

Abstract
The retinoblastoma tumor suppressor (RB) plays an important role in the regulation of cell cycle progression and terminal differentiation of many cell types. Rb–/– mouse embryos die at midgestation with defects in cell cycle regulation, control of apoptosis and terminal differentiation. However, chimeric mice composed of wild-type and Rb-deficient cells are viable and show minor abnormalities. To determine the role of Rb in development more precisely, we analyzed chimeric embryos and adults made with marked Rb–/– cells. Like their germline Rb–/– counterparts, brains of midgestation chimeric embryos exhibited extensive ectopic S-phase entry. In Rb-mutants, this is accompanied by widespread apoptosis. However, in chimeras, the majority of Rb-deficient cells survived and differentiated into neuronal fates. Rescue of Rb–/– neurons in the presence of wild-type cells occurred after induction of the p53 pathway and led to accumulation of cells with 4n DNA content. Therefore, the role of Rb during development can be divided into a cell-autonomous function in exit from the cell cycle and a non-cell-autonomous role in the suppression of apoptosis and induction of differentiation.
Keywords: apoptosis/cell cycle/central nervous system/differentiation/retinoblastoma
Introduction
The retinoblastoma gene (RB) is best known for its role in tumor suppression. In humans, inheritance of a mutated allele of the RB gene predisposes to familial retinoblastoma. RB is also mutated in many sporadic cancers, including retinoblastoma, sarcomas and various carcinomas (Goodrich and Lee, 1993). Mice heterozygous for germline mutation in the murine homolog of the retinoblastoma gene (Rb) are highly susceptible to pituitary and thyroid carcinomas, but do not develop retinoblastoma (Clarke et al., 1992; Jacks et al., 1992; Lee et al., 1992; Williams et al., 1994a).
Rb exerts its tumor suppressor function by controlling cell cycle progression from G1 into S-phase (Weinberg, 1995). Hypophosphorylated RB protein (pRB) prevents premature S-phase entry by binding to and inhibiting the E2F family of transcription factors. It can also recruit histone deacetylases to the E2F-bound promoters, thus actively inhibiting transcription of E2F target genes (Brehm and Kouzarides, 1999). When cells are stimulated to re-enter the cell cycle, pRB is phosphorylated by cyclin D–cdk4 and cyclin E–cdk2 complexes. Hyperphosphoryl ation of pRB reduces its affinity for E2Fs, leading to their release and the transcription of genes necessary for S-phase entry and progression (Nevins et al., 1997).
In addition, pRB also functions in the regulation of terminal differentiation of many tissue types (Lipinski and Jacks, 1999). An early indication of the role of Rb in differentiation was the phenotype of Rb–/– mouse embryos (Clarke et al., 1992; Jacks et al., 1992; Lee et al., 1992). These embryos die between 13 and 15 days of gestation (E13–15) with pronounced defects in erythroid, neuronal and lens development. These lineages are able to initiate the differentiation process, but fail to achieve a fully differentiated state. For example, erythrocytes do not enucleate efficiently, and expression of some of the late neuronal and lens differentiation markers is decreased or absent (Lee et al., 1994; Morgenbesser et al., 1994). Additionally, ectopic cell cycle entry and elevated apoptosis levels are apparent in the ocular lens and in the peripheral (PNS) and central nervous system (CNS). These results indicate that Rb might assist in achieving and/or maintaining the post-mitotic state associated with terminal differentiation of many tissue types, as well as in the protection from apoptosis and induction of late differentiation markers.
The phenotype in the nervous system of Rb–/– embryos has been most fully characterized. Extensive ectopic cell cycle entry and elevated apoptosis levels are apparent in both CNS and PNS by E12. The inappropriate cell cycle entry is accompanied by elevated activity of free E2F proteins and overexpression of E2F transcriptional targets, such as cyclin E (Macleod et al., 1996). Additionally, p53 protein levels and p53 DNA binding activity are enhanced in the brains of Rb–/– embryos, leading to increased expression of the p53 transcriptional target p21. In Rb–/– CNS, apoptosis has been shown to be p53 dependent, as cells in the CNS of Rb–/–p53–/– embryos continue to ectopically enter the S-phase, but do not die. In Rb–/– PNS, both p21 expression and apoptosis are p53 independent (Macleod et al., 1996).
The Rb–/– CNS/PNS phenotype initiates at the time in mouse embryo development when neuronal precursor cells normally initiate exit from the cell cycle and begin neuronal differentiation, suggesting that Rb function might be specifically required in the process of neurogenesis. This has been confirmed by following expression of a β-galactosidase reporter gene driven by an early pan-neuronal promoter, Tα1 α-tubulin (Slack et al., 1998). Expression of this reporter is correctly initiated in Rb–/– embryos at E12.5, but declines by E14.5, indicating that differentiating neurons are dying in the absence of functional pRB.
Cell-based studies have supported the importance of Rb in differentiation. For example, Rb–/– embryo lung bud fibroblasts can be induced to express early but not late adipocyte differentiation markers (Chen et al., 1996). Similarly, Rb–/– primary mouse fibroblasts induced to differentiate into muscle express early differentiation markers normally, but have attenuated expression of the late ones. Rb–/– myocytes are also able to re-enter the cell cycle following serum stimulation (Novitch et al., 1996). In this system, pRB has been shown to induce myogenesis through inhibition of E2F function to prevent cell cycle re-entry and through augmentation of muscle-specific transcription factor activity (Novitch et al., 1999). Neural precursor cells derived from Rb–/– embryos exhibit delayed exit from mitosis and deregulated E2F activity (Callaghan et al., 1999). Inactivation of pRB and the closely related p107 and p130 proteins, by expression of a mutant adenovirus E1A oncoprotein can prevent cell cycle withdraw and lead to death of in vitro differentiating cortical progenitor cells (Slack et al., 1998).
Because germline Rb–/– embryos die too early to achieve a terminally differentiated state in many tissues, we and others studied the role of Rb in development and differentiation using chimeric mice composed of wild-type and Rb–/– cells (Rb–/– chimeras) (Maandag et al., 1994; Williams et al., 1994b). Surprisingly, Rb–/– chimeras are viable and fertile even with extensive contribution of Rb–/– cells to all tissues, including brain, liver and blood. They develop pituitary tumors with reduced latency as compared with Rb+/– mice, but otherwise show only mild histopathological abnormalities, including hyperplasia of the adrenal medulla, abnormal lens architecture, cataracts, pleomorphic Purkinje cells and enlarged liver cells. Rb–/– chimeric embryos have delayed enucleation of erythrocytes, ectopic mitosis in the lens, and slightly increased pycnosis in the retina and the spinal ganglia. No abnormalities were observed in the developing CNS (Maandag et al., 1994; Williams et al., 1994b).
The lack of prominent developmental defects in Rb–/– chimeras contrasts with the pronounced phenotype of germline Rb–/– embryos and with the cell-based data suggesting an important role for pRB in differentiation. The data from chimeras have been interpreted to indicate that the defects in cell cycle control and apoptosis observed in the nervous system of Rb–/– embryos are non-cell autonomous in nature, as they are suppressed by the presence of wild-type cells. This contrasts with the known function of pRB in cell cycle regulation, where it is thought to restrict entry into S-phase in a cell-autonomous manner. In order to reconcile these discrepancies, as well as to characterize further the role of pRB in neuronal differentiation in vivo, we have performed a detailed analysis of the CNS phenotype in chimeric embryos and adults produced from marked Rb–/– cells.
