

Abstract
The protein kinase PKR (dsRNA-dependent protein kinase) phosphorylates the eukaryotic translation initiation factor eIF2α to downregulate protein synthesis in virus-infected cells. Two double-stranded RNA binding domains (dsRBDs) in the N-terminal half of PKR are thought to bind the activator double-stranded RNA, mediate dimerization of the protein and target PKR to the ribosome. To investigate further the importance of dimerization for PKR activity, fusion proteins were generated linking the PKR kinase domain to heterologous dimerization domains. Whereas the isolated PKR kinase domain (KD) was non-functional in vivo, expression of a glutathione S-transferase–KD fusion, or co-expression of KD fusions containing the heterodimerization domains of the Xlim-1 and Ldb1 proteins, restored PKR activity in yeast cells. Finally, coumermycin-mediated dimerization of a GyrB–KD fusion protein increased eIF2α phosphorylation and inhibited reporter gene translation in mammalian cells. These results demonstrate the critical importance of dimerization for PKR activity in vivo, and suggest that a primary function of double-stranded RNA binding to the dsRBDs of native PKR is to promote dimerization and activation of the kinase domain.
Keywords: dimerization/eIF2/PKR/protein kinase/Xlim-1
Introduction
Ligand-induced oligomerization of cell surface receptors for a number of cytokines, hormones and growth factors has been found to activate the serine/threonine or tyrosine kinase activity of the receptor (reviewed in Lemmon and Schlessinger, 1994; Heldin, 1995). Similarly, oligomerization of the endoplasmic reticulum transmembrane kinase IRE1 is required for its autophosphorylation/activation and signalling function (Shamu and Walter, 1996; Kaufman, 1999). As most, if not all, protein kinases are thought to undergo autophosphorylation during activation, mutual trans-autophosphorylation of dimerized kinases may be a common mechanism of activation. However, the role and requirement of dimerization for activation of cytoplasmic protein kinases has not been examined fully. The protein kinase PKR (dsRNA- dependent protein kinase) is a component in the interferon-induced antiviral defense mechanism in mammalian cells. The human PKR is composed of 551 amino acids with the kinase domain (KD) located in the C-terminal half of the protein (residues 265–551). The N-terminal half of PKR contains two double-stranded RNA (dsRNA) binding domains (dsRBDs) located between residues 6–79 and 96–169, respectively. The PKR is thought to exist in a latent form in cells, and following viral infection, double-stranded RNAs produced in the course of viral gene expression or during viral replication bind to the dsRBDs of PKR and activate the kinase (Kaufman, 2000). The only clearly verified substrate of PKR is the eukaryotic translation initiation factor eIF2α. The function of eIF2 is to bind the initiator Met-tRNAiMet to the small ribosomal subunit, and eIF2 does this by first forming a ternary complex with GTP and the Met-tRNAiMet. Phosphoryl ation of Ser51 on eIF2α converts eIF2 from a substrate to a competitive inhibitor of its guanine-nucleotide exchange factor eIF2B, and thereby blocks cellular protein synthesis (Dever, 1999). The activation of PKR by dsRNA is second order for enzyme concentration (Kostura and Mathews, 1989), and PKR purifies as a dimer in its activated state (Langland and Jacobs, 1992). However, inactive unphosphorylated PKR is a monomer (Langland and Jacobs, 1992). In addition, it has been observed that PKR can autophosphorylate in trans (Thomis and Samuel, 1995; Ortega et al., 1996). These findings have led to the hypothesis that dsRNA binding to PKR promotes dimerization and activation of the kinase.
A large number of studies have revealed that the N-terminus of PKR can dimerize (Cosentino et al., 1995; Patel et al., 1995; Ortega et al., 1996; Wu and Kaufman, 1996; Carpick et al., 1997); however, the importance of dimerization for PKR activity has not been directly investigated. Yeast two-hybrid experiments revealed that the dsRBDs of PKR mediate dimerization (Cosentino et al., 1995; Patel et al., 1995); however, it is debated whether dimerization is mediated by binding to the same molecule of dsRNA. High level expression of PKR impairs yeast cell growth due to phosphorylation of eIF2α and inhibition of translation (Chong et al., 1992; Dever et al., 1993; Romano et al., 1995). Consistent with the idea that dsRNA-mediated dimerization leads to activation of PKR, various mutations that impair dsRNA binding to PKR reduced or eliminated the toxicity of PKR in yeast (Romano et al., 1995; Patel and Sen, 1998). However, these results must be interpreted cautiously because dsRNA binding may also be necessary to relieve an autoinhibitory interaction in PKR, see below, and the lack of activity in these dsRBD mutants may reflect a failure to relieve this autoinhibition. Results from four studies further support the model that PKR dimerization is necessary for activation. (i) Biochemical studies on purified PKR revealed that PKR autophosphorylation displays second-order kinetics with respect to PKR concentration, consistent with an intermolecular mechanism of autophosphorylation (Kostura and Mathews, 1989). (ii) Two PKR mutant alleles, one lacking the first dsRBD and the other lacking the second dsRBD, functionally complement when co-expressed in yeast cells (Romano et al., 1995). (iii) An Ala67 to Glu (A67E) mutation in the first dsRBD of PKR was reported to impair dimerization and PKR activity without affecting dsRNA binding activity (Patel and Sen, 1998). However, more recent analysis indicates that this mutation reduces dsRNA binding to PKR (F.Zhang and A.G.Hinnebusch, personal communication), suggesting that the reduced activity of PKR-A67E may result from a defect in dsRNA binding and not dimerization. (iv) dsRNA binding proteins such as the N-terminal half of PKR (PKR-ΔK) or the vaccinia virus E3L protein have been reported to suppress PKR toxicity in yeast by forming inactive heterodimers with intact PKR (Romano et al., 1995, 1998b). However, this inhibition of PKR may also reflect sequestration of activating dsRNA molecules by PKR-ΔK or the E3L protein. While these various studies suggest that PKR dimerization is necessary for activity, there is little direct, unambiguous evidence supporting this idea.
Current models for PKR activation based on a number of genetic, biochemical and structural studies propose that dsRNA binding to the dsRBDs leads to a structural rearrangement of PKR relieving an autoinhibitory interaction between the dsRBDs and the KD, and facilitating kinase activation (Wu and Kaufman, 1997; Nanduri et al., 2000). Recently, it was reported that the second dsRBD interacted directly with the KD of PKR, and it was proposed that dsRNA binding to the first dsRBD initiates a structural rearrangement facilitating dsRNA binding to the second dsRBD. This binding of dsRNA to the second dsRBD was proposed to disrupt the autoinhibition of the KD by the dsRBD and lead to kinase activation (Nanduri et al., 2000). This model is supported by studies examining dimerization between full-length PKR and the isolated PKR-ΔK. Whereas a mutation that blocks dsRNA binding to the first dsRBD of intact PKR impairs binding to PKR-ΔK, the same mutation in the first dsRBD of PKR-ΔK does not affect binding to full-length PKR (Wu and Kaufman, 1997). This result is consistent with the theory that dsRNA binding to full-length PKR unmasks a dimerization domain. Supporting this theory, an additional dimerization domain was identified between residues 244 and 296 of PKR (Tan et al., 1998). This region includes the first two subdomains within the PKR kinase domain; however, it is not known whether these kinase subdomains contribute to the dimerization nor if this region that includes residues 244–296 of PKR is critical for dimerization of the full-length protein. Further support for the idea that the dsRBDs act as an autoinhibitory domain in PKR is the finding that a truncated form of PKR, consisting of residues 228–551 and lacking the two dsRBDs, is con stitutively active in mammalian cells (Wu and Kaufman, 1997). Accordingly, removal of the dsRBDs eliminates this auto-inhibition and unmasks a dimerization domain leading to kinase activation. One complication in interpreting the results of these studies and devising a model for PKR activation is the difficulty in distinguishing between effects of mutations on dsRNA binding versus dimerization. As dsRNA binding appears to relieve auto- inhibition, promote dimerization and stimulate kinase activation, it has been nearly impossible to assess directly the importance of dimerization for PKR activity.
