

Abstract
11S REGs (PA28s) are multimeric rings that bind proteasomes and stimulate peptide hydrolysis. Whereas REGα activates proteasomal hydrolysis of peptides with hydrophobic, acidic or basic residues in the P1 position, REGγ only activates cleavage after basic residues. We have isolated REGγ mutants capable of activating the hydrolysis of fluorogenic peptides diagnostic for all three active proteasome β subunits. The most robust REGγ specificity mutants involve substitution of Glu or Asp for Lys188. REGγ (K188E/D) variants are virtually identical to REGα in proteasome activation but assemble into less stable heptamers/hexamers. Based on the REGα crystal structure, Lys188 of REGγ faces the aqueous channel through the heptamer, raising the possibility that REG channels function as substrate-selective gates. However, covalent modification of proteasome chymotrypsin-like subunits by 125I-YL3-VS demonstrates that REGγ (K188E)’s activation of all three proteasome active sites is not due to relaxed gating. We propose that decreased stability of REGγ (K188E) heptamers allows them to change conformation upon proteasome binding, thus relieving inhibition of the CT and PGPH sites normally imposed by the wild-type REGγ molecule.
Keywords: affinity labeling/chimera/enzyme specificity/mutagenesis
Introduction
The proteasome is a large multisubunit protease present in archaebacteria, bacteria and eukaryotes (Bochtler et al., 1999). Quaternary structures for yeast and Thermoplasma proteasomes were initially obtained by electron microscopy (Dahlmann et al., 1989; Grziwa et al., 1991; Pühler et al., 1992) and more recently by X-ray crystallography (Löwe et al., 1995; Groll et al., 1997). These structural analyses reveal the proteasome to be a cylindrical particle composed of four stacked rings each containing seven subunits (Bochtler et al., 1999). The two end rings are composed of α subunits and the two central rings consist of β subunits. All Thermoplasma β subunits are proteolytically active, but only three mammalian β subunits possess the N-terminal threonine needed for peptide cleavage (Baumeister et al., 1998). The proteasome active sites face a large chamber buried in the center of the enzyme (Löwe et al., 1995; Groll et al., 1997). A 13 Å pore through the α ring of the archaebacterial proteasome connects the external solvent to antechambers that flank the central proteolytic chamber. In the yeast proteasome this narrow pore is occluded by N-terminal sequences from the α subunits, thereby enclosing the internal chambers.
The proteasome’s internal chambers are largely inaccessible from the external solvent, yet to be degraded substrates must somehow gain access to the central chamber. Serving this purpose are two protein complexes that bind and activate the proteasome. A regulatory complex containing 18 different subunits (Hoffman et al., 1992; Peters et al., 1993; Udvardy, 1993; DeMartino et al., 1994) associates with the 20S proteasome to produce the 26S proteasome responsible for the energy-dependent degradation of important regulatory proteins via the ubiquitin pathway (Hershko and Ciechanover, 1998; Rechsteiner, 1998; Voges et al., 1999). The other proteasome activator is a donut-shaped molecule called 11S REG (Dubiel et al., 1992) or PA28 (Ma et al., 1992). Red blood cell REG is composed of two subunits, REGα and REGβ, which are closely related to each other (Ahn et al., 1995; Realini et al., 1997) and to Ki, a major autoantigen in lupus patients (Nikaido et al., 1990). Recombinant Ki has been shown to activate the proteasome, so it is now called REGγ (Realini et al., 1997) or PA28γ (Tanahashi et al., 1997). Both REGα and REGγ self-associate into oligomeric rings that differ in their ability to activate specific proteasome β subunits. REGα enhances cleavage of peptides with basic, acidic or hydrophobic residues in the P1 position, whereas REGγ activates hydrolysis of peptide bonds following basic residues (Realini et al., 1997). Although recombinant human REGβ is monomeric, high concentrations of this subunit activate the proteasome in a manner similar to REGα (Realini et al., 1997; Zhang et al., 1998b). Moreover, REGβ readily associates with REGα, forming heteroheptamers that are presumed to be equivalent to the 11S REG molecule isolated from red blood cells (Zhang et al., 1999).
A crystal structure has been solved for the recombinant REGα heptamer (Knowlton et al., 1997). Each subunit contains four long helices that pack against one another. Mutagenesis studies have shown that the loop connecting helix 2 and helix 3 in the REGα subunit is important for proteasome activation. Thus, residues Arg141–Gly149 are called the activation loop (Zhang et al., 1998a). The sequences of the REGα and REGγ activation loops are identical, so it is unlikely that the activation loop per se accounts for the fact that REGγ only stimulates the proteasome’s trypsin-like subunit. For this reason, we asked whether sequences near the activation loop impart the restricted proteasome activation seen with REGγ versus broad activation by REGα. Characterization of REG chimeras involving exchange of sequences flanking the activation loops demonstrates that differential proteasome activation is not controlled by the divergent regions surrounding the conserved activation loop (Li and Rechsteiner, 2001). Although the last eight amino acids in REGα are disordered in the crystal, they originate next to the activation loop and differ in sequence from REGγ. Therefore, the C-terminal extensions were examined for their possible role in differential proteasome activation. Characterization of chimeras involving the last 8 or 12 amino acids in REGs α, β and γ demonstrated that C-terminal sequences are important for stabilizing REG heptamers and make major contributions to proteasome binding, but they do not affect the activation of specific proteasome subunits (Li et al., 2000).
In the experiments presented below, random mutagenesis was used to continue the search for REG structural elements controlling the differential activation of the proteasome’s catalytic subunits. Single-site mutations involving Lys188 enable REGγ to activate all three proteolytic subunits of the proteasome. We propose that proteasome activation by the mutant REGγ (K188E) results principally from increased substrate entry, and we attribute the restricted activation by REGγ to inhibitory conformational changes in the CT and PGPH subunits imparted by the wild-type proteasome activator.
Results
REGγ is a heptamer
Recombinant REGα has been shown to form heptamers by sedimentation (Johnston et al., 1997), X-ray crystallography (Knowlton et al., 1997) and mass spectrometry (MS) (Zhang et al., 1999). Although REGγ has been assumed also to form heptamers based on its elution from a Superdex 200 column (Realini et al., 1997), we felt that this assumption should be tested. Accordingly, we sedimented recombinant REGγ to equilibrium in a Beckman XL-A Optima analytical ultracentrifuge. Multiple data sets obtained from various speeds and loading concentrations were simultaneously fit to a single species model, returning a molecular weight of 214 ± 8 kDa (Figure 1). Since the molecular weight of a REGγ monomer is 29.5 kDa, these data indicate that there are seven subunits in the REGγ oligomer.
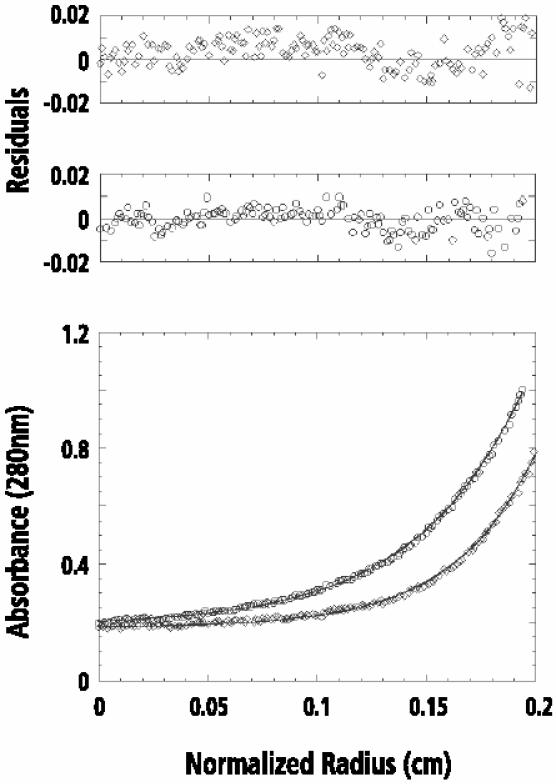
Fig. 1. Equilibrium sedimentation analysis of recombinant REGγ. Data collected at different concentrations of REGγ were fit simultaneously to a single species model. The lower panel shows the representative experimental data (circles, 110 µg/ml; diamonds, 55 µg/ml) of wild-type REGγ and the calculated curve fits (continuous lines) for a single species model. The resulting fit was good as shown by the randomly distributed residuals (upper panel). The returned molecular weight for REGγ is 214 ± 8 kDa.