Results
Contribution of Rb–/– cells to chimera CNS
A major limitation of earlier analyses of Rb–/– chimeras (Maandag et al., 1994; Williams et al., 1994b) was the inability to identify and follow the fate of individual mutant cells. To overcome this limitation, we have created male Rb–/– 129sv ES cells expressing β-galactosidase from a ubiquitous Rosa26 promoter (Zambrowicz et al., 1997). We injected these cells into wild-type C57BL/6 blastocysts, obtaining chimeric progeny with up to 90% Rb–/– cell contribution, as judged by coat color. Given the extensive analysis of the CNS of germline Rb–/– embryos, we concentrated our analysis on this tissue. To assess Rb–/– cell contribution, dissociated cells from E13.5 embryo brains were incubated with a fluorescent β-galactosidase substrate, 5-chloromethylfluorescein di-β-galactopyranoside (CMFDG), and analyzed by fluorescence-activated cell sorting (FACS). CMFDG-positive cells constituted up to 45% of all cells, with an average contribution of 20.5% (n = 10; Table I).
Table I. Summary of FACS data.
| E13.5 # | % Rb–/– | wt G1 | wt S | wt G2/M | Rb–/– G1 | Rb–/– S | Rb–/– G2/M |
|---|---|---|---|---|---|---|---|
| 1 | 17.8 | 86.4 | 5.1 | 8.3 | 67.4 | 9.7 | 22.5 |
| 2 | 45.6 | 98 | 1.2 | 0.4 | 85.1 | 6.6 | 8.4 |
| 3 | 22.5 | 95.7 | 3 | 1.6 | 79.1 | 6.4 | 14.9 |
| 4 | 27.7 | 96.9 | 2.3 | 1.2 | 82.5 | 7.2 | 10.3 |
| 5 | 11.3 | 86.3 | 5.8 | 8.1 | 59.6 | 14.9 | 25.3 |
| 6 | 6.8 | 92.1 | 3.8 | 4.2 | 66.2 | 16.7 | 16.9 |
| 7 | 14.3 | 93 | 5.5 | 1.5 | 53.9 | 23.9 | 22 |
| 8 | 27.8 | 96.6 | 3.4 | 0.2 | 71.2 | 19 | 9.8 |
| 9 | 21.4 | 77.3 | 14.7 | 7.6 | 65.1 | 15.4 | 19.6 |
| 10 | 9.6 | 91.8 | 4.5 | 3.9 | 72.4 | 9.5 | 17.8 |
| Average | 20.48 | 91.41 | 4.93 | 3.7 | 70.25 | 12.93 | 16.75 |
| SD | 11.43 | 6.43 | 3.73 | 3.24 | 9.92 | 5.95 | 3.83 |
| Rb/wt t-test | 2.3 × 10–5 | 0.0021 | 7.7 × 10–6 |
%Rb–/–, Rb–/– cell contribution; wt, wild-type cell population; Rb–/–, Rb–/– cell population; Rb/wt t-test, p value for two-tailed T-test for the difference between wild-type and Rb–/– populations.
In order to confirm the FACS data, we visualized Rb–/– cells in situ. Because staining with the chromogenic β-galactosidase substrate 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-gal) did not give satisfactory results, we determined the Rb–/– cell contribution to chimeric brains by visualizing these cells using fluorescent in situ hybridization (FISH). For approximately half of the embryos examined, we were able to distinguish injected Rb–/– male cells from the wild-type female host embryo cells by examining their sex chromosomes. Cy3-labeled XIST RNA probe was used to visualize the inactive X-chromosome in female (wild-type) cells, and biotinylated Y-paint DNA probe followed by avidin–fluorescein isothiocyanate (FITC) detection to mark the Y-chromosome in male (Rb–/–) cells (Figure 1). At E13.5, Rb–/– and wild-type cells in the chimeric embryo brains were intermixed or present in small patches without any obvious large cell clusters of homogeneous genotype. XIST- and Y-paint-positive cells were counted in 3–4 non-overlapping fields in the hindbrain (area around the fourth ventricle, including medulla, pons and cerebellar primordium) of chimeric embryos to estimate contribution. By this method, up to 72% of all cells were Rb–/–, with an average contribution of 55.9% (n = 8; Table II). The discrepancy between the levels of Rb–/– contribution obtained from FACS as compared with FISH analysis can be accounted for by differences between the two methods. By FACS analysis, the Rb–/– contribution was underestimated because of a high FDG fluorescence gate set to ensure exclusion of all wild-type cells from further analysis. In FISH experiments, only ∼80% of cells in female and male embryo controls stained with XIST probe or Y-paint, respectively (Figure 1). Therefore, this method can be used only to estimate the Rb–/– contribution. Additionally, for FACS analysis, we used cells from the entire embryo brain, while in the FISH experiments only the hindbrain was counted. The contribution of Rb–/– cells could vary in different regions of the chimeric brain. Cell-type-dependent variance in Rosa26-driven expression of β-galactosidase might also contribute to underestimation of Rb–/– contribution by FACS. Despite these differences, both methods confirmed that, unlike in germline Rb–/– embryos, Rb–/– cells were able to persist in chimeric CNS at E13.5. The Rb–/– cell contribution to chimeric brains continued at later stages of development. From FISH analysis, we have estimated the contribution at E15.5 to be on average 58.68% (n = 3; Figure 1). Since neither FACS nor FISH analysis gave satisfactory results in postnatal animals, Rb–/– cell contribution to adult brains was demonstrated by staining with X-gal (Figure 1). Owing to poor expression of β-galactosidase in adult brain, we were unable to quantitate the extent of Rb–/– cell contribution to the chimeric adult CNS. However, previous studies have used GPI analysis to demonstrate that Rb–/– cells contribute on average 20% to adult chimeric brains (Maandag et al., 1994; Williams et al., 1994b). This contribution is comparable to that obtained using wild-type instead of Rb-deficient cells for chimera generation.

Fig. 1. Rb–/– cells contribute to chimeric embryo and adult brain. (A) XIST and Y-chromosome paint FISH analysis on E13.5 and E15.5 embryo sections. In chimeras, male Rb–/– cells are marked with green Y-chromosome paint and female host cells with red XIST probe. Wild-type (wt) female and male control embryos are shown along with chimeras. Magnification 60×. (B) X-gal staining on 5-week-old (5wk) chimeric brain. Blue-staining, Rb–/– Rosa26 cells are evident. Magnification 40×.
Table II. Summary of immunohistochemistry data.
