

Abstract
Hepatitis C virus (HCV) is an important human pathogen that affects ∼100 million people worldwide. Its RNA genome codes for a polyprotein, which is cleaved by viral and cellular proteases to produce at least 10 mature viral protein products. We report here the discovery of a novel HCV protein synthesized by ribosomal frameshift. This protein, which we named the F protein, is synthesized from the initiation codon of the polyprotein sequence followed by ribosomal frameshift into the –2/+1 reading frame. This ribosomal frameshift requires only codons 8–14 of the core protein-coding sequence, and the shift junction is located at or near codon 11. An F protein analog synthesized in vitro reacted with the sera of HCV patients but not with the sera of hepatitis B patients, indicating the expression of the F protein during natural HCV infection. This unexpected finding may open new avenues for the development of anti-HCV drugs.
Keywords: HCV core protein/HCV F protein/hepatitis C virus/ribosomal frameshift
Introduction
Hepatitis C virus (HCV) is a positive-stranded RNA virus with a genome size of ∼9.6 kb. Infection by this virus frequently leads to chronic infection, which in turn may lead to severe liver diseases including liver cirrhosis and hepatocellular carcinoma. The first HCV genomic sequence, which was named the HCV-1 sequence, was isolated in 1989 (Choo et al., 1989). Since then, many more HCV sequences have been reported. Based on their sequence similarities, these HCV sequences have been grouped into six major types and many more subtypes (Smith and Simmonds, 1998).
The HCV genomic RNA contains a long open reading frame (ORF) that encodes a polyprotein slightly larger than 3000 amino acids. This polyprotein is proteolytically cleaved by viral and cellular proteases to generate at least 10 gene products. The core protein (p21c) is 191 amino acids in length and is located at the N-terminus of the polyprotein sequence. This protein packages the viral RNA. Its sequence in the HCV polyprotein sequence is followed by E1 and E2 envelope proteins, which are then followed by non-structural proteins. The translation of the HCV polyprotein sequence is regulated by a cap-independent mechanism that requires most of the 5′-non-coding region and the first nine codons of the polyprotein-coding sequence to serve as the internal ribosomal entry site (for a review see Rijnbrand and Lemmon, 2000).
Previous expression studies indicate that, besides the core protein, a 17 kDa protein is also expressed from the core protein-coding sequence of the HCV genomic RNA both in vitro and in mammalian cells (Lo et al., 1994, 1995; Ray et al., 1996). This expression is independent of the downstream E1 envelope protein sequence (Lo et al., 1994, 1995, 1996). The identity of this 17 kDa protein was unclear, and it was thought to be a truncated core protein. In this report, we demonstrate that this 17 kDa protein is synthesized by ribosomal frameshift and is derived mostly from the coding sequence that overlaps the core protein reading frame. This protein is highly conserved among different HCV isolates and is expressed during natural HCV infection, indicating that it may play an important role in the HCV life cycle.
Results
Synthesis of a 17 kDa protein from a coding sequence that overlaps the core protein reading frame
An inspection of the HCV-1 core protein-coding sequence revealed an overlapping coding sequence in the –2/+1 reading frame (Figure 1). This overlapping ORF, which lacks an AUG codon near its 5′ end, spans from nucleotide 5 to nucleotide 485 and has a coding capacity of ∼160 amino acids. To investigate whether the 17 kDa protein is derived from this overlapping ORF, nucleotide 432 of the HCV-1 sequence coding for the core protein was converted from U to A to create a premature termination codon in the overlapping ORF (Figure 1). This mutation removed the last 18 codons of the overlapping ORF without affecting the core protein-coding sequence. If the 17 kDa protein is derived from this overlapping ORF, then this mutation should reduce the size of the 17 kDa protein by ∼2 kDa without affecting the core protein. As shown in Figure 2A, the translation of the wild-type HCV-1 core protein RNA in vitro using rabbit reticulocyte lysates generated the 21 kDa core protein and the 17 kDa protein. In contrast, the translation of the HCV-1 RNA containing the U to A mutation at nucleotide 432 generated p21c and a smaller 15 kDa protein. This result strongly indicates that the 17 kDa protein is derived from the overlapping ORF.

Fig. 1. The HCV core protein sequence and its overlapping coding sequence. The amino acid sequences, shown by one-letter code, were deduced from the sequence of the HCV-1 isolate (Choo et al., 1991). The sequence shown ends at the C-terminus of the core protein-coding sequence. The amino acid sequence encoded by the overlapping coding sequence is shown in bold. Codons 8–14 of the core protein sequence are underlined and nucleotide 432 is boxed.
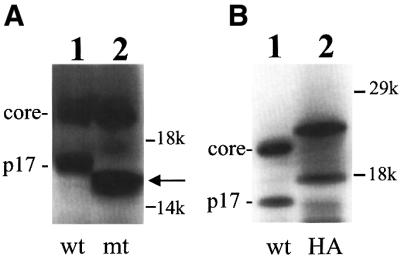
Fig. 2. Analysis of translational termination and initiation sites of the 17 kDa protein. (A) Termination of the 17 kDa protein sequence in the overlapping coding sequence. pCMV-CC contained the wild-type core protein-coding sequence (lane 1), and pCMV-CCmt contained the core protein-coding sequence with a premature termination codon in the alternative ORF (lane 2). The HCV sequences in these two DNA constructs were under the control of the T7 promoter and the immediate early promoter of cytomegalovirus. The RNA was synthesized using the T7 RNA polymerase and translated using the rabbit reticulocyte lysates or wheat germ extracts. The locations of p21c (core) and the 17 kDa (p17) protein bands are marked. The arrow denotes the location of the truncated 17 kDa protein. The locations of the molecular weight markers are also shown. (B) Translation initiation of the 17 kDa protein from the core protein initiation codon. Lane 1, translation of the wild-type HCV core protein-coding sequence; lane 2, translation of the core protein sequence that had been fused to the HA tag.
