

Abstract
There is increasing interest in elucidating the mechanisms involved in the negative regulation of lymphocyte activation. Herein, we show that the cytosolic protein tyrosine phosphatase PTP-PEST is expressed abundantly in a wide variety of haemopoietic cell types, including B cells and T cells. In a model B-cell line, PTP-PEST was found to be constitutively associated with several signalling molecules, including Shc, paxillin, Csk and Cas. The interaction between Shc and PTP-PEST was augmented further by antigen receptor stimulation. Overexpression studies, antisense experiments and structure–function analyses provided evidence that PTP-PEST is an efficient negative regulator of lymphocyte activation. This function correlated with the ability of PTP-PEST to induce dephosphorylation of Shc, Pyk2, Fak and Cas, and inactivate the Ras pathway. Taken together, these data suggest that PTP-PEST is a novel and unique component of the inhibitory signalling machinery in lymphocytes.
Keywords: inhibition/lymphocyte/phosphatase/PTP-PEST/Shc
Introduction
Antigen receptors on T and B cells play critical roles in lymphocyte development and activation (Weiss and Littman, 1994; Qian and Weiss, 1997; Benschop and Cambier, 1999). Depending on the biological context, their engagement can lead to cell activation, differentiation, anergy, tolerance or death. Accumulating evidence indicates that these various responses invariably are initiated by intracellular protein tyrosine phosphorylation. Whereas antigen receptors are devoid of intrinsic protein tyrosine kinase (PTK) activity, they are associated with subunits containing in their cytoplasmic region a short tyrosine-based signalling motif termed ITAM (for immunoreceptor tyrosine-based activation motif). This motif allows the recruitment of cytoplasmic PTKs.
Following antigen receptor stimulation, the ITAMs undergo rapid phosphorylation at two distinct tyrosines (Chan et al., 1994). Genetic and biochemical data show that this phosphorylation is mediated by the Src family of cytoplasmic PTKs (Peri and Veillette, 1994; Weiss and Littman, 1994; Chow and Veillette, 1995). In turn, ITAM tyrosine phosphorylation creates docking sites permitting the binding and activation of another group of PTKs, the Zap-70/Syk family (van Oers and Weiss, 1995). Through their capacity to activate phosphatidylinositol (PI) 3′ kinase, activated Src and Zap-70/Syk kinases also trigger the membrane recruitment and activation of a third class of PTKs, the Tec/Btk family (Yang et al., 2000). In combination, these three types of kinases are responsible for phosphorylation of several substrates during cell activation, including phospholipase C (PLC)-γ, Vav, Cbl, SLP-76, Blnk/SLP-65, LAT and Shc. These targets link antigen receptor stimulation to downstream pathways such as the Ras–MAPK cascade, calcium-regulated events, lipid metabolism and the actin cytoskeleton. Ultimately, they trigger effector functions.
Over the past few years, significant interest has been directed towards understanding the mechanisms involved in the negative regulation of lymphocyte activation. There is clear indication that protein tyrosine phosphatases (PTPs) such as SHP-1 and PEP play significant roles in this process (Cyster and Goodnow, 1995; Pani et al., 1995, 1996; Carter et al., 1999; Cloutier and Veillette, 1999; Gjorloff-Wingren et al., 1999; Zhang et al., 1999). SHP-1 and PEP apparently act by dephosphorylating proximal components of the antigen receptor signalling cascade, including Src kinases, ITAMs, Zap-70/Syk-related PTKs and/or central linker proteins such as Blnk/SLP-65 and SLP-76. Consequent to their actions, they induce a global reduction in immunoreceptor-mediated protein tyrosine phosphorylation, thereby preventing lymphocyte activation.
PTP-PEST is a 120 kDa cytosolic PTP belonging to the PEP family (Yang et al., 1993; Charest et al., 1995). Unlike its relatives PEP and PTP-HSCF, which are contained in selected haemopoietic cell types, PTP-PEST is expressed ubiquitously. However, higher levels of PTP-PEST are found in haemopoietic tissues (Davidson et al., 1997). Previous studies have revealed that PTP-PEST can physically associate with several signal transduction molecules, including the adaptor molecule Shc, the focal adhesion proteins paxillin, Hic-5, Cas, CasL (or HEF-1) and Sin, and the inhibitory PTK Csk (Habib et al., 1994; Charest et al., 1996; Davidson et al., 1997; Garton et al., 1997; Shen et al., 1998; Coté et al., 1999; Nishiya et al., 1999). These interactions occur via sequences positioned outside the catalytic domain of PTP-PEST (Figure 1A).

Fig. 1. Structure and expression pattern of PTP-PEST in haemopoietic cells. (A) The primary structure of PTP-PEST is depicted graphically. Two residues in the catalytic domain (Cys231 and Arg237), that are critical for phosphatase activity, are highlighted. The positions of the P1, P2, P3 and NPLH sequences in the C-terminal non-catalytic domain are also shown. (B) Expression pattern of PTP-PEST in mouse haemopoietic cells. The accumulation of PTP-PEST in various purified populations of mouse haemopoietic cells was examined by immunoblotting of equivalent amounts of cell lysates with a rabbit anti-PTP-PEST (PEST) serum. The identity of the higher molecular weight polypeptides found to react with the anti-PTP-PEST sera in this immunoblot is not known. The presence of comparable quantities of cellular proteins in each lane was verified by staining of the immunoblot membrane with Coomassie blue (data not shown). The position of PTP-PEST is indicated on the left. Exposure: 25 s (ECL).
A significant insight into the function of PTP-PEST has been provided by analyses of fibroblast cell lines having altered levels of PTP-PEST. Overexpression of PTP-PEST in rat fibroblasts was shown to cause a reduction in cell migration (Garton and Tonks, 1999). Conversely, whereas ablation of PTP-PEST expression in the mouse germline causes early embryonic lethality, embryo fibroblasts derived from these mice were demonstrated to exhibit an increase in cell spreading and a strong defect in motility (Angers-Loustau et al., 1999). Hence, PTP-PEST would appear to be a critical regulator of cytoskeletal organization in non-haemopoietic cells, possibly through its capacity to dephosphorylate focal adhesion proteins.
Here we have examined the possible involvement of PTP-PEST in the regulation of haemopoietic cell functions. We find that PTP-PEST is expressed abundantly in various haemopoietic cell types, including B cells and T cells. Through overexpression and antisense experiments, evidence is provided that PTP-PEST is an efficient negative regulator of antigen receptor-induced B- and T-cell activation. Furthermore, biochemical studies and structure–function analyses indicate that this function correlates with the ability of PTP-PEST to dephosphorylate a restricted set of tyrosine phosphorylation substrates in activated lymphocytes, including Shc, Cas, Pyk2 and Fak, and prevent activation of the Ras signalling cascade.
Results
PTP-PEST is broadly expressed in haemopoietic cells
Even though PTP-PEST is expressed in a wide range of tissues, it accumulates in greater amounts in spleen and thymus (Davidson et al., 1997). To examine further the distribution of PTP-PEST in haemopoietic cells, normal mouse haemopoietic cell types were purified as outlined in Materials and methods. Equivalent quantities of cell lysates were then probed by immunoblotting with an antiserum directed against PTP-PEST (Figure 1B). This analysis revealed that the PTP-PEST protein was present in splenic B cells (lane 1), thymocytes (lane 2), splenic T cells (lane 3), natural killer (NK) cells (lane 4) and bone marrow-derived mast cells (BMMCs) (lane 5). The abundance of PTP-PEST in these cells was in the same range as that observed in spleen (lane 6) and thymus (lane 7).
Association of PTP-PEST with signalling molecules in lymphocytes
Due to the importance of protein tyrosine phosphorylation for lymphocyte activation, we investigated whether PTP-PEST might have a regulatory role in this process. First, the capacity of PTP-PEST isolated from lymphocytes to interact with signalling proteins known to associate with PTP-PEST in non-haemopoietic cells was ascertained (Figure 2). To this end, we used the IgG+ mouse B-cell line A20. This cell line expresses PTP-PEST, but not the related phosphatases PEP and PTP-HCSF (our unpublished results). A20 cells were lysed in non-ionic detergent-containing buffer, and the ability of PTP-PEST to associate with these molecules was studied by immunoblotting of the appropriate immunoprecipitates with anti-PTP-PEST antibodies (Figure 2A). As documented for non-haemopoietic cells (Charest et al., 1996; Davidson et al., 1997; Garton et al., 1997; Shen et al., 1998; Coté et al., 1999), a significant proportion of PTP-PEST polypeptides expressed in A20 cells was found to be associated with Shc (lane 1), Csk (lane 2) and paxillin (lane 3). Small amounts were also observed in anti-Cas immunoprecipitates (lane 4), although their detection required longer autoradiographic exposures (data not shown). No PTP-PEST was detected in immunoprecipitates generated with normal rabbit serum (lane 5). Taking into account the total quantity of PTP-PEST in A20 cells, it was estimated that at least 25, 10, 5 and 2% was complexed with Shc, paxillin, Csk and Cas, respectively (data not shown).

