

Abstract
The transcriptional antiterminator protein LicT regulates the expression of Bacillus subtilis operons involved in β-glucoside metabolism. It belongs to a newly characterized family of bacterial regulators whose activity is controlled by the phosphoenolpyruvate:sugar phosphotransferase system (PTS). LicT contains an N-terminal RNA-binding domain (56 residues), and a PTS regulation domain (PRD, 221 residues) that is phosphorylated on conserved histidines in response to substrate availability. Replacement of both His207 and His269 with a negatively charged residue (aspartic acid) led to a highly active LicT variant that no longer responds to either induction or catabolite repression signals from the PTS. In contrast to wild type, the activated mutant form of the LicT regulatory domain crystallized easily and provided the first structure of a PRD, determined at 1.55 Å resolution. The structure is a homodimer, each monomer containing two analogous α-helical domains. The phosphorylation sites are totally buried at the dimer interface and hence inaccessible to phosphorylating partners. The structure suggests important tertiary and quaternary rearrangements upon LicT activation, which could be communicated from the protein C-terminal end up to the RNA-binding domain.
Keywords: B.subtilis PEP:sugar PTS/catabolite repression/PRD-containing regulator/structure/transcriptional antiterminator
Introduction
The expression of many catabolic operons in Gram-positive and Gram-negative bacteria is regulated by transcriptional antitermination (Rutberg, 1997; Henkin, 2000). BglG in Escherichia coli, and SacY and LicT in Bacillus subtilis are examples of a now well-characterized family of transcriptional antiterminator proteins involved in carbohydrate metabolism (Mahadevan and Wright, 1987; Crutz and Steinmetz, 1992; Schnetz et al., 1996). Operons regulated by these antiterminators are preceded by a terminator sequence whose transcription causes a detachment of the RNA polymerase from the mRNA matrix and leads to abortion of transcription. In the presence of an inducer, transcriptional antiterminator proteins become activated and consequently bind to an antiterminator RNA (called RAT) that is partially overlapping with the terminator. Binding of the antiterminator protein to the nascent mRNA prevents formation of the terminator stem–loop structure and allows transcription through the operon (Aymerich and Steinmetz, 1992).
Transcriptional antiterminators of the BglG/SacY family are modular proteins consisting of an N-terminal RNA-binding domain (CAT, ∼55 residues) and a C-terminal regulatory domain (PRD, residues 60–280). So far, only the structure of the CAT domains from SacY and LicT has been determined (Manival et al., 1997; van Tilbeurgh et al., 1997; Declerck et al., 1999). It was shown that isolated CAT domains possess constitutive antitermination activity in vivo and RNA-binding activity in vitro (Manival et al., 1997; Declerck et al., 1999). In the full-length proteins, the activity of CAT is modulated by the regulatory domain, which is subject to phosphorylations by the phosphoenolpyruvate(PEP):sugar phosphotransferase system (PTS). The PTS consists of the general enzymes EI and HPr, and the specific enzymes EII mediating the transport and concomitant phosphorylation of different sugars (Postma et al., 1993). Depending on substrate and phosphoryl group availability, HPr and/or EIIs (de)phosphorylate the PTS regulation domain (PRD) of the antitermination proteins and thereby control their RNA-binding activity (Amster-Choder and Wright, 1993; Tortosa et al., 1997; Bachem and Stülke, 1998; Knezevic et al., 2000). Other PRD-containing regulators such as the transcriptional activators LicR (Tobisch et al., 1999) and LevR (Martin-Verstraete et al., 1998) in B.subtilis, or MtlR (Henstra et al., 2000) in Bacillus stearothermophilus, which possess a helix–turn–helix DNA-binding motif at their N-terminus, are regulated similarly by the PTS. Sequence alignment shows that the PRDs of all transcriptional regulators can clearly be divided into two homologous subdomains, named PRD1 and PRD2 (Tortosa et al., 1997; Stülke et al., 1998), each comprising one and often two highly conserved histidines. In vivo and in vitro studies have shown that these histidines are the PTS-mediated phosphorylation sites, although no general rules seem to exist for the physiological response associated with their phosphorylation in different PRD-containing regulators (for a review see Stülke and Hillen, 2000).
