

Abstract
Ribonucleic acids (RNAs) play a central role in cellular processes by interacting with proteins, small molecules, and other RNAs. Accurate prediction of these interactions is critical for understanding post-transcriptional regulation and advancing RNA-targeted therapeutics. However, existing computational methods are limited by their reliance on hand-crafted features, modality-specific architectures, and often require structural or physicochemical data, which are experimentally challenging to obtain and unavailable for many RNA molecules. These constraints hinder generalizability and fail to capture the complex, context-dependent semantics of RNA interactions. We present BioLLMNet, a unified sequence-only framework that leverages pretrained biological language models to encode rich, contextualized representations for both RNA molecules and their interacting partners, including proteins, small molecules, and other RNAs. Our key innovation is the introduction of a novel learnable gating mechanism, which dynamically computes feature-wise weights to adaptively integrate multimodal embeddings based on input context. This mechanism, proposed here for the first time in RNA interaction modeling, enables the model to emphasize the most informative features from each partner and achieves seamless fusion of heterogeneous modalities. As a result, BioLLMNet represents a unified framework and can flexibly and consistently model all three types of interaction (RNA–protein, RNA–small molecule, and RNA–RNA) within a shared architecture, eliminating the need for modality-specific designs. Comprehensive evaluations on benchmark data sets demonstrate that BioLLMNet achieves state-of-the-art performance across all three types of interaction. Our results underscore the power of language model-based representations combined with dynamic feature fusion for generalizable, modality-aware RNA interaction prediction.
Keywords: RNA interaction prediction, biological language models, multimodal representation learning
Introduction
Ribonucleic acids (RNAs) are essential biomolecules that play diverse roles in cellular processes through interactions with other RNAs, proteins, and small molecules [1–8]. Understanding these RNA-associated interactions is crucial, as they regulate various physiological and pathological processes. Specifically, RNA–RNA interactions (RRIs) are involved in posttranscriptional processes, contributing to gene expression regulation [9, 10]. RNA–protein interactions are vital for maintaining cellular homeostasis, and disruptions in these interactions can lead to cellular dysfunctions or diseases such as cancer [11–14]. Furthermore, RNA-small molecule interactions have significant implications in therapeutic development, as RNAs can serve as potential drug targets, especially when conventional protein targets are less accessible [15, 16].
Despite the importance of these interactions, the discovery of RNA-associated interactions is often challenging due to the complexity of RNA structures and the limited availability of experimentally resolved RNA-containing structures [17, 18]. Existing computational methods have, therefore, been developed to predict such interactions and facilitate research in this area. These approaches typically involve encoding interacting molecules into computer-recognizable features, such as sequences, structures, and physicochemical properties, followed by integrating these features to predict interactions. For RRIs, tools like MD-MLI [19] and lncIBTP [20] convert RNA sequences and secondary structures into numerical vectors, then use machine learning algorithms to identify potential interaction partners. CORAIN [21] employs convolutional autoencoder-directed feature embedding and PMLIPred [10] utilizes CNN and BiGRU for RRIs. In the case of RNA–protein interactions, tools such as CatRAPID and PRPI–SC use both sequence and structural information to calculate interaction scores, combining features like hydrogen bonding potential, charge, and hydrophobicity [12, 22]. RPISeq-RF [23], IMPIner [24], CFRP [25], RPITER [26], LPI-CSFFR [27], and RNAincoder [28] are machine learning-based methods for predicting RNA–protein interactions.
For RNA–small molecule interactions, LigandRNA and dSPRINT extract molecular fingerprints and physicochemical properties of small molecules, then predict binding affinities with RNA using statistical models and similarity-based ranking [29, 30]. RSAPred [31] employs multivariate linear regression for RNA–small molecule interaction prediction.
While existing methods have made significant progress, they face several limitations. Most approaches rely on manually crafted features based on sequence motifs, structural properties, or physicochemical characteristics, which may not fully capture the complexity and dynamic nature of RNA interactions. Structural properties and physicochemical characteristics are challenging to obtain experimentally and are not always available for all RNA molecules, limiting their applicability in large-scale or novel settings. Additionally, these methods often lack flexibility in adapting to diverse RNA sequences and their interaction partners. While a few recent studies have started exploring RNA language models to capture sequence-level representations, their application remains limited [32]. In particular, existing approaches typically focus on RNA-only modeling and do not extend to integrating language model embeddings across different interaction modalities such as proteins and small molecules. No unified framework currently exists that systematically combines language model-derived features from both RNA and its diverse interaction partners. Incorporating modality-specific language model embeddings holds the potential to provide a richer, more contextual understanding of RNA-associated interactions, ultimately enhancing predictive performance and generalizability across diverse biological tasks.
To address the limitations of existing methods, we propose BioLLMNet, a unified sequence only novel deep learning framework for predicting RNA interactions with proteins, other RNAs, and small molecules. The name BioLLMNet reflects the core idea of our approach: employing biological large language models (BioLLM) across all three interacting modalities—RNA, protein, and small molecule—combined with a unified neural network (Net) for interaction prediction. To the best of our knowledge, BioLLMNet is the first method to leverage RNA language model embeddings for a comprehensive prediction of RNA-associated interactions. By utilizing pretrained biological language models, BioLLMNet captures rich contextual and structural information embedded in RNA sequences, features that are often overlooked by traditional handcrafted representations. Crucially, our framework is not limited to RNA alone; it employs language model-derived embeddings for both RNA and its interacting modality, whether proteins, small molecules, or other RNAs, thereby facilitating a unified, modality-agnostic representation space.