Fig. 2. Association of PTP-PEST with signalling molecules in mouse A20 B cells. IgG+ A20 B cells were lysed in NP-40-containing buffer, and the ability of PTP-PEST to associate with other polypeptides was determined by immunoblotting of the indicated immunoprecipitates with an anti-PTP-PEST (PEST) serum. The position of PTP-PEST is highlighted by an arrow on the left. (A) Association of PTP-PEST with signalling molecules in unstimulated A20 cells. NRS: normal rabbit serum. Exposure: 60 s (ECL). (B) Effects of BCR stimulation or PMA treatment on the association of PTP-PEST with signalling molecules. A20 cells were either left unstimulated or stimulated for 10 min with F(ab′)2 fragments of SAM IgG antibodies (20 µg/ml) or PMA (100 ng/ml), prior to cell lysis and immunoprecipitation. Exposures: first panel, 3 h; second, third and fourth panels, 10 s (ECL). (C) Time course of BCR stimulation. Cells were stimulated for the indicated times with F(ab′)2 fragments of SAM IgG antibodies. The accumulation of phosphotyrosine-containing proteins was monitored by immunoblotting of total cell lysates with anti-phosphotyrosine (P.tyr) antibodies (first panel), whereas the presence of Shc–PTP-PEST complexes was detected by immunoblotting of Shc immunoprecipitates with an anti-PTP-PEST serum (second panel). Tyrosine phosphorylation of total Shc (third and fourth panels) and PTP-PEST-bound Shc (fifth and sixth panels) was analysed in parallel. Note that whereas both the 52 and 46 kDa Shc isoforms were recovered in Shc immunoprecipitates (fourth panel), only the 52 kDa variant was associated with PTP-PEST (sixth panel). This presumably is because the 46 kDa Shc protein lacks part of the PTB domain, which mediates the binding to PTP-PEST. The migration of pre-stained molecular mass markers is indicated on the right. Exposures: first panel, 14 h; second panel, 4 h; third panel, 15 h; fourth panel, 2.5 h; fifth panel, 15 h; sixth panel, 2 h.
The impact of antigen receptor-induced activation on these interactions was characterized next (Figure 2B). A20 cells were stimulated for 10 min with F(ab′)2 fragments of sheep anti-mouse (SAM) IgG antibodies, and the associations were monitored as detailed for Figure 2A. B-cell receptor (BCR) stimulation was noted to provoke a 2- to 3-fold increase in the interaction of PTP-PEST with Shc (first panel, compare lanes 1 and 2). Between 50 and 60% of PTP-PEST became associated with Shc under these conditions. A similar effect was seen when cells were treated with phorbol 12-myristate 13-acetate (PMA) (lane 3), in agreement with an earlier report (Habib et al., 1994). In contrast, the degree of association of PTP-PEST with paxillin (second panel), Csk (third panel) and Cas (fourth panel) was not influenced by treatment with either anti-BCR antibodies (lane 2) or PMA (lane 3). In a time course of BCR stimulation (Figure 2C), it was observed that the induction of the Shc–PTP-PEST interaction was maximal after 10 min of activation (second panel, lane 3), and persisted for at least 40 min (lane 5). This increased stoichiometry was maintained at the later time points despite the progressive disappearance of overall protein tyrosine phosphorylation (first panel) and, in particular, tyrosine phosphorylation of Shc (third panel). It is noteworthy that the pool of Shc molecules that was associated specifically with PTP-PEST (fifth and sixth panels) underwent some degree of tyrosine phosphorylation in response to BCR stimulation. This observation implied that, in the event that PTP-PEST was responsible for Shc dephosphorylation (see below), there could be a window of co-existence of Shc tyrosine phosphorylation and association with PTP-PEST.
Inhibition of immunoreceptor-mediated cellular activation by PTP-PEST
In light of the high expression levels of PTP-PEST in lymphocytes and its ability to associate with molecules implicated in immunoreceptor signalling, we wanted to evaluate more specifically its effect on lymphocyte activation. Given the lethal impact of ptp-pest gene ablation in the mouse germline (Angers-Loustau et al., 1999), transfection experiments in model cell lines were used for this purpose. A20 B cells were stably transfected with a cDNA encoding wild-type mouse PTP-PEST, as detailed in Materials and methods. Monoclonal cell lines overexpressing PTP-PEST were generated by limiting dilution and identified by immunoblotting of total cell lysates with anti-PTP-PEST antibodies (data not shown). Clones expressing PTP-PEST at levels >5 times those observed in control cells were retained (Figure 3A, top). All the cell lines selected for further studies expressed unaltered levels of BCR, CD45, CD40 and FcγRIIB at the cell surface (data not shown).

Fig. 3. Effect of PTP-PEST on antigen receptor-induced activation. (A) Stable overexpression of PTP-PEST in A20 B cells. Stable transfectants overexpressing wild-type PTP-PEST (wt PEST) or expressing the neomycin resistance marker alone (Neo) were stimulated with the indicated concentrations of F(ab′)2 fragments of RAM IgG antibodies (abscissa), as detailed in Materials and methods. The accumulation of IL-2 in the supernatant was assessed by measuring tritiated thymidine incorporation in the IL-2-dependent indicator cell line HT-2 (ordinate). All assays were done in triplicate and repeated at least 10 times. The quantities of PTP-PEST contained in the various cell lines used in this report were determined by immunoblotting of total cell lysates with anti-PTP-PEST antibodies (top panel). Lane 1, Neo.1; lane 2, Neo.2; lane 3, Neo.3; lane 4, Neo.5; lane 5, Neo.6; lane 6, wtPEST.70 (18); lane 7, wtPEST.35 (4); lane 8, wtPEST.30 (9); lane 9, wtPEST.38 (11); lane 10, wtPEST.42 (15); lane 11, wtPEST.64 (16); lane 12, wtPEST.70 (18); lane 13, wtPEST.31 (4.5); lane 14, wtPEST.41 (5); lane 15, wtPEST.71 (6) (fold PTP-PEST overexpression is shown in parentheses). The position of PTP-PEST is indicated on the left. Exposure: 6 h. (B) Transient overexpression of PTP-PEST in A20 B cells. A20 cells were transiently transfected with the vector pXM139 alone or bearing a wild-type mouse ptp-pest cDNA, in the presence of an IL-2 promoter–luciferase reporter construct. Cells were then stimulated with F(ab′)2 fragments of SAM IgG antibodies for 6 h, and luciferase activity was measured in a luminometer as detailed in Materials and methods. Results are represented as the percentage of maximal stimulation obtained with PMA plus ionomycin. Expression levels of PTP-PEST are shown at the top. Exposure: 3 h.
Previous studies have shown that BCR-induced activation of A20 cells results in production of the lymphokine interleukin-2 (IL-2) (Justement et al., 1989; Muta et al., 1994). Hence, in order to study the impact of PTP-PEST in these cells, transfectants overexpressing PTP-PEST or expressing the neomycin resistance marker alone (Neo) were stimulated with various concentrations of F(ab′)2 fragments of rabbit anti-mouse (RAM) IgG antibodies. After 24 h, supernatants were harvested and assayed for production of IL-2 using the IL-2-dependent indicator cell line HT-2 (Figure 3A). This experiment showed that all A20 clones overexpressing PTP-PEST exhibited a pronounced reduction in BCR-induced IL-2 release, in comparison with control Neo clones.
To ensure that the effect of PTP-PEST was not due to compensatory modifications induced in stably transfected cells, transient transfection assays were also performed (Figure 3B). A20 cells were transfected by electroporation with the vector pXM139 alone or bearing a wild-type ptp-pest cDNA, in combination with pIL-2–luciferase, which contains the IL-2 promoter linked to a luciferase reporter. After 40 h, cells were stimulated for 6 h with F(ab′)2 fragments of SAM IgG and assayed for luciferase activity. We found that transient overexpression of PTP-PEST strongly inhibited (∼5-fold) BCR-induced IL-2 promoter activation in A20 cells. In similar experiments conducted using T-cell lines, we observed that PTP-PEST was also able to inhibit T-cell antigen receptor signalling (data not shown).
To help ensure that the inhibitory impact of PTP-PEST on immunoreceptor signalling was not an artefactual consequence of overexpression, the effect of ptp-pest antisense expression was also examined (Figure 4A–C). A similar approach has been used successfully by others to examine the function of endogenous molecules in immune cell signalling (Pimentel-Muinos and Seed, 1999; Suzu et al., 2000). For our studies, a cDNA fragment corresponding to the first 321 nucleotides of the coding region of mouse ptp-pest was cloned in the antisense or sense orientation in the expression vector pXM139. This portion of the ptp-pest cDNA encodes a short non-catalytic region at the N-terminus of PTP-PEST (residues 1–54) and the first 53 amino acids of the phosphatase domain. The short protein potentially encoded by the sense fragment lacks most of the catalytic domain and, thus, is not expected to possess any phosphatase activity.

Fig. 4. Expression of ptp-pest antisense in A20 B cells. (A) Effect of antisense ptp-pest expression on the abundance of PTP-PEST in A20 cells. A20 cells were transiently transfected with 40 µg of empty pXM139 or pXM139 bearing the first 321 nucleotides of the coding sequence of a mouse ptp-pest cDNA, in either the antisense or the sense orientation. After 48 h, equivalent numbers of viable cells were lysed and the abundance of PTP-PEST was examined by immunoblotting of total cell lysates with anti-PTP-PEST antibodies. The migration of PTP-PEST is indicated on the left. Exposure: 7 h. (B) Influence of antisense ptp-pest expression on BCR signalling. A20 cells were transfected as detailed in (A), in the presence of the IL-2 promoter–luciferase reporter construct. After 66 h, they were stimulated with F(ab′)2 fragments of SAM IgG antibodies for 6 h, and luciferase activity was measured as explained in Materials and methods. Data are shown as the percentage of maximal stimulation achieved with PMA plus ionomycin. (C) Impact of antisense syk expression on BCR signalling. As in (B), except that an antisense syk construct was also used. In keeping with the positive regulatory role of Syk in BCR signalling, a small decrease in activation of the reporter plasmid was observed in BCR-stimulated A20 cells expressing the antisense syk plasmid.
