in vitro and in vivo. In contrast to the GST–Tlg2p fusion protein containing the entire cytosolic tail of Tlg2p, a GST fusion protein containing the cytosolic domain of the truncated Tlg2p (GST–Tlg2p-Δ1–230) did not bind to Vps45p in vitro (Figure 1B). Similarly, in contrast to cells producing full-length Tlg2p, where the majority of Vps45p is membrane associated, in cells producing Tlg2p-Δ1–230, and not the full-length protein, Vps45p is found in the cytosol (Figure 6A). These data are in direct conflict with a recent report where a similar truncation of the vacuolar t-SNARE, Vam3p was found to have no effect on the ability of Vam3p to interact with its SM protein Vps33p (Dulubova et al., 2001). Our inability to detect Vps45p binding to the Tlg2p truncation mutant is unlikely to be due to improper folding because, as described below, this mutant was able to form SNARE complexes in vivo (Figure 6B). Thus, these data suggest that the Tlg2p–Vps45p complex possesses properties more like those of the syntaxin1a–Munc18a complex than those of the Vam3p–Vps33p complex. Further study is required to resolve and/or understand these differences.

Fig. 6. Truncation of Tlg2p by-passes the requirement for SNARE complex formation on VPS45. (A) Tlg2p-Δ1–230 does not recruit Vps45p to membranes. tlg2Δ cells (NozY3) expressing either full-length Tlg2p (from pNOz7) or Tlg2p-Δ1–230 (from pNOz8) were fractionated using differential centrifugation as in Figure 1. The amount of Vps45p contained within each fraction was determined by immunoblot analysis. (B) Tlg2p-Δ1–230 binds Tlg1p and Vti1p in the absence of VPS45. vps45Δ pre1-1 pre2-2 (NOzY13) cells harbouring a plasmid encoding either Tlg2p-Δ1–230 (pNOz8) or full-length Tlg2p (pNOz7) under control of the GAL1/10 promoter were grown using either glucose (SD) or galactose (Sgal/Raff) as a carbon source. Antibodies that recognize both the full-length Tlg2p and the truncated Tlg2p-Δ1–230 were used to precipitate Tlg2p-containing complexes from lysates of these cells. The amount of Tlg1p and Vti1p in these complexes was assessed through immunoblot analysis. (C) Tlg2p-Δ1–230 partially overcomes CPY missorting in vps45Δ cells. Proteins synthesized by wild-type cells (RPY10), vps45Δ cells (NozY2) and vps45Δ cells producing Tlg2p-Δ1–230 (NOzY2 transformed with pNOz8) were grown using galactose as a carbon source (Sgal/Raff) and metabolically labelled for a 10 min pulse using [35S]methionine. A chase period of 30 min was initiated by the addition of excess unlabelled methionine, after which time CPY was immunoprecipitated from both intracellular (I) and extracellular (E) fractions.
To test the ability of Tlg2p-Δ1–230 to form SNARE complexes, GAL-TLG2-Δ1–230 was transformed into proteasome-deficient cells lacking VPS45 (vps45Δ pre1-1 pre2-2). When these cells were grown using glucose as a carbon source, thus repressing expression from the GAL1/10 promoter, these cells produced only the endogenous, full-length, Tlg2p. Growth of these cells on galactose-containing media induced expression from the GAL1/10 promoter, and the cells now produced both the full-length Tlg2p and the truncated Tlg2p-Δ1–230 (Figure 6B; GAL-TLG2-Δ1–230, glucose versus galactose). We were unable to find an interaction between the full-length Tlg2p and either Tlg1p or Vti1p using cells grown on glucose, consistent with the data presented in Figure 4 (Figure 6B; GAL-TLG2-Δ1–230, glucose). In contrast, we observed efficient co-precipitation of both Tlg1p and Vti1p with Tlg2p-Δ1–230 (Figure 6B; GAL-TLG2-Δ1–230, galactose). To exclude the possibility that the co-precipitation of Tlg1p and Vti1p was non-specific, due to overproduction of a coiled-coil protein, we performed two sets of controls. First, we were unable to detect the presence of the unrelated SNARE Pep12p, Vam3p, Sed5p and Ufe1p in the Tlg2p-Δ1–230 immunoprecipitate (data not shown). In addition, overproduction of full-length Tlg2p did not lead to co-precipitation of Tlg1p and Vti1p in the absence of Vps45p (Figure 6B; GAL-TLG, galactose). Collectively, the data presented in Figure 6B indicate that removal of the N-terminal 230 residues from Tlg2p by-passes the requirement for Vps45p in SNARE complex assembly.