The dsRBDs of PKR were also reported to mediate ribosome association (Zhu et al., 1997; Wu et al., 1998) and it was proposed that targeting PKR to the ribosome provided the kinase better access to its substrate eIF2α. An internal deletion (Δ14–257) in PKR that removes the dsRBDs, as well as a portion of the dimerization domain located between residues 244–296, eliminated PKR activity in yeast cells and disrupted ribosome association (Zhu et al., 1997). However, this truncated protein was a potent eIF2α kinase in vitro (Zhu et al., 1997). It was proposed that loss of ribosome association eliminated PKR activity in vivo, whereas in vitro the isolated KD could access its substrate. In this model, ribosome binding and not dimerization is the critical function of the dsRBDs. In contrast, ribosome binding and specifically ribosomal protein L18 were reported to negatively regulate PKR in mammalian cells (reviewed in Raine et al., 1998; Kumar et al., 1999; Kaufman, 2000). Due to the complex role of the dsRBDs to both positively and negatively regulate PKR, we chose to address further the role and requirement of dimerization for PKR activity by generating fusion proteins linking the KD of PKR (residues 258–551) with various constitutive or regulated dimerization domains and then testing the activity of these proteins in yeast or mammalian cells. Our studies demonstrate that dimerization is essential for PKR activity in vivo, and indicate that a critical role of dsRNA binding to the dsRBDs of PKR is to mediate dimerization and activation of the kinase.
Results
Reconstitution of PKR activity in yeast through fusion of the PKR kinase domain to glutathione S-transferase
To examine the requirement of dimerization for PKR activity in vivo we constructed a set of plasmids to express in yeast cells under the control of a galactose-regulated promoter either full-length human PKR (residues 1–551), a catalytically dead PKR mutant protein in which the essential conserved lysine residue in kinase subdomain II was mutated to histidine (PKR-K296H), or the isolated KD of PKR (residues 258–551) (Figure 1A). As observed previously, high level expression of wild-type PKR was lethal in yeast (Figure 1B, upper panel). However, this toxicity was not observed in cells expressing the catalytically inactive PKR-K296H or the KD alone [PKR (258–551)] (Figure 1B, upper panel). As the dsRBDs in the N-terminal half of PKR are known to promote dimerization of PKR, we suspected that the lack of phenotype in cells expressing the isolated KD may be due to a failure of this truncated protein to dimerize. Structural analyses have revealed that glutathione S-transferase (GST) can form homodimers (Ji et al., 1992; Reinemer et al., 1992; Kaplan et al., 1997), so we fused the KD (residues 258–551) to GST (GST–PKR; Figure 1A) and expressed this protein in yeast under the control of a galactose-regulated promoter. High-level expression of GST–PKR, like wild-type full-length PKR, was lethal in yeast (Figure 1B, upper panel). Importantly, the toxicity associated with high-level expression of GST–PKR was alleviated in strains in which the Ser51 phosphorylation site in eIF2α was changed to Ala [Figure 1B, lower panel (eIF2α-S51A)], indicating that the toxic effects of GST–PKR were due to eIF2α phosphorylation and the resultant inhibition of translation initiation.

Fig. 1. Expression of a GST–PKR kinase domain fusion protein, but not the isolated kinase domain alone, is toxic in yeast. (A) Schematics of wild-type human PKR [PKR (wt)] showing two dsRNA binding domains (dsRBDs) in the N-terminal half of the protein and the kinase domain (residues ∼263–551) in the C-terminal half of the protein; the isolated PKR kinase domain [PKR (258–551)]; and a GST–PKR kinase domain fusion protein (GST–PKR). (B) Plasmids expressing wild-type PKR (p1420), PKR-K296H (p1421), the isolated PKR kinase domain [PKR (258–551); pC681], or the GST–PKR kinase domain fusion protein (pC661), as indicated, under the control of a yeast GAL-CYC1 hybrid promoter were introduced into strain J80 containing wild-type eIF2α and strain J82 containing eIF2α-S51A, as indicated. Trans formants were streaked on SGal medium (synthetic minimal medium containing 10% galactose) supplemented with essential nutrients, and the plates were incubated at 30°C for 6 days.
The lack of phenotype in yeast cells expressing the PKR KD alone (Figure 1B) could indicate that this kinase was inactive, poorly expressed, or unstable in vivo. Protein immunoblot analyses (Figure 2A) using antiserum raised against purified recombinant GST–PKR revealed that the KD [PKR(258–551)] was poorly expressed compared with the full-length protein [the PKR epitopes are shared between PKR(258–551) and native PKR]. Functionally active forms of PKR are known to be poorly expressed in yeast due to negative translational autoregulation (Dever et al., 1993; Romano et al., 1995); however, this autoregulation is abolished in eIF2α-S51A strains (Dever et al., 1993; Romano et al., 1995). The low expression of PKR(258–551) was observed in both the wild-type and eIF2α-S51A strains (Figure 2A and B), suggesting that the kinase was poorly expressed or unstable. However, as will be discussed later, the low expression of PKR(258–551) is not sufficient to explain the lack of phenotype in yeast cells expressing the protein. The inhibition of yeast cell growth in strains expressing GST–PKR indicated that this protein was a functional eIF2α kinase. Isoelectric focusing gels were used to examine eIF2α phosphorylation in yeast cells expressing wild-type PKR or GST–PKR. As shown in Figure 2C, expression of either native PKR (lanes 1 and 6) or GST–PKR (lane 3) resulted in a significant increase in the amount of the phosphorylated form of eIF2α in yeast. This eIF2α phosphorylation was on Ser51, as indicated by the absence of the phosphorylated form in eIF2α-S51A strains expressing GST–PKR (Figure 2C, lane 5).

Fig. 2. Immunoblot analysis of PKR expression and isoelectric focusing analysis of eIF2α phosphorylation in yeast strains expressing various forms of PKR. (A and B) Immunoblot analysis of PKR expression. The yeast strains J80 [(A), wild-type eIF2α] and J82 [(B), eIF2α-S51A] were transformed with plasmids to express the indicated forms of PKR or empty vector as follows: lane 1, empty vector (pEMBLyex4); lane 2, wild-type PKR (p1420); lane 3, PKR (258–551) (pC681); lane 4, GST–PKR (pC661); lane 5, Ldb–PKR (pC903) plus vector (pEMBLyex4); lane 6, Lim–PKR (pC901) plus vector (p2444); lane 7, Ldb–PKR (pC903) plus Lim–PKR (pC901). The various PKR proteins were expressed under the control of a yeast GAL-CYC1 promoter. Transformants were grown to exponential phase in SD medium, and then shifted to inducing conditions (SGR medium containing 10% galactose plus 2% raffinose) for ∼18 h. Whole-cell extracts were prepared and 100 µg aliquots were subjected to SDS–PAGE followed by immunoblot analysis using polyclonal antisera against GST–PKR or eIF2α, as indicated. Immune complexes were detected by enhanced chemiluminescence. The molecular mass of SDS size standards are shown on the left. The black or white dots identify the relevant PKR protein(s). (C) Isoelectric focusing analysis of eIF2α phosphorylation. The yeast strains J80 (S51) or J82 (S51A) were transformed with plasmids expressing the indicated PKR proteins and grown as described above. Whole-cell extracts were prepared and 20 µg aliquots were resolved by isoelectric focusing PAGE and then subjected to immunoblot analysis using polyclonal anti-eIF2α antiserum as described previously (Dever et al., 1992). Lane 7 is a darker and higher contrast exposure of lane 4. The positions of basally phosphorylated (eIF2α) and eIF2α phosphorylated on Ser51 (eIF2α–P) are indicated on the right. The percentage of total eIF2α that is phosphorylated on Ser51 was determined by quantitative densitometry and NIH Image software and is indicated below the lanes.