| E13.5 # | Genotype | Sex | % Rb–/– | TUNEL | Ect. BrdU | PH3 |
|---|---|---|---|---|---|---|
| 1 | chimera | m | nd | 53.5 | nd | 30 |
| 2 | chimera | f | 36.3 | 58.5 | nd | nd |
| 3 | chimera | f | 67.5 | 182 | 586.5 | 55 |
| 4 | chimera | f | 72.5 | 174.5 | 468.5 | 67 |
| 5 | chimera | m | nd | 178.5 | 652.5 | 46.3 |
| 6 | chimera | m | nd | 100 | 548 | 43 |
| 7 | chimera | f | 63.2 | 63.5 | nd | 51.3 |
| 8 | chimera | f | 39.8 | 28 | 320 | 48.5 |
| Average | 55.9 | 111.1 | 535.4 | 48.7 | ||
| SD | 16.6 | 64 | 127.8 | 11.3 | ||
| 9 | Rb–/– | nd | 100 | nd | nd | 154 |
| 10 | Rb–/– | nd | 100 | nd | nd | 148.5 |
| 11 | Rb–/– | nd | 100 | 526 | 585 | 141.5 |
| 12 | Rb–/– | nd | 100 | 492 | 484 | 183 |
| 13 | Rb–/– | nd | 100 | 444.5 | nd | nd |
| 14 | Rb–/– | nd | 100 | nd | 404 | nd |
| 15 | Rb–/– | nd | 100 | 247 | 277 | nd |
| Average | 427.4 | 437.5 | 156.7 | |||
| SD | 124.8 | 130.1 | 18.2 | |||
| 16 | wt | nd | 0 | 3.5 | 21 | 62 |
| 17 | wt | nd | 0 | 1.5 | 30 | 52.3 |
| 18 | wt | nd | 0 | 6 | 54 | 28.7 |
| 19 | wt | nd | 0 | nd | nd | 30.6 |
| 20 | wt | nd | 0 | 3 | nd | nd |
| 21 | wt | nd | 0 | 4.5 | nd | nd |
| 22 | wt | nd | 0 | 2.5 | nd | nd |
| Average | 3.5 | 31.7 | 43.4 | |||
| SD | 1.6 | 17.1 | 16.4 | |||
| wt/ch t-test | 0.0023 | 0.0007 | 0.5364 | |||
| Rb/ch t-test |
|
|
|
0.0001 |
0.3989 |
6.2 × 10–7 |
| E15.5 # |
Genotype |
Sex |
% Rb–/– |
TUNEL |
||
| 23 | chimera | f | nd | 20 | ||
| 24 | chimera | f | 31.3 | 19 | ||
| 25 | chimera | f | 70.9 | 28.5 | ||
| 26 | chimera | m | nd | 73 | ||
| 27 | chimera | m | nd | 108 | ||
| 28 | chimera | m | nd | 34 | ||
| 29 | chimera | m | nd | 57.5 | ||
| 30 | chimera | m | nd | 29 | ||
| 31 | chimera | f | nd | 36 | ||
| 32 | chimera | f | 58.7 | 46.5 | ||
| Average | 53.6 | 45.1 | ||||
| SD | 20.3 | 27.8 | ||||
| 33 | wt | nd | 0 | 10 | ||
| 34 | wt | nd | 0 | 18.5 | ||
| 35 | wt | nd | 0 | 17 | ||
| 36 | wt | nd | 0 | 12 | ||
| Average | 14.4 | |||||
| SD | 4 | |||||
| wt/ch t-test |
|
|
|
0.0521 |
||
| Neonate # |
Genotype |
Sex |
% Rb–/– |
TUNEL |
||
| 37 | chimera | nd | nd | 225 | ||
| 38 | chimera | nd | nd | 183 | ||
| 39 | chimera | m | nd | 377.5 | ||
| 40 | chimera | f | nd | 275.3 | ||
| 41 | chimera | m | nd | 485 | ||
| 42 | chimera | m | nd | 275 | ||
| Average | 303.5 | |||||
| SD | 110.2 | |||||
| 43 | wt | nd | nd | 133.5 | ||
| 44 | wt | nd | nd | 131.5 | ||
| Average | 132.5 | |||||
| SD | 1.4 | |||||
| wt/ch t-test |
|
|
|
0.0825 |
||
| Adult # |
Genotype |
Sex |
% Rb–/– |
TUNEL |
||
| 45 | chimera | m | 60 | 12.5 | ||
| 46 | chimera | f | 90 | 12.5 | ||
| 47 | chimera | m | 80 | 27.5 | ||
| 48 | chimera | m | 15 | 16.5 | ||
| Average | 61.2 | 17.2 | ||||
| SD | 33.3 | 7.1 | ||||
| 49 | wt | m | 0 | 15 | ||
| 50 | wt | f | 0 | 7.5 | ||
| Average | 11.2 | |||||
| SD | 5.3 | |||||
| wt/ch t-test | 0.2849 |
Ect. BrdU, ectopic BrdU incorporation; PH3, phospho-histone 3; f, female; m, male; nd, not determined; Rb/ch t-test, two-tailed T-test for the difference between Rb–/– and chimeric animals; wt/ch t-test, p-value for two-tailed T-test for the difference between wild-type and chimeric animals. For other abbreviations see Table I.
Low levels of apoptosis in chimeric CNS
The presence of Rb–/– cells in the CNS of Rb–/– chimeras contrasted with the extensive apoptosis in germline Rb–/– embryo brains (Clarke et al., 1992; Jacks et al., 1992; Lee et al., 1992, 1994). In order to examine specific levels of apoptosis in Rb–/– chimera brain, TdT biotin-16-dUTP nick end labeling (TUNEL) staining was performed on paraffin sections of E13.5 embryos. The total number of TUNEL-positive cells was counted in the hindbrain on 3–4 sagittal sections of every embryo (Figures 2 and 3; Table II). As previously described, the number of TUNEL-positive cells was greatly increased in germline Rb–/– embryos (average of 427.4 cells/section, n = 4) as compared with wild type (3.4 cells/section, n = 6). In chimeric embryos, TUNEL levels (111.1 cells/section, n = 8) were higher than in wild type, but despite the high contribution of Rb–/– cells to chimeric brains, were significantly lower than in Rb–/– embryos (p = 0.00012). For chimeras with known Rb–/– contribution, TUNEL levels were found to be significantly lower (p = 0.0045) than these calculated to be expected in an equivalent mixture of Rb–/– and wild-type cells (239 cells/section, n = 5) based on the average TUNEL levels observed in Rb–/– and wild-type embryos. Thus, the contribution of Rb–/– cells to the CNS of chimeras at midgestation can be accounted for by a reduction in the levels of apoptosis in the presence of wild-type cells. The levels of cell death in chimeric embryos decreased at later stages of embryonic development. At E15.5, there was only a small increase in the number of TUNEL-positive cells in the hindbrain of chimeric embryos (45.2 cells/section, n = 10) as compared with wild type (14.4 cells/section, n = 4). In neonates, because of low levels of apoptosis in the CNS, the number of TUNEL-positive cells per sagittal section was determined for all areas of the brain [303.5 (n = 6) and 132.5 cells/section (n = 2) in chimeras and wild type, respectively]. As the mice matured, total TUNEL levels gradually declined even further and the difference became insignificant [17.3 (n = 4) and 11.3 cells/section (n = 2), respectively].

Fig. 2. Apoptosis, but not ectopic cell cycle entry, is suppressed in Rb–/– chimeric brains. (A) S-phase activity and apoptosis levels in E13.5 chimeras and controls. BrdU incorporation demonstrates elevated ectopic S-phase entry in germline Rb–/– and chimeric embryos [brown-stained cells present in the intermediate zone (iz), white arrows]. Hindbrain area around the fourth ventricle (v) is shown, with the ventricular zone (vz) and iz indicated. TUNEL assay (brown-stained cells, black arrows) demonstrates reduced level of apoptosis in chimeric CNS as compared with germline Rb-mutant. (B) Analysis of older chimeric embryos, neonates and adults. At E15.5, ectopic cell cycle entry was present in the chimeric CNS, while levels of apoptosis remained low. In neonatal and adult chimeras, levels of S-phase entry declined in both wild-type and chimeric brains. Levels of TUNEL-positive cells were also low. Magnification 40×.

Fig. 3. Quantification of cell cycle and cell death analysis in the CNS of E13.5 germline Rb–/– and chimeric embryos. (A) S-phase activity (assessed by BrdU incorporation) and apoptosis (by TUNEL analysis) in hindbrain region on sagittal sections of germline Rb–/–, chimeric and wild-type (wt) E13.5 embryos. S-phase entry was increased in the brains of both germline Rb-deficient and chimeric embryos as compared with wild type. However, apoptosis was significantly suppressed in chimeras as compared with germline Rb–/– embryos. (B) FACS cell cycle profile analysis of Rb–/– and wild-type cells in chimeric CNS at E13.5. Rb–/– cells show reduced G1, and increased S-phase and G2/M fraction. (C) PH3 (a mitotic marker) expression in wild-type, germline Rb-deficient and chimeric embryo CNS at E13.5. M-phase entry was increased in germline Rb–/– embryos but not in chimeras as compared with wild type.