To effectively integrate the heterogeneous features from different biological entities, we introduce a learnable gating mechanism that dynamically learns to weigh the embeddings from each modality and fuses them for downstream prediction. This adaptive fusion enhances the model’s ability to capture nuanced interaction patterns and ensures robustness across interaction types. Through extensive experiments on benchmark datasets, we demonstrate that BioLLMNet consistently achieves state-of-the-art performance across all three interaction categories: RNA–RNA, RNA–protein, and RNA–small molecule. These results underscore the efficacy and generalizability of our approach, setting a new standard for RNA interaction prediction.
Materials and methods
Overview of BioLLMNet
BioLLMNet first encodes biological sequences from each modality using pretrained language models specifically designed for RNA, proteins, or small molecules. These embeddings, which differ in dimensionality, are then aligned through modality-specific transformation networks to enable consistent representations. The transformed features are subsequently fused using a learnable gated mechanism (a novel mechanism that we have introduced) that adaptively balances contributions from each modality. Finally, the combined representation is passed through a task-specific prediction head to determine the likelihood of interaction.
Central to our approach is the introduction of a novel learnable gating mechanism, which learns how to weigh and fuse language model embeddings from both the RNA and its interacting partner on a per-dimension basis. This allows the model to selectively emphasize modality-specific information depending on the interaction context, resulting in a more flexible and expressive representation. The full pipeline, including transformation and gating modules, is trained end-to-end using standard backpropagation. A schematic diagram of the architecture is shown in Fig. 1.
Figure 1.

Overall architecture of BioLLMNet: (a) Language model-based embedding generation for proteins, RNAs, and small molecules, where each modality is tokenized and encoded through its respective language model to produce a fixed-dimensional feature vector, and (b) we transform the feature space size of RNA to match the feature space size of the protein or molecules and for that, we employ a single layer MLP which performs this transformation, and (c) finally, we use a gated weight to first perform a weighted average of the two sequence embeddings (which are now in the same dimension after step 2), and then pass the joint embedding through a multilayer MLP network which is essentially the prediction head and these three MLPs (transformation, gated weight, and prediction head) are learned jointly with backpropagation.
Language model embeddings for biological modalities
We begin by independently encoding the input sequences of RNAs, proteins, and small molecules using pretrained language models specialized for each type of biological modality.
RNA language model embeddings
RNA sequences, composed of nucleotides adenine (A), uracil (U), cytosine (C), and guanine (G), are embedded using RiNALMo [33], a large-scale RNA foundation model. Given an input RNA sequence  , RiNALMo produces a global feature embedding of dimension 1280. These embeddings capture structural and contextual dependencies across the RNA sequence, enabling a rich representation for downstream tasks.
, RiNALMo produces a global feature embedding of dimension 1280. These embeddings capture structural and contextual dependencies across the RNA sequence, enabling a rich representation for downstream tasks.
Protein language model embeddings
Protein sequences  are encoded using ESM2 [34], a transformer-based model trained on millions of protein sequences. The model tokenizes the input amino acid sequence and outputs contextualized embeddings of dimension 1024. The representation corresponding to the [CLS] token is used as a global summary of the protein, serving as input for interaction modeling.
are encoded using ESM2 [34], a transformer-based model trained on millions of protein sequences. The model tokenizes the input amino acid sequence and outputs contextualized embeddings of dimension 1024. The representation corresponding to the [CLS] token is used as a global summary of the protein, serving as input for interaction modeling.
Small molecule language model embeddings
For small molecules, represented by SMILES strings 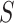 , we use MoleBERT [35] to generate molecular embeddings. MoleBERT parses the SMILES string into a molecular graph
, we use MoleBERT [35] to generate molecular embeddings. MoleBERT parses the SMILES string into a molecular graph  , where each atom
, where each atom  is encoded into a latent vector
is encoded into a latent vector  . These vectors are then quantized and embedded into a set of feature vectors
. These vectors are then quantized and embedded into a set of feature vectors  , capturing both atomic and topological features. The final molecular representation is obtained through graph-level pooling and has a dimension of 768.
, capturing both atomic and topological features. The final molecular representation is obtained through graph-level pooling and has a dimension of 768.
We select RiNALMo, ESM-2, and MoleBERT as backbone language models because they are state-of-the-art, domain-specific foundation models in their respective biological modalities. RiNALMo is the largest publicly available RNA foundation model, trained on a diverse corpus of RNA sequences and widely used for downstream RNA analysis [36, 37]. ESM-2 has demonstrated superior performance in protein structure and function prediction tasks [38, 39], and MoleBERT is a leading model for molecular property prediction with strong generalization across chemical space [40]. These models provide rich, modality-specific representations that are well suited for our unified interaction prediction framework.
Cross-modality feature transformation
The raw embeddings generated from different language models reside in vector spaces of varying dimensions (RNA: 1280, protein: 1024, and molecule: 768), making direct comparison or fusion infeasible. To address this, we project the RNA embeddings into the dimensionality of the corresponding interaction partner (protein or small molecule) using a learnable transformation.