In order to determine whether the complexes formed between Tlg2p-Δ1–230, Vti1p and Tlg1p were functional, we examined the ability of the truncated t-SNARE to rescue the membrane trafficking defects associated with loss of VPS45 function. The trafficking of the soluble vacuolar hydrolase CPY usually is followed to assess the integrity of the yeast endocytic system. In wild-type cells, proCPY (p2) is delivered efficiently from the trans-Golgi network (TGN) to the vacuole where it is processed proteolytically into its active, mature form (m). In vps mutants, this sorting process is perturbed and CPY is secreted from the cell. We reasoned that since the Tlg2p-Δ1–230 truncation mutant does not require Vps45p to form SNARE complexes, then it might be able to overcome the CPY sorting defect exhibited by cells lacking VPS45. Importantly, unlike full-length Tlg2p, Tlg2p-Δ1–230 was stable in cells lacking VPS45 (data not shown). Figure 6C shows that in contrast to wild-type cells, vps45 cells secrete the majority (70%) of their CPY into the extracellular media. In addition, the CPY that remains inside the cell is present in the p2 form, indicating that it does not reach the vacuole (Cowles et al., 1994; Piper et al., 1994). Expression of Tlg2p-Δ1–230 partially overcomes the CPY sorting defects exhibited by vps45 cells. CPY secretion from these cells is reduced to 40% and the CPY found intracellularly is processed into its mature form. The fact that we only observed partial rescue of the CPY sorting defect in these experiments is due possibly to mislocalization of the Tlg2p-Δ1–230 mutant, the majority of which is found in the vacuole (data not shown).
Discussion
SM proteins bind to t-SNAREs with high affinity (Pevsner et al., 1994a). In so doing, they reduce the affinity of t-SNAREs for their cognate v-SNAREs (Pevsner et al., 1994a). Thus, the simplest model for SM function is that they hold the t-SNARE in a fusion-incompetent state, acting as negative regulators of SNARE complex assembly. At the structural level, it has been shown that in the ternary complex, syntaxin undertakes a somewhat open conformation, whereas when complexed to Munc-18 it is held in a more closed conformation, in which the C-terminal H3 domain cannot interact with SNARE binding partners (Dulubova et al., 1999). Based on these data, it is conceivable that the mechanism by which SM proteins impart negative regulation is by stabilizing t-SNAREs in their closed conformation. This model, however, is inconsistent with the observation that deletion of SM proteins in both yeast and mammalian cells results in a complete block of vesicle transport (Schekman, 1992; Verhage et al., 2000). In the present study, we show that deletion of the SM protein Vps45p results in rapid proteasomal degradation of its cognate t-SNARE, Tlg2p. These data suggest that a major function of SM proteins is to act as chaperone-like molecules for their cognate t-SNAREs. These data also suggest that the levels of the t-SNARE in the cell are closely regulated by the relative expression of cognate SM proteins. One expectation from this is that the levels of the SM protein would either closely parallel those of the t-SNARE or exceed them. In agreement with this, we have been unable to overproduce Tlg2p significantly in wild-type cells, presumably because the levels of Vps45p become rate limiting (data not shown). Moreover, there is evidence from other systems that SM proteins and their t-SNAREs are expressed at a 1:1 stoichiometry (Hickson et al., 2000) and stable t-SNARE–SM complexes are readily detectable in a number of cell types (Carr et al., 1999; Yang et al., 2000). In fact, it seems likely that the majority of the t-SNARE is complexed to its SM protein at steady state. This interaction presumably is disrupted prior to SNARE complex formation, but the proportion of t-SNARE molecules participating in SNARE complexes at any one time is likely to be low under steady-state conditions as this is a very transient event. A quantitative analysis of such interactions is clearly warranted.
Based on the data that we have presented regarding the degradation of Tlg2p upon loss of Vps45p, it is tempting to speculate that the block in vesicle transport observed in cells lacking SM proteins may be an indirect effect due to a resultant loss of the cognate t-SNARE, rather than the absence of the SM protein per se. However, our studies involving the truncated version of Tlg2p, and studies using conditional alleles of SM proteins that suggest a direct involvement of these proteins in membrane docking/fusion events (Novick and Schekman, 1979; Piper et al., 1994), argue that this is not the case. We were able to stabilize the levels of Tlg2p in vps45Δ mutant cells by abolishing the activity of the proteasome. Surprisingly, the stabilized Tlg2p was localized correctly as determined both by subcellular fractionation and fluorescence microscopy. These data indicate that the stabilized protein is not completely unfolded because such a protein would probably have been targeted for degradation from the ER. Despite its correct localization, stabilized Tlg2p did not interact with its cognate SNARE binding partners Tlg1p and Vti1p. Collectively, these data strongly support the notion that SM proteins play a positive regulatory role in SNARE complex formation.