Subsequently, we also used electron microscopy to examine the oligomeric state of REGγ and REGγ (K188E), a mutant capable of activating all three proteasome catalytic subunits (see below). When REGγ and REGγ (K188E) were examined by negative staining, fields of roundish particles (diameter 11–13 nm) were observed for both samples (Figure 2A and D, respectively). The REGγ particles were consistently more uniform in size and appearance than REGγ (K188E). In two independent data sets of REGγ particles, 7-fold symmetry was detected, most strongly at a radius of 5–5.5 nm, around the outer rim of the particle (Figure 2, table). No other order of symmetry was found to be statistically significant. Correlation averaging of these data depicted a 7-fold symmetric particle, with a heavy accumulation of stain at the center surrounded by a thin annulus of protein density and then seven peripheral outcrops (Figure 2B and C). Thus visualized, REGγ is a heptamer with an outer diameter of ∼13 nm.

Fig. 2. Electron micrographs and averaged images of REGγ and REGγ (K188E) particles. Shown are fields of purified, negatively stained REGγ (A) and REGγ (K188E) (D), and the respective correlation averages of particles with 7-fold symmetry as judged by statistical analysis (B and E). (C and F) These images were explicitly symmetrized. (D, top inset) Correlation-average of image displaying 6-fold symmetry and (D, bottom inset) the corresponding symmetrized images. The averaged images (B), (E) and (D) (inset) represent 150, 133 and 100 particles, and have resolutions of 26, 28 and 28 Å, respectively. For REGγ (B and C), the inner ring of density is resolved into seven units whereas REGγ (K188E) (E and F) shows this feature as a continuous ring, presumably reflecting the lower resolution of the latter images. Bar, 40 nm (A) and 5 nm (B). Table: aindependent data sets from micrographs of different fields of the same grid in each sample; bthe spectral ratio product is shown only for the radial zones in which these symmetries were detected most strongly (at a radius of 5 nm).
As noted, REGγ (K188E) particles tended to be less regular in appearance (Figure 2D). Nevertheless, from statistical analysis of relatively well preserved molecules, we detected both 7- and 6-fold symmetry as statistically significant (Figure 2, table), and the data were partitioned accordingly (see Materials and methods). Correlation averaging of the 7-fold data depicted a heptamer that is similar in size and appearance to wild-type REGγ (Figure 2E and F). The 6-fold data yielded a hexamer that resembles the heptamer apart from its order of symmetry and being slightly smaller. The occurrence of hexamers as well as heptamers in the population of REGγ (K188E/D) oligomers correlates with the reduced stability of the mutant oligomers upon gel filtration (Table I). We note that the hexamer rings may not be completely closed, i.e. there may be a rift between one pair of neighboring subunits, which would tend to enlarge the diameter of this oligomer.
Table I. Properties of REGγ Lys188 mutants.
| Stimulation of cleavage (x-fold) |
Percentage heptamer | Relative affinity for proteasomea | |||
|---|---|---|---|---|---|
| LLVY | LLR | LLE | |||
| REGγ | 0.4 | 12 | 0.6 | >95 | 100 |
| REGα | 16 | 16 | 9 | ∼95 | 0 |
| γK188E | 14 | 15 | 9 | 50 | 90 |
| γK188D | 14 | 15 | 9 | 50 | 90 |
| γK188A | 6 | 13 | 6 | 60 | 60 |
| γK188C | 6 | 13 | 5 | 70 | 60 |
| γK188N | 5 | 14 | 5 | 80 | 75 |
| γK188Q | 5 | 14 | 5 | 80 | 75 |
| γK188H | 5 | 14 | 4 | >95 | 50 |
| γK188F | 4 | 13 | 4 | dimer | 40 |
| γK188S | 3 | 14 | 3 | >95 | 50 |
| γK188I | 2 | 12 | 3 | >95 | 60 |
| γK188P | 1 | 12 | 1 | 50 | 40 |
| γK188R | 0.7 | 10 | 0.7 | >95 | 75 |
This table summarizes the stability, proteasome activation and relative proteasomal affinity of twelve REGγ Lys188 mutants, compared with those of wild-type REGα and REGγ. Heptamer stability is the percentage of heptamer remaining after rechromatographing 1 mg of each REG heptamer on a Superdex 200 (26/60) column. The activities for proteasome activation are calculated as the fold stimulation: proteasome activity in the presence of 3 µg of each REG species divided by proteasome activity alone. Relative affinity for the proteasome is measured by the inhibition observed after addition of the first 1 µg of REGα(N146Y)/REGβ(N135Y) as shown in the Supplementary data.
aRelative proteasome binding affinity is determined in terms of REGα(N146Y)/REGβ(N135Y) competition and does not reflect the actual proteasomal binding by REG molecules.
Identification of REGγ mutants stimulating suc-LLVY-MCA cleavage
Random mutagenesis was used previously to identify the REG activation loop (Zhang et al., 1998a). This approach was modified slightly to screen for REGγ molecules capable of activating proteasomal cleavage of suc-LLVY-MCA (see Materials and methods). Four positive colonies were isolated from among 1400 colonies expressing mutagenized REGγ plasmids. DNA sequencing identified the four REGγ mutants as (L38V, K188N), (F102I, K188N), (L71P, K188E) and K188N. Because each positive variant was mutated at Lys188, we employed site-directed mutagenesis to generate a series of amino acid substitutions at this residue. In all, 12 mutants were constructed in which Lys188 of REGγ was changed to Ala, Arg, Asn, Asp, Cys, Gln, Glu, His, Ile, Phe, Pro or Ser. Wild-type REGγ and the Lys188 REGγ variants were expressed in Escherichia coli, purified, and the activation specificity of each protein was measured using the diagnostic peptides LLVY-MCA, LRR-MCA and LLE-βNA. Except for the K188R variant, all Lys188 mutants stimulated the hydrolysis of LRR-MCA by the proteasome to the same extent or slightly better than wild-type REGγ (Figure 3). With regard to stimulated hydrolysis of LLVY-MCA and LLE-βNA, the single-site variants could be placed, somewhat arbitrarily, into three groups. Substitution of negatively charged Glu or Asp for the positively charged Lys188 produced mutant REGγs with activation properties almost identical to REGα (Table I). Eight variants clearly stimulated hydrolysis of LLVY-MCA and LLE-βNA, but to extents ranging from 6-fold stimulation exhibited by K188A and K188C to 2-fold stimulation seen with K188I (Table I). Replacement of Lys188 by Pro or Arg did not enhance cleavage of peptides diagnostic for the CT-like or PGPH active sites of the proteasome. To determine the degree to which the activation specificity of REGγ (K188E/D) variants matches that of REGα, proteasomal cleavage of a series of fluorogenic peptides was measured in the presence of each activator. It is clear from the data in Table II that REGγ (K188E/D) are virtually identical to REGα in their ability to activate proteasomal hydrolysis of small fluorogenic peptides.