For each interaction type, we define a single-layer fully connected neural network (MLP) that maps the RNA embedding to the desired dimension. A ReLU activation is applied to introduce nonlinearity:
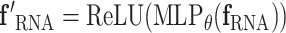 |
This transformation is modality-specific and is learned end-to-end with the rest of the network. In the case of RRIs, both embeddings are already in the same latent space (1280), so no dimensional transformation is required.

Gated fusion of modality embeddings
Once the embeddings are aligned to a common dimensional space, BioLLMNet uses a gated feature fusion mechanism to combine the representations of the RNA and the target modality. This mechanism allows the model to learn the relative importance of each modality at the feature level for each interaction instance.
Let  and
and  denote the transformed embeddings of the RNA and its interacting partner (protein, small molecule, or RNA), respectively. We introduce a learnable gate vector
denote the transformed embeddings of the RNA and its interacting partner (protein, small molecule, or RNA), respectively. We introduce a learnable gate vector  and compute the final fused representation as:
and compute the final fused representation as:
 |
where  denotes sigmoid activation and
denotes sigmoid activation and  represents element-wise multiplication. This allows the model to adaptively weigh and blend the contributions from each modality based on input-specific characteristics. A detailed step-by-step procedure for this gated fusion is described in Algorithm 1. Additionally, we provide a visualization of the learned partner contribution for each interaction type over all samples and embedding dimensions in Supplementary Section 2.
represents element-wise multiplication. This allows the model to adaptively weigh and blend the contributions from each modality based on input-specific characteristics. A detailed step-by-step procedure for this gated fusion is described in Algorithm 1. Additionally, we provide a visualization of the learned partner contribution for each interaction type over all samples and embedding dimensions in Supplementary Section 2.
Prediction head and optimization
The combined embedding  is passed through a three-layer multilayer perceptron (MLP) that serves as the prediction head. This network outputs the final interaction probability score. The entire architecture, including modality-specific transformers, transformation layers, gating mechanism, and prediction head, is trained end-to-end using standard backpropagation. Training and validation loss curves are presented in Supplementary Section 1.
is passed through a three-layer multilayer perceptron (MLP) that serves as the prediction head. This network outputs the final interaction probability score. The entire architecture, including modality-specific transformers, transformation layers, gating mechanism, and prediction head, is trained end-to-end using standard backpropagation. Training and validation loss curves are presented in Supplementary Section 1.
For binary interaction prediction tasks, we optimize the model using a binary cross-entropy loss:
 |
where  is the ground truth label and
is the ground truth label and  is the predicted probability. In multilabel or regression settings (e.g. binding affinity prediction), the loss function can be adapted accordingly.
is the predicted probability. In multilabel or regression settings (e.g. binding affinity prediction), the loss function can be adapted accordingly.
In cases where both modalities are in the RNA domain, such as prediction of miRNA–lncRNA interaction, BioLLMNet naturally extends without any architectural change. Since both sequences are encoded using RiNALMo, their embeddings lie in the same latent space. The same transformation and gated fusion mechanisms are applied, ensuring a unified handling of both cross-modality and intra-modality interaction prediction tasks.
Experiments
Dataset
To comprehensively evaluate BioLLMNet, we curated datasets from three major types of RNA-associated interactions: RNA–protein, RNA–small molecule, and RNA–RNA.
RNA–protein interactions
For RNA–protein interaction prediction, we use the RPI1460 dataset, which focuses on long noncoding RNA (lncRNA) binding with proteins. This dataset was collected from the recently published LPI-CSFFR benchmark [27], and includes 291 unique lncRNAs and 1460 unique proteins. we evaluate BioLLMNet on the RPI1460 dataset with five-fold cross validation similar to as RNAincoder [28]. It contains 1460 experimentally validated interacting lncRNA–protein pairs (positive examples) and an equal number of randomly sampled noninteracting pairs (negative examples), forming a balanced binary classification dataset with 2920 samples in total. The positive and negative data-points used in this task are extracted from LPI-CSFFR’s [27] official repository which is publicly available.
RNA–small molecule interactions
For RNA–small molecule interaction prediction, we use a dataset of binding affinity consisting of RNA-compound pairs with multiple measurements from different experimental protocols. The RNAs were manually categorized into six biologically relevant subtypes: Aptamers, miRNAs, Repeats, Ribosomal RNAs, Riboswitches, and Viral RNAs. Figure 2 summarizes the number of pairs of RNA compounds in these subtypes. Dataset is collected from the official server of ROBIN (Repository Of Binders to Nucleic acids) which are provided in a downloadable form in the RSAPred’s web server (https://web.iitm.ac.in/bioinfo2/RSAPred/Predict.html).
Figure 2.

Summary of the RNA–small molecule and RNA–RNA datasets, where the left panel shows the number of RNA–small molecule pairs stratified by RNA subtypes and the right panel shows the number of interacting and noninteracting miRNAs and lncRNAs across three species.
Following RSAPred, for the regression task, we employed a 10-fold cross-validation to predict the binding affinity values between RNA and small molecule pairs. To further assess the generalization of the model in a binary classification setting, we evaluated BioLLMNet on four additional held test sets that are publicly available on the RSAPred Web server (https://web.iitm.ac.in/bioinfo2/RSAPred/Predict.html). Following RSAPred, the predicted binding affinity values of the regression model were transformed into classification outputs by applying a threshold of 4.0, categorizing each RNA target sequence as active or inactive.
RNA–RNA interactions
To assess BioLLMNet’s generalizability in predicting RRIs, we use benchmark datasets from three different species: Arabidopsis thaliana (Ath), Glycine max (Gma), and Medicago truncatula (Mtr).