A20 B cells were transiently transfected with these plasmids, and the impact on the expression levels of endogenous PTP-PEST was monitored by immunoblotting of total cell lysates with anti-PTP-PEST antibodies (Figure 4A). This analysis revealed that introduction of antisense ptp-pest (lane 3) caused a decrease (∼2.5-fold) in the abundance of PTP-PEST in A20 cells, in comparison with pXM139 alone (lane 1). This effect was observed in several different experiments. As expected, though, transfection of the sense construct (lane 2) had no influence on the abundance of PTP-PEST. Staining of the immunoblot membrane with Coomassie blue confirmed that equivalent amounts of cellular proteins were loaded in each lane (data not shown).
Next, the effect of antisense ptp-pest on BCR signalling was evaluated (Figure 4B). Cells were transfected as outlined for Figure 4A, also in the presence of the IL-2 promoter reporter plasmid. After 3 days, they were stimulated or not with F(ab′)2 fragments of SAM IgG antibodies, and changes in IL-2 promoter-driven luciferase activity were monitored. We found that antisense ptp-pest, but not the sense construct, provoked an appreciable increase in BCR-induced IL-2 promoter activation (Figure 4B). This effect was seen at all concentrations of anti-BCR antibodies used; it was observed in seven independent experiments (data not shown). It is noteworthy that a small increase in IL-2 promoter activity was observed in unstimulated cells expressing antisense ptp-pest. This finding may suggest that PTP-PEST is also part of the machinery repressing the BCR-induced signalling cascade in unstimulated B cells. Note that an increase in BCR-induced lymphokine production was also observed in stable A20 transfectants expressing antisense ptp-pest (data not shown).
Lastly, to ensure that the stimulatory influence of antisense ptp-pest on BCR signalling was not a non-specific consequence of antisense RNA expression, the effect of transient transfection of another antisense construct was examined (Figure 4C). When constructing this vector, we ensured that it was directed against another polypeptide expressed in A20 cells (i.e. Syk), and that it covered a sequence equivalent in length and position to that used for the ptp-pest antisense construct. This experiment showed that, unlike the antisense ptp-pest, the antisense syk plasmid (corresponding to the first 321 nucleotides of the mouse syk cDNA sequence) had no stimulatory effect on BCR-induced activation of the IL-2 promoter. Therefore, in combination, the results shown in Figures 3 and 4 provided a compelling indication that PTP-PEST is a physiological negative regulator of antigen receptor signalling, and that this inhibitory function exists both in B cells and in T cells.
Selective inhibition of immunoreceptor-mediated protein tyrosine phosphorylation by PTP-PEST
To clarify the mechanism(s) by which PTP-PEST caused an inhibition of lymphocyte activation, its impact on BCR-induced protein tyrosine phosphorylation was examined (Figure 5). Stable A20 transfectants were stimulated with F(ab′)2 fragments of SAM IgG antibodies, and the accumulation of phosphotyrosine-containing proteins over time was monitored by immunoblotting of total cell lysates with anti-phosphotyrosine antibodies (Figure 5A). This evaluation showed that the extent and kinetics of tyrosine phosphorylation of most BCR-regulated substrates were not significantly affected by PTP-PEST overexpression (compare lanes 6–10 with lanes 1–5). Notably, however, the BCR-induced tyrosine phosphorylation of polypeptides of ∼125 (p125), 115 (p115) and 54 (p54) kDa were markedly reduced or abolished by augmented PTP-PEST expression. A partial diminution (∼50%) in the phosphotyrosine content of a 150 kDa polypeptide (p150) was also observed.

Fig. 5. Impact of PTP-PEST overexpression on BCR-induced protein tyrosine phosphorylation. (A) Overall protein tyrosine phosphorylation. Representative cell lines expressing the neomycin marker alone (Neo; lanes 1–5) or in combination with wild-type PTP-PEST (lanes 6–10) were stimulated for the indicated times with F(ab′)2 fragments of SAM IgG antibodies. Changes in intracellular protein tyrosine phosphorylation were assessed by immunoblotting of total cell lysates with anti-phosphotyrosine (P.tyr) antibodies. The migrations of pre-stained molecular mass markers are shown on the right, while those of p150, p125, p115 and p54 are indicated on the left. Exposure: 15 h. (B) Tyrosine phosphorylation of specific substrates. Cells expressing the neomycin marker alone (Neo; lane 1) or in combination with wild-type PTP-PEST (PEST; lane 2) were activated for 2.5 min with F(ab′)2 fragments of SAM IgG antibodies. Specific substrates were then immunoprecipitated from cell lysates using the appropriate antibodies, and their phosphotyrosine content was determined by anti-phospho tyrosine (P.tyr) immunoblotting. Equivalent amounts of the various substrates were immunoprecipitated in control cells and in cells overexpressing PTP-PEST (data not shown). The positions of the various polypeptides are indicated on the left. Exposures: SHIP, 9 h; PLCγ2, 30 h; Cas, 21 h; Fak, 30 h; Cbl, 9 h; Pyk2, 9 h; Vav, 9 h; Blnk, 30 h; Syk, 12 h; Dok, 12 h; and Shc, 12 h. (C) Effect of PTP-PEST overexpression on association of Shc with SHIP and Grb2. Cells were activated for the specified times. The extent of association of Shc with SHIP and Grb2 was established by immunoblotting of anti-Shc immunoprecipitates with anti-SHIP (first panel) or anti-Grb2 (second panel) antibodies. The abundance of Shc was also verified by anti-Shc immunoblotting of Shc immunoprecipitates (third panel). The migrations of SHIP, Grb2 and Shc are noted on the left. Exposures: first panel, 13 h; second panel, 39 h; third panel, 2.5 h.
These results implied that PTP-PEST prevented B-cell activation by inducing selective, rather than global, dephosphorylation of BCR-regulated substrates. To identify the targets of PTP-PEST more accurately, individual proteins were immunoprecipitated from activated cells and their phosphotyrosine content was measured by anti-phosphotyrosine immunoblotting (Figure 5B). This experiment demonstrated that enforced PTP-PEST expression abrogated BCR-induced tyrosine phosphorylation of four substrates: Cas (p130), Fak (p125), Pyk2 (p115) and Shc (p54). Furthermore, it abolished the binding of Shc to a 150 kDa tyrosine-phosphorylated protein representing SHIP, a 5′ inositol phosphatase (see below). Overall tyrosine phosphorylation of SHIP was also diminished by ∼50% in PTP-PEST-overexpressing cells. In contrast, PTP-PEST had little (<25% reduction) or no impact on the phosphotyrosine content of several other substrates, including phospholipase C-γ2, Cbl, Vav, Blnk/SLP-65, Syk and Dok. No tyrosine phosphorylation of paxillin was observed either in parental A20 cells or in its PTP-PEST-overexpressing variants (data not shown).
Previous reports have shown that tyrosine phosphorylation of Shc in activated B cells elicits its binding to the Src homology 2 (SH2) domains of SHIP and of the adaptor molecule Grb2 (Harmer and DeFranco, 1997, 1999; Pradhan and Coggeshall, 1997; Ingham et al., 1999). The interaction with SHIP is secured further by an association between the phosphotyrosine-binding (PTB) domain of Shc and sites of tyrosine phosphorylation on SHIP (Lamkin et al., 1997; Pradhan and Coggeshall, 1997). In view of the ability of PTP-PEST to inhibit tyrosine phosphorylation of Shc on the one hand, and associate with the Shc PTB domain on the other hand (Charest et al., 1996), the influence of PTP-PEST overexpression on the binding of Shc to SHIP and Grb2 was ascertained (Figure 5C). A20 transfectants were stimulated as detailed for Figure 5A, and the ability of Shc to interact with these two molecules was examined by immunoblotting of anti-Shc immunoprecipitates with anti-SHIP (top panel) or anti-Grb2 (bottom panel) antibodies. This assay indicated that PTP-PEST overexpression caused a pronounced reduction in the binding of Shc to SHIP and Grb2 in response to BCR stimulation (compare lanes 7–10 with lanes 2–5). It should be pointed out that Shc was also associated to some extent with SHIP and Grb2 in unstimulated A20 cells (lanes 1 and 6). This baseline interaction was not influenced appreciably by overexpression of PTP-PEST.
The ability of PTP-PEST to inhibit lymphocyte activation correlates with its capacity to bind Shc and paxillin
Our preliminary analyses demonstrated that the amounts of PTP-PEST associated with Shc, paxillin, Csk and Cas were greatly augmented in PTP-PEST-overexpressing A20 cells (data not shown). As indicated earlier, PTP-PEST binds to these polypeptides via non-overlapping sequences positioned in the C-terminal non-catalytic region (Figure 1A). Thus, to understand better the mechanism of action of PTP-PEST, the contribution of these individual domains to the inhibitory effect of PTP-PEST in B cells was assessed. Mutants carrying deletions in the P1 region (Cas-binding site), P2 region (Csk-binding site), P3 region (paxillin-binding site) or NPLH sequence (Shc-binding site) were engineered. Furthermore, to address the pertinence of the phosphatase activity of PTP-PEST, variants bearing mutations of critical residues in the catalytic domain (Arg237→ Met237 and Cys231→Ser231 mutants) were created. Monoclonal A20 cell lines stably expressing these mutants were produced according to the procedures detailed above. The expression levels of the various mutants were comparable with those of wild-type PTP-PEST (Figure 6A).