It has been reported that the dsRBDs facilitate PKR function in yeast by tethering the kinase to ribosomes where it can more readily access its substrate eIF2α (Zhu et al., 1997). Crude extracts were prepared from eIF2α-S51A strains expressing high levels of wild-type PKR or GST–PKR, and the association of the kinases to ribosomes was determined by fractionating the extracts on sucrose gradients to separate 40 and 60S ribosomal subunits, 80S monosomes and polysomes. Immunoblot analyses of individual fractions from the gradients demonstrated the expected association of wild-type PKR with large ribosomal complexes, with the peak amounts of PKR found in fractions between the 40 and 60S subunits as reported previously (see Figure 4A in Zhu et al., 1997); however, GST–PKR was found exclusively at the top of the gradient in fractions devoid of ribosomes (Figure 3). These results indicate that GST–PKR does not bind to ribosomes and suggest that ribosome association is not critical for PKR activity in cells.

Fig. 3. Ribosome association of wild-type PKR, but not the GST–PKR fusion protein. Transformants of strain J82 (expressing eIF2α-S51A) containing the PKR plasmid p1420 or the GST–PKR plasmid pC661 were grown in SGal medium to an OD600 ∼1.5. Whole-cell extracts were prepared in the presence of cycloheximide (50 mg/ml) and MgCl2 (10 mM), and then subjected to velocity sedimentation on 5–47% sucrose gradients as described previously (Zhu et al., 1997; Romano et al., 1998b). The gradients were fractionated while monitoring absorbance at 254 nm to identify the positions of free 40 and 60S subunits, and 80S monosomes (as indicated by the arrows). The distribution of PKR and GST–PKR along the gradients was visualized by SDS–PAGE and immunoblot analysis using polyclonal antiserum raised against the GST–PKR fusion protein. The first lane in each panel was loaded with 1/50 of the input (I) extracts fractionated on the gradients.
As PKR is an important component in the mammalian cell anti-viral defense mechanism, a number of viruses express inhibitors of PKR (Gale and Katze, 1998). To assess further the activity of GST–PKR we examined the sensitivity of GST–PKR to various inhibitors. The vaccinia virus K3L protein is a pseudosubstrate inhibitor that resembles eIF2α and binds to PKR and blocks kinase activity. In previous studies we found that a K3L-H47R mutant was a more potent inhibitor of PKR (Kawagishi-Kobayashi et al., 1997). As shown in Figure 4A, expression of the K3L or K3L-H47R protein alleviated the toxicity due to expression of wild-type PKR in yeast. As the K3L protein inhibits PKR through direct interaction with the KD, it is consistent that expression of K3L, and more significantly K3L-H47R, reversed the growth inhibition resulting from expression of GST–PKR in yeast (Figure 4A). The vaccinia virus E3L protein, a dsRNA binding protein, as well as PKR-ΔK, which contains the dsRBDs, dimerize with full-length PKR via the dsRBDs and block PKR function (Figure 4B; Romano et al., 1995, 1998b). However, yeast cell growth inhibition by GST–PKR is insensitive to expression of the E3L protein or PKR-ΔK (Figure 4B). These results are consistent with the absence of dsRBDs in GST–PKR, and indicate that GST can functionally replace the dsRBDs and mediate PKR activation.
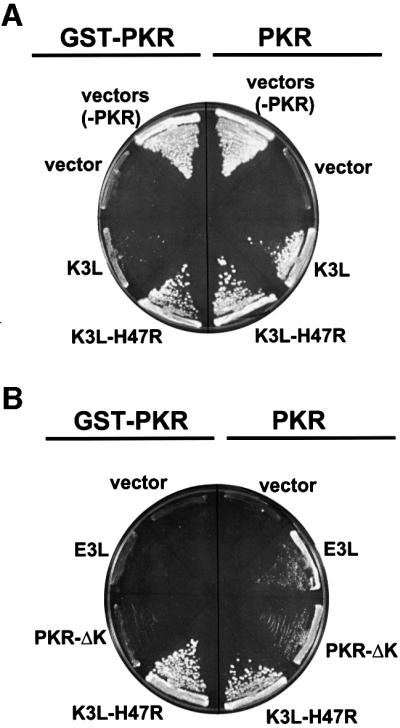
Fig. 4. GST–PKR and PKR (wt) show different sensitivities to the PKR inhibitors encoded by the vaccinia virus K3L and E3L genes. (A) Pseudosubstrate inhibition of PKR and GST–PKR. Yeast strain H1894 was transformed with a URA3 plasmid expressing GST–PKR (pC661), wild-type PKR (p1419), or the empty vector (pEMBLyex4) plus a LEU2 plasmid expressing the vaccinia virus K3L (pC365) or K3L-H47R (pC366) protein or the empty vector (pRS425). (B) Inhibition of PKR, but not GST–PKR, by the dsRNA-binding proteins E3L and PKR-ΔK. Yeast strain H1894 was transformed with a URA3 plasmid expressing GST–PKR (pC661) or wild-type PKR (p1419) plus a LEU2 plasmid expressing the vaccinia virus E3L (pC1315) or K3L-H47R (pC366) protein or the empty vector (pRS425), or a TRP1 plasmid expressing a truncated version of PKR lacking the kinase domain (PKR-ΔK, pC1316). All proteins in both (A) and (B) were expressed under the control of a yeast GAL-CYC1 promoter. Transformants were streaked on SGal minimal complete medium (synthetic minimal medium containing 10% galactose and supplemented with all amino acids), and the plates were incubated at 30°C for 8 days.
Reconstitution of PKR activity through heterodimerization of Lim–PKR and Ldb–PKR fusion proteins
The first 56 amino acids of the Xenopus LIM protein Xlim-1 and residues 300–338 near the C-terminus of the Xenopus protein Ldb1 (or NLI) are known to mediate the interaction between these binding partners (Jurata and Gill, 1997; Breen et al., 1998). In a modified version of the yeast two-hybrid assay, we fused the PKR KD (residues 258–551) to the heterodimerization domains from the Xenopus Xlim-1 and Ldb1 proteins (Figure 5A) and then expressed the fusion proteins in yeast. Expression of either Lim–PKR or Ldb–PKR alone in yeast (Figure 5B and C) had no effect on cell growth. However, when the Lim–PKR and Ldb–PKR fusions were co-expressed in the same yeast cell, growth was inhibited (Figure 5B and C). This growth inhibition was suppressed in eIF2α-S51A cells indicating that toxicity was due to phosphorylation of eIF2α (Figure 5B). Isoelectric focusing analyses revealed elevated levels of phosphorylated eIF2α in cells co-expressing Lim–PKR and Ldb–PKR (Figure 2C, lane 4) consistent with the growth inhibition observed in these cells (Figure 5B and C). Two results indicate that the slow growth of yeast co-expressing Lim–PKR and Ldb–PKR was not simply due to increased abundance of PKR in the cell. First, expression of the Lim–PKR fusion from two plasmids in the same cell did not impair cell growth (Figure 5B). Secondly, immunoblot analyses revealed that the total amount of PKR in cells co-expressing Lim–PKR and Ldb–PKR was less than in cells expressing just one of the fusion proteins (Figure 2A, lanes 5–7). As mentioned above, PKR expression is subject to negative translational autoregulation wherein inactive forms of PKR accumulate to higher levels in cells than functional forms of PKR. As co-expression of Lim–PKR and Ldb–PKR restores PKR activity, these proteins are subject to negative autoregulation when co-expressed, but not when expressed singly in cells. Consistent with the idea that co-expression of Lim–PKR and Ldb–PKR activated the negative translational autoregulation, the abundance of the fusion proteins expressed singly or together were equivalent in eIF2α-S51A strains where the autoregulation was abolished (Figure 2B). The PKR(258–551) KD fragment was expressed at levels only somewhat lower than Lim–PKR or Ldb–PKR, indicating, as mentioned above, that the lack of phenotype in cells expressing PKR(258–551) is probably not due to poor expression. Taken together, these results provide strong genetic evidence that PKR activity in vivo is dependent on the dimerization of PKR kinase domains.