We use three benchmarking datasets for the RRI prediction task compiled by YunxiaWang et al. [21]. For evaluation, one benchmarking dataset is used as the training set, and another dataset is used for validation following the strategy used in [21] and the dataset is publicly available at CORAIN web server. Thus, we have six train-test combinations and we report performance in all these combinations. The lengths of the sequences in the miRNA dataset are 10–50, whereas the lncRNA dataset ranges from 200 to 4000. Figure 2 shows the detailed statistics.
BioLLMNet shows strong performance in RNA–protein interaction prediction
For the RNA–protein interaction prediction task, we evaluate BioLLMNet on the RPI1460 dataset and compare its performance against six competitive baseline models: RPISeq-RF [23], IMPIner [24], CFRP [25], RPITER [26], LPI-CSFFR [27], and RNAincoder [28]. As shown in Table 1, BioLLMNet achieves the highest scores across all five evaluation metrics, demonstrating its superiority in modeling RNA–protein interactions.
Table 1.
Performance comparison of different methods on RPI1460 dataset, where percentage improvement over the second best model is shown alongside and best values are shown in bold.
| Method | MCC | ACC | F1 | Precision | Recall | AUC–ROC |
|---|---|---|---|---|---|---|
| RPISeq-RF [23] | 0.570 | 0.780 | 0.780 | 0.790 | 0.780 | 0.790 |
| IMPIner [24] | 0.520 | 0.760 | 0.770 | 0.720 | 0.830 | 0.801 |
| CFRP [25] | 0.630 | 0.810 | 0.820 | 0.830 | 0.780 | 0.834 |
| RPITER [26] | 0.412 | 0.690 | 0.510 | 0.610 | 0.480 | 0.720 |
| LPI-CSFFR [27] | 0.600 | 0.830 | 0.840 | 0.780 | 0.910 | 0.820 |
| RNAincoder [28] | 0.760 | 0.880 | 0.840 | 0.810 | 0.940 | 0.915 |
| BioLLMNet |
0.848 { 11.6%} 11.6%}
|
0.923 { 4.9%} 4.9%}
|
0.925 {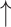 10.1%} 10.1%}
|
0.888 { 9.6%} 9.6%}
|
0.966 { 2.8%} 2.8%}
|
0.948 { 3.6%} 3.6%}
|
Specifically, BioLLMNet obtains a Matthews Correlation Coefficient (MCC) of 0.848, which represents an 11.6% improvement over the best-performing baseline (RNAincoder). In terms of accuracy, BioLLMNet achieves 0.923, improving upon RNAincoder by 4.9%. Similarly, it outperforms prior models with a 10.1% increase in F1 score, a 9.6% gain in precision, a 2.8% boost in recall, and 3.6% improvement in AUC–ROC. These improvements highlight the efficacy of leveraging language model-based representations for both RNA and proteins, as well as the effectiveness of the gated fusion mechanism employed by BioLLMNet. Overall, these results establish BioLLMNet as a new state-of-the-art method for RNA–protein interaction prediction.
BioLLMNet achieves state-of-the-art results in RNA–small molecule interaction prediction
We benchmarked BioLLMNet against two recent competitive baselines: RLaffinity [41] and RSAPred [31]. Following the evaluation protocol of RSAPred, we performed 10-fold cross-validation and report the Pearson correlation coefficient and mean absolute error (MAE) for six representative RNA subtypes: Aptamer, Repeats, Ribosomal, Riboswitch, Viral, and miRNA. For each metric and subtype, we identify the stronger of the two baselines as the reference, and compute BioLLMNet’s relative improvement over it. Table 2 presents the results. For Pearson correlation (higher is better), BioLLMNet outperforms the best baseline in five out of six subtypes, achieving gains ranging from +1.4% (Viral) to +5.1% (Ribosomal). The only case where BioLLMNet is not the top performer is miRNA, where RSAPred leads by a small margin. For MAE (lower is better), BioLLMNet establishes a clean sweep across all six subtypes, improving over the best baseline by +1.2% to +10.3%, with the largest margin again in the Ribosomal category. This consistent dominance across both metrics indicates that BioLLMNet not only achieves higher correlation with experimental measurements but also delivers systematically lower prediction errors. The improvements are particularly pronounced in structurally complex categories like Ribosomal RNAs, where the integration of pretrained language model embeddings with our gated cross-modal fusion appears to capture nuanced interaction determinants that elude existing methods. Furthermore, in a binary classification setup on four held-out test sets, regression outputs were thresholded at a binding affinity of 4.0 to produce binary interaction labels. As shown in Fig. 3, BioLLMNet achieves higher MCC scores across all RNA subtypes, notably attaining an MCC of 0.975 for Viral RNAs, while maintaining strong performance across Aptamers, miRNAs, and Riboswitches. This consistent advantage over both RSAPred and RLaffinity underscores the model’s ability to generalize effectively across diverse RNA families.
Table 2.