Ectopic cell cycle entry
Suppression of apoptosis of Rb–/– cells in chimeric brains could be due to a complete rescue of the Rb mutant phenotype in the presence of wild-type cells. Alternatively, wild-type cells could specifically suppress Rb–/– cell death without affecting other aspects of the mutant phenotype. Therefore, we analyzed chimeric brains at E13.5 for inappropriate S-phase entry. Pregnant females carrying chimeric or control embryos were injected with bromodeoxyuridine (BrdU) and killed after 1 h; embryo sections were examined for BrdU incorporation by immunohistochemistry. The number of BrdU-positive cells was counted in the hindbrain of every embryo in the same manner as for TUNEL analysis (Figures 2 and 3). We observed an increase in overall BrdU incorporation and especially in ectopic BrdU incorporation away from the ventricular area in both germline Rb–/– [437.5 intermediate zone (iz) cells/section, n = 4; Table II] and Rb–/– chimeric embryo brains (535.4 iz cells/section, n = 5) as compared with wild type (31.7 iz cells/section, n = 3). Levels of ectopic cell cycle entry in chimeric brains were comparable to those observed in germline Rb–/– embryos, and higher than those expected in wild type and Rb–/– cell mixture of the same composition (277 iz cells/section, n = 3). Although non-significant (p = 0.027), this increased BrdU incorporation could potentially suggest a non-cell-autonomous increase in cell cycle entry of Rb–/– cells in the presence of wild type. However, it is more likely to be due to the fact that many of the inappropriately cycling cells died in germline Rb–/– embryos, while in chimeras the majority of them survived. The increased and ectopic S-phase entry in chimeric CNS continued at E15.5, but eventually decreased to levels indistinguishable from wild type in neonates and adults (Figure 2).
In order to assess the cell cycle distribution of Rb–/– cells in chimeric brains, we performed a two-color FACS analysis. Dissociated brain cells from E13.5 Rb–/– Rosa26 chimeras were labeled with CMFDG, followed by paraformaldehyde fixation. Cells were than stained with propidium iodide (PI) to assess the DNA content. This allowed us to analyze cell cycle distribution separately among Rb–/– (CMFDG+) and wild-type (CMFDG–) cells within each chimeric embryo brain (n = 10). As shown in Table I, the average Rb–/– contribution was 20.5%. The S-phase cells comprised on average 4.9% of the wild type and 12.9% of the Rb–/– population (Figure 3). These data confirm that there is an increase in S-phase population in E13.5 Rb–/– chimeric embryo brains and that Rb–/– cells are, indeed, the cells that are inappropriately continuing to progress through the cell cycle. The cell cycle profile of wild-type cells from Rb–/– chimeric brains was comparable to that of cells from wild-type embryos, indicating that these cells were unaffected by the presence of their Rb–/– neighbors (see Supplementary data available at The EMBO Journal Online).
E2F and p53 activity
Inappropriate cell cycle entry in germline Rb–/– E13.5 embryos is accompanied by increased activity of the E2F transcription factors and induction of E2F target genes, such as cyclin E (Macleod et al., 1996). We investigated E2F activity in the brains of E13.5 chimeric embryos by in situ hybridization using 35S-labeled antisense RNA probe to cyclin E. cyclin E mRNA levels were increased in Rb–/– chimera embryos as compared with wild type (Figure 4), consistent with elevated E2F transcriptional activity.

Fig. 4. Analysis of cell cycle and p53 pathway in E13.5 chimeric embryos. (A) In situ hybridization with antisense cyclin E mRNA probe (green). Cyclin E expression was increased in chimeric embryo brain as compared with wild-type (wt) controls, suggesting elevated E2F activity. Hindbrain area around the fourth ventricle (v) is shown (false color, 40× magnification). (B) In situ hybridization with antisense p21 mRNA probe. Elevated levels of p21 expression in chimeric embryo CNS compared with wild-type controls suggest increased p53 transcriptional activity. (C) p53 gel shift analysis on chimeric embryo brain extracts demonstrates that p53 DNA binding activity correlates with the degree of Rb–/– chimerism. (D) Expression of the mitotic marker PH3 (brown-stained cells, black arrows) demonstrates increased and ectopic [in the intermediate zone (iz)] M-phase entry in CNS of germline Rb–/– but not chimeric embryos as compared with wild type.
In germline Rb–/– embryos, neuronal cell death in CNS is p53 dependent, and accompanied by elevated p53 protein levels and transcriptional activity (Macleod et al., 1996). We performed a p53 gel shift assay using E13.5 chimeric embryo brain extracts, and determined that p53 DNA binding activity was increased and correlated with the extent of Rb–/– contribution (determined by GPI analysis) in each embryo. Elevated p53 activity was confirmed in chimeras by in situ hybridization against p21, a transcriptional target of p53 (Figure 4). These data are consistent with the suppression of apoptosis by wild-type cells occurring at a point downstream of activation of the p53 pathway in the Rb–/– cells.
G2 arrest of Rb–/– cells
In germline Rb–/– embryos, aberrant cell cycle entry of neuronal precursor cells is believed to lead to their death. The fact that apoptosis was significantly suppressed in Rb–/– chimeras raised a question about the fate of the ectopically cycling Rb–/– cells. In addition to elevated S-phase, our two-color FACS analysis demonstrated an increase in the G2/M-phase population among Rb–/– cells (16.8% on average) as compared with wild type (3.7%). Importantly, there was no increase in the G2/M population in germline Rb–/– embryos as compared with wild type (see Supplementary data). These data suggest that Rb–/– cells in E13.5 chimeric embryos might not progress through M-phase, but instead arrest in G2 with 4n DNA content. Although the total number of Rb–/– cells with 4n DNA content in chimeric CNS remained relatively low, it has to be taken into account that only cells that have ectopically entered S-phase would be expected to arrest in G2. In order to confirm this result, we stained sections from E13.5 embryos with antibodies against the mitotic marker phospho-histone 3 (PH3). In wild-type hindbrain, levels of PH3 staining were low (43.4 cells/section, n = 4; Figures 3 and 4; Table I) and restricted to the ventricular area. In germline Rb–/– embryos, levels of PH3 were greatly elevated (156.8 cells/section, n = 4), especially in the intermediate zone, suggesting that cells that ectopically enter S-phase in germline Rb–/– embryos also enter mitosis. In Rb–/– chimeric embryos, despite high levels of ectopic BrdU incorporation, levels of PH3 staining were comparable to those seen in wild type (48.7 cells/section, n = 7), and significantly lower (p = 0.00019) than these expected in Rb–/– and wild-type cell mixture of equivalent composition (118.8 cells/section, n = 3). Together with the FACS data, this suggests that the majority of Rb–/– cells that inappropriately enter the cell cycle in chimeras never enter M-phase and instead arrest in late S- or G2-phase, with 4n DNA content.
Neuronal differentiation
Because Rb–/– cells in E13.5 chimera embryo brains inappropriately enter S-phase and possibly arrest at an incorrect stage of the cell cycle, we have investigated their long-term fate. In germline Rb–/– embryos, ectopic cell cycle entry and apoptosis initiate at a time in development when neuronal precursors are normally starting to exit the cell cycle and differentiate into neurons, suggesting that the dying cells were destined to become neurons. To determine whether the mutant cells in Rb–/– chimeric brains are able to differentiate into neuronal fates, E13.5 chimeric embryo sections were double stained with Y-paint FISH to mark Rb–/– cells, and with antibodies directed against neuronal differentiation markers, microtubule-associated protein 2 (MAP2) or neurofilament 165 kDa subunit. We observed expression of both MAP2 and neurofilament around Y-paint-positive Rb–/– nuclei (Figure 5). In order to determine whether Rb–/– cells themselves, and not only their wild-type neighbors, were able to express neuronal markers, cells from E13.5 chimeric embryo brains (n = 2) were dissociated, stained with CMFDG to mark Rb–/– cells, centrifuged onto a microscope slide and stained with an antibody against neurofilament (Figure 5). Neurofilament-positive cells comprised 8.1% of all cells (n = 257 total cells counted) and 8.2% of CMFDG-positive cells (n = 109). Therefore, at E13.5, equal proportions of wild-type and Rb–/– cells were able to differentiate into neuronal fates.