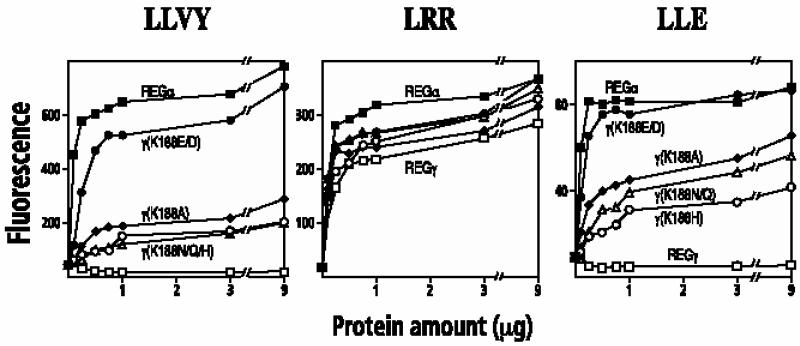
Fig. 3. Activation of fluorogenic peptide hydrolysis by REGγ Lys188 mutants. Human red blood cell proteasomes (170 ng) were mixed with increasing amounts of purified REG variants. The reaction was started by adding 50 µl of 200 µM Suc-LLVY-MCA (left), Boc-LRR-MCA (middle) or Boc-LLE-βNA (right) in 10 mM Tris pH 7.5. After a 10 min incubation, the reaction was quenched with 200 µl of cold 100% ethanol, and the released MCA or βNA was measured fluorometrically. Each data point represents the mean of three measurements from a single experiment; equivalent results were observed in at least two experiments using different preparations of the various REG proteins. Symbol representation: REGα, filled squares; REGγ, open squares; REGγ (K188E) or REGγ (K188D), filled circles; REGγ (K188H), open circles; REGγ (K188Q) or REGγ (K188N), open triangles; REGγ (K188A), filled diamonds. Data from REGγ (K188D) and REGγ (K188E) are combined since their activities are indistinguishable. The same is true for REGγ (K188N) and REGγ (K188Q).
Table II. Activated hydrolysis of fluorogenic peptides by REGα, REGγ and REGγ (K188D/E).
| Fluorogenic peptide | Stimulation of cleavage (x-fold) |
|||
|---|---|---|---|---|
| REGγ | γK188E | γK188D | REGα | |
| LLVY-MCA | 0.4 | 14 | 14 | 16 |
| LY-MCA | 2 | 3 | 3 | 5 |
| AAF-MCA | 1 | 4 | 4 | 6 |
| FSR-MCA | 7 | 7 | 7 | 10 |
| VLK-MCA | 5 | 6 | 6 | 7 |
| LRR-MCA | 11 | 15 | 15 | 16 |
| IEGR-MCA | 1 | 2 | 2 | 3 |
| IETD-MCA | 1 | 21 | 26 | 30 |
| LGHD-MCA | 1 | 10 | 11 | 11 |
| DEVD-MCA | 2 | 8 | 9 | 10 |
| YVAD-MCA | 0.9 | 7 | 8 | 9 |
| LLE-MCA | 0.6 | 9 | 9 | 9 |
Aliquots (3 µg) of wild-type REGα, REGγ and the two robust REGγ specificity mutants, REGγ (K188E/D), were mixed with 170 ng of red cell proteasome before the fluorogenic peptides listed above were added to start the reaction. Stimulation is calculated from MCA fluorescence produced by the proteasome in the presence of REGs divided by the fluorescence produced by the proteasome alone. All entries are the averages of three measurements.
There is some debate as to whether short fluorogenic peptides should be used as diagnostic reagents for determining proteasome cleavage specificity (Ustrell et al., 1995; Groettrup et al., 1996; Tanaka et al., 1997). For that reason, we also used two natural peptides as proteasome substrates in the presence of REGα, REGγ or the two robust REGγ specificity mutants (K188E/D). Cleavage products from either a 21-residue peptide (P21) or a 49-residue peptide (BBC1) were separated by HPLC and analyzed by MS. The profiles in Figure 4, which are representative of samples taken at six different times during the course of the reaction, show that wild-type REGγ decreased hydrolysis of P21 by the proteasome. In contrast, REGγ (K188E/D) and REGα markedly increased substrate consumption, producing more complicated patterns of cleavage products that are clearly different from the products produced by REGγ– proteasome complexes. MS analysis provided information on the cleavage sites (compare the open and filled arrows surrounding the sequence of P21 in Figure 4).
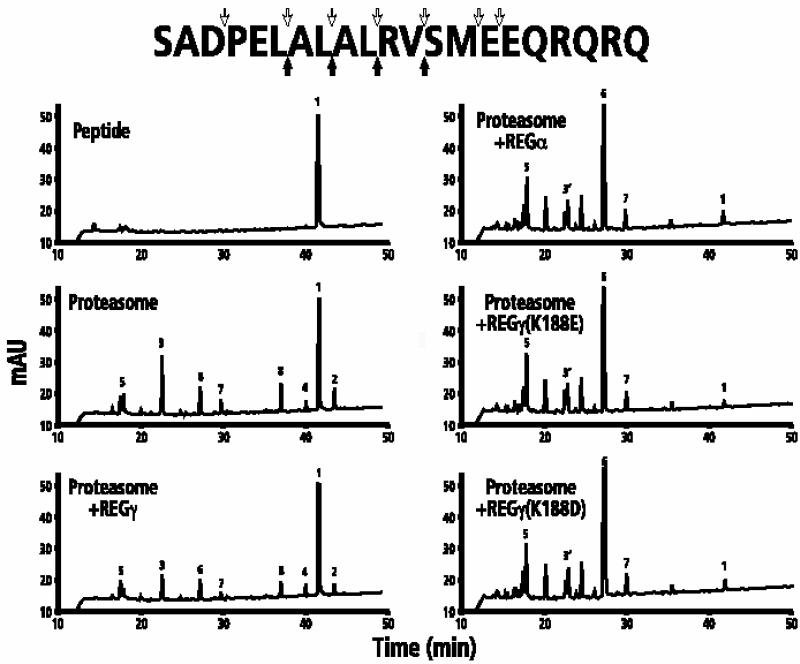
Fig. 4. HPLC/MS analysis of proteasomal cleavage products from P21. The upper left panel shows the HPLC profile of undigested P21; the remaining panels show the HPLC profiles of cleavage products generated after 12 h at 37°C by 600 ng of proteasome in the absence or presence of REGγ, REGα, REGγ (K188E) or REGγ (K188D). Major cleavage products were identified by MS as described in Materials and methods. Peak 1: the P21 substrate, SADPELALALRVSMEEQRQRQ; peak 2: SADPELALAL; peak 3: RVSMEEQRQRQ; peak 4: SADPELAL; peak 5: SMEEQRQRQ; peak 6: SADPEL; peak 7: ALRV; peak 8: ALAL; peak 3′: RVSM and RVSME. The undesignated peaks are those products whose sequence could not be unambiguously determined. The filled arrows beneath the P21 sequence reflect the primary cleavage sites by proteasome in the absence or presence of REGγ. The downward pointing open arrows are products generated in the presence of REGα or REGγ (K188E/D).
Somewhat different results were obtained upon hydrolysis of the longer peptide, BBC1. In the absence of proteasome activators, BBC1 was consumed with a half-life of 85 min, producing a series of peptides eluting between 34 and 38 min (Figure 5). With REGγ present, degradation was faster (t1/2 = 65 min), and peptides eluting between 34 and 38 min were largely absent due to their further digestion. The resulting smaller peptides ranged from 2 to 5 amino acids in length and eluted between 20 and 26 min. Substitution of REGα, REGγ (K188E) or REGγ (K188D) did not markedly alter the spectrum of peptides although these activators did speed BBC1 degradation even more (t1/2 = 45 min) and produced HPLC profiles that quantitatively, at least, could be distinguished from the REGγ profile as shown by the peaks labeled with slanted arrows in Figure 5. Taken together, assays using two natural peptides provide further evidence that REGγ (K188E/D) variants are equivalent to REGα in their activation properties.