One of the crucial questions that arises from these studies is, why is the stabilized Tlg2p molecule unable to interact with either Vti1p or Tlg1p despite its normal localization? This question is particularly pertinent since recombinant t-SNAREs can form ternary complexes in vitro even in the absence of an SM protein (Pevsner et al., 1994a). Perhaps the most obvious answer is that the SM protein is required to prime the t-SNARE so that it can form functional SNARE complexes in vivo. To test this idea, we attempted to construct a Tlg2p mutant that was unable to bind SM in vivo to determine if it could form SNARE complexes in the absence of Vps45p. It has been shown recently that deletion of the N-terminal Habc domain of either Sso1p or syntaxin1a enabled these proteins to form SNARE complexes more efficiently in vitro (Nicholson et al., 1998; Parlati et al., 1999). Because the N-terminus of the t-SNARE constitutes one of the major sites for interaction with the SM proteins, we reasoned that these mutants would be unable to bind the SM protein in vivo. We constructed a version of Tlg2p lacking 230 residues from the N-terminus and produced this in both vps45Δ and tlg2Δ cells. This protein failed to interact with Vps45p either in vitro or in vivo. Strikingly, this protein was able to form SNARE complexes, in vivo, with both Tlg1p and Vti1p in the absence of Vps45p, and partially overcame the membrane trafficking defects of a vps45Δ mutant. These data suggest that the interaction between a t-SNARE and its cognate SM protein is required subsequently to achieve SNARE complex formation. The stabilized Tlg2p in vps45Δ cells presumably is locked in its closed conformation (Dulubova et al., 1999). By inference, we suggest that Tlg2p is incapable of making the transition to the open conformation, which would allow it to interact with its SNARE partners, in the absence of Vps45p. The N-terminal truncation mutant of Tlg2p by-passes the requirement for SNARE complex formation on Vps45p since it cannot adopt the closed, and consequently permanently mimics the open, conformation with its core-binding domain available to bind Tlg1p and Vti1p.
Implications
A major finding of this study is that cells deploy specific mechanisms to dispose of the t-SNARE, Tlg2p, post-translationally if it is produced in the absence of its cognate SM protein, Vps45p. We have obtained preliminary data to suggest that a similar mechanism operates for other t-SNARE–SM partners in yeast, indicating that this is a general principle. This might explain, at least in part, why removal of SM proteins from cells causes a block in vesicle fusion. This degradative mechanism appears to be employed to dispose of excess t-SNARE rather than excess SM protein because the levels of Vps45p remained normal even in the absence of Tlg2p (Nichols et al., 1998; and Figure 1). One might imagine, therefore, that t-SNAREs rarely exist in the unbound form in the cell, otherwise they may be subject to rapid degradation. Presumably once Sec18p/NSF dissassembles the ternary complex (Jahn and Sudhof, 1999), the t-SNARE rapidly rebinds to its SM protein. Alternatively, it may remain bound to its t-SNARE, presumably in a non-inhibitory manner, even during ternary complex formation (Carr et al., 1999).
A second major finding here is that the primary function of the SM protein may be to facilitate SNARE complex formation in vivo rather than to repress it, as has been proposed based on in vitro binding studies (Pevsner et al., 1994a). The stabilized Tlg2p was localized correctly but failed to bind its SNARE partners. These data suggest that there are multiple layers of specificity in order to achieve correct cargo delivery in vivo. It has been shown previously that t-SNAREs can exist in either a closed or an open conformation (Dulubova et al., 1999). SM proteins preferentially bind t-SNAREs in the closed conformation (Dulubova et al., 1999). Based on the present data, we suggest that the major function for SM proteins is not to stabilize the t-SNARE in the closed conformation, because this is the default conformation of the free t-SNARE (Dulubova et al., 1999; Munson et al., 2000), but rather to induce a conformational change in the t-SNARE that primes it for either SNARE complex formation or for interaction with other modulators of SNARE complex assembly, such as members of the exocyst or Rab families (Munson et al., 2000). Such a model accommodates recent findings indicating that, in some instances, SM proteins may interact with their cognate t-SNAREs in a more open conformation (Dulubova et al., 2001), or even when present in a ternary SNARE complex (Carr et al., 1999).
Materials and methods
Reagents
Fixed Staphylococcus aureus cells (IgG Sorb) were obtained from The Enzyme Center (Malden, MA). [35S]Express label was obtained from New England Nuclear (Boston, MA). Oxalyticase was from Enzogenetics (Corvallis, OR) and zymolyase was obtained from Seikagaku Corporation (Tokyo, Japan). Enzymes used in DNA manipulations were from New England Biolabs (Beverly, MA) or Boehringer Mannheim Biochemicals (Indianapolis, IN). Rabbit polyclonal antibodies against Vti1p, Tlg1p, Sed5p and Ufe1p were a gift from John Coe and Wanjin Hong (Coe et al., 1999). Rabbit polyclonal antiserum that recognized Tlg2p was generously provided by Ben Nichols and Hugh Pelham (Holthuis et al., 1998). Additional Tlg2p-specific antiserum was raised in rabbits immunized using a fusion protein consisting of residues 100–276 of Tlg2p fused in-frame to GST, kindly provided by John Coe and Wanjin Hong (Coe et al., 1999). Rabbit polyclonal antibodies against Vps45p, Vam3p and Pgk1p have been described previously (Piper et al., 1994, 1997; Gerrard et al., 2000). Monoclonal antibodies that recognize Pep12p (2C3G4), Vph1p (10D7-A7-B2) and Dpm1p (5C5-A7) are available commercially from Molecular Probes (Eugene, OR).