Fig. 5. Neuronal differentiation of Rb-deficient cells in chimeric brains. (A) Double staining of E13.5 chimeric brain with Y-chromosome paint and antibodies against the neuronal differentiation marker MAP2. Male Rb–/– cells (marked with green Y-paint FISH probe) are present in CNS regions expressing MAP2 (red). (B) Dissociated Rb–/– cell from E13.5 chimeric CNS marked with CMFDG (green) and expressing neuronal differentiation marker neurofilament 165 kDa subunit (red). (C) Sections of 5-week-old (5wk) chimeric brain stained with X-gal and nuclear fast red (left panel) or cresyl echt violet (right panel). Rb–/–; Rosa26 cells (marked blue, arrows) in chimeric brain display normal neuronal morphology. (D) Section of cerebellum from the same chimera stained with X-gal and cresyl echt violet. Rb–/– cerebellar Purkinje neurons (arrow) are enlarged and display nuclear pleomorphism as compared with normal wild-type Purkinje cells (arrowhead). The internal granular (g) and molecular (m) cell layers of the cerebellum are marked. Magnification: (A, C and D) 60×; (B) 100×.
To determine the state of differentiation of Rb–/– cells in adult chimeric brains, we used X-gal staining followed by either hematoxylin–eosin, nuclear fast red or cresyl echt violet counterstain. Cortical and brain stem neurons are characterized by their large nuclei with open chromatin and prominent nucleoli, and by abundant cytoplasm with Nissl bodies. Neuronal processes are occasionally observed. Oligodendrocytes have small nuclei with dense chromatin, while nuclei of astrocytes are larger and oval in shape. We detected numerous Rb–/– cells with both neuronal and glial morphology present in the brain stem and cortex (Figure 5 and data not shown). The overall morphology of these cells appeared to be normal and the cerebral cortical architecture showed appropriate lamination. The overall cerebellar cortical architecture also appeared to be normal, but Rb–/– Purkinje cells were aberrant (Figure 5). They were considerably larger than wild-type Purkinje cells and displayed marked nuclear pleomorphism. The enlarged nuclei of Rb–/– Purkinje neurons suggested that these cells might be polyploid. Purkinje neurons have previously been reported to be enlarged in Rb–/– chimeras (Williams et al., 1994b). We have now confirmed that these abnormal neurons were indeed Rb deficient. Nevertheless, the majority of Rb–/– neurons appeared normal, establishing that pRB is not required in a cell-autonomous manner for neuronal differentiation.
Discussion
In germline Rb–/– embryos at midgestation, neuronal cells in the CNS show extensive ectopic cell cycle entry accompanied by widespread apoptosis. Detailed analysis of Rb–/– chimeric embryos reveals that in many aspects, including ectopic cell cycle entry and upregulated E2F and p53 activity, the fate of the Rb–/– neuronal cells in chimeras resembles that of Rb–/– cells in germline Rb-mutant embryos. However, other aspects of the germline Rb–/– CNS phenotype are not observed in the chimeric setting, as many of the aberrantly cycling Rb–/– cells in chimeric embryo CNS do not die and eventually differentiate into neurons. These findings demonstrate that while proper cell cycle regulation is dependent on the presence of functional pRB in a cell-autonomous manner, the requirement for pRB in neuronal cell survival and differentiation may be non-cell-autonomous (Figure 6). The pronounced cell cycle defects reported here contrast with previous studies (Maandag et al., 1994; Williams et al., 1994b), which reported no significant abnormalities in Rb–/– chimeric brains. These differences can be attributed to several factors. First, in earlier work it was not possible to distinguish between Rb–/– and wild-type cells within chimeric brain and, thus, to determine their exact fate. Secondly, the prior studies dealt mainly with adult CNS where the cell cycle defect is no longer apparent. Finally, in order to determine the cell cycle status of CNS cells in Rb–/– chimeras, previous studies relied on mitotic figures. We have now demonstrated that despite extensive ectopic cell cycle entry in chimeric embryo brains, there is no increase in M-phase entry. In fact, in agreement with previous studies, we have confirmed that mitotic figures in E13.5 Rb–/– chimeric embryos remained at wild-type level (see Supplementary data).
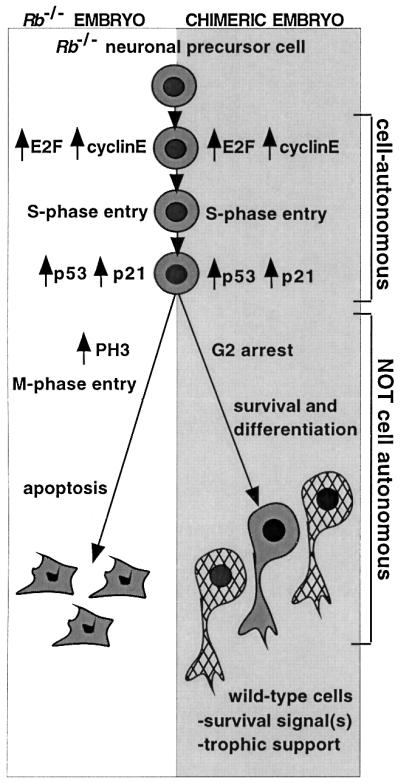
Fig. 6. Model of the fate of Rb–/– cells in CNS of germline Rb–/– and chimeric embryos. In both settings, Rb–/– neuronal precursors ectopically enter the cell cycle in a cell-autonomous fashion, with elevated E2F activity and high levels of expression of E2F transcriptional targets such as cyclin E. In both settings, inappropriate S-phase entry is accompanied by increased p53 activity and elevated expression of the p53 target p21. In germline Rb-mutant embryos, these events lead to ectopic M-phase entry and apoptosis. However, in chimeric CNS, ectopic M-phase progression and apoptosis of Rb–/– cells are suppressed in a non-cell-autonomous fashion. In the presence of wild-type neighbors, Rb–/– cells survive and differentiate into neuronal fates.
By showing an increase in the S-phase fraction among the Rb–/– but not wild-type cell populations, our FACS analysis demonstrated that the cell cycle defect observed in the CNS of chimeric embryos is confined to the Rb-deficient cell population and is cell-autonomous. Similar to the situation observed in germline Rb–/– embryos, ectopic cell cycle entry in chimeras appears to be accompanied by elevated E2F activity, supporting the view that the ectopic S-phase entry proceeds through the same mechanism in both germline Rb–/– and chimeric embryos.
In the chimeras, it would appear that an interaction of the Rb–/– cells with their wild-type neighbors enables the former to survive under conditions that in germline Rb-mutant embryos lead to their death. Therefore, pRB is not required in a cell-autonomous manner for the survival of differentiating CNS cells. Cell non-autonomy is usually thought to reflect the requirement for proper gene function in cells other than those exhibiting aberrant phenotype. Since differentiating neurons require the presence of glia and other cell types in order to survive, pRB could be necessary for the proper differentiation of some of these cells. Alternatively, the wild-type cells that allow survival of their Rb–/– neighbors in chimeras could be of the same type as the cells they rescue in a non-cell-autonomous but cell-type-intrinsic manner.