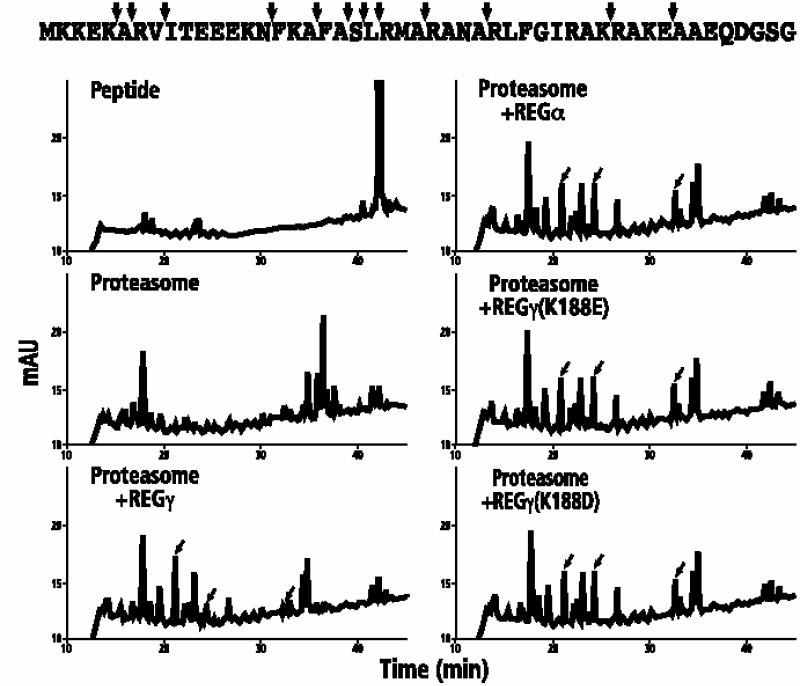
Fig. 5. HPLC/MS analysis of proteasomal cleavage products from BBC1. The upper left panel shows the HPLC profile of undigested BBC1; the remaining panels contain profiles of cleavage products generated by the proteasome alone or the proteasome in the presence of REGγ, REGα, REGγ (K188E) or REGγ (K188D). The arrows pointing to the BBC1 sequence identify major cleavage sites by the proteasome alone. The slanted arrows in the HPLC profiles denote cleavage products that are quantitatively distinct between proteasomal degradation in the presence of REGγ on one hand and in the presence of REGα or REGγ (K188E/D) on the other. Kinetic analyses (data not shown) revealed that the half-life of BBC1 was 85 min in the presence of the proteasome alone, 65 min in the presence of the proteasome plus REGγ, and 40 min in the presence of the proteasome and REGγ (K188E). Moreover, in each reaction mixture the pattern of cleavage products was essentially the same for samples taken at 10, 30, 135 and 600 min.
Physical properties of REGγ Lys188 variants
Previous studies have shown that proteasome activation is affected by the stability of REG heptamers and their affinities for the proteasome (Li et al., 2000). To examine the effect of Lys188 substitutions on REGγ heptamer stability, we rechromatographed wild-type and Lys188 variant heptamers on the Superdex 200 (26/60) size exclusion column used for purification (see Supple mentary data, available at The EMBO Journal Online). Wild-type REGγ remained fully heptameric, as did REGγ (K188H), REGγ (K188S), REGγ (K188I) and REGγ (K188R). Similar analyses showed that the percentage of REGγ (K188A), REGγ (K188C), REGγ (K188N) and REGγ (K188Q) that remained heptamers ranged from 60 to 80%, but more than half of the REGγ (K188D) and REGγ (K188E) heptamers dissociated during the second gel filtration (Table I). Replacement of Lys188 by Pro or Phe severely affected the stability of REGγ heptamers. Approximately 50% of REGγ (K188P) heptamers dissociated into monomers while REGγ (K188F) variants remained monomers/dimers.
As a measure of the relative affinities of REGγ and REGγ Lys188 mutants for the proteasome, we employed a competition assay (Li et al., 2000; see also Supplementary data). From the summary in Table I it can be seen that all REGγ Lys188 mutants were relatively resistant to REGα(N146Y)/REGβ(N135Y) competition with apparent proteasome affinities varying from 40 to 90% that of wild-type REGγ. The competition assays demonstrate that REGγ Lys188 mutants have lower, but comparable affinities for the proteasome, as does wild-type REGγ.
The presumed location of REGγ Lys188
Based on the fact that REGγ is 25% identical to REGα in sequence, we assume that the REGγ heptamer adopts a structure similar to that of REGα. In Figure 6, the hypothetical REGγ heptamer is compared with the known crystal structure of REGα. Lys188 resides in the third α-helix of the REGγ monomer and presumably faces the aqueous pore through the REGγ heptamer. The residue in REGα corresponding to REGγ Lys188 is Asp183. It is noteworthy that these two residues are oppositely charged and that the most robust REGγ activation specificity mutants (K188E/D) result in charge reversal at this position in helix 3. Comparison of helix 3 residues in REGα and the hypothetical REGγ heptamer also identifies other pore-lining residues differing in charge between the two homologs (see residues highlighted in Figure 6). To test whether charge differences at these positions might affect the activation specificities of REGγ and REGα, site-directed mutagenesis was used to generate a number of REGα Asp183 point mutants, and two sets of REGα and REGγ variants in which the charged, channel-lining residues were exchanged between the homologs. However, none of these mutations changed the activation specificity of the host REG molecules (see Supplementary data).

Fig. 6. Location of the identified mutation and other potential residues controlling REG activation specificity. (A) Arrangement of charged residues on the surface of the aqueous channel through REGα (left) and the assumed distribution of charged residues in the hypothetical REGγ (right) heptamer. The cut-away views of both REG heptamers are shown with the 7-fold symmetry axis vertical. One subunit is colored yellow and the others are shown in gray. The charged residues on the interior surface of both heptamers are shown in red (negatively charged: Asp or Glu) or blue (positively charged: Arg, Lys or His). (B) Sequence alignment of the third helices of REGα, REGβ and REGγ. The highlighted positions indicate the residues with charge properties that are similar or identical in REGα and REGβ but differ from those in REGγ. Lys188, the site of the identified REGγ mutation, is indicated with an arrow. The highlighted residues in REGα (or REGγ) were substituted with the corresponding residues in REGγ (or REGα) to generate single-site mutations in channel-lining residues. The numbers represent the positions of amino acids in the sequence of REGα (upper) or REGγ (lower).
Affinity labeling of proteasome β subunits in the presence of REGs
There are several possible mechanisms by which REGs α/β and REGγ might activate the proteasome differentially. Binding to the proteasome could induce conformational changes in the proteasome’s β subunits that alter catalysis. Such changes could, in principle, either increase or decrease catalysis. Alternatively, the REG homologs could open substrate selective channels into the proteasome. To obtain estimates of substrate access to the proteasome’s central chamber in the presence of REGγ and REGγ (K188E), we employed the active site-directed probe, 125I-YL3-VS, which covalently modifies all three of the active β subunits in an activity-dependent manner (Bogyo et al., 1997, 1998). The PhosphorImages presented in Figure 7A show increased labeling of CT/PGPH and T subunits in the presence of REGγ or REGγ (K188E). The initial labeling velocities for each reaction were quantified by graphical analysis of the PhosphorImages (Figure 7B), and Figure 7C summarizes the extents to which REGs γ and γ (K188E) stimulated initial labeling in six independent experiments. These data reveal that REGγ increased the rate of T subunit labeling 4.7-fold and the rate of CT/PGPH labeling 4.4-fold over that seen with the proteasome alone. Stimulation by the mutant REGγ (K188E) was greater, being 11-fold for the T subunit and 12-fold for the CT/PGPH subunits. The increased rate of modification induced by REGγ (K188E) is roughly comparable to its ability to stimulate sustained hydrolysis of fluorogenic peptides at the three active sites (see Figure 7C versus Table I). However, the finding that REGγ increased labeling of the CT and PGPH subunits was unexpected since the wild-type activator actually suppresses hydrolysis of the CT substrate sLLVY-MCA (Table I). Nonetheless, it is clear from the results presented in Figure 7 that REGγ promoted entry of the hydrophobic, suicide substrate 125I-YL3-VS to the central chamber of the proteasome. This argues against the possibility that the REGγ channel serves as a selective filter and leads us to propose that REGγ binding negatively regulates the proteasome’s CT/PGPH active sites (see Discussion).