Translation initiation of the 17 kDa protein from the initiation codon of the core protein
Although the overlapping ORF contains three AUG codons, none of them resides near the 5′ end of its coding sequence. Thus, the 17 kDa protein must be synthesized by initiation from a non-AUG codon or by frameshift after translation initiation from the AUG codon of the core protein sequence. To distinguish between these two possibilities, the coding sequence of the HA tag, a sequence derived from the influenza virus hemagglutinin gene, was fused in-frame to the 5′ end of the core protein-coding sequence. If the 17 kDa protein is synthesized from a non-AUG codon located near the 5′ end of the core protein-coding sequence, then its synthesis should not be affected by the presence of the HA tag. On the contrary, if the 17 kDa protein is synthesized from the AUG codon of the core protein-coding sequence, then the HA tag should increase its size correspondingly. As shown in Figure 2B, the HA tag increased the size of both p21c and the 17 kDa protein by a similar amount. The presence of the HA tag in both p21c and the 17 kDa protein was verified by immunoprecipitation using an anti-HA antibody (data not shown). Thus, the results shown in Figure 2 indicate that the 17 kDa protein is synthesized from the initiation codon of the core protein sequence and terminates in the overlapping ORF.
Ribosomal frameshift for the synthesis of the 17 kDa protein
The results shown in Figure 2 suggest a scenario of ribosomal frameshift for the synthesis of the 17 kDa protein. For convenience, we have named this protein ‘F’ protein for ‘frameshifting’. Since our previous results indicate that a mutation in codon 9 of the HCV-1 core protein-coding sequence significantly reduces the efficiency of F protein expression (Lo et al., 1994, 1995), the ribosomal frameshift for the synthesis of the F protein probably occurs in the vicinity of this codon. To investigate this possibility, we performed radiosequencing of p21c and F protein, which were synthesized in vitro in the presence of [35S]methionine and [3H]lysine. Few [35S]methionine counts were detected in the first sequencing cycle whether p21c or the F protein was sequenced (Figure 3A and B; see also below), indicating the absence of the initiator methionine residue, presumably caused by the N-terminal peptidase activity in the translation extracts. As shown in Figure 3A, p21c generated three distinct [3H]lysine peaks at sequencing cycles 5, 9 and 11. There are four lysine residues at amino acids 6, 9, 10 and 12 of the p21c sequence (including the initiating methionine as residue 1). Since Lys9 and Lys10 were not expected to be resolved into separate peaks in the sequencing reaction, the p21c sequencing result was in agreement with the predicted p21c sequence excluding the initiating methionine residue. The sequencing of the F protein generated [3H]lysine peaks resembling those generated from the sequencing of p21c, except that the peak at cycle 11 had largely disappeared (Figure 3B and C). Since the [3H]lysine peak detected at cycle 9 was not affected, this result indicates that the sequences of p21c and the F protein diverge at or in the vicinity of codon 11, which thus predicts the location of the ribosomal frameshift.



Fig. 3. Radiosequencing of p21c (A) and the F protein (B). A comparison of the [3H]lysine sequencing results of p21c and the F protein is shown in (C). The amino acid sequences of p21c and a portion of the F protein are aligned at the top of the sequencing cycles. The initiating methionine residue is shown in parentheses and the lysine residues are shown in bold letters. The arrow in (C) indicates the disappearance of the peak at cycle 11 when the F protein was sequenced. p21c and the F protein were synthesized using RNA derived from pCMV-CC and radiolabeled with [35S]methionine and [3H]lysine. These two proteins were then gel purified and subjected to radiosequencing as described in Materials and methods.
An A-rich sequence is located at codons 8–14 of the HCV core protein sequence. This sequence may mediate the ribosomal frameshift. To investigate the possible role of these codons in ribosomal frameshift, codons 2–7 and 15–50, which flank the putative ribosomal frameshift site, were deleted from the core protein-coding sequence. As shown in lane 2 of Figure 4A, the deletion of these flanking sequences resulted in the synthesis of two smaller proteins, which were presumably the deletion mutants of p21c and the F protein. These two proteins were then labeled with [3H]threonine and [35S]methionine and subjected to radiosequencing. As shown in Figure 4B and C, a strong [35S]methionine signal was detected in the first sequencing cycle when either of these two proteins was sequenced. This result indicates that loss of the deleted sequences prevented the cleavage of the initiator methionine from these two proteins. A single [3H]threonine peak was detected at sequencing cycle 10 when the larger protein was sequenced (Figure 4B). This result is consistent with the predicted sequence of the p21c deletion mutant, which contains a threonine residue at position 10 (Figure 4B). The sequencing of the smaller protein generated two [3H]threonine peaks at sequencing cycles 6 and 9 (Figure 4C). The peaks in this case were broader than that seen in Figure 4B, suggesting a possible sequence heterogeneity. As mentioned above, the ribosomal frameshift site is predicted at or in the vicinity of codon 11 (Figure 3). A +1 ribosomal frameshift at codon 9, 10 or 11 would generate the sequence MQKKTNVTR, and at codon 12 would generate the sequence MQKKNNVTR at the N-terminus of the F protein mutant. The latter scenario does not appear likely since it would generate only one threonine residue at amino acid 8. In contrast, a –2 ribosomal frameshift at codons 9, 10 or 11 would generate the sequence MQKKKTNVTR, and at codon 12 would generate the sequence MQKKNTNVTR, for the F protein mutant. Both sequences contain two threonine residues at amino acids 6 and 9. Thus, the [3H]threonine sequencing result is consistent with the F protein being generated by a –2 ribosomal frameshift. However, due to the possible sequence heterogeneity observed in Figure 4B, some F protein may also be generated by +1 ribosomal frameshift.