In either case, the wild-type cells could alter the fate of their Rb–/– neighbors by providing them with a specific survival or anti-apoptotic signal(s), or by creating an overall better growth environment able to support continued survival of Rb-deficient cells. If the survival signal is specific, it could be either a secreted factor (or factors) able to act at a distance, or it could require direct cell–cell contact. If cell–cell contact between Rb–/– and wild-type cells were required, one would expect to see more death in the middle of large patches of Rb–/– cells. However, Rb–/– and wild-type cells appeared to be intermixed in the chimeric CNS, making it impossible to determine whether direct contact is required for the rescue. Candidates for possible secreted rescue signals include neurotrophins, a family of neuronal cell-specific survival, growth and differentiation factors demonstrated to be essential for survival of differentiating neurons (Conover and Yancopoulos, 1997). mRNAs for several of the neurotrophins and their receptors have been shown to be expressed at approximately normal levels in the CNS of germline Rb–/– embryos (Lee et al., 1994). However, even small variations in levels of these factors could lead to pronounced differences in neuronal survival and differentiation. Several of the non-neuron-specific growth and survival factors, such as insulin-like growth factor-I (D’Ercole et al., 1996), have also been shown to be expressed in the CNS and to play a role in neuronal survival and differentiation. These factors might also influence survival of Rb–/– cells in chimeric CNS. Another interesting possibility is that apoptosis in germline Rb–/– embryos occurs in response to a secreted factor produced by the mutant cells. If this were the case, the rescue from apoptosis in Rb–/– chimeras could be due to the dilution of such a factor to below a critical threshold level.
In germline Rb–/– embryos, ectopic cell cycle entry and increased E2F activity in the CNS are accompanied by increased p53 protein levels and activity, and p53 has been shown to be necessary for the death of Rb–/– CNS cells (Macleod et al., 1996). In Rb–/–E2F-1–/– embryos, apoptosis and increased p53 activity are also suppressed. Therefore, activation of the p53 pathway in germline Rb–/– embryos proceeds through an E2F-1-dependent pathway (Tsai et al., 1998). Similarly, E2F and p53 activity were upregulated in Rb–/– chimeric embryos. However, despite activation of p53 function, the fate of the Rb–/– cells was altered from apoptosis to survival. In response to cellular stress, activation of the p53 pathway can induce either cell cycle arrest or apoptosis, depending on the cell type involved, nature of the stress and the environmental conditions, including the presence of growth and survival factors. In the case of germline Rb–/– embryos and Rb–/– chimeras, the first two factors remain constant. It is possible that wild-type cells present in the chimeric CNS supply a survival signal(s) able to alter the p53 response of their Rb–/– neighbors and prevent activation of the p53-dependent apoptotic pathway. This could be mediated by specific down-regulation of pro-apoptotic targets of p53. Alternatively, the apoptotic pathway could be activated, but blocked at a later point. In fact, expression of p53 in cultured cerebellar granule neurons has been shown to lead to caspase-3 activation and apoptosis (Cregan et al., 1999). Bax-deficient neurons were resistant, and caspase-3 null neurons showed a significant delay and partial suppression of p53-induced apoptosis. Modulation of activity of these molecules could also be involved in protection from p53-dependent apoptosis in vivo.
We have shown that inhibition of the p53-dependent apoptotic pathway by the presence of wild-type cells in Rb–/– chimera CNS occurred without affecting p53-dependent expression of p21. Increased levels of p21 protein have been demonstrated to lead to preferential arrest of Rb–/– cells in G2/M and to either endoreduplication or apoptosis (Niculescu et al., 1998; Qiu et al., 1998). The cell cycle profiles of Rb–/– cells from chimeric embryos also show increased G2 fraction, which could be attributed to increased p21 levels in these cells. Since we observed elevated levels of expression of PH3 (a mitotic marker) only in germline Rb–/–, but not in chimeric embryo CNS, one possibility consistent with our data is that survival of Rb–/– cells in chimeras may involve specific inhibition of cell cycle progression from G2 into M-phase. Differentiation-associated G2 arrest of Rb–/– cells has been seen previously in cultured Rb–/– myocytes. These cells are unable to maintain G0 arrest upon re-stimulation with serum, and eventually arrest in the G2-phase with 4n DNA content (Novitch et al., 1996). Although Rb-deficient myocytes do not usually progress past G2, or die, they can be triggered by caffeine treatment to enter the M-phase, leading to mitotic catastrophe. Similar to the situation observed in the CNS of germline Rb–/– and chimeric embryos, p21 is upregulated in serum-stimulated Rb–/– myocytes.
In addition to its role in negative regulation of cell cycle entry and apoptosis, pRB has been proposed to promote terminal differentiation of many cell types, including myocytes, adipocytes and neurons (Lipinski and Jacks, 1999). In the CNS, increased levels of pRB activity in wild type (Kastner et al., 1998; Callaghan et al., 1999) and onset of the cell cycle defect in germline Rb–/– and chimeric embryos correlate with the initiation of cell cycle exit of neuronal precursor cells, suggesting that this is the cell population affected by the loss of Rb. In addition, although expression of early neuronal differentiation markers is correctly initiated in Rb–/– embryos, it quickly declines and fully differentiated neurons are never observed (Lee et al., 1994; Slack et al., 1998). In vitro, functional ablation of the pRB-family proteins by expression of viral oncoproteins in differentiating cortical progenitor cells prevents exit from the cell cycle and leads to death (Slack et al., 1998). Nevertheless, Rb–/– chimeric adults had normal overall brain architecture, and Rb-deficient cells in chimeric CNS had normal neuronal or glial morphology. Therefore, any requirement for pRB function in neuronal differentiation and survival does not appear to be cell autonomous. Given the increased G2 cell population present in chimeric brains, it is possible that at least some of the differentiated Rb–/– neurons are arrested in G2 instead of the G1-phase of the cell cycle. This would suggest that G2 arrest, with 4n DNA content, is compatible with neuronal differentiation. In addition, pRB has been proposed to coordinate the initiation of the differentiation program with terminal cell cycle withdrawal. In chimeric embryos, we observed initiation of expression of neuronal differentiation markers concurrent with S-phase progression of Rb–/– cells, suggesting that in Rb-deficient neuronal precursors, induction of the neuronal differentiation program might occur without prior exit from the cell cycle.
Within the chimeric CNS, the only exception to the differentiation rescue seemed to be the Rb–/– Purkinje neurons, which were abnormally enlarged and showed nuclear pleomorphism. It has been reported that inactivation of the Rb-family proteins by expression of viral oncoproteins leads to cell cycle re-entry and death of mature Purkinje cells, but not of cortical neurons (Feddersen et al., 1995; Slack et al., 1998). Our findings support the notion that Purkinje neurons are uniquely dependent on the presence of functional pRB for normal development in a cell-autonomous fashion. Another tissue that appears to require the presence of functional pRB in a cell-autonomous manner is the ocular lens. We have seen no suppression of either cell cycle defect or cell death in the lens (data not shown), and adult Rb–/– chimeras develop bilateral cataracts (Maandag et al., 1994; Williams et al., 1994b). These findings suggest that pRB is required in a cell-autonomous manner for cell cycle regulation in all tissue types, while the requirement for suppression of apoptosis and induction of differentiation is either cell-autonomous or non-cell-autonomous, depending on the tissue type involved.
Although the underlying molecular mechanisms remain to be elucidated, we have demonstrated that in the CNS of Rb–/– chimeras, cells expressing wild-type pRB are able to rescue their Rb-deficient neighbors from apoptosis and possibly cause them to arrest in the G2-phase of the cell cycle. This suggests that cell–cell interactions might play a much larger role in the regulation of aberrant cell cycle and cell death than previously recognized. Although our data are restricted to the study of pRB in a developmental setting, similar interactions between wild-type and Rb-deficient cells might be expected to occur during tumorigenesis, where a mixture of cells with different genotypes is also present. Thus, tumor development and progression could be affected by the responsiveness of tumor cells mutant in the pRB pathway to factors produced by genetically normal cells.
Materials and methods
Rb–/–; Rosa26+/– ES cells and generation of chimeric mice
129sv Rb+/– mice (Jacks et al., 1992) were crossed with mice of the same genetic background heterozygous for a transgene expressing β-galactosidase from ubiquitous Rosa26 promoter (Zambrowicz et al., 1997). Rb+/–; Rosa26 mice were intercrossed and ES cells were generated as described (Robertson, 1987).