Comparison of BioLLMNet, RLaffinity, and RSAPred (Pearson and MAE), where improvements for BioLLMNet are computed against the best baseline in each column and best values are shown in bold.
| Metric | Method | Aptamer | Repeats | Ribosomal | Riboswitch | Viral | miRNA |
|---|---|---|---|---|---|---|---|
| Pearson | BioLLMNet |
0.7713 ( 4.2%) 4.2%) |
0.9340 ( 2.6%) 2.6%) |
0.8933 ( 5.1%) 5.1%) |
0.9462 ( 2.3%) 2.3%) |
0.8004 ( 1.4%) 1.4%) |
0.8712 |
| RLaffinity | 0.7400 | 0.9100 | 0.8500 | 0.9250 | 0.7890 | 0.8750 | |
| RSAPred [31] | 0.7180 | 0.8940 | 0.8360 | 0.9090 | 0.7840 | 0.8810 | |
| MAE | BioLLMNet |
0.5726 ( 2.9%) 2.9%) |
0.3572 ( 1.6%) 1.6%) |
0.5743 ( 10.3%) 10.3%) |
0.5236 ( 1.2%) 1.2%) |
0.5582 ( 7.1%) 7.1%) |
0.3827 ( 4.3%) 4.3%) |
| RLaffinity | 0.5900 | 0.3630 | 0.6400 | 0.5300 | 0.6010 | 0.4000 | |
| RSAPred | 0.6070 | 0.3670 | 0.6870 | 0.5370 | 0.6070 | 0.4340 |
Figure 3.

Comparison of MCC Scores on RNA–small molecule interaction.
BioLLMNet retains its superiority in transformation along same language: lncRNA–miRNA interaction prediction
The core idea of BioLLMNet is combining sequences from different modalities, requiring feature space transformation. We explore whether gated combinations work for the same modality by applying it to miRNA–lncRNA interaction prediction, where both sequences belong to the RNA domain but differ significantly in length. We use three RRI datasets for evaluation with cross-validation across six train-test combinations similar to as CORAIN [21]. miRNA sequences range from 10 to 50 nucleotides, while lncRNA sequences vary from 200 to 4000 nucleotides. As shown in Table 3, BioLLMNet achieves the highest accuracy in four out of six settings. Specifically, it matches the top performance on ATH-GMA, surpasses both baselines on GMA-ATH with a 3.0% improvement, and significantly outperforms CORAIN on MTR-ATH with a 17.2% gain. However, BioLLMNet underperforms in two cases (ATH-MTR and GMA-MTR) compared to CORAIN but maintains overall competitive performance, matching or surpassing existing methods in the majority of settings. These findings highlight the ability of BioLLMNet to generalize across species and RNA types, making it a robust and scalable framework for RRI prediction. Finally, Table 4 presents the statistical significance of BioLLMNet’s performance improvements compared to baseline methods across all RNA interaction tasks
Table 3.
Comparison of BioLLMNet, CORAIN, and PMLIPred on miRNA–lncRNA interaction datasets based on accuracy (%), where percentage improvement over the second best model is shown alongside and best values are shown in bold.

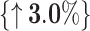


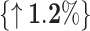
Table 4.
Statistical significance ( -values) of BioLLMNet compared to the best baseline methods across different RNA interaction tasks
-values) of BioLLMNet compared to the best baseline methods across different RNA interaction tasks
| Task | Baseline |
 -value
-value
|
|---|---|---|
| RNA–Protein | RNAincoder | .000542 |
| RNA–small Molecule | RSAPred | .007923 |
| RNA–RNA | CORAIN | .003200 |
Case study
We conducted a case study to evaluate BioLLMNet’s ability to identify RNA–protein interactions, focusing on the RNA sequence “2QEX-0,” which interacts with 37 different proteins. We selected it for the case study because it is the RNA in our dataset with the largest number of known interaction partners. This makes it a representative and informative example, as it covers a broad range of interaction patterns. The large number of partners allows us to meaningfully assess model performance across diverse interaction contexts within a single example. As shown in Fig. 4, BioLLMNet correctly predicts all interactions with proteins from the 2QEX family (29 proteins) but makes two incorrect predictions with 4GD1-G and 2WH1-Y. Upon examining the confidence scores, the incorrect predictions show significantly lower scores, demonstrating BioLLMNet’s robustness with 100% recall and 75% specificity. Confidence scores are extracted by taking the output of the sigmoid layer of the prediction head which provides the probability score for each corresponding class. To contextualize the case study results, we compared BioLLMNet with the two best-performing baselines, RNAincoder and LPI-CSFFR, using the same 2QEX-0 RNA instance. Figure 5 presents the corresponding confusion matrices, where BioLLMNet achieves the highest accuracy with 29 true positive and 6 true negative predictions, while making only 2 false positive errors and no false negatives. In contrast, RNAincoder and LPI-CSFFR show reduced recall and precision, with both models producing more false positives and false negatives. This analysis confirms that BioLLMNet not only achieves perfect recall but also maintains high specificity, resulting in the most balanced and reliable predictions for diverse interaction partners in this representative case.
Figure 4.

Case study of interactions of the 2QEX-0 RNA complex with different protein complexes, where (a) Actual interactions: 2QEX-0 interacts will all 29 proteins from the 2QEX protein family, however, it does not interact with the other eight proteins and (b) Predicted interaction result with BioLLMNet and BioLLMNet predicts the true interactions perfectly, however, it predicts two noninteracting edges as interacting edges.
Figure 5.

Confusion matrices for the 2QEX-0 case study, comparing (left) BioLLMNet, (middle) RNAincoder, and (right) LPI-CSFFR. BioLLMNet achieves the highest accuracy and perfect recall, while RNAincoder and LPI-CSFFR show increased false positives and false negatives.