LicT is involved in the transcriptional regulation of the bglS gene and the bgl operon in B.subtilis. In the presence of lichenan or aryl-β-glucosidic substrates, LicT provokes antitermination at RAT sequences situated between licT and bglS, which encodes an extracellular β-glucanase (Schnetz et al., 1996), and upstream of the bglPH operon (Krüger et al., 1996). BglP encodes EIIbgl, the specific β-glucoside phosphotransferase, and bglH encodes a phospho-β-glucosidase (Le Coq et al., 1995). To be activated, LicT requires not only the presence of inducing sugars, but also the absence of glucose or other rapidly metabolized carbon sources (Krüger et al., 1996). Recent in vivo and in vitro studies have shown that the regulation of LicT activity by the PTS is mediated by antagonistically acting phosphorylations of the four highly conserved histidines: His100 and His159 in PRD1, and His207 and His269 in PRD2. Up to three of these residues can be phosphorylated in vitro from PEP by EI and HPr, His207 being the major site of phosphorylation (Lindner et al., 1999). In vitro phosphorylation of His100 was not established, but genetic studies have suggested that it is phosphorylated by BglP in the absence of incoming sugar, resulting in LicT inhibition under non-inducing conditions. His159 would also be involved in this induction mechanism, whereas His207 and His269 would be involved in the carbon catabolite control of LicT activity by the PTS (P.Tortosa, submitted). HPr-dependent phosphorylation of these histidines would occur in the absence of rapidly metabolized sugar and serve as the signal for catabolite repression relief required for the activation of LicT. The activated form of LicT should thus be dephosphorylated on PRD1 and phosphorylated on PRD2.
Although it is now well established that PTS-mediated phosphorylation of transcriptional regulators tunes the functional response, the molecular and structural mechanisms of these regulatory phenomena remain largely unknown. So far, no three-dimensional structure of PRD has been reported, and very few biochemical and biophysical studies have been performed on purified proteins. Based on genetic studies with the BglG antiterminator, phosphorylation-induced modification of the monomer–dimer equilibrium has been proposed as the general mechanism regulating the activity of PRD- containing proteins (Amster-Choder and Wright, 1992), but no in vitro experiments have supported this model so far. Biochemical studies on the phosphorylated forms of antiterminator proteins are hampered by the poor efficiency of the phosphotransferase reaction, the difficulty of separating differently phosphorylated molecules and the low stability of the phosphohistidyl linkage. To overcome these problems, we have constructed LicT variants in which one or two of the phosphorylatable histidines were replaced by an aspartate supposed to mimic a phosphorylated residue. Here we have tested the effect of His→Asp mutations at position 207 and/or 269 on the carbon catabolite control of LicT activity and found that the LicT variant carrying the H207D/H269D double mutation was highly and constitutively active in vivo. In contrast to wild type, the PTS regulation domain of this activated mutant form readily crystallized and allowed us to solve the first structure of a PRD, at 1.55 Å resolution.
Results and discussion
His207 and His269→Asp mutations activate LicT in vivo
Our genetic analyses have suggested that LicT-PRD2 is the target of the positive regulation exerted by EI and HPr, the general components of the PTS (P.Tortosa, N.Declerck, H.Dutartre, J.Deutscher and D.Le Coq, submitted). Phosphorylation of His207 and His269 would be required for the activation of LicT and the eliciting of its antitermination activity in the presence of the inducing sugar and in the absence of other carbon source such as glucose. In order to test this hypothesis, we have constructed LicT variants in which His207 and/or His269 is replaced by an aspartic acid whose negatively charged side chain is supposed to mimic a phosphoryl group. Indeed, replacement of a phosphorylatable amino acid with an aspartate has previously been shown to lead to structural changes similar to those observed after phosphorylation (Wittekind et al., 1989).
The licT genes harbouring one or both His→Asp mutations were introduced into the chromosome of a B.subtilis strain containing a bglP′–lacZ fusion gene. Expression of this reporter cassette is under the control of LicT, therefore the amount of β-galactosidase (β-gal) produced by the cells under different growth conditions reflects the modulation of LicT transcriptional antitermination activity in vivo. As seen in Figure 1, a high antitermination activity is observed in the LicT-H207D/H269D double mutant strain, especially under non-inducing conditions where production of β-gal is increased 42-fold compared with the wild-type level. This constitutively active phenotype seems to be the result of a synergetic effect of the two His→Asp mutations since the antitermination activity measured in the LicT-H207D or -H269D single mutant strains remains at the wild-type level under non-inducing conditions and is stimulated in the presence of the inducer. The single mutant strains as well as the double mutant strain are less sensitive than the wild-type strain to carbon catabolite repression: the repression factor (β-gal activity of the induced strain/β-gal activity of the induced strain in the presence of glucose) associated with the H207D, H269D and H207D/H267D mutations is 1.7, 2.0 and 1.7, respectively, compared with 8 for the wild type. Repression by glucose is not completely abolished in any of the mutant strains, due to the fact that expression of the bglP′–lacZ reporter gene is also subjected to a catabolite control by the PTS at the level of transcription initiation (Krüger et al., 1996). The mutant proteins have also been found to be constitutively active in a Δpts genetic context (P.Tortosa, submitted), demonstrating that phosphorylation of LicT by HPr is no longer required for the activation of the LicT variants. These results confirm that His207 and His269 are involved in the catabolite regulation of LicT activity by the PTS and that the negative charge brought about by the aspartate side chain introduced at these positions generates functional modifications similar to those expected for the phosphorylation of LicT-PRD2.