Plasmids
All DNA manipulations were performed using routine procedures (Sambrook et al., 1989). PNOz13 (VPS45Δ::KanMX4) was constructed by firstly subcloning the 2.3 kb SmaI–SphI fragment containing the VPS45 open reading frame (ORF) from pTS45 (Piper et al., 1994) into pUC19 (Yanisch-Perron et al., 1985). This construct was digested with NheI and Bsu36I to drop out 1.3 kb of the coding region which was replaced with the KANMX4 module (Wach et al., 1994). pNOz7 expresses full-length TLG2 from the GAL1/10 promoter and was constructed by using PCR to amplify the TLG2 ORF and 500 bp downstream sequence. The oligonucleotides used for this contained BamHI restriction endonuclease recognition sites to facilitate subcloning of the PCR product, and were as follows: upstream primer 5′-CGGGATCCATGTTTAGAGATAGAACTAATTTATTTTTAT-3′; downstream primer 5′-CGGGATCCCAAACCAAGTGCCACAGCACAGAAATCC-3′. The resulting PCR product was cloned into the BamHI site of pNOz2. pNOz8 expresses a truncated version of TLG2 (missing bases 1–690 of the ORF). This plasmid was constructed in a manner similar to that used to create pNOz7 except that the upstream primer used in the PCR was as follows: 5′-CGGGATCCATGACGTTGCAGAGACAGCAACAGC-3′. This introduces an initiating methionine just before residue 231 of Tlg2p. pNOz2 was constructed by removing the polylinker region from pRS316 (Sikorski and Hieter, 1989) through a PvuII digest, with the same from YCpGAL (Steel et al., 2000). This introduced the GAL1/10 promoter and polylinker region from YCpGAL into pRS316. Plasmids encoding the cytosolic tails of Tlg2p and Sed5p fused to GST were kindly provided by John Coe and Wanjin Hong (Coe et al., 1999). pRC39 encoding the cytosolic tail of Pep12p fused to GST has been described previously (Tellam et al., 1997). The plasmid encoding GST–Tlg2p-Δ1–230 (the cytosolic tail of Tlg2p lacking the putative Habc domain, residues 1–230 fused to GST) is encoded by pNoz9. pNOz9 was constructed by using the upstream primer described above for the construction of pNOz8 and the downstream primer 5′-GCACCTAGGGGCCCTTAAGTAGCA-3′ to amplify the desired coding region from the GST–Tlg2p-encoding plasmid E204. For in vitro transcription/translation, VPS45 was amplified using PCR and cloned into the BamHI and XbaI sites of pBluecript (Promega).
Strains
All yeast strains used in these studies are described in Table I. Yeast strains were constructed using standard genetic techniques and grown in rich media (YEPD; 1% yeast extract, 1% peptone, 2% dextrose) or standard minimal medium using either glucose (SD), or raffinose and galactose (SGal/Raff) as carbon sources [all to 2% (w/v)], lacking appropriate amino acids (Sherman et al., 1986). NOzY1, NOzY2, NOzY11, NOzY13 and NOzY8 were constructed by disrupting VPS45 in SF838-9D, RPY10, LHY55, LHY58 and LHY913, respectively. In all cases, VPS45 was disrupted using SmaI–SphI-digested pNOz13. NOzY3 and NOzY4 were derived from SF838-9D and RPY10, respectively. In both cases, the TLG2 locus was replaced with the KANMX4 module using the disruption cassette kindly provided by Howard Reizman (Seron et al., 1998).
Table I. Yeast strains used in this study.
| Strain | Genotype | Source |
|---|---|---|
| RPY10 | MATα ura3-52 leu2-3 112 his4-519 ade6 gal2 | Piper et al. (1994) |
| SF838-9D | MATα ura3-52 leu2-3 112 his4-519 ade6 gal2 pep4-3 | Rothman and Stevens (1986) |
| SGY39 | MATα ura3-52 leu2-3 112 his4-519 ade6 gal2 pep4-3 pep12Δ::Kanr | Gerrard et al. (2000) |
| NOzY3 | MATα ura3-52 leu2-3 112 his4-519 ade6 gal2 pep4-3 tlg2Δ::Kanr | this study |
| NOzY4 | MATα ura3-52 leu2-3 112 his4-519 ade6 gal2 tlg2Δ::Kanr | this study |
| NOzY1 | MATa ura3-52 leu2-3 112 his4-519 ade6 gal2 pep4-3 vps45Δ::Kanr | this study |
| NOzY2 | MATα ura3-52 leu2-3 112 his4-519 ade6 gal2 vps45Δ::Kanr | this study |
| LHY55 | MATa ura3-52 leu2-3 112 his3-11,15 | Linda Hicke |
| LHY58 | MATa ura3-52 leu2-3 112 his3-11,15 pre1-1 pre2-2 | Linda Hicke |
| NOzY11 | MATa ura3-52 leu2-3 112 his3-11,15 vps45Δ::Kanr | this study |
| NOzY13 | MATa ura3-52 leu2-3 112 his3-11,15 pre1-1 pre2-2 vps45Δ::Kanr | this study |
| LHY913 | MATa ura3-52 leu2Δ-1 his3Δ-200 cim3-1 | Linda Hicke |
| NOzY8 | MATa ura3-52 leu2Δ-1 his3Δ-200 cim3-1 vps45Δ::Kanr | this study |
| RPY15 | MATα ura3-52 leu2-3 112 his4-519 ade6 gal2 pep4-3 vps45-13 | Piper et al. (1995) |
SF838-9D, SGY39, NOzY1, NOzY2, NOzY3, NOzY4 and NozY14 are congenic to RPY10; LHY58, NOzY11 and NOzY13 are congenic to LHY55; and NOzY8 is congenic to LHY913.