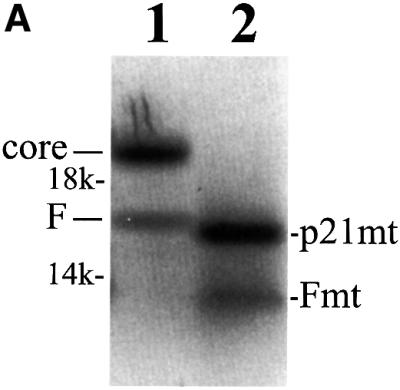


Fig. 4. Expression and radiosequencing of p21c and the F protein deletion mutants. (A) Expression of the deletion mutants of p21c and the F protein. Lane 1, the translation of the wild-type core protein sequence; lane 2, the translation of the core protein sequence with the deletions of codons 2–7 and 15–50. (B) Radiosequencing of the p21c deletion mutant that had been labeled with [3H]threonine and [35S]methionine. The predicted sequence of the p21c mutant is aligned at the top of the sequencing cycles. Codons 8–14 are underlined and the sequence following these codons represents the p21c sequence starting from codon 51. (C) Radiosequencing of the F deletion mutant that had been labeled with [3H]threonine and [35S]methionine. The translation and the radiosequencing were conducted as described in Materials and methods. The sequence of the F protein mutant, predicted based on a –2 ribosomal frameshift, is also aligned at the top of the sequencing cycles.
Besides ribosomal frameshift, it is also possible that the production of the F protein was caused by the deletion of one nucleotide or the insertion of two nucleotides at or near codon 11 during RNA synthesis by the T7 RNA polymerase. There is a stretch of 10 As at codons 8–11 of the core protein sequence (Figure 1). The slippage of T7 RNA polymerase at this region during transcription could fuse the F protein-coding sequence to the core protein-coding sequence. To rule out this possibility, we have used the enzymatic method to sequence directly a T7 transcript containing codons 8–14 of the HCV sequence. As shown in Figure 5, no apparent nucleotide deletion or insertion that would lead to the synthesis of the F protein was visible in this HCV sequence, indicating that the F protein was not likely to have been generated as a result of the sequence heterogeneity of the T7 RNA transcript.
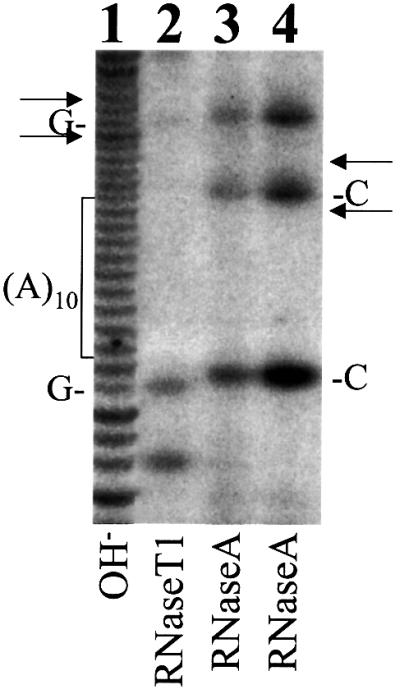
Fig. 5. Enzymatic sequencing of the T7 transcript containing codons 8–14 of the HCV sequence. Lane 1, partial alkaline (OH–) hydrolysis of the HCV RNA; lane 2, partial digestion with RNase T1, which cuts after G; lanes 3 and 4, partial digestion with RNase A, which cuts after U and C. The locations of the 10 A stretch and its two flanking C residues and G residues are indicated. Arrows indicate the locations of RNase T1 and RNase A bands that would be produced if there was a one nucleotide deletion or a two nucleotide insertion in the 10 A stretch. Note that there was no apparent sequence heterogeneity flanking the 10 A stretch.
Regulation of ribosomal frameshift by codons 1–14 of the core protein-coding sequence in Huh7 cells
The results shown in Figure 4 indicate that codons 8–14 of the core protein sequence were sufficient to mediate ribosomal frameshift. To investigate further whether this sequence can also mediate ribosomal frameshift in liver cells, we have fused the first 14 codons of the core protein sequence to the zero frame, the –2/+1 frame and the –1/+2 frame of the luciferase-coding sequence. The expression of the fused sequence was under the control of the EF1α gene promoter. These DNA constructs, named FS(0)Luc, FS(–2/+1) and FS(–1/+2), are illustrated in Figure 6A. While the FS(0)Luc construct is expected to express the luciferase reporter as a fusion protein to the core sequence, FS(–2/+1)Luc can express the luciferase sequence only by –2/+1 ribosomal frameshift. The FS(–1/+2)Luc construct cannot express the luciferase sequence with or without ribosomal frameshift due to the presence of a termination codon in the –1/+2 reading frame immediately upstream of the luciferase reporter sequence. This construct serves as a negative control. These DNA plasmids were then transfected into Huh7 cells, a well differentiated human hepatoma cell line. A human growth hormone (hGH) reporter plasmid was also co-transfected to serve as an internal control for monitoring the transfection efficiency. As shown in Figure 6B, 1–2% luciferase activity was expressed by the FS(–2/+1)Luc construct relative to that expressed by the FS(0)Luc construct. In contrast, the FS(–1/+2)Luc negative control produced ∼0.05% luciferase activity, which was near the background level. This result supports our observation that codons 1–14 of the core protein sequence can indeed mediate the –2/+1 ribosomal frameshift in Huh7 cells. This 1–2% efficiency of ribosomal frameshift is not unusual as it is similar to that which was reported previously for a number of genes (Grentzmann et al., 1998; Ivanov et al., 2000).