Fig. 1. In vivo effect of His→Asp mutations on LicT regulation. The antitermination activity of the wild-type (LicT-wt) or mutant proteins carrying the His207→Asp (H207D) or His269→Asp (H269D) single or double mutation was determined by measuring the β-gal activity of B.subtilis transformants harbouring the wild-type or mutant licT alleles. Strains were grown in glucitol minimal medium (non-inducing conditions, white bars), supplemented with salicin (inducing conditions, grey bars) or with salicin and glucose (repressing conditions, black bars). The β-gal activities are expressed relative to the value measured for the strain expressing the wild-type gene under non-inducing conditions. The activity of the wild-type protein under non-inducing conditions is 24 ± 2 Miller units/mg protein.
The fact that LicT-H207D/H269D no longer requires induction by salicin to be activated suggests that the double mutation in PRD2 impedes the inactivation mechanism mediated by EIIbgl (BglP). This seems to contradict the current regulation model in which the phosphorylation of PRD2 (upon carbon catabolite repression relief) is not supposed to prevent additional phosphorylation of PRD1 (in the absence of the inducer). However, there are no experimental data so far regarding the in vivo behaviour of LicT doubly phosphorylated on PRD2. We therefore do not know whether LicT-H207D/H269D responds to induction signals in the same way as doubly phosphorylated LicT. Another possible explanation for the constitutive phenotype observed here is that the double mutation may alter the structural integrity of the PRD, which would no longer be recognized by BglP and/or prevent interaction with RNA. This possibility has been ruled out by the finding that the purified LicT-H207D/H269D variant is in fact properly folded, as demonstrated herein. We believe that the strong dominant effect induced by the double mutation is due to the fact that the mutations, in contrast to the phosphorylations, are not reversible; therefore, the LicT variant behaves as an activated form permanently phosphorylated on PRD2. It is very unlikely that in the wild-type context under activating conditions, 100% of the antiterminator molecules would ever be doubly phosphorylated on PRD2; therefore, the level of activation observed with the double mutant form will never be reached. This might explain why we measured higher antitermination activity in the LicT-H207D/H269D mutant strain than in the induced wild-type strain.
Structure determination
So far, our attempts to solve the crystal structure of the wild-type regulatory domain of LicT have failed due to the poor reproducibility of the crystallizing conditions and the low quality of the diffraction data (H.van Tilbeurgh, unpublished results). In contrast, the activated double mutant form we constructed, LicT-PRD-H207D/H269D, reproducibly yielded well-diffracting crystals overnight. The structure was determined by multiple anomalous dispersion (MAD) diffraction measurements on seleno-methionine (Se-Met)-substituted crystals at 2.0 Å resolution. The quality of the maps was excellent and allowed the straightforward construction of the whole model. The structure was refined against data collected at 1.55 Å on native crystals. The structure is well defined except for two residues in a connecting loop (residues 222–223), for which no electron density could be observed. Refinement statistics are gathered in Table I.
Table I. Data collection and refinement statistics.
| Se-Met λ1 | Se-Met λ2 | Se-Met λ3 | Native | |
|---|---|---|---|---|
| Diffraction data statistics | ||||
| Resolution (Å) | 30–1.8 | 30–1.8 | 30–1.8 | 20–1.55 |
| Wavelength (Å) | 0.979 | 0.979 | 0.8855 | 1.00 and 1.54 |
| Unique reflections | 46 340 | 46 347 | 46 342 | 37 674 |
| Redundancy | 2.6 | 2.6 | 2.6 | 2 3.1 |
| Completeness (%) | 99.7 (98.1) | 99.7 (98.2) | 99.7 (97) | 99.3 (98.2) |
| Rmerge(%)a | 7 (13.2) | |||
| Rsym (%)b | 3.7 (27.7) | 3.5 (25.2) | 4.1 (29.2) | |
| <I>/<σ(I)> | 22.4 (3.9) | 22.4 (3.7) | 21.4 (2.0) | 15.6 (4.4) |
| No. of sites | 7 | |||
| Refinement statistics | ||||
| Resolution range (Å) | 20–1.55 | |||
| Total no. of non-hydrogen atoms | 1903 | |||
| Water molecules | 191 | |||
| R-factor (%) | 23.2 | |||
| Free R-factor (%)c | 25.5 | |||
| Average B-factor (Å2) | 20.4 | |||
| R.m.s.d. bond lengths (Å) | 0.002 | |||
| R.m.s.d. bond angles (°) | 0.98 | |||
Values in parentheses concern the highest resolution shell.