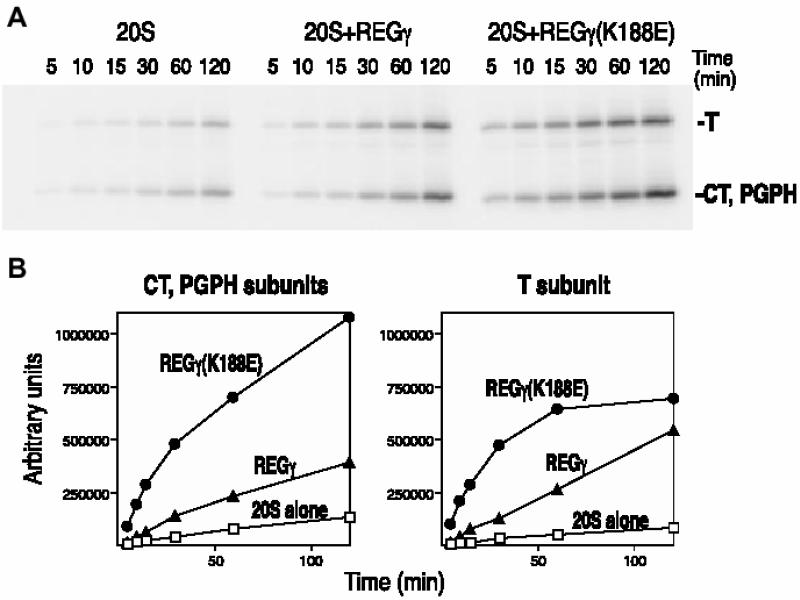
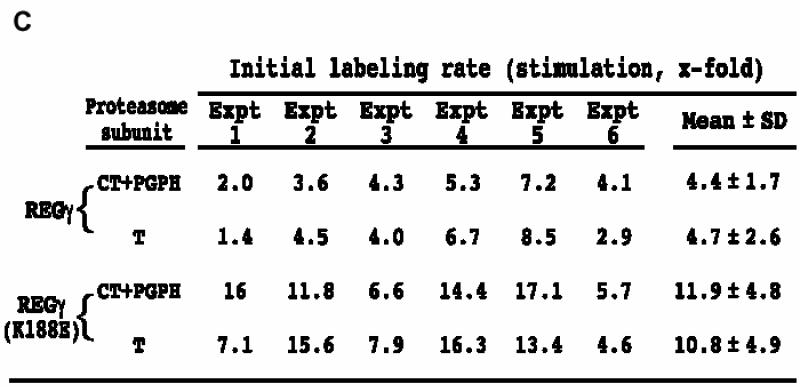
Fig. 7. Affinity labeling of proteasome catalytic subunits in the presence of REGγ and REGγ (K188E). (A) A representative PhosphorImage of proteasome subunits labeled by 125I-YL3-VS. (B) Quantitation of proteasome subunit labeling. Bands of activity corresponding to the T-like subunit and a combination of the unresolved CT and PGPH subunits were quantitated with the ImageQuant software (Molecular Dynamics). Total counts for each band were plotted as a function of time (CT+PGPH subunits, left panel; T subunit, right panel). (C) Stimulation of the initial labeling velocity by mutant and wild-type REGγ. The affinity labeling experiment has been repeated six times and the average stimulation by REG molecules is calculated as following: initial velocity in the presence of REG/initial velocity of proteasome.
Discussion
REGγ (K188E/D) and REGα activate the proteasome almost identically
A major finding from the studies presented above is that substitution of Glu or Asp for Lys188 converts the proteasome activation properties of REGγ to that of REGα. Several assays demonstrate this essential point. For example, REGα and REGγ (K188E/D) activate proteasome hydrolysis of the diagnostic peptides LLVY- MCA, LRR-MCA and LLE-βNA over the same concentration range and to almost the same extent (Figure 3). Cleavage of each fluorogenic peptide listed in Table II was stimulated almost identically by REGγ (K188E/D) and REGα, indicating that the activation specificity of the mutant REGγ is virtually equivalent to that of REGα. Analysis of P21 and BBC1 digests (Figures 4 and 5) extends equivalent activation by REGγ (K188E/D) and REGα to natural peptides as well. Despite the strikingly similar activation properties of REGγ (K188E/D) and REGα, the two molecules do not activate the proteasome identically since REGα consistently produces 10–20% more hydrolysis of fluorogenic peptides with hydrophobic residues in the P1 positions (Figure 3; Table II).
Lys188 lines the aqueous channel through REGγ
Perhaps it is not surprising that amino acid substitutions at a single site, Lys188, change the proteasome activation properties of REGγ to those of REGα. However, the apparent location of Lys188 was unexpected. Sequence alignments show that REGγ Lys188 corresponds to REGα Asp183, a residue that faces the aqueous channel through the REGα heptamer and is located almost 60 Å from the surface of REGα that binds the proteasome. The assumption that REGγ Lys188 and REGα Asp183 occupy equivalent positions is based on the substantial similarities in sequence and structural properties of the two REG homologs as well as on experiments involving site-specific mutations, deletion or exchange of sequences between them. REGα has been shown to be a heptamer by sedimentation (Johnston et al., 1997), MS (Zhang et al., 1999) and X-ray crystallography (Knowlton et al., 1997). REGγ is likewise a heptamer as demonstrated by velocity sedimentation (Figure 1) and electron microscopy (Figure 2). REGα and REGγ both remain active upon deletion of their homolog-specific inserts (Zhang et al., 1998b) and REGα–γ or REGγ–α chimeras involving C-terminal sequence exchanges are fully active (Zhang et al., 1998c; Li et al., 2000). Moreover, three REGγ–α variants chimeric for activation loop flanking sequences form heptamers, with two of them being fully active (Li and Rechsteiner, 2001). Finally, the activation loops of REGs α and γ are identical and specific substitutions in either homolog result in a REG molecule that binds the proteasome but does not activate it (Zhang et al., 1998a). Thus, REGs α and γ are, for the most part, unaffected by sequence exchanges, and corresponding mutations in each protein produce similar effects. These observations provide strong evidence that REGα and REGγ adopt identical folds.
The mechanism of REG activation: selective channels or conformational changes in proteasome β subunits?
The crystal structure of the yeast proteasome shows that α subunit N-terminal sequences form a barrier between the enzyme’s internal chambers and the external solvent (Groll et al., 1997). The crystal structure of REGα reveals a 20–30 Å-wide aqueous channel through a donut-shaped heptamer (Knowlton et al., 1997), and electron microscopic images suggest that REG heptamers bind the proteasome with their aqueous channels normal to and centered over the proteasome α subunit ring (Gray et al., 1994). This led to the proposal that REG binding would displace proteasome α subunit N-terminal sequences opening a continuous channel to the enzyme’s central proteolytic chamber (Knowlton et al., 1997). A crystal structure of the yeast proteasome complexed with a Trypanosoma REG (PA26) has recently been solved and in fact, a continuous channel does exist from the upper surface of REG to the proteasome’s central catalytic chamber (Whitby et al., 2000). Thus, there is little doubt that REG binding creates a channel through which substrates and products should exchange more readily between the external solvent and the enzyme’s buried catalytic sites.
In principle, formation of a continuous channel could account for the broad activation of peptide hydrolysis by REG α and β molecules (Groll et al., 2000; Whitby et al., 2000). To explain the differential activation by REGs α/β and γ, however, it is necessary to propose that either REG channels act as selective filters or REGγ binding inhibits catalysis by the CT and PGPH active sites. The location of Lys188 on the channel surface and the dramatic effects of reversing its charge by substitution of Asp or Glu would seem to support the idea that REG channels function as selective filters. However, we do not favor this hypothesis for several reasons. First, proteasomal cleavage of small fluorogenic tripeptides of comparable dimensions, AAF-MCA, LLE-βNA and LRR-MCA, is affected to markedly different extents by REGγ and REGγ (K188E/D) (Table II). Secondly, if our structural model for REGγ is correct, a ring of positive charge at the entrance to the REGγ channel (Figure 6) should present a barrier to the diffusion of positively charged, fluorogenic peptides into the proteasome. Yet these are the very substrates whose hydrolysis is stimulated by REGγ (Table II).