Immunoblot analysis
Steady-state levels of SNARE proteins were assessed using immunoblot analysis of whole-cell extracts (WCEs). These were prepared by vortexing cells harvested from 10 OD600 equivalents in early log phase in 200 µl of Laemmli sample buffer in the presence of glass beads (425–600 µm, acid washed). Glass beads were allowed to settle and 10 µl of the lysate was loaded per lane of an SDS–polyacrylamide gel. Proteins were next transferred to nitrocellulose and standard immunoblot analyses carried out using the antibodies described.
Subcellular fractionation
Cells were fractionated using differential centrifugation following osmotic lysis as previously described (Horazdovsky and Emr, 1993). Cells were grown in rich media (10 ml) to OD600 = 1 and then incubated in 1 ml of 50 mM Tris–HCl, pH 8, 1% 2-mercaptoethanol for 10 min at 30°C. Cells were then converted to spheroplasts by treatment with zymolyase (150 mg/ml) in 1 ml of 1.2 M sorbitol, 50 mM potassium phosphate pH 7.5, 1 mM magnesium chloride at 30°C for 45 min. Spheroplasts were washed once in 1.2 M sorbitol prior to osmotic lysis in 1.3 ml of cold 0.2 M sorbitol, 50 mM Tris–HCl pH 7.5, 1 mM EDTA. Unbroken cells were removed by centrifugation for 5 min at 500 g (this and all subsequent centrifugation steps were performed at 4°C), yielding 1.2 ml of WCE. A 1.0 ml aliquot of WCE was subjected to centrifugation at 13 000 g for 10 min to yield a P13 (pellet) fraction. The resulting supernatant fraction was subjected to centrifugation at 100 000 g for 30 min, yielding P100 (pellet) and S100 (supernatant) fractions. The P13 and P100 pellets were resuspended in 200 µl of 8 M urea, 5% SDS, 40 mM Tris–HCl pH 6.8, 0.1 mM EDTA, 0.4 mg/ml bromophenol blue, 5% 2-mercaptoethanol. Proteins were precipitated from the S100 and 0.2 ml of the WCE using 10% trichloroacetic acid (TCA), and resuspended in 200 and 40 µl of 8 M urea, 5% SDS, 40 mM Tris–HCl pH 6.8, 0.1 mM EDTA, 0.4 mg/ml bromophenol blue, 5% 2-mercaptoethanol, respectively.
For gradient fractionation of membranes, 200 OD600 equivalents of mid-log cells were harvested and lysed as described above in the presence of protease inhibitors [0.1 mM phenylmethylsulfonyl fluoride (PMSF), 0.1 µg/ml each of pepstatin, leupeptin and chymostatin]. Following removal of unbroken cells by centrifugation for 5 min at 500 g, total membranes were collected by centrifugation at 100 000 g for 30 min. Membranes were resuspended in 40% sorbitol, 10 mM triethanolamine pH 7.2, 10 mM sodium azide, 1 mM EDTA plus protease inhibitors and layered onto a self-forming 40–65% sorbitol gradient (Cleves et al., 1991). After centrifugation at 200 000 g for 40 h, the gradient was fractionated and the presence of various marker proteins in each of the fractions was determined by immunoblot analysis.
In vitro binding assays
VPS45 was transcribed and translated in vitro using a coupled reticulocyte lysate system (Promega) supplemented with [35S]methionine. The ability of the labelled Vps45p to bind to the GST fusion proteins was determined as described by Nielsen et al. (2000).
Pulse–chase radiolabelling and denaturing immunoprecipitation of Tlg2p and CPY
The fate of newly synthesized Tlg2p was followed by immunoprecipitation of the protein from cells that had incorporated radiolabelled methionine into proteins synthesized during a 5 min time period as previously described for other integral membrane proteins (Nothwehr et al., 1995). Briefly, yeast cultures were grown overnight at 30°C in synthetic minimal medium lacking methionine to OD600 = 1. The 0.5 OD600 of cells per time point to be analysed were transferred to fresh medium and incubated at 30°C for 10 min prior to labelling. Labelling was initiated by the addition of 100 µCi of [35S]Express label per 0.5 OD600 of cells. After 5 min, the chase was initiated by the addition of excess unlabelled methionine and cysteine (final concentration of 100 µg/ml). The chase was terminated by addition of sodium azide to 10 mM and chilling the cells on ice. Cells were converted to spheroplasts and lysed using 1% SDS, 8 M urea. Lysates were then adjusted to 0.1% SDS, 0.1% Triton X-100, 0.8 M urea and 20 mM Tris–HCl pH 8.0 prior to the addition 1 µl of antiserum that specifically recognized Tlg2p. The immunoprecipitation of radiolabelled CPY from intracellular and extracellular fractions was performed as previously described (Piper et al., 1994).