aRmerge = (Σ|I – <I>|/ΣI), where I is the observed intensity and <I> is the average intensity of equivalent reflections in the various data sets.
bRsym = (Σ|I – <I>|/ΣI), where I is the observed intensity and <I> is the average intensity of symmetry-related reflections.
cRfree, crystallographic R-factor for 8% of reflections set aside during the refinement (Brünger, 1992).
Architecture of the PRD and structure of the dimer
The 1.55 Å resolution crystal structure (Figure 2) reveals that the regulatory region of LicT consists of two structurally similar domains corresponding to PRD1 (residues 67–170) and PRD2 (residues 171–277) as previously identified from sequence alignments (Tortosa et al., 1997; Stülke et al., 1998). In Figure 3, the backbone structure of the LicT-PRD dimer is displayed. In the monomer, PRD1 and PRD2 are rotated by ∼90° relative to each other and make few interdomain contacts. Structurally, PRD1 and PRD2 belong to the globin fold family; the closest structural relatives are the phycoerythrins (Schirmer et al., 1985) as detected by the DALI program (Holm and Sander, 1993). Each domain forms a compact bundle comprising five helices (α1–α5). Sequence alignment suggests that the location and length of these helices are very well conserved among PRD modules (Figure 4A). The core of the PRD module consists of two pairs of antiparallel helices, making an angle of ∼60°. The first pair contains the antiparallel helices α1 and α4, while the second pair contains α2 and α5. The third helix (α3) is oriented perpendicularly to α5 at the periphery of the bundle. The helices are connected by loops of varying length.

Fig. 2. View of the weighted 2Fo – Fc electron density map of the refined LicT-PRD-H207D/H269D structure. The region around the phosphorylatable PRD1 histidines (His100 and His159) is represented.

Fig. 3. (A) Ribbon drawing using the Molscript package of the LicT-PRD dimer perpendicular to the 2-fold symmetry axis (Carson, 1991; Kraulis, 1991). The individual monomers are coloured yellow and pink. The subdomains PRD1 and PRD2 and their individual α-helices (α1–α5) are indicated. The phosphorylatable histidines (His100 and His159) in PRD1 and the mutated aspartates in PRD2 (Glu207 and Glu269) are shown in ball-and-stick representation. (B) The LicT-PRD dimer viewed in the same direction to that shown in (A) but rotated over 90° along the 2-fold symmetry axis.


Fig. 4. (A) Alignment (Thompson et al., 1994) of the PRD1 and PRD2 sequences in transcriptional antiterminators (LicT, SacY, SacT, BglG and GlcT) and activators (MtlR, LicR and LevR). All sequences are from B.subtilis except BglG (E.coli) and MtlR (B.stearothermophilus). The alignment was edited using ESPript (Gouet et al., 1999). Secondary structure elements, as derived from the PRD1 crystal structure, are superimposed. Numbering follows the sequence of LicT-PRD1. Equivalent residues in regions of high similarities (boxed in blue) are shown in red. The most conserved residues are boxed in red: His100, Arg107, Glu152 and His159 in PRD1; His207, Arg214, Glu262 and His269 in PRD2. (B) Simplified stereo views of the conserved residues (ball-and-stick representation) at the PRD1–PRD1 (lower) and PRD2–PRD2 (upper) interfaces. Only the supporting helices (coloured red and yellow for the two individual monomers) are shown. Hydrogen bonds are represented by dashed lines.