Ablation study
To assess the impact of BioLLMNet’s fusion strategy, we perform an ablation study focused on how representations from RNA and its interaction partner are integrated. Specifically, we compare three fusion mechanisms:
(i) Concatenation + MLP, where the RNA and partner embeddings are simply concatenated and passed through a multilayer perceptron for prediction. This baseline does not model any interaction between the embeddings beyond what the MLP can learn. (ii) Element-wise Average, which computes a direct average of the two embeddings. This variant assumes equal importance of both modalities and removes any learnable fusion. (iii) Gated Fusion (BioLLMNet), our proposed method, where a learnable gate assigns per-dimension weights to the RNA and partner embeddings, enabling the model to dynamically emphasize relevant features depending on the input context.
As shown in Table 5, the gated fusion strategy consistently outperforms concatenation and element-wise averaging across all three interaction prediction tasks. For RNA–protein, it achieves the highest MCC (0.848), along with top accuracy (92.3%) and F1-score (92.5%), surpassing the element-wise average and concatenation. On the RNA–molecule (Aptamer) task, gated fusion attains an MCC of 0.952, accuracy of 96.4%, and F1-score of 94.8%, clearly outperforming the next best element-wise average. For RNA–RNA (MTR–ATH), gated fusion also leads with an MCC of 0.710, accuracy of 68.0%, and F1-score of 70.0%, showing consistent superiority across metrics and modalities. These results clearly demonstrate that the gated mechanism contributes significantly to performance by allowing the model to adaptively weigh modality-specific features, rather than treating them uniformly.
Table 5.
Fusion mechanism comparison on RNA–Protein, RNA–Molecule, and RNA–RNA (MCC, Accuracy, and F1-Score; mean  SD over five runs), where best values are shown in bold.
SD over five runs), where best values are shown in bold.
| Method | RNA–protein | RNA–molecule (Aptamer) | RNA–RNA (MTR–ATH) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| MCC | Acc (%) | F1 (%) | MCC | Acc (%) | F1 (%) | MCC | Acc (%) | F1 (%) | |
| Concatenation | 0.780  0.002 0.002 |
91.2  0.2 0.2 |
91.5  0.2 0.2 |
0.870  0.001 0.001 |
94.7  0.1 0.1 |
93.5  0.1 0.1 |
0.690  0.003 0.003 |
66.1  0.3 0.3 |
68.2  0.3 0.3 |
| Element-wise Avg | 0.800  0.001 0.001 |
91.9  0.2 0.2 |
92.0  0.2 0.2 |
0.890  0.002 0.002 |
95.2  0.1 0.1 |
93.9  0.1 0.1 |
0.702  0.002 0.002 |
67.4  0.2 0.2 |
69.1  0.2 0.2 |
| Gated (BioLLMNet) |
0.848  0.001 0.001 |
92.3  0.1 0.1 |
92.5  0.1 0.1 |
0.952  0.001 0.001 |
96.4  0.1 0.1 |
94.8  0.1 0.1 |
0.710  0.002 0.002 |
68.0  0.2 0.2 |
70.0  0.2 0.2 |
Conclusion
In this work, we presented BioLLMNet, a unified sequence only deep learning framework that leverages pretrained biological language models for RNA interaction prediction across three major modalities: proteins, small molecules, and other RNAs. Importantly, our framework extends beyond RNA alone, incorporating language model-derived embeddings for all interacting partners (proteins, small molecules, and RNAs) within a unified, modality-independent representation space. Unlike prior approaches that typically focus on a single interaction type and rely on handcrafted features, structural properties, or physicochemical characteristics, BioLLMNet utilizes only the sequences and their learned embeddings from RNA, protein, and molecular language models, offering a scalable and generalizable solution. Through a modality-agnostic transformation and a learnable gating mechanism, BioLLMNet adaptively fuses heterogeneous embeddings, leading to consistent improvements in predictive performance across RNA–RNA, RNA–protein, and RNA–small molecule benchmarks.
Our experiments demonstrate that BioLLMNet not only outperforms existing methods across all evaluated tasks, but also remains robust across species, RNA subtypes, and interaction scenarios. Rigorous ablation study also validates our proposed approach. These results establish the viability of using cross-modality language model representations as a foundation for RNA interaction modeling. We note that existing domain-specific tool and classical algorithms for RNA interactions [42, 43] may provide complementary perspectives, which is yet to be tested, and the use of BioLLMNet alongside the domain-specific tools could be valuable for addressing specific questions or focused applications. While the framework supports heterogeneous modalities, it does not yet exploit cross-modal attention or joint multimodal pretraining, which could further enhance interpretability and performance. We leave this for the future iteration of this work. Furthermore, future extensions could adopt strategies akin to LLAVA [44], where joint embeddings from natural language and visual modalities are learned, enabling the seamless incorporation of diverse feature types such as 3D molecular structures and other high-dimensional bio-sequence features.
Key Points
BioLLMNet is the first sequence-only method to unify RNA–RNA, RNA–protein, and RNA–small molecule interaction prediction within a single architecture.
Our method leverages pretrained biological language models for all modalities, capturing rich contextual information without requiring handcrafted features or structural data.
We introduce a novel learnable gating mechanism that adaptively weighs and fuses heterogeneous embeddings from different biological modalities.
BioLLMNet consistently outperforms existing methods across benchmark datasets in all three interaction types, demonstrating strong generalizability and robustness.