Tlg2p complex immunoprecipitations
Tlg2p-containing complexes were immunoprecipitated as previously described (Abeliovich et al., 1998). Briefly, sodium azide was added to 10 mM to 50 OD600 equivalents of cells in early log phase cultures. The cells were then washed using 10 ml of HKDNE buffer (50 mM HEPES pH 7.4, 150 mM KCl, 1 mM dithiothreitol, 0.5% NP-40, 1 mM EDTA) supplemented with 1 mM PMSF, and resuspended in 500 µl of the same. Cell lysis was achieved by vortexing for 3 min in the presence of glass beads (425–600 µm, acid washed). Lysates were diluted to 1 mg/ml protein and then cleared by centrifugation for 15 min at 16 000 g. A 1 ml aliquot of lysate was added to 20 µl of protein A–Sepharose beads coupled to IgG from either anti-Tlg2p antiserum or pre-immune serum (antibodies were covalently cross-linked to protein A–Sepharose beads using dimethyl pimelimidate⋅2HCl: 5 µl of serum bound to 20 µl of beads). Following incubation at 4°C for 6 h, the protein A–Sepharose beads were washed five times with HKDNE buffer. Specifically bound proteins were eluted from the beads using 100 mM glycine pH 2.8, 150 mM NaCl. Samples were neutralized using 1 M Tris–HCl pH 8 prior to boiling in Laemmli sample buffer and separation using SDS–PAGE, and immunoblot analysis using antibodies that specifically recognize Tlg1p and Vti1p. For experiments involving a temperature shift, cells were grown overnight at 25°C to early-log phase. Then 50 OD600 equivalents were harvested from these cultures and resuspended in YEPD medium that had been pre-warmed to 37°C, before incubation at 37°C was initiated.
Acknowledgments
Acknowledgements
The authors would like to thank John Coe, Wanjin Hong, Ben Nichols, Hugh Pelham, Linda Hicke, Howard Reizman and Tom Stevens for plasmids, antibodies and strains, Suzie Verma and Charlotte Widberg for their technical assistance, and Sonja Gerrard, Sally Martin, Rob Parton, Rohan Teasdale and members of the James’ lab for discussions during the course of this work. This work was supported by a project grant from the National Health and Medical Research Council of Australia. The IMB is a special Research Centre of the Australian Research Council.
References
- Abeliovich H., Grote,E., Novick,P. and Ferro-Novick,S. (1998) Tlg2p, a yeast syntaxin homolog that resides on the Golgi and endocytic structures. J. Biol. Chem., 273, 11719–11727. [DOI] [PubMed] [Google Scholar]
- Abeliovich H., Darsow,T. and Emr,S.D. (1999) Cytoplasm to vacuole trafficking of aminopeptidase I requires a t-SNARE–Sec1p complex composed of Tlg2p and Vps45p. EMBO J., 18, 6005–6016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burd C.G., Peterson,M., Cowles,C.R. and Emr,S.D. (1997) A novel Sec18p/NSF-dependent complex required for Golgi-to-endosome transport in yeast. Mol. Biol. Cell, 8, 1089–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr C.M., Grote,E., Munson,M., Hughson,F.M. and Novick,P.J. (1999) Sec1p binds to SNARE complexes and concentrates at sites of secretion. J. Cell Biol., 146, 333–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleves A.E., McGee,T.P., Whitters,E.A., Champion,K.M., Aitken,J.R., Dowhan,W., Goebl,M. and Bankaitis,V.A. (1991) Mutations in the CDP–choline pathway for phospholipid biosynthesis bypass the requirement for an essential phospholipid transfer protein. Cell, 64, 789–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coe J.G., Lim,A.C., Xu,J. and Hong,W. (1999) A role for Tlg1p in the transport of proteins within the Golgi apparatus of Saccharomyces cerevisiae. Mol. Biol. Cell, 10, 2407–2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowles C.R., Emr,S.D. and Horazdovsky,B.F. (1994) Mutations in the VPS45 gene, a SEC1 homologue, result in vacuolar protein sorting defects and accumulation of membrane vesicles. J. Cell Sci., 107, 3449–3459. [DOI] [PubMed] [Google Scholar]
- Dulubova I., Sugita,S., Hill,S., Hosaka,M., Fernandez,I., Sudhof,T.C. and Rizo,J. (1999) A conformational switch in syntaxin during exocytosis: role of munc18. EMBO J., 18, 4372–4382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulubova I., Yamaguchi,T., Wang,Y., Sudhof,T.C. and Rizo,J. (2001) Vam3p structure reveals conserved and divergent properties of syntaxins. Nature Struct. Biol., 8, 258–264. [DOI] [PubMed] [Google Scholar]