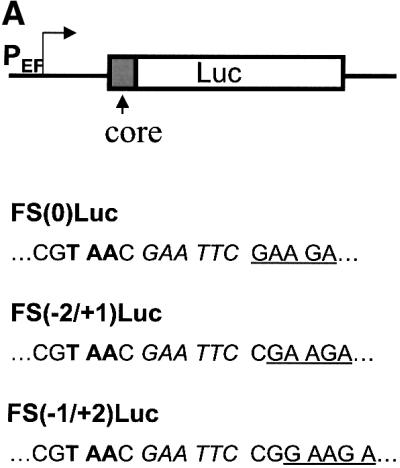
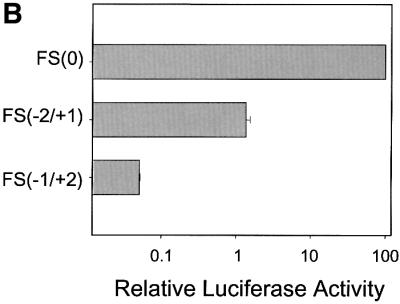
Fig. 6. Analysis of the ribosomal frameshift efficiency in Huh7 cells. (A) Schematic illustration of the luciferase reporter constructs. PEF indicates the EF1α promoter, Luc indicates the luciferase reporter, and core indicates codons 1–14 of the core protein sequence. Sequences located at the fusion junction of the core protein and the luciferase reporter (underlined) are also shown. Italic letters indicate the EcoRI site that was used to fuse the two sequences. Bold letters denote the termination codon located in the –1/+2 reading frame. (B) The luciferase reporter assay. Huh7 cells were transfected with various luciferase reporter constructs, and the relative frameshift efficiencies were determined by the procedures described in Materials and methods. The experiments were carried out in triplicate and repeated at least twice. Data represent the average of the results.
Expression of the F protein during natural HCV infection
Although the F protein is produced by the HCV-1 sequence in vitro and in transfected mammalian cells (Lo et al., 1994, 1995, 1996; Ray et al., 1996), whether it can be synthesized during natural HCV infection in patients was unclear. For that reason, we decided to look for F protein-reactive antibodies in HCV patients. As mentioned above, a stretch of 10 As is located between codons 8 and 11 of the core protein-coding sequence. A single A nucleotide was deleted from this stretch to generate an F protein analog composed of the first 10 codons of the core protein sequence fused to the overlapping ORF. This F protein analog was synthesized in the presence of [35S]methionine in vitro, and immunoprecipitated with the sera isolated from six HCV patients and five hepatitis B virus (HBV) patients. As shown in Figure 7A, while none of the HBV sera immunoprecipitated the F protein analog, five of the six HCV sera (i.e. from patients 1, 4, 7, 10 and 11) clearly reacted with the F protein, while the sixth (from patient 3) showed weak reactivity. Since the F protein analog contains a 10 residue sequence from the core protein, it was possible that the immunoreactivity seen in Figure 7A was due to the presence of anti-core antibodies that recognized these 10 residues. To rule out this possibility, an AUG initiation codon was generated at the beginning of the overlapping ORF for the expression of a truncated F protein lacking the preceding core protein sequence. This coding sequence was then transcribed and translated in vitro in the presence of [35S]methionine. The radiolabeled truncated F protein was immunoprecipitated with the same panel of patient sera. As shown in Figure 7B, weak but nevertheless detectable signals of the truncated F protein could be precipitated by the sera of all of the HCV patients except patient number 1. In a separate immunoprecipitation experiment, the serum of patient number 1 was also found to react weakly with the truncated F protein (data not shown). In contrast, all of the HBV sera generated no significant signals. To examine further the prevalence of anti-F antibodies in different HCV patients, we have also performed an enzymatic immunoassay on eight other HCV patients whose HCV genotypes are known. Two non-HCV serum samples were also tested to serve as negative controls. As shown in Table I, five of the eight HCV patients showed reactivities to the truncated F protein. Note that patients 6 and 8 displayed a negative response to the F protein prior to seroconversion. Thus, the results shown in Figure 7 and Table I strongly indicate that the F protein is produced during natural HCV infection in patients. Moreover, the results shown in Table I indicate that the expression of the F protein is not limited to genotype 1a.

Fig. 7. Analysis of F protein-reactive antibodies in patients. (A) Immunoprecipitation of the F protein analog. The F RNA was synthesized from pGEM-core9a with T7 RNA polymerase and translated using rabbit reticulocyte lysates. The protein was radiolabeled with [35S]methionine. A 10 µl aliquot of the translational mixture was then incubated with 10 µl of the sera isolated from HCV or HBV patients for radioimmunoprecipitation analysis. ‘+’ and ‘–’ indicate HCV and HBV sera, respectively. (B) Immunoprecipitation of the truncated F protein. The truncated F protein was radiolabeled with [35S]methionine and radioimmunoprecipitated as in (A). The protein without immunoprecipitation was run in parallel in lane M to serve as a marker.