Two experimental findings pose more direct problems for the selective channel model. It is apparent from the HPLC profiles in Figure 5 that the BBC1 peptide was fully consumed by the proteasome with or without added REGs. If REGs simply increase entry of BBC1 into the proteasome, one would expect the same set of products in the presence or absence of REGs; the products would just appear sooner. The increased frequency of multiple cleavages in BBC1 indicates that REGs expand the number of peptide bonds susceptible to hydrolysis by the proteasome once BBC1 enters the central proteolytic chamber. There are two mechanisms by which this could occur. REGs could activate or change the specificity of proteasome β subunits, thereby increasing the probability of multiple cleavages in BBC1. Or REGs could slow the exit of initial BBC1 cleavage products thereby promoting further cleavages (Dolenc et al., 1998). The latter possibility is inconsistent with the fact that REGγ and γ (K188E) speed destruction of BBC1 (see Results) presumably by promoting entry of the peptide to the proteasome’s central chamber.
Secondly, use of 125I-YL3-VS clearly demonstrates that REGγ does not exclude hydrophobic peptides since the wild-type proteasome activator increased β subunit modification almost 5-fold (Figure 7). Labeling by 125I-YL3-VS requires the compound to enter the proteasome’s central chamber, bind to β subunit active sites and form a covalent bond to the N-terminal threonines. The initial labeling rate is a composite of these three steps so we can not strictly conclude that REGγ promotes 125I-YL3-VS entry. It could, in principle, speed either or both of the latter two steps. Still, given the recent finding that binding of PA26 or deletion of proteasome α subunit sequences activates all three β subunits (Groll et al., 2000; Whitby et al., 2000), it is very likely that REGγ opens a non-selective channel into the proteasome.
Initial labeling of proteasome β subunits in the presence of REGγ (K188E) is 2.3- or 3-fold faster than in the presence of REGγ (Figure 7C). For the T site, this difference approximates the slightly greater ability of REGγ (K188E) to increase cleavage of LRR-MCA (Table I). However, the 3-fold greater CT/PGPH labeling by REGγ (K188E) relative to REGγ is an order of magnitude lower than its ability to stimulate sustained cleavage of sLLVY-MCA by the CT site. The complete proteolytic reaction requires two additional steps besides substrate entry, binding and covalent bond formation. The covalent adduct to the N-terminal threonine of active β subunits must be hydrolyzed and the product released. We propose that at the T site these two steps proceed at similar rates when either REGγ or REGγ (K188E) is bound; however, at the CT site they are 10-fold slower in the presence of REGγ. That is, REGγ inhibits hydrolysis of the tetrahedral intermediate and/or product release from the CT site. Alternatively, REGγ (K188E) stimulates one or both of these steps (see Table III).
Table III. Differential effects of mutant and wild-type REGγ on specific steps during catalysis.
| Steps in bond cleavage by proteasome | Differential stimulation (REGγ (K188E)/REGγ) |
|
|---|---|---|
| T site | CT site | |
| Entry of S to central chamber | ||
| Binding of S to active sites | 2.3 | ∼3 |
| Covalent adduct between threonine and S | ||
| Hydrolysis of covalent adduct | ||
| Product release | ? | ? |
| Sustained hydrolysis of fluorogenic peptides | 1.5 | 35 |
Substrate (S) cleavage by proteasomes can be divided into five steps: entry of substrate to the proteasome’s central catalytic chamber, binding of substrate to active β subunits, formation of covalent adduct between nucleophile threonine and substrate, hydrolysis of covalent adduct and product release. Activity assays using fluorogenic peptides reflect the complete reaction. Compared with REGγ, the mutant REGγ (K188E) stimulates catalysis at T and CT subunits 1.5- and 35-fold faster, respectively (calculated from Table I). The affinity labeling assay using 125I-YL3-VS is only diagnostic for the first three steps, where REGγ (K188E) is 2.3- and 3-fold more efficient than REGγ in stimulating the labeling of T and CT subunits, respectively (calculated from Figure 7C). The marked difference (wild type versus mutant) between their stimulation of catalysis by the CT subunit and their stimulation in CT labeling suggests that REGγ and REGγ (K188E) have markedly differential effects on the last two steps, hydrolysis and product release, at the CT/PGPH sites.
The following considerations strongly favor inhibition by REGγ. First, it is clear from the hydrolysis of fluorogenic peptides that REGγ does inhibit the CT and PGPH activities (Table II). Secondly, Groll et al. (2000) have recently shown that removing N-terminal sequences from the α3 subunit of the yeast 20S proteasome stimulates hydrolysis of LLE-βNA by 10-fold and hydrolysis of LRR-MCA or LLVY-MCA by 20-fold. Their results are in good agreement with the enhanced hydrolysis of the very same peptides induced by REGγ (K188E) as shown in Table II. Since removing α3 N-terminal sequences opens a gate to the central chamber (Groll et al., 2000), it seems reasonable to interpret the stimulation by REGγ (K188E) mainly in terms of increased substrate access. Stohwasser et al. (2000) came to much the same conclusion from recent kinetic analyses of proteasome activation by REG homologs. If increased catalysis of LRR-MCA reflects gate opening by REGγ (K188E) and REGγ, it follows that REGγ must inhibit catalysis at the CT and PGPH active sites.
Proposed mechanism for differential proteasome activation by REGs
REGγ (K188E/D) differ from wild-type REGγ in an important physical property; they are less stable heptamers. In fact, they appear to form hexamers as well as heptamers (Figure 2). We suspect that this may be the very property that prevents REGγ (K188E/D) from inhibiting the CT and PGPH active sites. Our hypothesis is based on the data in Table I, where, with two exceptions (K188P and K188F), a trend is present between the heptamer instability of Lys188 variants and activation of LLVY-MCA and LLE-βNA hydrolysis (see Supplementary data). We assume that REG–proteasome interaction involves conformational adjustments that could take place either in the REG heptamers or in the proteasome or most likely in both. We speculate that wild-type REGγ subunits bind each other so tightly that they resist conformational adjustment upon association with the proteasome. As a consequence most changes occur within the proteasome, and some of these conformational adjustments inhibit the CT and PGPH subunits. In contrast, when REGγ (K188E) binds, adjustments occur both in the bound REG and in the proteasome. As a result, the CT and PGPH subunits remain active (see Figure 8). For reasons unknown, the T-like subunit escapes inhibition by REGγ. However, it is clearly affected upon REGγ and REGγ (K188E/D) binding, as shown by the HPLC patterns in Figure 5. All REGs, α, γ and γK188E/D, induced additional cleavages in BBC1, and these are likely to result from altered specificity of the T subunit, since BBC1 is such a basic peptide. Furthermore, surveys using fluorogenic peptide libraries indicate that there are specificity changes upon REGγ binding (J.L.Harris, P.B.Alper, J.Li, M.Rechsteiner and B.J.Backes, in preparation).

Fig. 8. A model for differential proteasome activation by REG proteins. The yeast 20S proteasome (Groll et al., 1997) and human 11S REGα (Knowlton et al., 1997) are altered to illustrate the REG–proteasome interaction. The three catalytic β subunits are marked with asterisks and labeled. In this model, we propose that the REG–proteasome interaction involves conformational adjustments in both the REG heptamers and the proteasome (shown as arrows). The wild-type REGγ heptamer is rigid so that conformational adjustments occur only within the proteasome, and these conform ational changes inhibit the CT and PGPH subunits (left, shown as smaller asterisks). In contrast, REGα/β and REGγ (K188E/D) heptamers are relatively flexible (right, shown in an open form). Upon binding to these heptamers, the proteasome undergoes less conformational adjustment and the three catalytic subunits remain active.