- Gerrard S.R., Bryant,N.J. and Stevens,T.H. (2000) VPS21 controls entry of endocytosed and biosynthetic proteins into the yeast prevacuolar compartment. Mol. Biol. Cell, 11, 613–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghislain M., Udvardy,A. and Mann,C. (1993) S.cerevisiae 26S protease mutants arrest cell division in G2/metaphase. Nature, 366, 358–362. [DOI] [PubMed] [Google Scholar]
- Halachmi N. and Lev,Z. (1996) The Sec1 family: a novel family of proteins involved in synaptic transmission and general secretion. J. Neurochem., 66, 889–897. [DOI] [PubMed] [Google Scholar]
- Hata Y., Slaughter,C.A. and Sudhof,T.C. (1993) Synaptic vesicle fusion complex contains unc-18 homologue bound to syntaxin. Nature, 366, 347–351. [DOI] [PubMed] [Google Scholar]
- Heinemeyer W., Kleinschmidt,J.A., Saidowsky,J., Escher,C. and Wolf,D.H. (1991) Proteinase yscE, the yeast proteasome/multicatalytic–multifunctional proteinase: mutants unravel its function in stress induced proteolysis and uncover its necessity for cell survival. EMBO J., 10, 555–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinemeyer W., Gruhler,A., Mohrle,V., Mahe,Y. and Wolf,D.H. (1993) PRE2, highly homologous to the human major histocompatibility complex-linked RING10 gene, codes for a yeast proteasome subunit necessary for chrymotryptic activity and degradation of ubiquitinated proteins. J. Biol. Chem., 268, 5115–5120. [PubMed] [Google Scholar]
- Hickson G.R., Chamberlain,L.H., Maier,V.H. and Gould,G.W. (2000) Quantification of SNARE protein levels in 3T3-L1 adipocytes: implications for insulin-stimulated glucose transport. Biochem. Biophys. Res. Commun., 270, 841–845. [DOI] [PubMed] [Google Scholar]
- Holthuis J.C., Nichols,B.J., Dhruvakumar,S. and Pelham,H.R. (1998) Two syntaxin homologues in the TGN/endosomal system of yeast. EMBO J., 17, 113–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horazdovsky B.F. and Emr,S.D. (1993) The VPS16 gene product associates with a sedimentable protein complex and is essential for vacuolar protein sorting in yeast. J. Biol. Chem., 268, 4953–4962. [PubMed] [Google Scholar]
- Jahn R. and Sudhof,T.C. (1999) Membrane fusion and exocytosis. Annu. Rev. Biochem., 68, 863–911. [DOI] [PubMed] [Google Scholar]
- Munson M., Chen,X., Cocina,A.E., Schultz,S.M. and Hughson,F.M. (2000) Interactions within the yeast t-SNARE Sso1p that control SNARE complex. Nature Struct. Biol., 7, 894–902. [DOI] [PubMed] [Google Scholar]
- Nichols B.J., Holthuis,J.C. and Pelham,H.R. (1998) The Sec1p homologue Vps45p binds to the syntaxin Tlg2p. Eur. J. Cell Biol., 77, 263–268. [DOI] [PubMed] [Google Scholar]
- Nicholson K.L., Munson,M., Miller,R.B., Filip,T.J., Fairman,R. and Hughson,F.M. (1998) Regulation of SNARE complex assembly by an N-terminal domain of the t-SNARE Sso1p. Nature Struct. Biol., 5, 793–802. [DOI] [PubMed] [Google Scholar]
- Nielsen E., Christoforidis,S., Uttenweiler-Joseph,S., Miaczynska,M., Dewitte,F., Wilm,M., Hoflack,B. and Zerial,M. (2000) Rabenosyn-5, a novel Rab5 effector, is complexed with hVPS45 and recruited to endosomes through a FYVE finger domain. J. Cell Biol., 151, 601–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nothwehr S.F., Conibear,E. and Stevens,T.H. (1995) Golgi and vacuolar membrane proteins reach the vacuole in vps1 mutant yeast cells via the plasma membrane. J. Cell Biol., 129, 35–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novick P. and Schekman,R. (1979) Secretion and cell-surface growth are blocked in a temperature-sensitive mutant of Saccharomyces cerevisiae. Proc. Natl Acad. Sci. USA, 76, 1858–1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parlati F., Weber,T., McNew,J.A., Westermann,B., Sollner,T.H. and Rothman,J.E. (1999) Rapid and efficient fusion of phospholipid vesicles by the α-helical core of a SNARE complex in the absence of an N-terminal regulatory domain. Proc. Natl Acad. Sci. USA, 96, 12565–12570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pevsner J., Hsu,S.C., Braun,J.E., Calakos,N., Ting,A.E., Bennett,M.K. and Scheller,R.H. (1994a) Specificity and regulation of a synaptic vesicle docking complex. Neuron, 13, 353–361. [DOI] [PubMed] [Google Scholar]