Fig. 6. Structural requirements for inhibition of antigen receptor signalling by PTP-PEST. (A) Expression of PTP-PEST mutants in A20 cells. The expression levels of the various PTP-PEST mutants were determined by immunoblotting of total cell lysates from representative cell lines with anti-PTP-PEST (PEST) antibodies. The migration of PTP-PEST is shown on the left. Exposures: lanes 1–5, 3 h; lanes 6–8, 3 h; lanes 9–12, 6 h. (B–E) Impact of mutations on the ability of PTP-PEST to associate with signalling molecules. The capacity of the PTP-PEST mutants to bind other polypeptides was assessed by immunoblotting of the appropriate immunoprecipitates with anti-PTP-PEST antibodies. The associations of the PTP-PEST mutants with other binding proteins not shown here were not affected (data not shown). The migration of PTP-PEST is indicated on the left. Exposures: (B) 8 h; (C) 3 h; (D) 6 h; and (E) 4 h.
As expected, deletion of the P2 region drastically reduced the ability of PTP-PEST to be co-immunoprecipitated with Csk (Figure 6C, compare lanes 2 and 3), while that of P3 abrogated its interaction with paxillin (Figure 6D, compare lanes 2 and 3). Likewise, removal of the NPLH motif abolished the binding of PTP-PEST to Shc (Figure 6E, compare lanes 2 and 3). Surprisingly, though, deletion of P1 had no appreciable effect on the association of PTP-PEST with Cas (Figure 6B, compare lanes 2 and 3). While this observation may appear inconsistent with earlier reports indicating that Cas interacts with P1 (Garton et al., 1997), it probably indicates that Cas can associate with PTP-PEST through alternative mechanisms. An immune complex phosphatase assay demonstrated that R237M and C231S PTP-PEST lacked measurable catalytic activity towards an exogenous substrate. In contrast, all other mutants had normal phosphatase activity (data not shown).
The influence of the mutations on the ability of PTP-PEST to provoke dephosphorylation of target substrates was assessed. After stimulation of A20 clones with F(ab′)2 fragments of anti-mouse IgG antibodies, individual substrates were recovered by immunoprecipitation and probed by immunoblotting with anti-phosphotyrosine antibodies. The results obtained in several independent experiments were quantitated using a phosphoimager (data not shown), and are represented graphically in Figure 7A. This analysis revealed that deletion of the P1 region (putative Cas-binding site) had only a small effect on the ability of PTP-PEST to cause dephosphorylation of Cas and Fak. Removal of the P2 sequence (Csk-binding site) had no apparent consequence. In contrast, deletion of the P3 region (paxillin-binding site) strongly reduced the ability of PTP-PEST to promote dephosphorylation of Pyk2, Fak and, to a lesser extent, Cas, but not Shc. Conversely, removal of the NPLH motif (Shc-binding site) interfered with the dephosphorylation of Shc, whereas it did not affect the capacity of PTP-PEST to dephosphorylate Fak and Cas. A small defect in the ability of ΔNPLH PTP-PEST to promote dephosphorylation of Pyk2 was also noted. Finally, we found that the phosphatase-inactive R237M PTP-PEST mutant had a greatly attenuated capacity to induce dephosphorylation of Pyk2, Fak and Cas. Surprisingly, however, its proficiency at decreasing the phosphorylation of Shc was only slightly reduced. This last result implied that part of the effect of PTP-PEST on Shc tyrosine phosphorylation was mediated independently of its catalytic activity (see Discussion). Similar results were obtained with the other phosphatase-inactive variant of PTP-PEST (C231S PTP-PEST) (data not shown).

Fig. 7. Influence of PTP-PEST mutants on BCR-induced protein tyrosine phosphorylation and IL-2 production. (A) BCR-induced protein tyrosine phosphorylation. The extent of BCR-induced tyrosine phosphorylation of individual substrates in A20 derivatives expressing the various PTP-PEST polypeptides was compared with that observed in Neo cells. Data from several independent experiments were quantitated with a phosphoimager, and are represented graphically. (B–D) BCR-triggered lymphokine production. Cells were tested as detailed in Figure 3A. In some cases, pools of transfectants were assayed. All assays were done in triplicate and repeated a minimum of three times.
The various transfectants were tested subsequently for their ability to produce lymphokines in response to stimulation with increasing concentrations of F(ab′)2 fragments of RAM IgG antibodies (Figure 7B–D). These analyses revealed that the ability of PTP-PEST to inhibit BCR-induced lymphokine secretion was compromised by deletion of the P3 region or the NPLH motif, but not by removal of the P1 or P2 region. The inhibitory effect of PTP-PEST was also severely attenuated by the R237M and C231S mutations. Thus, taken together, these data showed that the ability of PTP-PEST to inhibit B-cell activation was dependent on the domains required for its association with Shc (NPLH) and paxillin (P3), but not Csk (P2). Moreover, it correlated with the capacity of PTP-PEST to promote dephosphorylation of Shc, Pyk2, Fak and Cas.
The Ras pathway is a potential target of PTP-PEST in lymphocytes
In an attempt to elucidate the mechanism by which PTP-PEST inhibited lymphocyte activation, we investigated the capacity of constitutively activated versions of several molecules to bypass the signalling defect in PTP-PEST-overexpressing cells (Figure 8A–C). Briefly, A20 cells stably overexpressing PTP-PEST were transiently transfected with various cDNA constructs. After 18 h, cells were stimulated for 24 h with F(ab′)2 fragments of RAM IgG antibodies (10 µg/ml) and the accumulation of IL-2 in the supernatant was measured utilizing a bioassay. This study showed that an activated version of Ras (V12 Myc-Ras) significantly restored BCR-induced lymphokine production in PTP-PEST-overexpressing A20 cells (Figure 8A). In contrast, introduction of equivalent amounts of an inactive Ras (data not shown), or of activated Rac, Rho or cdc42 (known regulators of cytoskeletal organization) (Figure 8A), had no effect. Likewise, expression of constitutively active forms of calcineurin (a downstream effector of calcium-mediated signals) (Figure 8A) or of the Src kinases Lyn and FynT (Figure 8B), or introduction of a membrane-targeted version of the protein tyrosine kinase Btk (Figure 8C) had little or no influence on the inhibitory impact of PTP-PEST. Similar results were obtained with overexpression of the Syk kinase (Figure 8C). Collectively, these findings suggested that a significant component of the inhibitory effect of PTP-PEST on B-cell activation may be caused by inhibition of the Ras pathway.

Fig. 8. Selective rescue of PTP-PEST-mediated inhibition by activated Ras. (A–C) Rescue of the PTP-PEST-induced block by activated Ras. Pools of A20 transfectants stably overexpressing wild-type PTP-PEST were transiently transfected with the indicated constructs. Cells subsequently were activated with F(ab′)2 fragments of RAM IgG antibodies, and IL-2 release in the supernatant was measured using a bioassay. Cells expressing the neomycin phosphotransferase alone (Neo) were transfected with empty vector alone, and were used as control. All assays were done in triplicate. Expression of all constructs was confirmed by immunoblotting of total cell lysates with the appropriate antibodies (data not shown).
Given these results, we examined whether PTP-PEST overexpression interfered with activation of MAPK, the major downstream target of the Ras pathway. For this purpose, A20 clones expressing the neomycin resistance marker alone or in combination with PTP-PEST were stimulated with suboptimal concentrations of F(ab′)2 fragments of SAM IgG antibodies, and MAPK activation was monitored by immunoblotting of total cell lysates with antibodies specific for the activated form of MAPK (Figure 9, first panel). We observed that PTP-PEST overexpression reduced the activation of p42MAPK (Erk-2) in response to BCR stimulation (compare lanes 5 and 6 with lanes 3 and 4). Inhibition of p44MAPK (Erk-1) activation was also detected upon longer autoradiographic exposure of the immunoblot (data not shown). Similar findings were made when the activity of p42MAPK was measured in an immune complex kinase reaction using myelin basic protein (MBP) as exogenous substrate (data not shown). In contrast to the effect on MAPK activation, PTP-PEST had no influence on the activation of the serine-threonine kinase Akt, a downstream target of the PI 3′ kinase pathway in activated B cells (third and fourth panels).
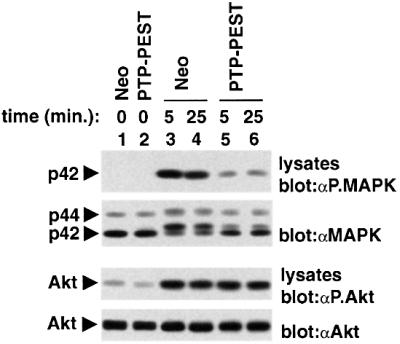
Fig. 9. Impact of PTP-PEST overexpression on MAPK activation. Cells expressing the neomycin resistance marker alone (Neo) or in combination with wild-type PTP-PEST were stimulated for the indicated times with F(ab′)2 fragments of SAM IgG antibodies (0.37 µg/ml). Activation of MAPK (Erk) was assayed by immuno blotting of total cell lysates with antibodies specific for the activated form of MAPK (first panel). Levels of expression of MAPK were confirmed by immunoblotting of parallel lysates with anti-MAPK antibodies (second panel). Activation of Akt was studied as control, using an antibody specific for activated Akt (third panel). Levels of Akt were determined by immunoblotting with an anti-Akt antibody (fourth panel). The positions of p42MAPK (Erk-2), p44MAPK (Erk-1) and Akt are indicated on the left. Exposures: first panel, 4 h; second panel, 4 h; third panel, 48 h; fourth panel, 3 min (ECL).