Table I. EIA of the antibodies that react with the truncated F proteina.
| Patient no. | Before seroconversionb | After seroconversionb | Genotype |
|---|---|---|---|
| 1 | 0.523c | 1a | |
| 2 | 0.499c | 1a | |
| 3 | 0.139c | 4 | |
| 4 | 0.043 | 6a | |
| 5 | 0.009 | 0.014 | 3a |
| 6 | 0.024 | 0.555c | 2b/3 |
| 7 | 0.020 | 0.012 | 2b |
| 8 | 0.038 | 0.167c | 1b |
| NC | 0.036 | ||
| NC | 0.019 |
aPatients 1–8 were determined to be HCV positive based on the Chiron c22 assay. NC indicates the negative control serum samples.
bDetails of the serological assay are described in Materials and methods. The OD was read at 492 nm. The serum samples of patients 5–8 collected prior to seroconversion were also tested.
cThese samples have a P/N ratio of >3 and are considered positive for the antibodies that react with the F protein.
Conservation of the F protein-coding sequence in the genomes of different HCV isolates
The results shown in Table I indicate that the F protein is conserved in different HCV genotypes. A survey of the HCV sequences compiled in the database indicates that the F protein ORF, which has a length of 162 codons, is conserved in 100% of the HCV genotype 1a sequences (Table II). Shorter forms ranging from 126 to 155 codons in length are also conserved in the great majority of the other HCV sequences reported. As shown in Table II, most HCV genomes of genotypes 1b and 2a have the capacity to code for the F protein with a length of 144 and 126 residues, respectively. Thus, the length of the F protein appears to be genotype specific. It is noteworthy that all of the infectious HCV clones that have been generated so far contain the intact F protein ORF (Kalykhalov et al., 1997; Yanagi et al., 1997, 1998, 1999; Beard et al., 1999; Lohmann et al., 1999), with genotype 1a encoding a 162 residue F protein (Kalykhalov et al., 1997; Yanagi et al., 1997), genotype 1b encoding a 144 residue F protein (Yanagi et al., 1998; Beard et al., 1999; Lohmann et al., 1999), and genotype 2a encoding a 126 residue F protein (Yanagi et al., 1999).
Table II. The length of the F protein encoded by the genomes of different HCV genotypesa,b.
| Genotypes | 126 aac | 131 aac | 140 aac | 144 aac | 155 aac | 162 aac | Othersd |
|---|---|---|---|---|---|---|---|
| 1a [16] | 100% [16] | ||||||
| 1b [102] | 1% [1] | 3% [3] | 80% [82] | 7% [7] | 9% [9] | ||
| 2a [24] | 80% [19] | 4% [1] | 8% [2] | 8% [2] | |||
| 2b [7] | 71% [5] | 29% [2] | |||||
| 3a [11] | 36% [4] | 46% [5] | 18% [2] | ||||
| 3b [12] | 42% [5] | 50% [6] | 8% [1] | ||||
| 4 [24] | 58% [14] | 13% [3] | 8% [2] | 17% [4] | 4% [1] | ||
| 5 [2] | 50% [1] | 50% [1] | |||||
| 6 [10] | 30% [3] | 30% [3] | 40% [4] |
aA total of 235 HCV core protein-coding sequences compiled in the http://s2as02.genes.nig.ac.jp website were analyzed. The genotyping was conducted using the procedures of Ohno and Mizokami (1998). Several of the HCV sequences could be grouped into more than one genotype. For these sequences, their grouping was arbitrary. Twenty-seven HCV sequences could not be genotyped by the procedures of Ohno and Mizokami, and were not considered.
bNumbers in brackets indicate the total number of HCV sequences.
cThe initiation codon of the core protein was considered as residue 1, and the total length was calculated based on a –2 ribosomal frameshift at codons 11–12.
dThe termination codon was found at other non-conserved locations.
Discussion
Previous studies have demonstrated that the long ORF in the HCV genomic RNA codes for a polyprotein, which is proteolytically cleaved by cellular and viral proteases to generate at least 10 viral protein products. In this report, we demonstrate that an additional protein is expressed from a coding sequence that overlaps the core protein-coding sequence. The constraint imposed by two overlapping coding sequences is probably the reason why the HCV core protein-coding sequence has an unusually low synonymous substitution rate (Ina et al., 1994).
The protein sequencing results shown in Figures 3 and 4 indicate that the F protein is synthesized by a –2/+1 ribosomal frameshift. It is unlikely that the F protein was synthesized as a result of the RNA sequence heterogeneity generated by the T7 RNA polymerase, as direct sequencing of the HCV RNA did not reveal a one nucleotide deletion or a two nucleotide insertion, which is required for the synthesis of the F protein. We previously reported that the F protein could be expressed from the HCV-1 core protein sequence in Escherichia coli (Lo et al., 1995). During the course of this study, we found that no detectable amount of the F protein could be expressed from the HCV-1 sequence in E.coli, and our previous result was caused by an unintended deletion of nucleotide 80, which fused the first 26 codons of the core protein sequence to the overlapping ORF to produce a protein resembling the F protein. The inability of the F protein to be expressed in E.coli also argues against a role for T7 RNA polymerase in the expression of the F protein, since the expression of the core protein in E.coli in our studies was also controlled by the T7 promoter (Lo and Ou, 1998).
The ribosomal frameshift for the synthesis of the F protein is independent of the HCV internal ribosomal entry site (data not shown; see also Selby et al., 1993). This efficiency is ∼30% in vitro. In Huh7 cells, this efficiency was determined to be ∼1–2% using only codons 1–14 of the core protein sequence. In our previous expression studies in mammalian cells, we found that the expressing efficiency for the F protein was substantially higher than 2% (Lo et al., 1995, 1996). Thus, it is likely that the nucleotide sequence downstream of codon 14 is important for increasing the efficiency of ribosomal frameshift in cell cultures. Nevertheless, this 1–2% efficiency is not unusual. The ribosomal frameshifting efficiency can range from 1 to 40% depending on the genes and the expressing systems used (Grentzmann et al., 1998; Ivanov et al., 2000).