Chimeric mice were created as described (Bradley, 1987) with 8–10 Rb–/–; Rosa26 129sv ES cells injected per wild-type C57BL/6 blastocyst.
Embryos and adult brain tissues
Embryos were harvested at the equivalent of E13.5 (11 days after blastocyst transfer), E15.5 and E18.5–19.5. Chimeras were identified by PCR genotyping (Jacks et al., 1992). Embryos were fixed in 4% paraformaldehyde, embedded in paraffin and sectioned at 4 µm. Adult chimeras were identified by coat color. Following killing, brains were surgically removed and processed as above.
TUNEL assay
TUNEL was carried out as described (Macleod et al., 1996).
BrdU labeling and detection
Adult mice were injected intraperitoneally (i.p.) with 100 µg of BrdU per gram body weight 1 h before killing. Embryos were labeled in utero by i.p. injecting pregnant females. Immunohistochemistry was performed as described (Morgenbesser et al., 1994).
X-gal staining
Adult brains were dissected, fixed in 4% paraformaldehyde for 1 h at room temperature, rinsed in phosphate-buffered saline (PBS), incubated in X-gal solution (Boehringer) up to 1 week at 4°C and post-fixed in 4% paraformaldehyde. Sections were counterstained with hematoxylin–eosin, nuclear fast red or cresyl echt violet.
Immunohistochemistry
PH3 staining was performed as described (Pan et al., 1998). Anti-PH3 antibody (Upstate) was incubated overnight at 4°C. For neurofilament and MAP2 double staining with Y-paint FISH, general FISH protocol was followed, except that proteinase K treatment was omitted and the antigen was unmasked in 0.01 M sodium citrate buffer at 80°C for 2 h. Sections were cooled in buffer for 30 min at room temperature, and washed twice in PBS and once in dH2O. XIST probe was omitted and rhodamine red-conjugated goat anti-mouse secondary antibody (Jackson ImmunoResearch) was added to the detection buffer at 1:200 dilution. The anti-neurofilament antibody (2H3; Developmental Studies Hybridoma Bank, University of Iowa, Iowa City, IA) was used at 1:100 dilution and anti-MAP2 antibody (HM-2; Sigma) at 1:400.
Y-paint and XIST FISH
For FISH, double-stranded DNA XIST probe was generated as described (Panning and Jaenisch, 1996). Y-chromosome paint template was a generous gift from Barbara Panning (UCSF, San Francisco, CA). It was labeled by PCR incorporation of biotinylated dUTP (Boehringer). Deparaffinized and dehydrated slides were treated with 0.5% Triton X-100 in PBS for 10 min, washed in PBS, and incubated in 10 µg/ml proteinase K in 50 mM Tris pH 8.0, 5 mM EDTA for 20 min at 37°C. Slides were washed twice in PBS and once in dH2O, and transferred into 0.1 M triethanolamine solution. Acetic anhydride was added dropwise with gentle agitation to 0.25%, followed by 10 min incubation. Slides were washed twice in PBS, dehydrated in an alcohol gradient, allowed to air dry and pre-warmed to 85°C. Genomic DNA was denatured in 70% formamide, 2× SSC at 85°C for 25 min, followed by dehydration in an ice-cold alcohol gradient. Sections were allowed to air dry at room temperature. Y-paint and XIST probes were mixed at 1:1 ratio. Sections were overlaid with the probe mixture, coverslipped and incubated overnight at 37°C in a 50% formamide, 2× SSC humidifying chamber. Coverslips were removed and slides were washed at 40°C in 50% formamide, 2× SSC three times for 5 min, 2× SSC three times for 5 min, 1× SSC twice for 5 min and at room temperature in 4× SSC for 5 min. Sections were overlaid with detection buffer [4× SSC, 2 mg/ml bovine serum albumin, 1:200 avidin–FITC (Vector)], coverslipped and incubated for 1 h at 37°C in 4× SSC in a humidifying chamber. Slides were washed at room temperature for 5 min in 4× SSC, 5 min in 4× SSC with 0.1% Tween-20 and 1:50 000 4′,6-diamidino-2-phenylindole (DAPI), and 5 min in 4× SSC, then coverslipped.
p21 and cyclin E in situ hybridization
p21 and cyclin E RNA probe preparation and in situ hybridization were carried out as described (Macleod et al., 1996).
FACS analysis
E13.5 embryo brains were dissected, minced with a razor blade and dissociated for 20 min at 37°C in 1% trypsin/PBS solution with 200 U/ml DNase I. Cells were washed with 5 ml of MEM– 5% inactivated fetal calf serum (IFS), pelleted, resuspended in 0.5 ml of MEM– 5% IFS, triturated and strained through a 70 µm nylon cell strainer. LacZ expression was detected using the DetectaGene Green CMFDG LacZ gene expression kit (Molecular Probes) according to the manufacturer’s instructions. Following CMFDG staining, cells were pelleted and resuspended in 200 µl of PBS. Cell suspension (50 µl) was removed for cytospin analysis. Remaining cells were fixed in 1% paraformaldehyde. Cells were washed, treated with RNase A, and incubated overnight at 4°C in 0.5% Tween-20 and 35 µl of 5 mg/ml PI solution. Cells were examined using a Beckton-Dickinson FACScan flow cytometer (Beckton Dickinson Immunocyto metry Sys.) and analyzed using CellQuest software.
Cytospin and immunofluorescence
E13.5 embryo brain cells were prepared and CMFDG stained as for FACS analysis. Cells were spun onto microscope slides coated with Vectabond (Vector) according to the manufacturer’s instructions. Cells were fixed in 2% paraformaldehyde, washed twice in PBS and incubated with 0.25% Triton X-100 for 10 min. Slides were blocked in 5% goat serum, 0.1% Triton X-100, followed by 1 h incubation with primary antibody in blocking solution. Immune complexes were detected using rhodamine-conjugated secondary antibody (Jackson ImmunoResearch). Cells were counterstained with DAPI.
p53 gel shift
Chimeric embryos were harvested at E13.5. Brains were dissected and p53 gel shift analysis was performed as described (Macleod et al., 1996). The Rb–/– contribution to the brains was assessed by GPI analysis (Williams et al., 1994b).
Supplementary data
Supplementary data for this paper are available at The EMBO Journal Online.
Acknowledgments
Acknowledgements
The Y-paint probe template was a generous gift from Barbara Panning (UCSF, San Francisco, CA). The authors thank Gyorgi Csankovski, Glen Paradis and Margaret McLaughlin for technical advice, and Julien Sage and Elsa Flores for critical reading of the manuscript. This work was funded in part by grants from the NCI. T.J. is an Associate Investigator at the HHMI.
References
- Bradley A. (1987) Production and analysis of chimeric mice. In Robertson,E.J. (ed.), Teratocarcinomas and Embryonic Stem Cells: A Practical Approach. IRL Press, Oxford, UK, pp. 113–152.