Discussion
In this study, we have demonstrated that the cytosolic protein tyrosine phosphatase PTP-PEST is expressed abundantly in diverse haemopoietic cell types, including B and T cells. Using a model B-cell line, PTP-PEST was shown to associate with several signalling molecules, including Shc (25% of PTP-PEST), paxillin (10%), Csk (5%) and Cas (2%). While these interactions were constitutive in nature, the stoichiometry of the association between Shc and PTP-PEST was augmented further (up to 60%) by BCR stimulation. Through overexpression studies and antisense experiments, we obtained compelling evidence that PTP-PEST is an efficient negative regulator of immunoreceptor signalling in B and T cells. This effect correlated with the ability of PTP-PEST to associate with Shc and paxillin, and promote selective dephosphorylation of Shc, Cas and the focal adhesion-associated PTKs Pyk2 and Fak. Lastly, our data suggested that the inhibitory impact of PTP-PEST on lymphocyte activation may be caused in significant part by a down-regulation of the Ras pathway.
Several lines of evidence indicate that the inhibitory impact of PTP-PEST on lymphocyte activation is not merely a consequence of its overexpression. (i) The inhibitory effect of PTP-PEST was observed both in B- (this report) and T-cell lines (our unpublished results), and both in stable and in transient transfection assays. (ii) The diminution in antigen receptor-induced protein tyrosine phosphorylation effected by PTP-PEST overexpression concerned a selective subset of antigen receptor-regulated substrates, known to associate directly or indirectly with PTP-PEST. (iii) Mutations or deletions in PTP-PEST capable of abolishing its association with these partners compromised the ability to inhibit antigen receptor-induced protein tyrosine phosphorylation and lymphokine production. (iv) Overexpression of another phosphatase, the PTP-PEST-related enzyme PEP, had no measurable impact on BCR-induced signal transduction in A20 B cells (J.-F.Cloutier and A.Veillette, unpublished results). (v) Transient reduction of endogenous PTP-PEST expression by antisense, but not sense, transfection resulted in enhanced BCR-triggered IL-2 promoter activation. (vi) Our preliminary data indicate that, in response to BCR stimulation, PTP-PEST translocates with Shc in the cell fractions corresponding to lipid rafts (where a significant part of BCR signalling is thought to occur).
Thus, collectively, these findings strongly suggest that PTP-PEST is a novel component of the inhibitory machinery regulating antigen receptor signalling in lymphocytes. Whereas the data at hand already provide convincing evidence that this is likely to be the case, we are planning to attempt to support this idea further by generating PTP-PEST-deficient normal lymphocytes through inducible gene ablation in the mouse.
Even though PTP-PEST appears to be akin to SHP-1 and PEP, the mechanism by which it regulates immunoreceptor signalling is unique. In the case of SHP-1 and PEP, their effect seemingly is mediated by dephosphorylation of the most central components of the antigen receptor signalling cascade, including the Src kinases, the ITAMs and Syk/Zap-70. In contrast, PTP-PEST orchestrates the dephosphorylation of a restricted set of downstream elements in the antigen receptor signalling cascade, i.e. Shc, Pyk2, Fak and Cas. Interestingly, this function correlates with the singular ability of PTP-PEST to recruit its targets via non-catalytic sequences, thereby serving as a ‘scaffold’ for the nucleation of its inhibitory signal.
While Shc, Pyk2, Fak and Cas have long been reported to undergo tyrosine phosphorylation in response to BCR stimulation, there has been no experimental evidence that they are required for B-cell activation. Through our biochemical studies and structure–function analyses of PTP-PEST, we can certainly argue that at least some of these substrates are likely to be crucial for antigen receptor-triggered cell activation. The mechanism by which the reduction of Shc tyrosine phosphorylation by PTP-PEST effected a decrease in B-cell activation is reasonably clear, since we observed that the attenuated Shc tyrosine phosphorylation was accompanied by a diminution in the extent of association of Shc with Grb2 and SHIP during B-cell activation. As will be discussed below, the reduced recruitment of Grb2 by Shc probably contributed to the inhibition of the Ras–MAPK cascade noted in PTP-PEST-overexpressing cells. In contrast, the functional significance of the compromised Shc–SHIP interaction is less clear. As SHIP is also a negative regulator of BCR signalling (Ono et al., 1997; Okada et al., 1998; Hashimoto et al., 1999), its displacement would not necessarily be expected to result in a net inhibition of B-cell activation. Nonetheless, evidence was published recently that SHIP can behave as a positive regulator in IL-4-induced cell proliferation (Giallourakis et al., 2000). Hence, it is possible that SHIP also has as yet unappreciated positive effects in immunoreceptor signalling, and that these effects are inhibited by PTP-PEST.
Despite the fact that paxillin was not tyrosine phosphorylated detectably in activated A20 cells, the paxillin-binding region of PTP-PEST (i.e. P3) was required for the ability of PTP-PEST to dephosphorylate Pyk2, Fak and Cas. Because paxillin can associate with Pyk2 and Fak, it seems reasonable to postulate that these substrates are recruited to PTP-PEST via paxillin. While we have had difficulties in further supporting this view experimentally, some evidence agreeing with this notion has been published by others (Shen et al., 1998). Moreover, as both Pyk2 and Fak can bind Cas (Manie et al., 1997), it is plausible that the two PTKs in turn help recruit Cas in the vicinity of PTP-PEST. Thus, paxillin would appear to link PTP-PEST and tyrosine-phosphorylated Pyk2, Fak and Cas.
The precise impact of tyrosine phosphorylation of Pyk2, Fak and Cas in activated B lymphocytes is not established. As all three substrates have been implicated in the reorganization of the cytoskeleton in non-haemopoietic cells, their tyrosine phosphorylation may mediate cytoskeletal changes needed for optimal antigen receptor-induced cell activation. However, we observed that expression of constitutively activated forms of Rac, Rho or cdc42 were unable to correct the inhibitory influence of PTP-PEST on BCR-induced lymphokine secretion. In light of the capacity of these small G proteins to bypass the requirement for protein tyrosine phosphorylation to provoke cytoskeletal reorganization, this observation raised the possibility that dephosphorylation of Pyk2, Fak and Cas influenced other BCR-regulated pathways. Along these lines, it has been reported that Pyk2 and Fak can activate the Ras–MAPK pathway in response to integrin stimulation in non-haemopoietic cells (Sieg et al., 1998; Renshaw et al., 1999). Hence, it is possible that the impact of these substrates on BCR-induced cellular activation converges with that of Shc, thereby further amplifying the activation of the Ras–MAPK cascade.
Rescue experiments with PTP-PEST-overexpressing A20 cells showed that activated Ras, but not activated Rac, Rho, cdc42, calcineurin, Lyn, FynT or Btk, was able to correct the inhibitory impact of PTP-PEST on BCR-induced lymphokine secretion. On the basis of this finding, it is attractive to speculate that the PTP-PEST-mediated inhibition may be mediated in significant part by an inactivation of the Ras pathway. Along these lines, stimulation of the Ras–Raf–MAPK pathway during B-cell activation has been studied extensively (Campbell, 1999). Current models indicate that this cascade is a point of intersection of several early mediators of BCR signalling, including PLC-γ, Blnk/SLP-65 and Shc. As presented above, it is possible that Pyk2, Fak and/or Cas are also involved in this response. Although the Ras pathway is thought to play a critical role in antigen receptor-mediated B-cell activation, the published experimental evidence supporting this idea is limited. For example, McMahon and Monroe (1995) reported that expression of dominant-negative variants of Ras or Raf blocked the induction of the primary response gene egr-1 upon BCR stimulation. In a similar way, it has been published that the ability of FcγRIIB to repress B-cell activation correlated with the capacity to inhibit the Ras–MAPK cascade (Tridandapani et al., 1997; Tamir et al., 2000). Nonetheless, the participation of the Ras pathway in the biological consequences of B-cell activation has not been clearly addressed. We believe that the data presented herein constitute additional evidence that this pathway is crucial for BCR-induced lymphokine production.
As expected, the ability of PTP-PEST to inhibit lymphocyte activation was dependent on its catalytic activity. Nonetheless, it is of note that the two phosphatase-inactive versions of PTP-PEST (R237M and C231S PTP-PEST) were still able to inhibit partially certain BCR-induced events, in particular Shc tyrosine phosphorylation. As these mutants lacked measurable PTP activity, this observation implied that part of the inhibitory impact of PTP-PEST is mediated through non-catalytic mechanisms. By remaining able to associate with Shc, inactive PTP-PEST molecules may compromise its ability to be phosphorylated by PTKs, perhaps through steric hindrance. Alternatively, PTP-PEST may recruit other PTPs participating in the dephosphorylation of Shc. The residual capacity of the phosphatase-inactive variants to inhibit protein tyrosine phosphorylation also probably explains the fact that they did not behave as frank ‘dominant-negative’ mutants, capable of enhancing BCR-induced lymphokine secretion.