Although –1 and +1 ribosomal frameshifts are well documented, the –2 ribosomal frameshift is rare in eukaryotic cells and has only been documented once for the expression of ornithine decarboxylase antizyme (Matsufuji et al., 1996). For antizyme, this frameshift can be –2, +1 or a mixture of both, depending on the host cells used for the expression studies (Ivanov et al., 1998). The molecular mechanism by which the –2 frameshift is accomplished to synthesize F protein remains to be determined. It appears to take place at or near codon 11. The sequence of codons 8–14 is A rich and highly conserved among the HCV sequences reported (Rijnbrand and Lemon, 2000; Figure 1). This conservation may be due to its dual functions in coding and in directing the ribosomal frameshift.
Although ribosomal frameshift for gene expression has been demonstrated for RNA viruses of several different families including retroviruses (Jacks and Varmus, 1985), coronaviruses (Brierley et al., 1989) and astroviruses (Marczinke et al., 1994), HCV is the first example that uses this mechanism to express a gene that is totally embedded in another coding sequence. This strategy allows HCV to increase the coding potential of its genome and provides a means to produce a protein that may be needed in only relatively small amounts in the HCV life cycle.
The function of the F protein in the life cycle of HCV is unclear. It is interesting that a similar overlapping ORF is also detected in the core protein gene of the related GB virus type B (GBV-B) (Bukh et al., 1999). GBV-B has a shorter core protein-coding sequence, and its overlapping ORF, which also resides in the –2/+1 reading frame, can encode a protein that is 109 amino acids in length. The conservation of the F protein in the distantly related GBV-B indicates that this protein probably plays an important role in the HCV life cycle. The F protein displays no clear sequence homologies to other proteins of known function except that it is highly basic with a pI of ∼12. The F protein does not appear to be essential for viral RNA replication, as its absence did not abolish the replication of an HCV RNA replicon in Huh7 hepatoma cells (Lohmann et al., 1999; Blight et al., 2000). It may play a role in viral entry and/or viral morphogenesis or, alternatively, regulate a cellular function important for the viral life cycle.
In summary, our results show that a novel protein product is synthesized from the HCV genome by a ribosomal frameshift mechanism. The conservation of this gene in different HCV isolates and its expression during natural HCV infection, as evidenced by the presence of its antibodies in patients, suggest that this protein plays an important role in the HCV life cycle. Our studies thus identify a new potential target for the development of anti-HCV therapies. Such therapies may take the form of molecules that prevent the function of the F protein itself, or that prevent the ribosomal frameshift event (Dinman et al., 1998).
Materials and methods
Construction of DNA plasmids
For the construction of pCMV-CC, the HCV-1 core protein-coding sequence was isolated by PCR using the following two primers: sense, CGTGCCCCCGCAAGCTTGCTAG; and antisense, CCGTGGAGTTCTAGACTTGGTTAGGCCGAAGC. The antisense primer contained a termination codon (underlined) at the end of the core protein-coding sequence for translation termination. The PCR product was cloned into the XbaI–HindIII site of the pRc/CMV vector (Invitrogen). pCMV-CCmt was identical to pCMV-CC, except that nucleotide 432 of the core protein-coding sequence was mutated from U to A and the XbaI–NarI fragment of the vector was removed during the construction. pCMV-HACC contained the HA tag-coding sequence. This plasmid was constructed by inserting the HA-coding sequence TACCCATACGACGTCCCAGACTACGCT between the first and the second codons of the core protein-coding sequence in pCMV-CC. pCMV-CCΔ2 was identical to pCMV-CC, except that codons 2–7 and 15–50 had been deleted from the HCV-1 core protein-coding sequence by PCR. pGEM-core9a contained core protein-coding sequence with one nucleotide deletion in the 10 A stretch spanning from codons 8 to 11. This core protein-coding sequence was inserted in the BamHI–HindIII site of the pGEM vector (Promega). For the construction of pET-orf, which expresses the truncated F protein, a C to G mutation was introduced at nucleotide 13 of the core protein-coding sequence to create a translation initiation codon for the overlapping ORF. This coding sequence was then isolated by PCR and cloned into the NdeI–BamHI site of the pET-3a vector (Lo and Ou, 1998). The HCV sequences in all of the PCR-derived DNA constructs were verified by direct nucleotide sequencing.
The luciferase reporter constructs were created by joining the firefly luciferase-coding sequence to the 3′ end of the following sequence via the EcoRI site: 5′AACCTCAAACAGACACCATGAGCACGAATCCTAAACCTCAAAAAAAAAACAAACGTAACGAATTC3′. The sequence underlined corresponds to codons 1–14 of the core protein sequence and the sequence in italics indicates the EcoRI site. The hybrid sequence was cloned into the pCDEF vector under the expressing control of the EF1α promoter (Lu et al., 1999). In pEF-FS(0)Luc, the luciferase reporter was fused to the same reading frame as the core protein sequence (i.e. the zero reading frame). In pEF-FS(–2/+1)Luc, the luciferase reporter was fused to the +1 reading frame by the insertion of one nucleotide, C, after the EcoRI site. In pEF-FS(–1/+2)Luc, two nucleotides, CG, were inserted next to the EcoRI site.