- Brehm A. and Kouzarides,T. (1999) Retinoblastoma protein meets chromatin. Trends Biochem. Sci., 24, 142–145. [DOI] [PubMed] [Google Scholar]
- Callaghan D.A., Dong,L., Callaghan,S.M., Hou,Y.X., Dagnino,L. and Slack,R.S. (1999) Neural precursor cells differentiating in the absence of Rb exhibit delayed terminal mitosis and deregulated E2F 1 and 3 activity. Dev. Biol., 207, 257–270. [DOI] [PubMed] [Google Scholar]
- Chen P.L., Riley,D.J., Chen,Y. and Lee,W.H. (1996) Retinoblastoma protein positively regulates terminal adipocyte differentiation through direct interaction with C/EBPs. Genes Dev., 10, 2794–2804. [DOI] [PubMed] [Google Scholar]
- Clarke A.R., Maandag,E.R., van Roon,M., van der Lugt,N.M., van der Valk,M., Hooper,M.L., Berns,A. and te Riele,H. (1992) Requirement for a functional Rb-1 gene in murine development. Nature, 359, 328–330. [DOI] [PubMed] [Google Scholar]
- Conover J.C. and Yancopoulos,G.D. (1997) Neurotrophin regulation of the developing nervous system: analyses of knockout mice. Rev. Neurosci., 8, 13–27. [DOI] [PubMed] [Google Scholar]
- Cregan S.P., MacLaurin,J.G., Craig,C.G., Robertson,G.S., Nicholson,D.W., Park,D.S. and Slack,R.S. (1999) Bax-dependent caspase-3 activation is a key determinant in p53-induced apoptosis in neurons. J. Neurosci., 19, 7860–7869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Ercole A.J., Ye,P., Calikoglu,A.S. and Gutierrez-Ospina,G. (1996) The role of the insulin-like growth factors in the central nervous system. Mol. Neurobiol., 13, 227–255. [DOI] [PubMed] [Google Scholar]
- Feddersen R.M., Clark,H.B., Yunis,W.S. and Orr,H.T. (1995) In vivo viability of postmitotic Purkinje neurons requires pRb family member function. Mol. Cell. Neurosci., 6, 153–167. [DOI] [PubMed] [Google Scholar]
- Goodrich D.W. and Lee,W.H. (1993) Molecular characterization of the retinoblastoma susceptibility gene. Biochim. Biophys. Acta, 1155, 43–61. [DOI] [PubMed] [Google Scholar]
- Jacks T., Fazeli,A., Schmidt,E., Bronson,R., Goodell,M. and Weinberg,R. (1992) Effects of an Rb mutation in the mouse. Nature, 359, 295–300. [DOI] [PubMed] [Google Scholar]
- Kastner A., Espanel,X. and Brun,G. (1998) Transient accumulation of retinoblastoma/E2F-1 protein complexes correlates with the onset of neuronal differentiation in the developing quail neural retina. Cell Growth Differ., 9, 857–867. [PubMed] [Google Scholar]
- Lee E.Y.-H.P., Chang,C.-Y., Hu,N., Wang,Y.-C.J., Lai,C.-C., Herrup,K. and Lee,W.-H. (1992) Mice deficient for Rb are nonviable and show defects in neurogenesis and haematopoiesis. Nature, 359, 288–295. [DOI] [PubMed] [Google Scholar]
- Lee E.Y., Hu,N., Yuan,S.S., Cox,L.A., Bradley,A., Lee,W.H. and Herrup,K. (1994) Dual roles of the retinoblastoma protein in cell cycle regulation and neuron differentiation. Genes Dev., 8, 2008–2021. [DOI] [PubMed] [Google Scholar]
- Lipinski M.M. and Jacks,T. (1999) The retinoblastoma gene family in differentiation and development. Oncogene, 18, 7873–7882. [DOI] [PubMed] [Google Scholar]
- Maandag E.C.R., van der Valk,M., Vlaar,M., Feltkamp,C., O’Brien,J., van Roon,M., van der Lugt,N., Berns,A. and te Riele,H. (1994) Developmental rescue of an embryonic-lethal mutation in the retinoblastoma gene in chimeric mice. EMBO J., 13, 4260–4268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macleod K.F., Hu,Y. and Jacks,T. (1996) Loss of Rb activates both p53-dependent and independent cell death pathways in the developing mouse nervous system. EMBO J., 15, 6178–6188. [PMC free article] [PubMed] [Google Scholar]
- Morgenbesser S.D., Williams,B.O., Jacks,T. and DePinho,R.A. (1994) p53-dependent apoptosis produced by Rb-deficiency in the developing mouse lens. Nature, 371, 72–74. [DOI] [PubMed] [Google Scholar]
- Nevins J.R., Leone,G., DeGregori,J. and Jakoi,L. (1997) Role of the Rb/E2F pathway in cell growth control. J. Cell Physiol., 173, 233–236. [DOI] [PubMed] [Google Scholar]
- Niculescu A.R., Chen,X., Smeets,M., Hengst,L., Prives,C. and Reed,S.I. (1998) Effects of p21Cip1/Waf1 at both the G1/S and the G2/M cell cycle transitions: pRb is a critical determinant in blocking DNA replication and in preventing endoreduplication. Mol. Cell. Biol., 18, 629–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novitch B.G., Mulligan,G.J., Jacks,T. and Lassar,A.B. (1996) Skeletal muscle cells lacking the retinoblastoma protein display defects in muscle gene expression and accumulate in S and G2 phases of the cell cycle. J. Cell Biol., 135, 441–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novitch B.G., Spicer,D.B., Kim,P.S., Cheung,W.L. and Lassar,A.B. (1999) pRb is required for MEF2-dependent gene expression as well as cell-cycle arrest during skeletal muscle differentiation. Curr. Biol., 9, 449–459. [DOI] [PubMed] [Google Scholar]
- Pan H., Yin,C., Dyson,N.J., Harlow,E., Yamasaki,L. and Van Dyke,T. (1998) Key roles for E2F1 in signaling p53-dependent apoptosis and in cell division within developing tumors. Mol. Cell, 2, 283–292. [DOI] [PubMed] [Google Scholar]
- Panning B. and Jaenisch,R. (1996) DNA hypomethylation can activate Xist expression and silence X-linked genes. Genes Dev., 10, 1991–2002. [DOI] [PubMed] [Google Scholar]
- Qiu X.B., Schonthal,A.H. and Cadenas,E. (1998) Anticancer quinones induce pRb-preventable G2/M cell cycle arrest and apoptosis. Free Radic. Biol. Med., 24, 848–854. [DOI] [PubMed] [Google Scholar]
- Robertson E.J. (1987) Embryo-derived stem cell lines. In Robertson,E.J. (ed.), Teratocarcinomas and Embryonic Stem Cells: A Practical Approach. IRL Press, Oxford, UK, pp. 72–112.
- Slack R.S., El-Bizri,H., Wong,J., Belliveau,D.J. and Miller,F.D. (1998) A critical temporal requirement for the retinoblastoma protein family during neuronal determination. J. Cell Biol., 140, 1497–1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai K.Y., Hu,Y., Macleod,K.F., Crowley,D., Yamasaki,L. and Jacks,T. (1998) Mutation of E2f-1 suppresses apoptosis and inappropriate S phase entry and extends survival of Rb-deficient mouse embryos. Mol. Cell, 2, 293–304. [DOI] [PubMed] [Google Scholar]
- Weinberg R.A. (1995) The retinoblastoma protein and cell cycle control. Cell, 81, 323–330. [DOI] [PubMed] [Google Scholar]
- Williams B.O., Remington,L., Albert,D.M., Shizuo,M., Bronson,R.T. and Jacks,T. (1994a) Cooperative tumorigenic effects of germline mutations in Rb and p53. Nature Genet., 7, 480–484. [DOI] [PubMed] [Google Scholar]
- Williams B.O., Schmitt,E.M., Remington,L., Bronson,R.T., Albert,D.M., Weinberg,R.A. and Jacks,T. (1994b) Extensive contribution of Rb-deficient cells to adult chimeric mice with limited histopathological consequences. EMBO J., 13, 4251–4259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zambrowicz B.P., Imamoto,A., Fiering,S., Herzenberg,L.A., Kerr,W.G. and Soriano,P. (1997) Disruption of overlapping transcripts in the ROSA β geo 26 gene trap strain leads to widespread expression of β-galactosidase in mouse embryos and hematopoietic cells. Proc. Natl Acad. Sci. USA, 94, 3789–3794. [DOI] [PMC free article] [PubMed] [Google Scholar]