Surprisingly, the inhibitory effect of PTP-PEST on B-cell activation did not require the interaction with Csk (via the P2 region) (Davidson et al., 1997). Through its ability to inactivate Src-related PTKs, Csk is a potent negative regulator of T-cell receptor signalling (Chow et al., 1993; Schmedt et al., 1998). However, we have found previously that overexpression of Csk in a B-cell line had no noticeable consequence on B-cell activation (L.M.L.Chow and A.Veillette, unpublished results). Moreover, introduction of activated versions of Lyn and FynT failed to correct the suppressive effect of PTP-PEST on BCR-induced lymphokine production (this report). It is possible that the difference in the impact of Csk in B and T cells relates to the differential expression of Syk and Zap-70 in the two cell types (van Oers and Weiss, 1995). B cells contain Syk, while T cells typically express Zap-70. Previous reports have shown that Syk, but not Zap-70, can substitute for Src kinases during immunoreceptor signalling, possibly as a consequence of its ability to phosphorylate the ITAMs (Chu et al., 1996; Latour et al., 1997; Zoller et al., 1997).
Given its broad distribution in haemopoietic cells, PTP-PEST is likely to be a general negative regulator of immunoreceptor signalling. In keeping with this notion, we found that PTP-PEST overexpression inhibited antigen receptor-induced activation not only in B cells, but also in T cells. Obviously, though, it is possible that PTP-PEST also inhibits signalling through other types of receptors expressed in haemopoietic cells, including cytokine receptors, chemokine receptors and integrins. Future studies will be necessary to address these possibilities.
Taken together, our data provide a strong indication that PTP-PEST is an efficient and unique negative regulator of lymphocyte activation. Unlike other PTPs, it targets a highly selective subset of substrates in the antigen receptor signalling cascade. Given this property, it is interesting to speculate that PTP-PEST may actually serve to modify qualitatively the outcome of antigen receptor stimulation. It may prevent some of the biological outcomes of antigen receptor stimulation such as lymphokine secretion, proliferation or differentiation, while favouring others such as tolerance, anergy or apoptosis. In this context, it is intriguing that anergic T lymphocytes exhibit a selective down-regulation of the Ras–MAPK pathway (Kang et al., 1992). The possibility that PTP-PEST is actively involved in this phenomenon or other types of antigen receptor-induced biological responses deserves consideration.
Materials and methods
Tissues, cells and stable transfections
Unless specified, mouse cells and tissues were obtained from C57BL/6 mice. Resting splenic B and T cells were isolated from 6- to 8-week-old mice, using columns purchased from Cytovax Biotechnology Inc. More than 90% of cells obtained possessed markers specific for B or T cells, respectively (data not shown). IL-2-activated NK cells were obtained from spleens of Nude CD1 mice, as described elsewhere (Cao et al., 1999). BMMCs were derived from mouse femur bone marrow by prolonged growth in IL-3-containing medium. The surface IgG+ mouse B-cell line A20 was maintained in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS), penicillin, streptomycin and β-mercaptoethanol. Stable derivatives expressing the neomycin phosphotransferase alone, or in combination with wild-type or mutant PTP-PEST molecules, were generated by electroporation (260 V, 960 µF) followed by selection in medium containing G418 (0.5 mg/ml). Monoclonal cell lines were isolated by limiting dilution. Cos-1 cells were propagated in α-minimal essential medium supplemented with 10% FBS and antibiotics.
Antibodies
Polyclonal rabbit antibodies directed against PTP-PEST were described elsewhere (Davidson et al., 1997). These antibodies do not react with the related phosphatases PEP and PTP-HSCF (data not shown). Rabbit antisera specifically reacting with Shc, Pyk2, Syk, Dok, SHIP, Csk, Vav, Cbl or phosphotyrosine were generated in our laboratory (our unpublished data). When necessary, affinity purification was achieved by passing the crude serum over a column containing the immunogen immobilized on Affigel (Biorad Laboratories, Hercules, CA). Antibodies directed against Blnk were provided by Dr A.Chan, Washington University, St Louis, MO, whereas those reacting against p42MAPK were provided by Dr S.Meloche, IRCM, Montréal, Québec, Canada. Additional antibodies were purchased from Transduction Laboratories, Lexington, KY (Cas, paxillin and Grb2), Santa Cruz Biotechnology, Inc., Santa Cruz, CA (Fak, Btk and PLC-γ2), New England Biolabs, Inc., Beverly, MA (Akt, phosphoS473-Akt and phospho-MAPK), Chemicon International, Inc., Temecula, CA (calcineurin) and Upstate Biotechnology, Lake Placid, NY (MAPK). A mouse monoclonal antibody reacting against Myc (mAb 9E10) was described elsewhere (Cloutier and Veillette, 1996). Anti-phosphotyrosine mouse mAb 4G10 was purchased from Upstate Biotechnology.
cDNAs and constructs
The wild-type mouse ptp-pest cDNA and a variant lacking the P2 region (amino acids 674–690) were described elsewhere (Davidson et al., 1997). Mutants carrying deletions of the P1 region (amino acids 332–348), P3 region (amino acids 355–374) or NPLH sequence (amino acids 599–602), or single point mutations in the phosphatase domain (C231S and R237M PTP-PEST) were created by PCR. cDNA fragments used for antisense expression were produced by PCR utilizing the appropriate mouse cDNAs as template. All cDNA variants were verified by sequencing, to ensure that no unwanted mutation had been introduced in the process of their creation (data not shown). Constructs encoding Myc-tagged versions of constitutively activated Ras, Rac, Rho or cdc42, or of inactive Ras (N17 Ras), were provided by Dr N.Lamarche, McGill University, Montréal, Canada (Lamarche et al., 1996). cDNAs coding for activated Lyn (Tyr506→Phe506 Lyn) and FynT (Tyr528→Phe528 FynT) were generated in our laboratory. The constructs producing the constitutively activated form of calcineurin (O’Keefe et al., 1992) and the membrane-bound variant of Btk (CD16-Btk; Bolland et al., 1998) were obtained from Drs G.Crabtree (Stanford University, Palo Alto, CA) and J.Ravetch (Rockefeller University, New York, NY), respectively. pIL-2–luciferase was kindly provided by Dr A.Altman (La Jolla Institute for Allergy and Immunology, La Jolla, CA). For stable expression in A20 cells, ptp-pest cDNAs were cloned in the mammalian expression vector pNT-Neo, which possesses an SRα-based promoter and the neo gene. For transient expression in A20 B cells and Cos-1 cells, cDNAs were cloned in pXM139, which contains the SV40 origin of replication and the adenovirus major late promoter.
Immunoprecipitations and immunoblots
Cells were lysed in TNE buffer (1× TNE: 50 mM Tris pH 8.0, 1% NP-40 and 2 mM EDTA) supplemented with protease and phosphatase inhibitors, as outlined elsewhere (Davidson et al., 1992). After pre-clearing, post-nuclear lysates were immunoprecipitated for 1.5 h with the antibodies specified in the text. Immune complexes were recovered with protein A–Sepharose (Amersham Pharmacia Biotech, Baie d’Urfé, Québec, Canada) pre-coupled, if necessary, to RAM IgG (Jackson Immunoresearch Laboratories). After several washes, proteins were eluted in SDS-containing sample buffer, boiled and resolved by electrophoresis. Immunoblots were performed according to a previously described protocol (Veillette et al., 1988). Immunoreactive products were detected using [125I]protein A (Amersham Pharmacia Biotech) or [125I]goat anti-mouse IgG (ICN Biomedicals). Where specified, protein A–horseradish peroxidase and enhanced chemiluminescence (ECL) reagents (Amersham Pharmacia Biotech) were utilized. Radioactivity was quantitated using a phosphoimager (BAS2000, Fuji).
Immune complex phosphatase assays
Wild-type and mutated versions of the PTP-PEST proteins were expressed by transient transfection in Cos-1 cells. Proteins were then recovered by immunoprecipitation with affinity-purified anti-PTP-PEST antibodies and assayed for phosphatase activity using the Protein Tyrosine Phosphatase Assay System (New England Biolabs, Inc.), as described elsewhere (Cloutier and Veillette, 1999). The expression of the various PTP-PEST mutants was verified by immunoblotting of parallel immunoprecipitates with anti-PTP-PEST antibodies. All experiments were conducted under linear assay conditions (data not shown).
Cell stimulation
A20 cells (1 × 107 cells/ml) were stimulated for the indicated times at 37°C with F(ab′)2 fragments of SAM IgG (20 µg/ml; Jackson Immunoresearch Laboratories, West Grove, PA). After stimulation, cells were lysed in 1× TNE buffer as outlined above. Lysates were used subsequently for immunoprecipitations or immunoblots.
BCR-induced lymphokine production
Individual clones or pools of at least three independent transfectants of A20 cells (105 cells in 200 µl) were stimulated for 24 h at 37°C in 96-well plates, in the presence of various concentrations of F(ab′)2 fragments of RAM IgG (Jackson Immunoresearch Laboratories, West Grove, PA). After this period, supernatants were harvested, frozen at –70°C to destroy carry-over cells and assayed for IL-2 content using the IL-2-dependent cell line HT-2.