In vitro translation
The plasmids pCMV-CC, pCMV-CCmt, pCMV-HACC and pCMV-CCΔ2 were linearized with XbaI for RNA synthesis. The translation was carried out at 30°C for 1 h using the following conditions: 0.5–1 µg of RNA, 10 µl of rabbit reticulocyte lysates (Promega), 0.5 µl of 1 mM amino acid mixture without methionine, and 50 µCi of [35S]methionine (>1000 Ci/mmol; ICN). The proteins synthesized were then analyzed by gel electrophoresis and autoradiography. The replacement of reticulocyte lysates with wheat germ extracts in the translation reaction generated the same results.
Radiosequencing
A 10 µg aliquot of RNA derived from pCMV-CC (or pCMV-CCΔ2), 50 µl of wheat germ extracts (Promega), 150 µCi of [3H]lysine (or [3H]threonine), 5 µCi of [35S]methionine and 1 mM amino acid mixture minus methionine and lysine (or methionine and threonine) were mixed and incubated at 30°C for 1 h. The protein samples were then purified on a 12.5% SDS–polyacrylamide gel and eluted in 10 mM ammonium bicarbonate containing 75 µg/ml bovine serum albumin (New England Biolabs) at 37°C overnight. The samples were then subjected to Edman degradation and the sequencing cycles were analyzed by scintillation counting.
Enzymatic sequencing of the HCV RNA
The HCV RNA with deletions of codons 2–7 and the sequence downstream of codon 14 was synthesized with the T7 RNA polymerase in the presence of [γ-32P]GTP. This results in the end-labeling of the RNA. The RNA was partially hydrolyzed in 50 mM NaHCO3 pH 9, containing 1.6 mM EDTA at 90°C for 6 min, or digested with RNase T1 or RNase A at 56°C for 4 min in a buffer containing 10 mM Tris–HCl pH 7, 1 mM EDTA and 7 M urea. The samples were then analyzed on a 12% sequencing gel.
Cell transfection and the luciferase assay
Huh7 cells were maintained in Dulbecco’s modified essential medium containing 10% fetal bovine serum. The luciferase reporter constructs were transfected into Huh7 cells by the calcium phosphate precipitation method. Luciferase assay was conducted ∼48 h post-transfection, using a kit supplied by Promega. To monitor the transfection efficiency, pXGH, an hGH-expressing plasmid (Lu et al., 1999), was co-transfected into Huh7 cells, and the level of hGH released into the medium was determined. Relative frameshift efficiencies were calculated using the following equation:
![]()
where Lucx is luciferase activity, LucB is background luciferase activity, Luc0 is luciferase activity of FS(0)Luc, TEx is transfection efficiency, and TE0 is transfection efficiency of FS(0)Luc. Background luciferase activity represents the luciferase activity determined using pCDEF-transfected Huh7 lysates.
Enzymatic immunoassay
This truncated F protein sequence was expressed in E.coli using the pET-3a vector following our previous procedures (Lo and Ou, 1998). The cells were lysed by sonication in phosphate-buffered saline (PBS), and the F protein was solubilized with 6 M guanidine–HCl. The F protein sample was dialyzed against PBS, followed by a brief centrifugation in a microfuge. The protein was resuspended in 0.9 M acetic acid containing 6.25 M urea and purified on an acid–urea gel (McNabb and Courtney, 1992). For the serological assay, 100–125 ng of F protein were coated in a well of a microtiter plate and incubated with 20 µl of serum and 180 µl of diluent at 37°C for 1 h. The well was then washed and subsequently incubated with 200 µl of horseradish peroxidase-conjugated anti-human antibody at 37°C for another hour. The sample was then washed again for color development.
Acknowledgments
Acknowledgements
We wish to thank Drs Jim and Ellen Strauss and Michael Lai for critical reading of the manuscript, Dr Ralph Reid of the UCSF Biomolecular Resource Center for advice and performance of the radiosequencing experiments, and members of J.-h.Ou’s laboratory for helpful discussions throughout the entire project. This work was supported by Research Scholar Grant #PF-01-037-01-MBC from the American Cancer Society (to J.C.) and research grants from the National Institutes of Health (U019AI40038 to J.H.O. and R03AI45873 to T.S.B.Y.).
References
- Beard M.R. et al. (1999) An infectious molecular clone of a Japanese genotype 1b hepatitis C virus. Hepatology, 30, 316–324. [DOI] [PubMed] [Google Scholar]
- Blight K.J., Kolykhalov,A.A. and Rice,C.M. (2000) Efficient initiation of HCV RNA replication in cell culture. Science, 290, 1972–1974. [DOI] [PubMed] [Google Scholar]
- Brierley I., Digard,P. and Inglis,S.C. (1989) Characterization of an efficient coronavirus ribosomal frameshifting signal: requirement for an RNA pseudoknot. Cell, 57, 537–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bukh J., Apgar,C.L. and Yanagi,M. (1999) Toward a surrogate model for hepatitis C virus: an infectious molecular clone of the GB virus-B hepatitis agent. Virology, 262, 470–478. [DOI] [PubMed] [Google Scholar]
- Choo Q.-L., Kuo,G., Weiner,A.J., Overby,L.R., Bradley,D.W. and Houghton,M. (1989) Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science, 244, 359–362. [DOI] [PubMed] [Google Scholar]