In summary, the experiments presented above illustrate two important features of the proteasome activators known as 11S REG or PA28. First, a single amino acid change converts the activation specificity of REGγ to that of REGs α and β. Substitution of Glu or Asp for REGγ Lys188 is attended by a decrease in heptamer stability, and this may allow conformational changes in REGγ (K188E/D) that prevent inactivation of the CT and PGPH subunits of the proteasome. The second, more important finding has to do with the mechanism of activation by REGs. Use of the active site probe, 125I-YL3-VS, clearly demonstrates that REGs do not simply open gates to the proteasome’s central chamber. Rather, REGγ (K188E/D) either permits unimpaired catalysis by the proteasome’s CT and PGPH subunits or actually activates them. Conversely, REGγ either inhibits these two subunits or fails to activate them. We clearly favor the idea that REGγ inhibits the CT/PGPH active sites, but in either case, the results presented above provide compelling evidence that REGs induce conformational changes in proteasome active sites, or possibly, in non-catalytic modifier sites (Schmidtke et al., 2000; Myung et al., 2001).
Materials and methods
Construction of an expression library encoding random REGγ mutants
Error-prone PCR was used to introduce random mutations into the REGγ cDNA (Zhang et al., 1998a). The PCR products were inserted into pET26(b) through NdeI–BamHI sites. The resulting plasmid library was transformed into BL21(DE3) cells. Approximately 60% of the isolated colonies contained a single-site mutation.
Isolation of REGγ mutants stimulating the chymotrypsin-like activity of the proteasome
Transformants were picked and grown at 37°C in LB containing 25 µg/ml kanamycin. Protein expression was induced with 0.8 mM isopropyl-β-d-thiogalactopyranoside for 2 h at 30°C. Cells were collected by centrifugation and lysed with 10 mM Tris–HCl pH 7.5, 0.5% Triton X-100 and 0.3 mg/ml polymixin B sulfate. Aliquots were incubated with 170 ng of proteasome and 100 µM LLVY-MCA. After a 10 min incubation, reactions were terminated with 200 µl of ethanol and fluorescence was measured as described (Li et al., 2000). Highly active colonies were rescreened as above and plasmids were then purified and sequenced.
Site-directed mutagenesis
Oligonucleotide-directed mutagenesis was used to introduce point mutations into REGs α or γ as described (Zhang et al., 1998a). All constructs were sequenced to confirm the absence of unintended mutations.
Purification of REG mutants and determination of their activation specificity
REG mutants were expressed and purified to homogeneity as described (Li et al., 2000). Each REG protein (3 µg) was mixed with 170 ng of proteasome and 100 µM substrate (LLVY-MCA, LRR-MCA or LLE-βNA) to measure their activation specificity as described (Li et al., 2000).
Digestion of natural synthetic peptides
Proteasomal hydrolysis of P21 and BBC1 peptides was carried out as described (Zhang et al., 1998b). Digestion products were applied to a C18 HPLC column and separated with a gradient of 0–45% acetonitrile containing 0.1% trifluoroacetic acid. To identify the products derived from P21, fractions were collected manually, concentrated, and subjected to MS.
Equilibrium sedimentation
Sedimentation equilibrium was used to assess the solution states of wild-type and chimeric REGγ proteins as recently described (Li et al., 2000). Briefly, samples ranging in concentration from 27.5 to 110 µg/ml were centrifuged at 20°C at speeds ranging from 11 000 to 13 000 r.p.m. until sedimentation and chemical equilibria were attained. Data were collected by radial scanning of the centrifuge cell with 10 absorbance measurements at 280 nm taken every 0.001 cm. Values of 0.741 ml/g for the partial specific volume and 1.016 g/ml for the solvent density were used.
Electron microscopy
Grids bearing carbon-coated nitrocellulose films were glow-discharged prior to being floated for 2 min on 10 µl drops of sample at a protein concentration of 30 µg/ml. The sample drop was then blotted away, and the grid was negatively stained by floating on a drop of 1% uranyl acetate for 10 s. Specimens were observed in a Philips EM400T transmission electron microscope, and micrographs were recorded at a nominal magnification of 46 000×. Details of the image analysis are presented as Supplementary data.
Active site affinity labeling
Purified human red blood cell proteasomes (1 µg) were diluted into 100 µl of reaction buffer (50 mM Tris pH 7.4, 5 mM MgCl2, 2 mM dithiothreitol). Purified recombinant REGγ and REGγ (K188E) were diluted to a final concentration of 20 µg/ml from a 1 mg/ml stock solution into the buffer containing proteasomes, and the mixture was incubated for 10 min at 37°C to induce complex formation. The radiolabeled probe, 125I-YL3-VS, was then added (∼106 c.p.m.), the reactions were incubated at room temperature for 5, 10, 15, 30, 60 and 120 min, and quenched by addition of one-fourth volume of 4× SDS sample buffer. Samples were analyzed by SDS–PAGE (12.5%) and the resulting gels fixed, dried and exposed to a PhosphorImaging screen (Molecular Dynamics). Bands of activity corresponding to the T-like subunit and combination of CT and PGPH subunits (both run as a single band and can not be resolved) were quantitated using the ImageQuant software (Molecular Dynamics).
Supplementary data
Supplementary data for this paper are available at The EMBO Journal Online.
Acknowledgments
Acknowledgements
We thank Chris Hill, Carlos Gorbea, Goeff Goellner, Patrick Young and Vicença Ustrell for critical reading of the manuscript. We thank Su Li and Frank Whitby for figure preparation. These studies were supported by National Institutes of Health grant GM60334.
References
- Ahn J.Y. et al. (1995) Primary structures of two homologous subunits of PA28, a γ-interferon-inducible protein activator of the 20S proteasome. FEBS Lett., 366, 37–42. [DOI] [PubMed] [Google Scholar]
- Baumeister W., Walz,J., Zuhl,F. and Seemuller,E. (1998) The proteasome: paradigm of a self-compartmentalizing protease. Cell, 92, 367–380. [DOI] [PubMed] [Google Scholar]
- Bochtler M., Ditzel,L., Groll,M., Hartmann,C. and Huber,R. (1999) The proteasome. Annu. Rev. Biophys. Biomol. Struct., 28, 295–317. [DOI] [PubMed] [Google Scholar]
- Bogyo M., McMaster,J.S., Gaczynska,M., Tortorella,D., Goldberg,A.L. and Ploegh,H. (1997) Covalent modification of the active site threonine of proteasomal β subunits and the Escherichia coli homolog HslV by a new class of inhibitors. Proc. Natl Acad. Sci. USA, 94, 6629–6634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogyo M., Shin,S., McMaster,J.S. and Ploegh,H.L. (1998) Substrate binding and sequence preference of the proteasome revealed by active-site-directed affinity probes. Chem. Biol., 5, 307–320. [DOI] [PubMed] [Google Scholar]
- Dahlmann B., Kopp,F., Kuehn,L., Niedel,B., Pfeifer,G., Hegerl,R. and Baumeister,W. (1989) The multicatalytic proteinase (prosome) is ubiquitous from eukaryotes to archaebacteria. FEBS Lett., 251, 125–131. [DOI] [PubMed] [Google Scholar]
- DeMartino G.N., Moomaw,C.R., Zagnitko,O.P., Proske,R.J., Ma,C.P., Afendis,S.J., Swaffield,J.C. and Slaughter,C.A. (1994) PA700, an ATP-dependent activator of the 20S proteasome, is an ATPase containing multiple members of a nucleotide-binding protein family. J. Biol. Chem., 269, 20878–20884. [PubMed] [Google Scholar]