Fig. 5. Reconstitution of PKR activity in yeast by co-expression of Lim–PKR and Ldb–PKR fusion proteins. (A) Schematics of Lim–PKR and Ldb–PKR. In Lim–PKR the first 56 amino acids of the Xenopus Xlim-1 protein (black box) are fused to the PKR kinase domain residues 258–551 (gray box). In Ldb–PKR residues 290–350 of the 375 amino acid Xenopus Ldb1 protein (cross-hatched box) are fused to the PKR kinase domain residues 258–551 (gray box). As indicated by the double-headed arrow, the Xenopus Lim and Ldb domains are known to heterodimerize. (B) The yeast strains J80 (eIF2α) and J82 (eIF2α-S51A) were transformed with the Lim–PKR expression plasmid (pC901) plus plasmids expressing Ldb–PKR (pC903) or Lim–PKR (pC944) or the empty vector (p2444), as indicated. (C) Yeast strain J80 was co-transformed with the Ldb–PKR plasmid (pC903) plus the Lim–PKR (pC901) or Lim–PKR-K296H (pC1097) plasmid or empty vector (pEMBLyex4), as indicated. All Lim–PKR and Ldb–PKR fusion proteins in (B) and (C) were expressed under the control of a yeast GAL-CYC1 promoter. Transformants were streaked on SGal minimal complete medium (synthetic minimal medium containing 10% galactose and supplemented with all amino acids), and the plates were incubated at 30°C for 7 days.
One hypothesis to explain the requirement for dimerization for PKR activity is that kinase activation requires mutual trans-autophosphorylation of the PKR monomers within the dimer. This idea is supported by the observation that PKR can autophosphorylate in trans (Thomis and Samuel, 1995; Ortega et al., 1996). Alternatively, dimerization of PKR kinase domains may be required to assemble the active site necessary for activation, and autophosphorylation then occurs either in cis or in trans. To determine whether both monomers within the PKR dimer must be functional kinases, we introduced the inactivating K296H mutation into the Lim–PKR construct. As shown in Figure 1B, the PKR-K296H mutation abolished PKR toxicity in yeast and previous work demonstrated that this mutation eliminates the ability of PKR to phosphorylate eIF2α (see Materials and methods; Dever et al., 1993). When the Ldb–PKR fusion was co-expressed with Lim–PKR-K296H, yeast cell growth was inhibited significantly (Figure 5C). The growth inhibition in yeast co-expressing Ldb–PKR and Lim–PKR-K296H was only slightly less than that observed when both the Ldb and Lim fusion proteins contained the wild-type KD (Figure 5C). These results demonstrate that only one KD in the dimer needs to be functional to generate an active kinase, and that the catalytically defective KD of PKR-K296H can provide the complementary contacts required for activation of wild-type PKR. Furthermore, these results suggest that kinase activation may involve autophosphorylation in cis. Alternatively, the substitution of His for Lys296 in PKR may severely reduce, but not eliminate, PKR kinase activity. In this latter model, PKR-K296H can phosphorylate and activate the wild-type KD, which can then phosphorylate eIF2α; however, a PKR-K296H homodimer is inactive.
Coumermycin-dependent activation of GyrB–PKR kinase domain fusion protein in mammalian cells
Having obtained genetic evidence demonstrating a requirement for dimerization for PKR activity in yeast, we next wanted to test the importance of dimerization for PKR activity in mammalian cells. For this analysis we chose the chemical-induced dimerization of the Escherichia coli GyrB protein by the drug coumermycin. It was previously demonstrated that coumermycin-mediated dimerization of a Raf–GyrB fusion protein results in activation of the Raf kinase (Farrar et al., 1996). For the experiments with PKR, constructs were generated to express fusion proteins consisting of the first 220 amino acids of E.coli GyrB fused to the wild-type PKR kinase domain (residues 258–551) or the same domain containing the inactivating K296H mutation (Figure 6A). Plasmids to express wild-type full-length PKR, GyrB–PKR, or an empty vector were transfected into NIH 3T3 cells along with a luciferase reporter construct. In transient transfectants containing an empty vector, high luciferase expression was detected independent of coumermycin concentration, whereas in cells expressing wild-type PKR, luciferase expression was inhibited in the absence or presence of coumermycin (Figure 6B). In cells expressing GyrB–PKR, a dose-dependent inhibition of luciferase expression was observed upon addition of coumermycin with effective inhibition observed at coumermycin concentrations as low as 10 ng/ml (Figure 6B). The coumermycin-dependent inhibition of luciferase expression required both GyrB and a functional PKR kinase domain as no inhibition was observed in cells expressing the GyrB–PKR-K296H mutant or the PKR kinase domain (residues 258–551) alone (Figure 6C, and data not shown).

Fig. 6. Coumermycin-induced activation of GyrB–PKR fusion proteins in mammalian cells. (A) Schematics of wild-type PKR [PKR(wt)]; GyrB–PKR fusion protein consisting of the N-terminal 220 amino acids of E.coli GyrB fused to the human PKR kinase domain residues 258–551 (GyrB–PKR); and coumermycin (black dumbbell)-mediated dimerization of GyrB–PKR fusion proteins (GyrB–PKR + coumermycin). (B) NIH 3T3 cells were co-transfected with the luciferase reporter plasmid pGL3-Control (Promega) and either empty vector (vector, pC869), or plasmids to express either wild-type PKR [PKR (wt), pC882] or the GyrB–PKR fusion protein [GyrB–PKR (258–551), pC939], as indicated. Twenty-four hours following transfection, cells were treated with dimethylsulfoxide (DMSO) alone or the indicated concentration of coumermycin dissolved in DMSO. After another 24 h, cells were harvested, lysed and samples of the whole-cell extracts were assayed for luciferase activity. The results are the average and standard deviation from three independent experiments. (C) NIH 3T3 cells were co-transfected with the luciferase reporter plasmid pGL3-Control (Promega) and either empty vector (vector, pC869), or plasmids to express wild-type PKR [PKR (wt), pC882], GyrB–PKR (pC939), or GyrB–PKR-K296H (pC940), as indicated. Twenty-four hours following transfection, cells were treated with DMSO alone (–coumermycin) or with 100 ng/ml coumermycin dissolved in DMSO. Following 24 h stimulation, cells were harvested, lysed and samples of the whole-cell extracts were assayed for luciferase activity. The results are the average and standard deviation from three independent experiments.