Supplementary Material
Contributor Information
Abrar Rahman Abir, Department of Computer Science and Engineering, Bangladesh University of Engineering and Technology, Dhaka 1000, Bangladesh.
Md Toki Tahmid, Department of Computer Science and Engineering, Bangladesh University of Engineering and Technology, Dhaka 1000, Bangladesh.
Md Shamsuzzoha Bayzid, Department of Computer Science and Engineering, Bangladesh University of Engineering and Technology, Dhaka 1000, Bangladesh.
Conflict of interest: None declared.
Funding
None declared.
Data availability
All datasets used in this study can be found at https://drive.google.com/drive/folders/1qDX5u_5BgptB0Ah5o4-uEF91oSV4-7ci?usp=sharing. Source code is available at https://github.com/abrarrahmanabir/BioLLMNet.git.
References
- 1. Palcau AC, Canu V, Donzelli S. et al. CircPVT1: a pivotal circular node intersecting long non-coding-PVT1 and c-MYC oncogenic signals. Mol Cancer 2022; 21:33. 10.1186/s12943-022-01514-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mou X, Liew SW, Kwok CK. Identification and targeting of g-quadruplex structures in MALAT1 long non-coding. Nucleic Acids Res 2022; 50:397–410. 10.1093/nar/gkab1208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chen LL. The expanding regulatory mechanisms and cellular functions of circular RNAs. Nat Rev Mol Cell Biol 2020; 21:475–90. 10.1038/s41580-020-0243-y [DOI] [PubMed] [Google Scholar]
- 4. Goodall GJ, Wickramasinghe VO. RNA in cancer. Nat Rev Cancer 2021; 21:22–36. 10.1038/s41568-020-00306-0 [DOI] [PubMed] [Google Scholar]
- 5. Ramanathan M, Porter DF, Khavari PA. Methods to study RNA–protein interactions. Nat Methods 2019; 16:225–34. 10.1038/s41592-019-0330-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhang Z, Sun W, Shi T. et al. Capturing RNA–protein interaction via CRUIS. Nucleic Acids Res 2020; 48:e52. 10.1093/nar/gkaa143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gainza P, Sverrisson F, Monti F. et al. Deciphering interaction fingerprints from protein molecular surfaces using geometric deep learning. Nat Methods 2020; 17:184–92. 10.1038/s41592-019-0666-6 [DOI] [PubMed] [Google Scholar]
- 8. Zhang S, Amahong K, Sun X. et al. The miRNA: a small but powerful RNA for covid-19. Brief Bioinform 2021; 22:1137–49. 10.1093/bib/bbab062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Van Treeck B, Parker R. Emerging roles for intermolecular RNA–RNA interactions in RNP assemblies. Cell 2018; 174:791–802. 10.1016/j.cell.2018.07.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kang Q, Meng J, Cui J. et al. PmliPred: a method based on hybrid model and fuzzy decision for plant miRNA-lncRNA interaction prediction. Bioinformatics 2020; 36:2986–92. 10.1093/bioinformatics/btaa074 [DOI] [PubMed] [Google Scholar]
- 11. Weidmann CA, Mustoe AM, Jariwala PB. et al. Analysis of RNA–protein networks with RNP-map defines functional hubs on RNA. Nat Biotechnol 2021; 39:347–56. 10.1038/s41587-020-0709-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bellucci M, Agostini F, Masin M. et al. Predicting protein associations with long noncoding RNAs. Nat Methods 2011; 8:444–5. 10.1038/nmeth.1611 [DOI] [PubMed] [Google Scholar]
- 13. Lanjanian H, Nematzadeh S, Hosseini S. et al. High-throughput analysis of the interactions between viral proteins and host cell RNAs. Comput Biol Med 2021; 135:104611. 10.1016/j.compbiomed.2021.104611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Duan N, Arroyo M, Deng W. et al. Visualization and characterization of RNA–protein interactions in living cells. Nucleic Acids Res 2021; 49:e107. 10.1093/nar/gkab614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dang CV, Reddy EP, Shokat KM. et al. Drugging the ‘undruggable’ cancer targets. Nat Rev Cancer 2017; 17:502–8. 10.1038/nrc.2017.36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Llombart V, Mansour MR. Therapeutic targeting of “undruggable” myc. EBioMedicine 2022; 75:103756. 10.1016/j.ebiom.2021.103756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mahmud SMH, Chen W, Liu Y. et al. Predtis: prediction of drug-target interactions based on multiple feature information using gradient boosting framework with data balancing and feature selection techniques. Brief Bioinform 2021; 22:bbab046. 10.1093/bib/bbab046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hwang B, Lee JH, Bang D. Single-cell RNA sequencing technologies and bioinformatics pipelines. Exp Mol Med 2018; 50:1–14. 10.1038/s12276-018-0071-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Song J, Tian S, Yu L. et al. MD-MLI: prediction of miRNA-lncRNA interaction by using multiple features and hierarchical deep learning. IEEE/ACM Trans Comput Biol Bioinform 2020; 19:1724–33. [DOI] [PubMed] [Google Scholar]
- 20. Zhang Y, Jia C, Kwoh CK. Predicting the interaction biomolecule types for lncrna: an ensemble deep learning approach. Brief Bioinform 2021; 22:bbaa228. [DOI] [PubMed] [Google Scholar]