- Pevsner J., Hsu,S.C. and Scheller,R.H. (1994b) n-Sec1: a neural-specific syntaxin-binding protein. Proc. Natl Acad. Sci. USA, 91, 1445–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piper R.C., Whitters,E.A. and Stevens,T.H. (1994) Yeast Vps45p is a Sec1p-like protein required for the consumption of vacuole-targeted, post-Golgi transport vesicles. Eur. J. Cell Biol., 65, 305–318. [PubMed] [Google Scholar]
- Piper R.C., Bryant,N.J. and Stevens,T.H. (1997) The membrane protein alkaline phosphatase is delivered to the vacuole by a route that is distinct from the VPS-dependent pathway. J. Cell Biol., 138, 531–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riento K., Kauppi,M., Keranen,S. and Olkkonen,V.M. (2000) Munc18-2, a functional partner of syntaxin 3, controls apical membrane trafficking in epithelial cells. J. Biol. Chem., 275, 13476–13483. [DOI] [PubMed] [Google Scholar]
- Rothman J.E. (1994) Mechanisms of intracellular protein transport. Nature, 372, 55–63. [DOI] [PubMed] [Google Scholar]
- Rothman J.H. and Stevens,T.H. (1986) Protein sorting in yeast: mutants defective in vacuole biogenesis mislocalize vacuolar proteins into the late secretory pathway. Cell, 47, 1041–1051. [DOI] [PubMed] [Google Scholar]
- Sambrook J.E., Fritsch,E.F. and Maniatis,T. (1989) Molecular Cloning: A Laboratory Manual, 2nd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 53–105.
- Schekman R. (1992) Genetic and biochemical analysis of vesicular traffic in yeast. Curr. Opin. Cell Biol., 4, 587–592. [DOI] [PubMed] [Google Scholar]
- Schimmoller F., Simon,I. and Pfeffer,S.R. (1998) Rab GTPases, directors of vesicle docking. J. Biol. Chem., 273, 22161–22164. [DOI] [PubMed] [Google Scholar]
- Seron K. et al. (1998) A yeast t-SNARE involved in endocytosis. Mol. Biol. Cell, 9, 2873–2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman F., Fink,G.R. and Hicks,J. (1986) Methods in Yeast Genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 146–153.
- Sikorski R.S. and Hieter,P. (1989) A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics, 122, 19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steel G.J., Harley,C., Boyd,A. and Morgan,A. (2000) A screen for dominant negative mutants of SEC18 reveals a role for the AAA protein consensus sequence in ATP hydrolysis. Mol. Biol. Cell, 11, 1345–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tellam J.T., James,D.E., Stevens,T.H. and Piper,R.C. (1997) Identific ation of a mammalian Golgi Sec1p-like protein, mVps45. J. Biol. Chem., 272, 6187–6193. [DOI] [PubMed] [Google Scholar]
- Thurmond D.C., Ceresa,B.P., Okada,S., Elmendorf,J.S., Coker,K. and Pessin,J.E. (1998) Regulation of insulin-stimulated GLUT4 translocation by Munc18c in 3T3L1 adipocytes. J. Biol. Chem., 273, 33876–33883. [DOI] [PubMed] [Google Scholar]
- Verhage M. et al. (2000) Synaptic assembly of the brain in the absence of neurotransmitter secretion. Science, 287, 864–869. [DOI] [PubMed] [Google Scholar]
- Wach A., Brachat,A., Pohlmann,R. and Philippsen,P. (1994) New heterologous modules for classical or PCR-based gene disruptions in Saccharomyces cerevisiae. Yeast, 10, 1793–1808. [DOI] [PubMed] [Google Scholar]
- Webb G.C., Hoedt,M., Poole,L.J. and Jones,E.W. (1997) Genetic interactions between a pep7 mutation and the PEP12 and VPS45 genes: evidence for a novel SNARE component in transport between the Saccharomyces cerevisiae Golgi complex and endosome. Genetics, 147, 467–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolford C.A., Daniels,L.B., Park,F.J., Jones,E.W., Van Arsdell,J.N. and Innis,M.A. (1986) The PEP4 gene encodes an aspartyl protease implicated in the posttranslational regulation of Saccharomyces cerevisiae vacuolar hydrolases. Mol. Cell. Biol., 6, 2500–2510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang B., Steegmaier,M., Gonzalez,L.C.,Jr and Scheller,R.H. (2000) nSec1 binds a closed conformation of syntaxin1A. J. Cell Biol., 148, 247–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanisch-Perron C., Vieira,J. and Messing,J. (1985) Improved M13 phage cloning vectors and host strains: nucleotide sequences of the M13mp18 and pUC19 vectors. Gene, 33, 103–119. [DOI] [PubMed] [Google Scholar]