To confirm that coumermycin treatment specifically activated GyrB–PKR leading to an inhibition of luciferase mRNA translation, control experiments were conducted to examine GyrB–PKR expression, luciferase mRNA levels and eIF2α phosphorylation. Protein immunoblot analyses revealed that the expression of GyrB–PKR and GyrB– PKR-K296H in NIH 3T3 cells was unaffected by coumermycin treatment (Figure 7A). The slightly elevated expression of the mutant GyrB–PKR-K296H versus GyrB–PKR is consistent with the negative translational autoregulation of functional PKR kinases, as has been reported previously for yeast and mammalian cells (Barber et al., 1993; Dever et al., 1993; Romano et al., 1995). Interestingly, the mobility of GyrB–PKR in SDS– PAGE was slightly lower in extracts from cells treated with coumermycin (Figure 7A, lane 3 versus lane 2). Previously it has been observed that autophosphorylation reduces the mobility of PKR in SDS–PAGE (Romano et al., 1998a). Accordingly, the data in Figure 7A suggests that coumermycin-mediated dimerization of GyrB–PKR leads to kinase activation and autophosphorylation. Whereas addition of coumermycin to cells expressing GyrB–PKR led to a 90% reduction in luciferase activity (Figure 6C), RT–PCR analyses demonstrated that luciferase mRNA levels were unaffected by drug treatment (Figure 7B). In addition, luciferase mRNA levels were independent of the PKR construct in the cell (Figure 7B). Finally, protein immunoblot analyses using antibodies that specifically recognize the Ser51 phosphorylated form of eIF2α (DeGracia et al., 1997) were used to quantify eIF2α phosphorylation in the transfected cells. Expression of wild-type PKR or GyrB–PKR in the absence of drug led to a 2- to 3-fold increase in the ratio of the Ser51 phosphorylated form to total eIF2α (Figure 8, compare lanes 7 and 3 to lane 1). Consistent with the coumermycin-dependent activation of GyrB–PKR, an additional 3-fold increase in eIF2α phosphorylation was observed in cells expressing GyrB–PKR following treatment with the drug (Figure 8, lane 4 versus lane 3). In contrast, eIF2α phosphorylation was insensitive to coumermycin addition in cells expressing wild-type PKR or inactive forms of the kinase (Figure 8). These results support the conclusion that coumermycin-mediated dimerization of GyrB–PKR leads to activation of the kinase, increased eIF2α phosphorylation, and the resulting inhibition of translation initiation.

Fig. 7. Coumermycin treatment does not alter GyrB–PKR expression or luciferase reporter mRNA levels in NIH 3T3 cell transfectants. (A) Analysis of PKR expression. NIH 3T3 cells were co-transfected with the luciferase reporter plasmid pGL3-Control (Promega) and either empty vector (lane 1, vector, pC869), or plasmids to express GyrB–PKR (lanes 2 and 3, pC939) or GyrB–PKR-K296H (lanes 4 and 5, pC940), as indicated. Twenty-four hours following transfection, cells were treated with DMSO alone (– coumermycin) or with 100 ng/ml coumermycin dissolved in DMSO (+ coumermycin). Following 24 h stimulation with the drug, cells were harvested, lysed and samples of the whole-cell extracts were subjected to SDS–PAGE and then immunoblotted with anti-PKR (upper panel) or anti-TFIIB (lower panel) antisera as indicated. (B) Analysis of luciferase mRNA levels. NIH 3T3 cells were co-transfected with the luciferase reporter plasmid pGL3-Control (pGL3-Luc) or empty vector (pC869), as indicated, and either empty vector (no label, pC869) or plasmids to express wild-type PKR [PKR (1–551), pC882], GyrB–PKR (pC939), or GyrB–PKR-K296H (pC940), as indicated. Twenty-four hours following transfection, cells were treated with DMSO alone (– coumermycin) or with 100 ng/ml coumermycin dissolved in DMSO (+ coumermycin). Following 24 h stimulation, cells were harvested, lysed and the amount of luciferase reporter and β-actin mRNAs was determined by RT–PCR, as described previously (Kawagishi-Kobayashi et al., 2000). Lanes 1 and 2 are control experiments for the RT–PCR analysis in which either cellular RNA was omitted (lane 1) or the RNA was obtained from non-transfected cells (lane 2).

Fig. 8. Increased phosphorylation of eIF2α on Ser51 in NIH 3T3 cells expressing GyrB–PKR and treated with coumermycin. NIH 3T3 cells were co-transfected with the luciferase reporter plasmid pGL3-Control and either empty vector (lanes 1 and 2, vector, pC869), or plasmids to express GyrB–PKR (lanes 3 and 4, pC939), GyrB–PKR-K296H (lanes 5 and 6, pC940), or wild-type PKR [lanes 7 and 8, PKR (wt), pC882], as indicated. Twenty-four hours following transfection, cells were treated with DMSO alone (– coumermycin) or with 100 ng/ml coumermycin dissolved in DMSO (+ coumermycin). Following 24 h stimulation, cells were harvested, lysed and samples of the whole-cell extracts were subjected to SDS–PAGE and then immunoblotted with phosphospecific antibodies raised against an eIF2α peptide containing phosphoserine-51 (DeGracia et al., 1997) (upper and middle panel). Subsequently, the blot was stripped and probed with anti-eIF2α monoclonal antibodies (Scorsone et al., 1987) (lower panel). As indicated, the middle panel is a longer exposure (60 s) of the same blot presented in the top panel (5 s exposure). The relative level of eIF2α phosphorylation in comparison with the untreated vector transfectant (lane 1) was determined by quantitative densitometry and NIH Image software, and is indicated below the lanes.
Discussion
In this report we have demonstrated that dimerization of the PKR KD is both necessary and sufficient for activation of its eIF2α kinase activity in both yeast and mammalian cells. Removal of the N-terminal dsRBDs from PKR yielded a non-functional KD; however, PKR activity was restored by fusing heterologous dimerization domains to the KD. These results indicate that a primary role of dsRNA in the activation of wild-type PKR is to promote dimerization of the protein. Previous studies demonstrated that the dsRBDs as well as a second region located between residues 242–296 of PKR could mediate dimerization of the protein (Cosentino et al., 1995; Patel et al., 1995; Ortega et al., 1996; Wu and Kaufman, 1996; Carpick et al., 1997; Tan et al., 1998); however, the importance of dimerization and subcellular localization for activation of the PKR kinase was not clear. PKR has been reported to bind to ribosomes in both yeast (Zhu et al., 1997) and mammalian cells (Raine et al., 1998; Wu et al., 1998). In yeast, a PKR mutant containing a deletion of residues 14–257, and thus lacking the dsRBDs, lost the ability to bind to ribosomes and also was not a functional kinase in vivo (Zhu et al., 1997). These results suggested that ribosome binding was necessary for PKR activity in vivo; however, studies in mammalian cells suggested that ribosome binding interfered with PKR activity (Raine et al., 1998; Kumar et al., 1999). Our results demonstrate that ribosome association is not crucial for PKR to phosphorylate eIF2α and regulate translation in yeast cells. The GST–PKR fusion protein functionally substituted for native PKR to downregulate protein synthesis in yeast cells; however, this protein did not bind to ribosomes (Figure 3). Supporting this idea that ribosome association is not critical for PKR activity in yeast, it was found the vaccinia virus E3L protein could displace PKR from ribosomes; however, ribosome displacement did not correlate with loss of PKR activity (Romano et al., 1998b). A mutant version of the E3L protein that failed to inhibit PKR activity in yeast was still able to displace PKR from ribosomes. Therefore, dimerization and not ribosome association appears to be crucial for PKR function in vivo.
Previously, it was reported that a truncated form of PKR consisting of residues 228–551, and thus lacking the dsRBDs, was able to downregulate reporter gene expression in mammalian cells, and that the protein was an active kinase in vitro (Wu and Kaufman, 1997). However, deletion of an additional 36 residues resulted in an inactive kinase, PKR(264–551), which is similar to the non-functional, truncated mutant, PKR(258–551) examined in this study (Wu and Kaufman, 1997). These results suggest that a stimulatory domain resides within residues 228–257 of PKR. It is likely that this region mediates dimerization as our data demonstrate that heterologous dimerization domains can functionally substitute for residues 1–257 of PKR, and interestingly, a dimerization domain was mapped between residues 242–296 of PKR (Tan et al., 1998). Taken together, the results of these studies suggest that a dimerization element, residing between residues 242–258 of PKR, is responsible for the functional activity of PKR(228–551). At odds with this model, we found that PKR(242–551), like PKR(258–551), was non-functional in yeast, and that its activity was restored when fused to GST (data not shown). These results indicate that the dimerization domain mapped between residues 242–296 is insufficient to mediate PKR activation in vivo, and by extension, they suggest that an additional element located between residues 228–242 of PKR facilitates dimerization. The role of this putative dimerization region for the activity of full-length PKR remains to be resolved.