- Dolenc I., Seemuller,E. and Baumeister,W. (1998) Decelerated degradation of short peptides by the 20S proteasome. FEBS Lett., 434, 357–361. [DOI] [PubMed] [Google Scholar]
- Dubiel W., Pratt,G., Ferrell,K. and Rechsteiner,M. (1992) Purification of an 11S regulator of the multicatalytic protease. J. Biol. Chem., 267, 22369–22377. [PubMed] [Google Scholar]
- Gray C.W., Slaughter,C.A. and DeMartino,G.N. (1994) PA28 activator protein forms regulatory caps on proteasome stacked rings. J. Mol. Biol., 236, 7–15. [DOI] [PubMed] [Google Scholar]
- Groettrup M., Soza,A., Kuckelkorn,U. and Kloetzel,P.M. (1996) Peptide antigen production by the proteasome: complexity provides efficiency. Immunol. Today, 17, 429–435. [DOI] [PubMed] [Google Scholar]
- Groll M., Ditzel,L., Löwe,J., Stock,D., Bochtler,M., Bartunik,H.D. and Huber,R. (1997) Structure of 20S proteasome from yeast at 2.4 Å resolution. Nature, 386, 463–471. [DOI] [PubMed] [Google Scholar]
- Groll M., Bajorek,M., Kohler,A., Moroder,L., Rubin,D.M., Huber,R., Glickman,M.H. and Finley,D. (2000) A gated channel into the proteasome core particle. Nature Struct. Biol., 11, 1062–1067. [DOI] [PubMed] [Google Scholar]
- Grziwa A., Baumeister,W., Dahlmann,B. and Kopp,F. (1991) Localization of subunits in proteasomes from Thermoplasma acidophilum by immunoelectron microscopy. FEBS Lett., 290, 186–190. [DOI] [PubMed] [Google Scholar]
- Hershko A. and Ciechanover,A. (1998) The ubiquitin system. Annu. Rev. Biochem., 67, 425–479. [DOI] [PubMed] [Google Scholar]
- Hoffman L., Pratt,G. and Rechsteiner,M. (1992) Multiple forms of the 20S multicatalytic and the 26S ubiquitin/ATP-dependent proteases from rabbit reticulocyte lysate. J. Biol. Chem., 267, 22362–22368. [PubMed] [Google Scholar]
- Johnston S.C., Whitby,F.G., Realini,C., Rechsteiner,M. and Hill,C.P. (1997) The proteasome 11S regulator subunit REGα (PA28α) is a heptamer. Protein Sci., 6, 2469–2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowlton J.R., Johnston,S.C., Whitby,F.G., Realini,C., Zhang,Z., Rechsteiner,M. and Hill,C.P. (1997) Structure of the proteasome activator REGα (PA28α). Nature, 390, 639–643. [DOI] [PubMed] [Google Scholar]
- Li J. and Rechsteiner,M. (2001) Molecular dissection of the 11S REG (PA28) proteasome activators. Biochimie, 83, 373–383. [DOI] [PubMed] [Google Scholar]
- Li J., Gao,X., Joss,L. and Rechsteiner,M. (2000) The proteasome activator 11S REG or PA28: chimeras implicate carboxyl-terminal sequences in oligomerization and proteasome binding but not in the activation of specific proteasome catalytic subunits. J. Mol. Biol., 299, 641–654. [DOI] [PubMed] [Google Scholar]
- Löwe J., Stock,D., Jap,B., Zwickl,P., Baumeister,W. and Huber,R. (1995) Crystal structure of the 20S proteasome from the archaeon T. acidophilum at 3.4 Å resolution. Science, 268, 533–539. [DOI] [PubMed] [Google Scholar]
- Ma C.P., Slaughter,C.A. and DeMartino,G.N. (1992) Identification, purification, and characterization of a protein activator (PA28) of the 20S proteasome (macropain). J. Biol. Chem., 267, 10515–10523. [PubMed] [Google Scholar]
- Myung J., Kim,K.B., Lindsten,K., Dantuma,N.P. and Crews,C.M. (2001) Lack of proteasome active site allostery as revealed by subunit-specific inhibitors. Mol. Cell, 7, 411–420. [DOI] [PubMed] [Google Scholar]
- Nikaido T., Shimada,K., Shibata,M., Hata,M., Sakamoto,M., Takasaki,Y., Sato,C., Takahashi,T. and Nishida,Y. (1990) Cloning and nucleotide sequence of cDNA for Ki antigen, a highly conserved nuclear protein detected with sera from patients with systemic lupus erythematosus. Clin. Exp. Immunol., 79, 209–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters J.M., Cejka,Z., Harris,J.R., Kleinschmidt,J.A. and Baumeister,W. (1993) Structural features of the 26S proteasome complex. J. Mol. Biol., 234, 932–937. [DOI] [PubMed] [Google Scholar]
- Pühler G., Weinkauf,S., Bachmann,L., Muller,S., Engel,A., Hegerl,R. and Baumeister,W. (1992) Subunit stoichiometry and three-dimensional arrangement in proteasomes from Thermoplasma acidophilum. EMBO J., 11, 1607–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Realini C., Jensen,C.C., Zhang,Z., Johnston,S.C., Knowlton,R., Hill,C.P. and Rechsteiner,M. (1997) Characterization of recombinant REGα, REGβ, and REGγ proteasome activators. J. Biol. Chem., 272, 25483–25492. [DOI] [PubMed] [Google Scholar]
- Rechsteiner M. (1998) The 26S proteasome. In Peters,J.M., Harris,J.R. and Finley,D. (eds), Ubiquitin and the Biology of the Cell. Plenum, New York, NY, pp. 147–189.
- Schmidtke G., Emch,S., Groettrup,M. and Holzhutter,H.G. (2000) Evidence for the existence of a non-catalytic modifier site of peptide hydrolysis by the 20S proteasome. J. Biol. Chem., 275, 22056–22063. [DOI] [PubMed] [Google Scholar]
- Stohwasser R., Salzmann,U., Giesebrecht,J., Kloetzel,P.M. and Holzhütter,H.G. (2000) Kinetic evidences for facilitation of peptide channelling by the proteasome activator PA28. Eur. J. Biochem., 267, 6221–6230. [DOI] [PubMed] [Google Scholar]
- Tanahashi N. et al. (1997) Molecular properties of the proteasome activator PA28 family proteins and γ-interferon regulation. Genes Cells, 2, 195–211. [DOI] [PubMed] [Google Scholar]
- Tanaka K., Tanahashi,N., Tsurumi,C., Yokota,K.Y. and Shimbara,N. (1997) Proteasomes and antigen processing. Adv. Immunol., 64, 1–38. [DOI] [PubMed] [Google Scholar]
- Udvardy A. (1993) Purification and characterization of a multiprotein component of the Drosophila 26S (1500 kDa) proteolytic complex. J. Biol. Chem., 268, 9055–9062. [PubMed] [Google Scholar]
- Ustrell V., Realini,C., Pratt,G. and Rechsteiner,M. (1995) Human lymphoblast and erythrocyte multicatalytic proteases: differential peptidase activities and responses to the 11S regulator. FEBS Lett., 376, 155–158. [DOI] [PubMed] [Google Scholar]
- Voges D., Zwickl,P. and Baumeister,W. (1999) The 26S proteasome: a molecular machine designed for controlled proteolysis. Annu. Rev. Biochem., 68, 1015–1068. [DOI] [PubMed] [Google Scholar]
- Whitby F.G., Masters,E.I., Kramer,L., Knowlton,J.R., Yao,Y., Wang,C.C. and Hill,C.P. (2000) Structural basis for the activation of 20S proteasomes by 11S regulators. Nature, 408, 115–120. [DOI] [PubMed] [Google Scholar]
- Zhang Z., Clawson,A., Realini,C., Jensen,C., Knowlton,J.R., Hill,C.P. and Rechsteiner,M. (1998a) Identification of an activation region in the proteasome activator REGα. Proc. Natl Acad. Sci. USA, 95, 2807–2811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z., Realini,C., Clawson,A., Endicott,S. and Rechsteiner,M. (1998b) Proteasome activation by REG molecules lacking homolog-specific inserts. J. Biol. Chem., 273, 9501–9509. [DOI] [PubMed] [Google Scholar]
- Zhang Z., Clawson,A. and Rechsteiner,M. (1998c) The proteasome activator 11S regulator or PA28. Contribution by both α and β subunits to proteasome activation. J. Biol. Chem., 273, 30660–30668. [DOI] [PubMed] [Google Scholar]
- Zhang Z., Krutchinsky,A., Endicott,S., Realini,C., Rechsteiner,M. and Standing,K.G. (1999) Proteasome activator 11S REG or PA28: recombinant REGα/REGβ hetero-oligomers are heptamers. Biochemistry, 38, 5651–5658. [DOI] [PubMed] [Google Scholar]