- Choo Q.L. et al. (1991) Genetic organization and diversity of the hepatitis C virus. Proc. Natl Acad. Sci. USA, 88, 2451–2455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinman J.D., Ruiz-Echevarria,M.J. and Peltz,S.W. (1998) Translating old drugs into new treatments: ribosomal frameshifting as a target for antiviral agents. Trends Biotechnol., 16, 190–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grentzmann G., Ingram,J.A., Kelly,P.J., Gesteland,R.A. and Atkins,J.F. (1998) A dual-luciferase reporter system for studying recoding signals. RNA, 4, 479–486. [PMC free article] [PubMed] [Google Scholar]
- Ina Y., Mizokami,M. Ohba,K. and Gojobori,T. (1994) Reduction of synonymous substitutions in the core protein gene of hepatitis C virus. J. Mol. Evol., 38, 50–56. [DOI] [PubMed] [Google Scholar]
- Ivanov I.P., Gesteland,R.F., Matsufuji,S. and Atkins,J.F. (1998) Programmed frameshifting in the synthesis of mammalian antizyme is +1 in mammals, predominantly +1 in fission yeast, but –2 in budding yeast. RNA, 4, 1230–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov I.P., Matsufuji,S., Murakami,Y., Gesteland,R.F. and Atkins,J.F. (2000) Conservation of polyamine regulation by translational frameshifting from yeast to mammals. EMBO J., 19, 1907–1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacks T. and Varmus,H.E. (1985) Expression of the Rous sarcoma virus pol gene by ribosomal frameshifting. Science, 230, 1237–1242. [DOI] [PubMed] [Google Scholar]
- Kalykhalov A.A., Agapov,E.V., Blight,K.J., Mihalik,K., Feinstone,S.M. and Rice,C.M. (1997) Transmission of hepatitis C by intrahepatic inoculation with transcribed RNA. Science, 277, 570–574. [DOI] [PubMed] [Google Scholar]
- Lo S.-Y. and Ou,J.H. (1998) Expression and dimerization of hepatitis C virus core protein in E.coli. In Lau,J.Y.-N. (ed.), Hepatitis C Protocols. Humana Press, Totowa, NJ, pp. 325–330. [DOI] [PubMed]
- Lo S.-Y., Selby,M., Tong,M. and Ou,J.H. (1994) Comparative studies of the core gene products of two different hepatitis C virus isolates: two alternative forms determined by a single amino acid substitution. Virology, 199, 124–131. [DOI] [PubMed] [Google Scholar]
- Lo S.-Y., Masiarz,F., Hwang,S.B., Lai,M.M.C. and Ou,J.H. (1995) Differential subcellular localization of hepatitis C virus core gene products. Virology, 213, 455–461. [DOI] [PubMed] [Google Scholar]
- Lo S.-Y., Selby,M.J. and Ou,J.H. (1996) Interaction between hepatitis C virus core protein and E1 envelope protein. J. Virol., 70, 5177–5182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohmann V., Korner,F., Koch,J.-O., Herian,U., Theilmann,L. and Bartenschlager,R. (1999) Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science, 285, 110–113. [DOI] [PubMed] [Google Scholar]
- Lu W., Lo,S.-Y., Chen,M., Wu,K.-J., Fung,Y.K.T. and Ou,J.-H. (1999) Activation of p53 tumor suppressor by hepatitis C virus core protein. Virology, 264, 134–141. [DOI] [PubMed] [Google Scholar]
- Marczinke B., Bloys,A.J., Brown,T.D.K., Willcocks,M.M., Carter,M.J. and Brierley,I. (1994) The human astrovirus RNA-dependent RNA polymerase coding region is expressed by ribosomal frameshifting. J. Virol., 68, 5588–5595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsufuji S., Matsufuji,T., Wills,N.M., Gesteland,R.F. and Atkins,J.F. (1996) Reading two bases twice: mammalian antizyme frameshifting in yeast. EMBO J., 15, 1360–1370. [PMC free article] [PubMed] [Google Scholar]
- McNabb D.S. and Courtney,R.J. (1992) Posttranslational modification and subcellular localization of the p12 capsid protein of herpes simplex virus type 1. J. Virol., 66, 4839–4847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno T. and Mizokami,M. (1998) Genotyping by type-specific primers that can type HCV types 1–6. In Lau,J.Y.-N. (ed.), Hepatitis C Protocols. Humana Press, Totowa, NJ, pp. 159–164.
- Ray R.B., Lagging,L.M., Meyer,K. and Ray,R. (1996) Hepatitis C virus core protein cooperates with ras and transforms primary rat embryo fibroblasts to tumorigenic phenotype. J. Virol., 70, 4438–4443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rijnbrand R.C.A. and Lemon,S.M. (2000) Internal ribosomal entry site-mediated translation in hepatitis C virus replication. Curr. Top. Microbiol. Immunol., 242, 85–116. [DOI] [PubMed] [Google Scholar]
- Selby M.J. et al. (1993) Expression, identification and subcellular localization of the proteins encoded by the hepatitis C viral genome. J. Gen. Virol., 74, 1103–1113. [DOI] [PubMed] [Google Scholar]
- Smith D.B. and Simmonds,P. (1998) Hepatitis C virus: types, subtypes and beyond. In Lau,J.Y.-N. (ed.), Hepatitis C Protocols. Humana Press, Totowa, NJ, pp. 133–146.
- Yanagi M., Purcell,R.H., Emerson,S.U. and Bukh,J. (1997) Transcripts from a single full-length cDNA clone of hepatitis C virus are infectious when directly transfected into the liver of a chimpanzee. Proc. Natl Acad. Sci. USA, 94, 8738–8743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagi M., St Claire,M., Shapiro,M., Emerson,S.U., Purcell,R.H. and Bukh,J. (1998) Transcripts of a chimeric cDNA clone of hepatitis C virus genotype 1b are infectious in vivo. Virology, 244, 161–172. [DOI] [PubMed] [Google Scholar]
- Yanagi M., Purcell,R.H., Emerson,S.U. and Bukh,J. (1999) Hepatitis C virus: an infectious molecular clone of a second major genotype (2a) and lack of viability of intertypic 1a and 2a chimeras. Virology, 262, 250–263. [DOI] [PubMed] [Google Scholar]