Luciferase assays
A20 (10 × 106 cells) was transfected by electroporation (300 V, 975 µF) with 20 µg of pIL-2–luciferase plus 10 µg of pXM139 or pXM139-ptp-pest wt, or 40 µg of pXM139-antisense ptp-pest. After 40–66 h, A20 cells (2 × 106 viable cells) were stimulated for 6 h with 10 µg/ml of F(ab′)2 fragments of SAM IgG. As a control, cells were activated with the combination of PMA (100 ng/ml ) and ionomycin (1 µM). They were then lysed and assayed for luciferase activity, using the luciferase reporter assay system (Promega Co., Madison, WI) and a luminometer (EG&G Berthold). Results are presented as the percentage of luciferase activity induced by PMA plus ionomycin. Equivalent numbers of viable cells were also lysed in parallel with boiling sample buffer, for immunoblotting of total cell lysates with anti-PTP-PEST sera.
Rescue of PTP-PEST-mediated inhibition by transient transfection.
Pools of A20 derivatives stably expressing the neomycin resistance marker alone (Neo) or in combination with wild-type PTP-PEST were transiently transfected with the specified cDNAs. After 18 h, cells were harvested and stimulated for 24 h with F(ab′)2 fragments of RAM IgG antibodies (10 µg/ml). BCR-induced IL-2 secretion was tested as detailed above. Equivalent numbers of viable cells were also analyzed in parallel by immunoblotting of total cell lysates with the appropriate antibodies, to ensure that the transfected cDNAs were expressed adequately.
Immune complex kinase assays
p42MAPK was recovered from total cell lysates using a rabbit antiserum specific for p42MAPK. After several washes, MAPK activity was measured in an immune complex kinase assay using MBP as exogenous substrate. Radiolabelled products of these reactions were resolved by gel electrophoresis and detected by autoradiography. Levels of p42MAPK were determined by immunoblotting of parallel immunoprecipitates with anti-p42MAPK antibodies. All reactions were conducted under linear assay conditions (data not shown).
Acknowledgments
Acknowledgements
We thank Sylvain Latour and Jeff Robson for critical reading of the manuscript. We also acknowledge Isabelle Gamache for technical help, and Amnon Altman, Andy Chan, Gerry Crabtree, Nathalie Lamarche, Sylvain Meloche and Jeff Ravetch for gifts of reagents. This work was supported by grants from the Medical Research Council of Canada and the National Cancer Institute of Canada to A.V. A.V. is a Senior Scientist of the Medical Research Council of Canada.
References
- Angers-Loustau A., Coté,J.F., Charest,A., Dowbenko,D., Spencer,S., Lasky,L.A. and Tremblay,M.L. (1999) Protein tyrosine phosphatase-PEST regulates focal adhesion disassembly, migration and cytokinesis in fibroblasts. J. Cell Biol., 144, 1019–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benschop R.J. and Cambier,J.C. (1999) B cell development: signal transduction by antigen receptors and their surrogates. Curr. Opin. Immunol., 11, 143–151. [DOI] [PubMed] [Google Scholar]
- Bolland S., Pearse,R.N., Kurosaki,T. and Ravetch,J.V. (1998) SHIP modulates immune receptor responses by regulating membrane association of Btk. Immunity, 8, 509–516. [DOI] [PubMed] [Google Scholar]
- Campbell K.S. (1999) Signal transduction from the B cell antigen-receptor. Curr. Opin. Immunol., 11, 256–264. [DOI] [PubMed] [Google Scholar]
- Cao M.Y., Davidson,D., Yu,J., Latour,S. and Veillette,A. (1999) Clnk, a novel SLP-76-related adaptor molecule expressed in cytokine-stimulated hemopoietic cells. J. Exp. Med., 190, 1527–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter J.D., Neel,B.G. and Lorenz,U. (1999) The tyrosine phosphatase SHP-1 influences thymocyte selection by setting TCR signaling thresholds. Int. Immunol., 11, 1999–2014. [DOI] [PubMed] [Google Scholar]
- Chan A.C., Desai,D.M. and Weiss,A. (1994) The role of protein tyrosine kinases and protein tyrosine phosphatases in T cell antigen receptor signal transduction. Annu. Rev. Immunol., 12, 555–592. [DOI] [PubMed] [Google Scholar]
- Charest A., Wagner,J., Muise,E.S., Heng,H.H. and Tremblay,M.L. (1995) Structure of the murine MPTP-PEST gene: genomic organization and chromosomal mapping. Genomics, 28, 501–507. [DOI] [PubMed] [Google Scholar]
- Charest A., Wagner,J., Jacob,S., McGlade,C.J. and Tremblay,M.L. (1996) Phosphotyrosine-independent binding of SHC to the NPLH sequence of murine protein-tyrosine phosphatase-PEST. Evidence for extended phosphotyrosine binding/phosphotyrosine interaction domain recognition specificity. J. Biol. Chem., 271, 8424–8429. [DOI] [PubMed] [Google Scholar]
- Chow L.M. and Veillette,A. (1995) The Src and Csk families of tyrosine protein kinases in hemopoietic cells. Semin. Immunol., 7, 207–226. [DOI] [PubMed] [Google Scholar]
- Chow L.M., Fournel,M., Davidson,D. and Veillette,A. (1993) Negative regulation of T-cell receptor signalling by tyrosine protein kinase p50csk. Nature, 365, 156–160. [DOI] [PubMed] [Google Scholar]
- Chu D.H., Spits,H., Peyron,J.F., Rowley,R.B., Bolen,J.B. and Weiss,A. (1996) The Syk protein tyrosine kinase can function independently of CD45 or Lck in T cell antigen receptor signaling. EMBO J., 15, 6251–6261. [PMC free article] [PubMed] [Google Scholar]
- Cloutier J.F. and Veillette,A. (1996) Association of inhibitory tyrosine protein kinase p50csk with protein tyrosine phosphatase PEP in T cells and other hemopoietic cells. EMBO J., 15, 4909–4918. [PMC free article] [PubMed] [Google Scholar]
- Cloutier J.F. and Veillette,A. (1999) Cooperative inhibition of T-cell antigen receptor signaling by a complex between a kinase and a phosphatase. J. Exp. Med., 189, 111–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coté J.F., Turner,C.E. and Tremblay,M.L. (1999) Intact LIM 3 and LIM 4 domains of paxillin are required for the association to a novel polyproline region (Pro 2) of protein-tyrosine phosphatase-PEST. J. Biol. Chem., 274, 20550–20560. [DOI] [PubMed] [Google Scholar]
- Cyster J.G. and Goodnow,C.C. (1995) Protein tyrosine phosphatase 1C negatively regulates antigen receptor signaling in lymphocytes and determines thresholds for negative selection. Immunity, 2, 13–24. [DOI] [PubMed] [Google Scholar]
- Davidson D., Chow,L.M., Fournel,M. and Veillette,A. (1992) Differential regulation of T cell antigen responsiveness by isoforms of the src-related tyrosine protein kinase p59fyn. J. Exp. Med., 175, 1483–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson D., Cloutier,J.F., Gregorieff,A. and Veillette,A. (1997) Inhibitory tyrosine protein kinase p50csk is associated with protein-tyrosine phosphatase PTP-PEST in hemopoietic and non-hemopoietic cells. J. Biol. Chem., 272, 23455–23462. [DOI] [PubMed] [Google Scholar]
- Garton A.J. and Tonks,N.K. (1999) Regulation of fibroblast motility by the protein tyrosine phosphatase PTP-PEST. J. Biol. Chem., 274, 3811–3818. [DOI] [PubMed] [Google Scholar]
- Garton A.J., Burnham,M.R., Bouton,A.H. and Tonks,N.K. (1997) Association of PTP-PEST with the SH3 domain of p130cas; a novel mechanism of protein tyrosine phosphatase substrate recognition. Oncogene, 15, 877–885. [DOI] [PubMed] [Google Scholar]
- Giallourakis C., Kashiwada,M., Pan,P.Y., Danial,N., Jiang,H., Cambier,J., Coggeshall,K.M. and Rothman,P. (2000) Positive regulation of interleukin-4-mediated proliferation by the SH2-containing inositol-5′-phosphatase. J. Biol. Chem., 275, 29275–29282. [DOI] [PubMed] [Google Scholar]
- Gjorloff-Wingren A., Saxena,M., Williams,S., Hammi,D. and Mustelin,T. (1999) Characterization of TCR-induced receptor-proximal signaling events negatively regulated by the protein tyrosine phosphatase PEP. Eur. J. Immunol., 29, 3845–3854. [DOI] [PubMed] [Google Scholar]
- Habib T., Herrera,R. and Decker,S.J. (1994) Activators of protein kinase C stimulate association of Shc and the PEST tyrosine phosphatase. J. Biol. Chem., 269, 25243–25246. [PubMed] [Google Scholar]