In addition to the results in this paper, two other reports provide data supporting the importance of dimerization for PKR activity. First, the work of Tan et al. (1998) identified a dimerization region located between residues 242–296 of PKR. Interestingly, this same region of PKR has been found to interact with the PKR inhibitors P58IPK and the NS5A protein from hepatitis C virus (Gale et al., 1996, 1998). Importantly, it was shown that P58IPK could disrupt the interaction between PKR(242–296) and PKR(242–551) (Tan et al., 1998). Thus, P58IPK may prevent PKR activation by blocking kinase domain dimerization. Secondly, the PK2 protein encoded by the baculovirus Autographa californica nuclear polyhedrosis virus is an eIF2α kinase inhibitor that resembles the C-terminal half of an eIF2α kinase domain (Dever et al., 1998). The PK2 protein was found to interact with the PKR kinase domain in yeast two-hybrid and co-immunoprecipitation assays, and expression of PK2 blocked the activity of both PKR and GCN2 in yeast cells as well as endogenous eIF2α kinases in cells from Spodoptera frugiperda, an insect host for baculovirus (Dever et al., 1998). Interestingly, PK2 expression appeared to block autophosphorylation of PKR co-expressed in yeast cells, consistent with the notion that PK2 inhibits PKR activation by forming inactive heterodimers with PKR and preventing formation of PKR homodimers. Finally, it is interesting to note that the other members of the eIF2α kinase family, HRI, GCN2 and PERK, have also been reported to dimerize, and despite differences in the nature of the activating signal for these kinases, dimerization appears to be critical for kinase activation (see Qiu et al., 1998; Bertolotti et al., 2000; Chen, 2000).
Several reagents generated in these studies may be useful for future analyses of dimerization and PKR function. The heterodimerization domains in Xlim-1 and Ldb1 are compact elements composed of 50–60 residues. We anticipate that they could be useful reagents to test the effects of generating heterodimers among any two proteins of interest. A recent advance of the yeast two-hybrid system is the reverse two-hybrid in which mutations are sought that disrupt the interaction between two proteins (Leanna and Hannink, 1996; Vidal et al., 1996). As dimerization of the PKR KD via heterologous homo- or heterodimerization domains generates sufficient PKR activity to severely inhibit or block yeast cell growth, the PKR KD could prove to be a useful reagent for reverse two-hybrid assays. Mutations that impair a two-hybrid interaction between dimerization domains would be expected to alleviate the growth inhibition caused by expression of fusion proteins containing the dimerization domain fused to the PKR KD. Thus, the use of the PKR KD in the two-hybrid fusions would allow for a positive screen for mutations that reduced the interaction between the binding partners. Finally, with the GyrB–PKR fusion we can activate PKR in the absence of dsRNA treatment. High level expression of PKR in mammalian cells is known to promote apoptosis (reviewed in Kaufman, 2000); however, the mechanism for how this occurs is not fully understood. We have observed some signs of apoptosis upon activation of GyrB–PKR in cells treated with coumermycin. As the dsRNA treatments required to activate native PKR may also activate other cellular stress pathways leading to apoptosis, the coumermycin-dependent activation of PKR offers the opportunity to separate PKR activation from dsRNA treatment of cells. We anticipate that the GyrB–PKR fusion will be a useful tool to selectively activate PKR in mammalian cells in order to monitor the gene expression changes resulting from enhanced eIF2α phosphorylation.
Materials and methods
Plasmids
The low (p1419) and high (p1420) copy-number URA3 plasmids to express PKR in yeast under the control of the GAL-CYC1 promoter have been described previously (Dever et al., 1993). The high copy-number URA3 plasmid p1421 contains PKR-K296H (and not PKR-K296R as reported previously; Dever et al., 1993) under the control of the GAL-CYC1 promoter. Plasmids to express GST–PKR in yeast (pC661) and bacteria (pC676) were described previously (Kawagishi-Kobayashi et al., 2000). A SacI–HindIII fragment encoding the PKR kinase domain (residues 258–551) was obtained by PCR and inserted into the yeast expression vector pEMBLyex4 (Cesareni and Murray, 1987) under the control of the GAL-CYC1 promoter creating the plasmid pC681. The Lim–PKR expression plasmid pC901 was generated by inserting both a SacI–BamHI fragment obtained by PCR encoding Xlim-1 residues 1–58 and a BamHI–HindIII fragment containing the PKR kinase domain from plasmid pC661 into the vector pEMBLyex4. The Ldb–PKR expression plasmid was generated by first subcloning a SacI–BamHI fragment obtained by PCR encoding residues 290–350 of Ldb1 and the BamHI– HindIII fragment encoding the PKR kinase domain from pC661 into pBluescript (Stratagene, Inc.). Plasmid pC903 was created by subcloning a SacI–SalI Ldb–PKR fragment from the resulting construct into p2444, a modified version of pEMBLyex4 in which TRP1 is inserted into the URA3 marker (a kind gift of Graham Pavitt). The Lim–PKR allele from plasmid pC901 was isolated as a SacI–HindIII fragment and inserted into p2444 creating pC944. The plasmid pC1097 encoding Lim–PKR-K296H was generated by replacing the BamHI–HindIII fragment encoding the PKR kinase domain in plasmid pC901 with a PCR fragment encoding PKR residues 258–551 and containing the K296H mutation.
The high copy-number LEU2 plasmids expressing K3L (pC365), K3L-H47R (pC366) and E3L (pC1315) under the control of the GAL-CYC1 promoter were a kind gift of Makiko Kawagishi-Kobayashi and were generated by transferring ApaI–BamHI fragments from the plasmids pC140 (K3L; Kawagishi-Kobayashi et al., 1997), pC407 (K3L-H47R; Kawagishi-Kobayashi et al., 1997) and pC178 (E3L; Romano et al., 1998b) to the vector pRS425 (Christianson et al., 1992). Similarly, an ApaI–BamHI fragment from p1766, encoding PKR-ΔK under the control of the GAL-CYC1 promoter (Romano et al., 1995, 1998b), was transferred to the high copy-number TRP1 vector pRS424 (Christianson et al., 1992) creating pC1316.
For mammalian cell expression, proteins were expressed under the control of an SV40 promoter in plasmids derived from the vector pSG5 (Stratagene, Inc.). The plasmids pC869, a derivative of pSG5 with a modified polylinker, and pC882, a derivative of pC869 that expresses wild-type PKR (residues 1–551), were described previously (Kawagishi-Kobayashi et al., 2000). A SacI–BamHI fragment encoding E.coli GyrB residues 1–220 was obtained by PCR using as a template the plasmid pKS-GyrB (a kind gift of Michael Farrar and Roger Perlmutter). The GyrB–PKR expression vector pC939 was generated by inserting this fragment along with the BamHI–HindIII PKR kinase domain fragment from pC661 into pC869. The GyrB–PKR-K296H expression vector pC940 was generated by replacing the BamHI–HindIII fragment encoding the PKR kinase domain in pC939 with the corresponding fragment encoding the PKR-K296H kinase domain in pC1097.
Strains
The yeast strain H1894 (MATa ura3-52 leu2-3 leu2-112 trp1-Δ63 gcn2Δ) was described previously (Kawagishi-Kobayashi et al., 1997). The strains J80 (MATa ura3-52 leu2-3 leu2-112 trp1-Δ63 gcn2Δ sui2Δ p[SUI2, LEU2]) and J82 (MATa ura3-52 leu2-3 leu2-112 trp1-Δ63 gcn2Δ sui2Δ p[SUI2-S51A, LEU2]) are derivatives of H1645 in which chromosomal GCN2 has been replaced with an unmarked gcn2Δ allele as described previously (Dever et al., 1992).