- 21. Wang Y, Pan Z, Mou M. et al. A task-specific encoding algorithm for RNAs and RNA-associated interactions based on convolutional autoencoder. Nucleic Acids Res 2023; 51:e110–0. 10.1093/nar/gkad929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhou H, Wekesa JS, Luan Y. et al. PRPI-SC: an ensemble deep learning model for predicting plant lncRNA–protein interactions. BMC Bioinform 2021; 22:415. 10.1186/s12859-021-04328-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Muppirala UK, Honavar VG, Dobbs D. Predicting RNA-protein interactions using only sequence information. BMC Bioinform 2011; 12:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pan X, Yang Y, Xia C-Q. et al. Recent methodology progress of deep learning for RNA–protein interaction prediction. Wiley Interdisc Rev RNA 2019; 10:e1544. 10.1002/wrna.1544 [DOI] [PubMed] [Google Scholar]
- 25. Dai Q, Guo M, Duan X. et al. Construction of complex features for computational predicting ncRNA-protein interaction. Front Genet 2019; 10:18. 10.3389/fgene.2019.00018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Peng C, Han S, Zhang H. et al. RPITER: a hierarchical deep learning framework for ncRNA–protein interaction prediction. Int J Mol Sci 2019; 20:1070. 10.3390/ijms20051070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Huang X, Shi Y, Yan J. et al. LPI-CSFFR: combining serial fusion with feature reuse for predicting lncRNA-protein interactions. Comput Biol Chem 2022; 99:107718. 10.1016/j.compbiolchem.2022.107718 [DOI] [PubMed] [Google Scholar]
- 28. Wang Y, Chen Z, Pan Z. et al. RNAincoder: a deep learning-based encoder for RNA and RNA-associated interaction. Nucleic Acids Res 2023; 51:W509–19. 10.1093/nar/gkad404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Philips A, Milanowska K, Lach G. et al. LigandRNA: computational predictor of RNA-ligand interactions. RNA 2013; 19:1605–16. 10.1261/rna.039834.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Etzion-Fuchs A, Todd DA, Singh M. dSPRINT: predicting DNA, RNA, ion, peptide and small molecule interaction sites within protein domains. Nucleic Acids Res 2021; 49:e78. 10.1093/nar/gkab356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Krishnan SR, Roy A, Michael M. et al. Reliable method for predicting the binding affinity of RNA-small molecule interactions using machine learning. Brief Bioinform 2024; 25:bbae002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Liu H, Jian Y, Zeng C. et al. RNA-protein interaction prediction using network-guided deep learning. Communications Biology 2025; 8:247. 10.1038/s42003-025-07694-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Penić RJ, Vlašić T, Huber RG. et al. Rinalmo: General-purpose rna language models can generalize well on structure prediction tasks. Nature Communications,2025;16:5671. Nature Publishing Group UK London. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lin Z, Akin H, Rao R. et al. Language models of protein sequences at the scale of evolution enable accurate structure prediction. BioRxiv 2022, preprint: not peer reviewed.:500902 (https://www.biorxiv.org/content/10.1101/2022.07.20.500902v1). [Google Scholar]
- 35. Xia J, Zhao C, Bozhen H. et al. Mole-bert: rethinking pre-training graph neural networks for molecules. The Eleventh International Conference on Learning Representations, 2023, https://openreview.net/forum?id=jevY-DtiZTR.
- 36. Abir AR, Tahmid MT, Rahman MS. LOCAS: multi-label mRNA localization with supervised contrastive learning. Briefings in Bioinformatics, 2025;26:bbaf441. Oxford University Press. [Google Scholar]
- 37. Abir AR, Tahmid MT, Rayan RI. et al. DeepRNA-twist: language-model-guided RNA torsion angle prediction with attention-inception network. Brief Bioinform 2025; 26:bbaf199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ahmed KT, Ansari MI, Zhang W. DTI-LM: language model powered drug–target interaction prediction. Bioinformatics 2024; 40:btae533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cordoves-Delgado G, Garcia-Jacas CR. Predicting antimicrobial peptides using ESMfold-predicted structures and ESM-2-based amino acid features with graph deep learning. J Chem Inf Model 2024; 64:4310–21. 10.1021/acs.jcim.3c02061 [DOI] [PubMed] [Google Scholar]
- 40. Sultan A, Rausch-Dupont M, Khan S. et al. Transformers for molecular property prediction: domain adaptation efficiently improves performance. arXiv, arXiv:2503.03360. 2025, preprint: not peer reviewed (https://arxiv.org/html/2503.03360v3).
- 41. Sun S, Gao L. Contrastive pre-training and 3d convolution neural network for RNA and small molecule binding affinity prediction. Bioinformatics 2024; 40:btae155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Armaos A, Colantoni A, Proietti G. et al. cat rapid omics v2. 0: going deeper and wider in the prediction of protein–RNA interactions. Nucleic Acids Res 2021; 49:W72–9. 10.1093/nar/gkab393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. John B, Enright AJ, Aravin A. et al. Human microRNA targets. PLoS Biol 2004; 2:e363. 10.1371/journal.pbio.0020363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Liu H, Li C, Qingyang W. et al. Visual instruction tuning. Adv Neural Inform Process Syst 2023; 36:34892–916. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All datasets used in this study can be found at https://drive.google.com/drive/folders/1qDX5u_5BgptB0Ah5o4-uEF91oSV4-7ci?usp=sharing. Source code is available at https://github.com/abrarrahmanabir/BioLLMNet.git.