- Harmer S.L. and DeFranco,A.L. (1997) Shc contains two Grb2 binding sites needed for efficient formation of complexes with SOS in B lymphocytes. Mol. Cell. Biol., 17, 4087–4095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harmer S.L. and DeFranco,A.L. (1999) The src homology domain 2-containing inositol phosphatase SHIP forms a ternary complex with Shc and Grb2 in antigen receptor-stimulated B lymphocytes. J. Biol. Chem., 274, 12183–12191. [DOI] [PubMed] [Google Scholar]
- Hashimoto A., Hirose,K., Okada,H., Kurosaki,T. and Iino,M. (1999) Inhibitory modulation of B cell receptor-mediated Ca2+ mobilization by Src homology 2 domain-containing inositol 5′-phosphatase (SHIP). J. Biol. Chem., 274, 11203–11208. [DOI] [PubMed] [Google Scholar]
- Ingham R.J., Okada,H., Dang-Lawson,M., Dinglasan,J., van der Geer,P., Kurosaki,T. and Gold,M.R. (1999) Tyrosine phosphorylation of shc in response to B cell antigen receptor engagement depends on the SHIP inositol phosphatase. J. Immunol., 163, 5891–5895. [PubMed] [Google Scholar]
- Justement L.B., Kreiger,J. and Cambier,J.C. (1989) Production of multiple lymphokines by the A20.1 B cell lymphoma after cross-linking of membrane Ig by immobilized anti-Ig. J. Immunol., 143, 881–889. [PubMed] [Google Scholar]
- Kang S.M., Beverly,B., Tran,A.C., Brorson,K., Schwartz,R.H. and Lenardo,M.J. (1992) Transactivation by AP-1 is a molecular target of T cell clonal anergy. Science, 257, 1134–1138. [DOI] [PubMed] [Google Scholar]
- Lamarche N., Tapon,N., Stowers,L., Burbelo,P.D., Aspenstrom,P., Bridges,T., Chant,J. and Hall,A. (1996) Rac and Cdc42 induce actin polymerization and G1 cell cycle progression independently of p65PAK and the JNK/SAPK MAP kinase cascade. Cell, 87, 519–529. [DOI] [PubMed] [Google Scholar]
- Lamkin T.D., Walk,S.F., Liu,L., Damen,J.E., Krystal,G. and Ravichandran,K.S. (1997) Shc interaction with Src homology 2 domain containing inositol phosphatase (SHIP) in vivo requires the Shc-phosphotyrosine binding domain and two specific phosphotyrosines on SHIP. J. Biol. Chem., 272, 10396–10401. [DOI] [PubMed] [Google Scholar]
- Latour S., Fournel,M. and Veillette,A. (1997) Regulation of T-cell antigen receptor signalling by Syk tyrosine protein kinase. Mol. Cell. Biol., 17, 4434–4441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manie S.N. et al. (1997) Involvement of p130(Cas) and p105(HEF1), a novel Cas-like docking protein, in a cytoskeleton-dependent signaling pathway initiated by ligation of integrin or antigen receptor on human B cells. J. Biol. Chem., 272, 4230–4236. [DOI] [PubMed] [Google Scholar]
- McMahon S.B. and Monroe,J.G. (1995) Activation of the p21ras pathway couples antigen receptor stimulation to induction of the primary response gene egr-1 in B lymphocytes. J. Exp. Med., 181, 417–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muta T., Kurosaki,T., Misulovin,Z., Sanchez,M., Nussenzweig,M.C. and Ravetch,J.V. (1994) A 13-amino-acid motif in the cytoplasmic domain of FcγRIIB modulates B-cell receptor signalling. Nature, 368, 70–73. [DOI] [PubMed] [Google Scholar]
- Nishiya N., Iwabuchi,Y., Shibanuma,M., Coté,J.F., Tremblay,M.L. and Nose,K. (1999) Hic-5, a paxillin homologue, binds to the protein-tyrosine phosphatase PEST (PTP-PEST) through its LIM 3 domain. J. Biol. Chem., 274, 9847–9853. [DOI] [PubMed] [Google Scholar]
- O’Keefe S.J., Tamura,J., Kincaid,R.L., Tocci,M.J. and O’Neill,E.A. (1992) FK-506- and CsA-sensitive activation of the interleukin-2 promoter by calcineurin. Nature, 357, 692–694. [DOI] [PubMed] [Google Scholar]
- Okada H., Bolland,S., Hashimoto,A., Kurosaki,M., Kabuyama,Y., Iino,M., Ravetch,J.V. and Kurosaki,T. (1998) Role of the inositol phosphatase SHIP in B cell receptor-induced Ca2+ oscillatory response. J. Immunol., 161, 5129–5132. [PubMed] [Google Scholar]
- Ono M., Okada,H., Bolland,S., Yanagi,S., Kurosaki,T. and Ravetch,J.V. (1997) Deletion of SHIP or SHP-1 reveals two distinct pathways for inhibitory signaling. Cell, 90, 293–301. [DOI] [PubMed] [Google Scholar]
- Pani G., Kozlowski,M., Cambier,J.C., Mills,G.B. and Siminovitch,K.A. (1995) Identification of the tyrosine phosphatase PTP1C as a B cell antigen receptor-associated protein involved in the regulation of B cell signaling. J. Exp. Med., 181, 2077–2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pani G., Fischer,K.D., Mlinaric-Rascan,I. and Siminovitch,K.A. (1996) Signaling capacity of the T cell antigen receptor is negatively regulated by the PTP1C tyrosine phosphatase. J. Exp. Med., 184, 839–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peri K.G. and Veillette,A. (1994) Tyrosine protein kinases in T lymphocytes. Chem. Immunol., 59, 19–39. [PubMed] [Google Scholar]
- Pimentel-Muinos F.X. and Seed,B. (1999) Regulated commitment of TNF receptor signaling: a molecular switch for death or activation. Immunity, 11, 783–793. [DOI] [PubMed] [Google Scholar]
- Pradhan M. and Coggeshall,K.M. (1997) Activation-induced bi-dentate interaction of SHIP and Shc in B lymphocytes. J. Cell. Biochem., 67, 32–42. [DOI] [PubMed] [Google Scholar]
- Qian D. and Weiss,A. (1997) T cell antigen receptor signal transduction. Curr. Opin. Cell Biol., 9, 205–212. [DOI] [PubMed] [Google Scholar]
- Renshaw M.W., Price,L.S. and Schwartz,M.A. (1999) Focal adhesion kinase mediates the integrin signaling requirement for growth factor activation of MAP kinase. J. Cell Biol., 147, 611–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmedt C., Saijo,K., Niidome,T., Kuhn,R., Aizawa,S. and Tarakhovsky,A. (1998) Csk controls antigen receptor-mediated development and selection of T-lineage cells. Nature, 394, 901–904. [DOI] [PubMed] [Google Scholar]
- Shen Y., Schneider,G., Cloutier,J.F., Veillette,A. and Schaller,M.D. (1998) Direct association of protein-tyrosine phosphatase PTP-PEST with paxillin. J. Biol. Chem., 273, 6474–6481. [DOI] [PubMed] [Google Scholar]
- Sieg D.J., Ilic,D., Jones,K.C., Damsky,C.H., Hunter,T. and Schlaepfer,D.D. (1998) Pyk2 and Src-family protein-tyrosine kinases compensate for the loss of FAK in fibronectin-stimulated signaling events but Pyk2 does not fully function to enhance FAK cell migration. EMBO J., 17, 5933–5947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzu S., Tanaka-Douzono,M., Nomaguchi,K., Yamada,M., Hayasawa,H., Kimura,F. and Motoyoshi,K. (2000) p56(dok-2) as a cytokine-inducible inhibitor of cell proliferation and signal transduction. EMBO J., 19, 5114–5122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamir I., Stolpa,J.C., Helgason,C.D., Nakamura,K., Bruhns,P., Daeron,M. and Cambier,J.C. (2000) The RasGAP-binding protein p62dok is a mediator of inhibitory FcγRIIB signals in B cells. Immunity, 12, 347–358. [DOI] [PubMed] [Google Scholar]
- Tridandapani S., Chacko,G.W., Van Brocklyn,J.R. and Coggeshall,K.M. (1997) Negative signaling in B cells causes reduced Ras activity by reducing Shc–Grb2 interactions. J. Immunol., 158, 1125–1132. [PubMed] [Google Scholar]
- van Oers N.S. and Weiss,A. (1995) The Syk/ZAP-70 protein tyrosine kinase connection to antigen receptor signalling processes. Semin. Immunol., 7, 227–236. [DOI] [PubMed] [Google Scholar]
- Veillette A., Bookman,M.A., Horak,E.M. and Bolen,J.B. (1988) The CD4 and CD8 T cell surface antigens are associated with the internal membrane tyrosine-protein kinase p56lck. Cell, 55, 301–308. [DOI] [PubMed] [Google Scholar]
- Weiss A. and Littman,D.R. (1994) Signal transduction by lymphocyte antigen receptors. Cell, 76, 263–274. [DOI] [PubMed] [Google Scholar]
- Yang Q., Co,D., Sommercorn,J. and Tonks,N.K. (1993) Cloning and expression of PTP-PEST. A novel, human, nontransmembrane protein tyrosine phosphatase. J. Biol. Chem., 268, 6622–6628. [PubMed] [Google Scholar]
- Yang W.C., Collette,Y., Nunès,J.A. and Olive,D. (2000) Tec kinases: a family with multiple roles in immunity. Immunity, 12, 373–382. [DOI] [PubMed] [Google Scholar]
- Zhang J., Somani,A.K., Yuen,D., Yang,Y., Love,P.E. and Siminovitch,K.A. (1999) Involvement of the SHP-1 tyrosine phosphatase in regulation of T cell selection. J. Immunol., 163, 3012–3021. [PubMed] [Google Scholar]
- Zoller K.E., MacNeil,I.A. and Brugge,J.S. (1997) Protein tyrosine kinases Syk and ZAP-70 display distinct requirements for Src family kinases in immune response receptor signal transduction. J. Immunol., 158, 1650–1659. [PubMed] [Google Scholar]