Immunoblot analysis of PKR expression
Whole-cell yeast extracts were prepared and proteins were resolved by SDS–PAGE and blotted to nitrocellulose membranes as described previously (Romano et al., 1995, 1998b). Polyclonal anti-PKR antiserum was raised against a GST–PKR kinase domain fusion protein consisting of PKR residues 258–551. The GST–PKR expression vector pC676 was introduced into the E.coli strain DH5α, and the fusion protein was purified as described previously (Kawagishi-Kobayashi et al., 2000). Antibody production in New Zealand White rabbits was conducted by Hazelton Laboratories. For immunoblot analyses the anti-GST–PKR antiserum was diluted 1:1000.
Mammalian cell experiments
For transient transfections, NIH 3T3 (4 × 104 cells per well in a 24-well plate) were seeded the day before transfection. Cells were incubated with a mixture of DNA and lipofectamine reagents (Life Technologies, Inc.) for 6 h. The DNA mixtures contained 350 ng of the pGL3-Control plasmid (Promega), which expresses luciferase mRNA under the control of the SV40 promoter and enhancer, and 50 ng of the various PKR expression vectors. Cells were incubated for 24 h, treated with coumermycin, and then incubated an additional 24 h. Cells were harvested, lysed and 20% of the extract was assayed for luciferase activity. Quantitation of mRNA levels by RT–PCR was performed as described previously (Kawagishi-Kobayashi et al., 2000).
Acknowledgments
Acknowledgements
We thank Minerva Garcia-Barrio, Fan Zhang, Alan Hinnebusch, Graham Pavitt, Makiko Kawagishi-Kobayashi, Pat Romano, Orna Elroy-Stein and members of the LEGR and LMGR for advice, reagents and useful discussions. We thank Blaine White and Gary Krause for the anti-eIF2α-phosphoserine-51 antibodies, Joe Breen and Igor Dawid for the Xlim-1 and Ldb1 plasmids, Michael Farrar and Roger Perlmutter for the GyrB plasmid, Minerva Garcia-Barrio for help with the sucrose gradient experiments, and Alan Hinnebusch for comments on the manuscript.
References
- Barber G.N., Wambach,M., Wong,M.L., Dever,T.E., Hinnebusch,A.G. and Katze,M.G. (1993) Translational regulation by the interferon-induced double-stranded RNA-activated 68-kDa protein kinase. Proc. Natl Acad. Sci. USA, 90, 4621–4625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertolotti A., Zhang,Y., Hendershot,L., Harding,H. and Ron,D. (2000) Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nature Cell Biol., 2, 326–332. [DOI] [PubMed] [Google Scholar]
- Breen J.J., Agulnick,A.D., Westphal,H. and Dawid,I.B. (1998) Interactions between LIM domains and the LIM domain-binding protein Ldb1. J. Biol. Chem., 273, 4712–4717. [DOI] [PubMed] [Google Scholar]
- Carpick B.W., Graziano,V., Schneider,D., Maitra,R.K., Lee,X. and Williams,B.R.G. (1997) Characterization of the solution complex between the interferon-induced, double-stranded RNA-activated protein kinase and HIV-1 trans-activating region RNA. J. Biol. Chem., 272, 9510–9516. [DOI] [PubMed] [Google Scholar]
- Cesareni G. and Murray,J.A.H. (1987) Plasmid vectors carrying the replication origin of filamentous single-stranded phages. In Setlow,J.K. and Hollaender,A. (eds), Genetic Engineering: Principles and Methods. Vol. 9. Plenum Press, New York, NY, pp. 135–154.
- Chen J.-J. (2000) Heme-regulated eIF2α kinase. In Sonenberg,N., Hershey,J.W.B. and Mathews,M.B. (eds), Translational Control of Gene Expression. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 529–546.
- Chong K.L., Feng,L., Schappert,K., Meurs,E., Donahue,T.F., Friesen,J.D., Hovanessian,A.G. and Williams,B.R.G. (1992) Human p68 kinase exhibits growth suppression in yeast and homology to the translational regulator GCN2. EMBO J., 11, 1553–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christianson T.W., Sikorski,R.S., Dante,M., Shero,J.H. and Hieter,P. (1992) Multifunctional yeast high-copy-number shuttle vectors. Gene, 110, 119–122. [DOI] [PubMed] [Google Scholar]
- Cosentino G.P., Venkatesan,S., Serluca,F.C., Green,S.R., Mathews,M.B. and Sonenberg,N. (1995) Double-stranded-RNA-dependent protein kinase and TAR RNA-binding protein form homo- and heterodimers in vivo. Proc. Natl Acad. Sci. USA, 92, 9445–9449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeGracia D.J., Sullivan,J.M., Neumar,R.W., Alousi,S.S., Hikade,K.R., Pittman,J.E., White,B.C., Rafols,J.A. and Krause,G.S. (1997) Effect of brain ischemia and reperfusion on the localization of phosphorylated eukaryotic initiation factor 2α. J. Cereb. Blood Flow Metab., 17, 1291–1302. [DOI] [PubMed] [Google Scholar]
- Dever T.E. (1999) Translation initiation: adept at adapting. Trends Biochem. Sci., 24, 398–403. [DOI] [PubMed] [Google Scholar]
- Dever T.E., Feng,L., Wek,R.C., Cigan,A.M., Donahue,T.D. and Hinnebusch,A.G. (1992) Phosphorylation of initiation factor 2α by protein kinase GCN2 mediates gene-specific translational control of GCN4 in yeast. Cell, 68, 585–596. [DOI] [PubMed] [Google Scholar]
- Dever T.E., Chen,J.J., Barber,G.N., Cigan,A.M., Feng,L., Donahue,T.F., London,I.M., Katze,M.G. and Hinnebusch,A.G. (1993) Mammalian eukaryotic initiation factor 2α kinases functionally substitute for GCN2 in the GCN4 translational control mechanism of yeast. Proc. Natl Acad. Sci. USA, 90, 4616–4620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dever T.E., Sripriya,R., McLachlin,J.R., Lu,J., Fabian,J.R., Kimball,S.R. and Miller,L.K. (1998) Disruption of cellular translational control by a viral truncated eukaryotic translation initiation factor 2α kinase homolog. Proc. Natl Acad. Sci. USA, 95, 4164–4169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrar M.A., Alberol,I. and Perlmutter,R.M. (1996) Activation of the Raf-1 kinase cascade by coumermycin-induced dimerization. Nature, 383, 178–181. [DOI] [PubMed] [Google Scholar]
- Gale M. Jr and Katze,M.G. (1998) Molecular mechanisms of interferon resistance mediated by viral-directed inhibition of PKR, the interferon-induced protein kinase. Pharmacol. Ther., 78, 29–46. [DOI] [PubMed] [Google Scholar]
- Gale M., Tan,S.-L., Wambach,M. and Katze,M.G. (1996) Interaction of the interferon-induced PKR protein kinase with inhibitory proteins P58IPK and vaccinia virus K3L is mediated by unique domains: implications for kinase regulation. Mol. Cell. Biol., 16, 4172–4181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gale M. et al. (1998) Control of PKR protein kinase by hepatitis C virus nonstructural 5A protein: molecular mechanisms of kinase regulation. Mol. Cell. Biol., 18, 5208–5218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heldin C.-H. (1995) Dimerization of cell surface receptors in signal transduction. Cell, 80, 213–223. [DOI] [PubMed] [Google Scholar]
- Ji X., Zhang,P., Armstrong,R.N. and Gilliland,G.L. (1992) The three-dimensional structure of a glutathione S-transferase from the µ gene class. Structural analysis of the binary complex of isoenzyme 3-3 and glutathione at 2.2-Å resolution. Biochemistry, 31, 10169–10184. [DOI] [PubMed] [Google Scholar]
- Jurata L.W. and Gill,G.N. (1997) Functional analysis of the nuclear LIM domain interactor NLI. Mol. Cell. Biol., 17, 5688–5698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan W., Husler,P., Klump,H., Erhardt,J., Sluis-Cremer,N. and Dirr,H. (1997) Conformational stability of pGEX-expressed Schistosoma japonicum glutathione S-transferase: a detoxification enzyme and fusion-protein affinity tag. Protein Sci., 6, 399–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman R.J. (1999) Stress signaling from the lumen of the endoplasmic reticulum: coordination of gene transcriptional and translational controls. Genes Dev., 13, 1211–1233. [DOI] [PubMed] [Google Scholar]
- Kaufman R. (2000) Double-stranded RNA-activated protein kinase PKR. In Sonenberg,N., Hershey,J.W.B. and Mathews,M.B. (eds), Translational Control of Gene Expression. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 503–527.
- Kawagishi-Kobayashi M., Silverman,J.B., Ung,T.L. and Dever,T.E. (1997) Regulation of the protein kinase PKR by the vaccinia virus pseudosubstrate inhibitor K3L is dependent on residues conserved between the K3L protein and the PKR substrate eIF2α. Mol. Cell. Biol., 17, 4146–4158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawagishi-Kobayashi M., Cao,C., Lu,J., Ozato,K. and Dever,T. (2000) Pseudosubstrate inhibition of protein kinase PKR by swine pox virus C8L gene product. Virology, 276, 424–434. [DOI] [PubMed] [Google Scholar]
- Kostura M. and Mathews,M.B. (1989) Purification and activation of the double-stranded RNA-dependent eIF-2 kinase DAI. Mol. Cell. Biol., 9, 1576–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar K.U., Srivastava,S.P. and Kaufman,R.J. (1999) Double-stranded RNA-activated protein kinase (PKR) is negatively regulated by 60S ribosomal subunit protein L18. Mol. Cell. Biol., 19, 1116–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langland J.O. and Jacobs,B.L. (1992) Cytosolic double-stranded RNA-dependent protein kinase is likely a dimer of partially phosphorylated Mr = 66 000 subunits. J. Biol. Chem., 267, 10729–10736. [PubMed] [Google Scholar]
- Leanna C. and Hannink,M. (1996) The reverse two-hybrid system: a genetic scheme for selection against specific protein/protein interactions. Nucleic Acids Res., 24, 3341–3347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmon M.A. and Schlessinger,J. (1994) Regulation of signal transduction and signal diversity by receptor oligomerization. Trends Biochem. Sci., 19, 459–463. [DOI] [PubMed] [Google Scholar]
- Nanduri S., Rahman,F., Williams,B.R. and Qin,J. (2000) A dynamically tuned double-stranded RNA binding mechanism for the activation of antiviral kinase PKR. EMBO J., 19, 5567–5574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega L.G., McCotter,M.D., Henry,G.L., McCormack,S.J., Thomis,D.C. and Samuel,C.E. (1996) Mechanism of interferon action. Biochemical and genetic evidence for the intermolecular association of the RNA-dependent protein kinase PkR from human cells. Virology, 215, 31–39. [DOI] [PubMed] [Google Scholar]
- Patel R.C. and Sen,G.C. (1998) Requirement of PKR dimerization mediated by specific hydrophobic residues for its activation by double-stranded RNA and its antigrowth effects in yeast. Mol. Cell. Biol., 18, 7009–7019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel R.C., Stanton,P., McMillan,N.M.J., Williams,B.R.G. and Sen,G.C. (1995) The interferon-inducible double-stranded RNA-activated protein kinase self-associates in vitro and in vivo. Proc. Natl Acad. Sci. USA, 92, 8283–8287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu H., Garcia-Barrio,M.T. and Hinnebusch,A.G. (1998) Dimerization by translation initiation factor 2 kinase GCN2 is mediated by interactions in the C-terminal ribosome-binding region and the protein kinase domain. Mol. Cell. Biol., 18, 2697–2711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raine D.A., Jeffrey,I.W. and Clemens,M.J. (1998) Inhibition of the double-stranded RNA-dependent protein kinase PKR by mammalian ribosomes. FEBS Lett., 436, 343–348. [DOI] [PubMed] [Google Scholar]
- Reinemer P., Dirr,H.W., Ladenstein,R., Huber,R., Lo Bello,M., Federici,G. and Parker,M.W. (1992) Three-dimensional structure of class pi glutathione S-transferase from human placenta in complex with S-hexylglutathione at 2.8 Å resolution. J. Mol. Biol., 227, 214–226. [DOI] [PubMed] [Google Scholar]
- Romano P.R., Green,S.R., Barber,G.N., Mathews,M.B. and Hinnebusch,A.G. (1995) Structural requirements for double-stranded RNA binding, dimerization and activation of the human eIF-2α kinase DAI in Saccharomyces cerevisiae. Mol. Cell. Biol., 15, 365–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romano P.R. et al. (1998a) Autophosphorylation in the activation loop is required for full kinase activity in vivo of human and yeast eukaryotic initiation factor 2α kinases PKR and GCN2. Mol. Cell. Biol., 18, 2282–2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romano P.R., Zhang,F., Tan,S.L., Garcia-Barrio,M.T., Katze,M.G., Dever,T.E. and Hinnebusch,A.G. (1998b) Inhibition of double-stranded RNA-dependent protein kinase PKR by vaccinia virus E3: role of complex formation and the E3 N-terminal domain. Mol. Cell. Biol., 18, 7304–7316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scorsone K.A., Panniers,R., Rowlands,A.G. and Henshaw,E.C. (1987) Phosphorylation of eukaryotic initiation factor 2 during physiological stresses which affect protein synthesis. J. Biol. Chem., 262, 14538–14543. [PubMed] [Google Scholar]
- Shamu C.E. and Walter,P. (1996) Oligomerization and phosphorylation of the Ire1p kinase during intracellular signaling from the endoplasmic reticulum to the nucleus. EMBO J., 15, 3028–3039. [PMC free article] [PubMed] [Google Scholar]
- Tan S.L., Gale,M.J. and Katze,M.G. (1998) Double stranded RNA-independent dimerization of the interferon-induced protein kinase, PKR and inhibition of dimerization by the cellular P58 IPK inhibitor. Mol. Cell. Biol., 18, 2431–2443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomis D.C. and Samuel,C.E. (1995) Mechanism of interferon action: characterization of the intermolecular autophosphorylation of PKR, the interferon-inducible, RNA-dependent protein kinase. J. Virol., 69, 5195–5198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal M., Brachmann,R., Fattaey,A., Harlow,E. and Boeke,J. (1996) Reverse two-hybrid and one-hybrid systems to detect dissociations of protein–protein and DNA–protein interactions. Proc. Natl Acad. Sci. USA, 93, 10315–10320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S. and Kaufman,R.J. (1996) Double-stranded (ds) RNA binding and not dimerization correlates with the activation of the dsRNA-dependent protein kinase (PKR). J. Biol. Chem., 271, 1756–1763. [DOI] [PubMed] [Google Scholar]
- Wu S. and Kaufman,R.J. (1997) A model for the double-stranded RNA (dsRNA)-dependent dimerization and activation of the dsRNA-activated protein kinase PKR. J. Biol. Chem., 272, 1291–1296. [DOI] [PubMed] [Google Scholar]
- Wu S., Kumar,K.U. and Kaufman,R.J. (1998) Identification and requirement of three ribosome binding domains in dsRNA-dependent protein kinase (PKR). Biochemistry, 37, 13816–13826. [DOI] [PubMed] [Google Scholar]
- Zhu S., Romano,P.R. and Wek,R.C. (1997) Ribosome targeting of PKR is mediated by two double-stranded RNA-binding domains and facilitates in vivo phosphorylation of eukaryotic initiation factor-2. J. Biol. Chem., 272, 14434–14441. [DOI] [PubMed] [Google Scholar]