Gel filtration chromatography, solution X-ray scattering and analytical ultracentrifugation clearly demonstrated that LicT-PRD is present as a dimer in solution (N.Declerck, submitted). Although there is only one molecule per asymmetric unit in our crystals, analysis of the packing shows that an extensive dimer is generated by a crystallographic dyad (Figure 3). An identical dimer was also present in a different crystal form obtained under similar crystallization conditions (H.van Tilbeurgh, unpublished results). The considerable interaction surface and the numerous protein contacts established by this dimer are much more important than those observed in any other crystal contact, suggesting that this crystallographic dimer corresponds to the biologically relevant solution dimer. The dimer has the overall shape of a cylinder with dimensions 66 Å × 41 Å × 39 Å. An incursion is observed between PRD1 and PRD2 in one direction, giving the structure a bi-lobal aspect. The dimer is formed by a head-to-head (PRD1) and tail-to-tail (PRD2) association of two PRD monomers. The PRD1 and PRD2 domains in one monomer, connected by a short peptide (residues 166–169), are wrapped around the equivalent domains of the other monomer.
The main contributions to dimer formation come from the PRD1–PRD1 and PRD2–PRD2 contact surfaces. The dimer interfaces of both PRD1 and PRD2 involve their α2, α3 and α5 helices, facing their identical counterparts in the other monomer. The considerable shifts in the relative orientations of these helices between PRD1 and PRD2 result in significantly differing interfaces for the two domains. The orientation of helices α2 and α5 at the PRD2 interface is antiparallel to that of α2 and α5 in the other monomer in such a way that they contact each other over the whole length. The α2 helices at the PRD1 interface, however, are at an angle of 30° and they contact each other merely through their respective N- and C-terminal residues. The α5 helices at the PRD1 interface are tilted by 30° around their centre. The α3 helices are antiparallel at both the PRD1 and PRD2 dimer interfaces.
Dimer formation buries ∼3600 Å2 of each monomer, which corresponds to 35% of the solvent-accessible surface as calculated by CNS (Brünger et al., 1998). This surface area is considerable for a dimer of this molecular mass (Jones and Thornton, 1995), but close inspection of this interface reveals the presence of important occluded cavities at both the PRD1 and PRD2 interfaces. These cavities are filled with 18 well-defined water molecules in total, but only one of these is involved in bridging dimer interactions (between Asn90 OD1 and Arg107 NH1). The calculated buried surface may therefore not directly reflect dimer strength.
The dimer interface has both polar and hydrophobic components. The PRD1 and PRD2 interfaces each contain six direct hydrogen bonds (Table II). The majority of the residues involved in these hydrogen bonds are not conserved among the antiterminator sequences although alternative hydrogen bonds are usually possible with the residues at homologous positions in the other proteins of the PRD family. Besides polar interactions, hydrophobic packing is also observed at the PRD1 (Leu125, Tyr126, Leu118, Val161, Leu165 and Phe155) and PRD2 (Tyr236, Ile268, Phe227, Leu228 and Ile264) interfaces. The majority of hydrophobic residues that are in van der Waals contact are carried by helices α3 and α5.
Table II. Polar dimer interactionsa.
| Monomer A | Monomer B | Distance (Å) |
|---|---|---|
| Linker | ||
| Lys60-O | Leu62-N | 2.71 |
| PRD1 | ||
| Ser92-Oγ | Asp99-Oδ2 | 2.90 |
| His100-Nδ1 | Asn162-Oδ1 | 2.95 |
| Lys110-Nζ | Asn166-O | 2.68 |
| PRD2 | ||
| Arg203-Nδ1 | Asp207-Οδ1 | 2.83 |
| Asp225-Ο1 | Tyr236-Oη | 2.96 |
| Asp261-Oδ2 |
Arg272-Nε |
2.84 |
| PRD1 |
PRD2 |
|
| Glu121-Οε2 | Lys209-Nζ | 3.2 |
aSymmetrical dimeric interactions are not indicated.
Although the PRD1–PRD1 and PRD2–PRD2 interfaces provide the majority of the dimer contact surface, a few additional contacts are made between PRD1 and PRD2 from the opposing monomer at the centre of the molecule. This contact region involves the packing of helices α3 and α2 of the opposing monomer but involves very few direct protein contacts. This interaction is stabilized by one salt bridge between the very well conserved Glu121 (α3 PRD1) and Lys209 (α2 PRD2) and may play a role in the communication between the two domains.
The only regulator for which structural information on dimer formation is available at the sequence level is the antiterminator BglG from E.coli. Using truncated constructs, it was shown that its dimerization domain was situated in the 104 C-terminal residues (Boss et al., 1999). This region matches the PRD2 in LicT exactly. However, the prediction that the corresponding region between Gly175 and Thr207 in BglG would be involved in a coiled-coil leucine zipper dimerization motif is not supported by our structure. This region encompasses the α1–α2 helical motif and only the second helix is involved in dimerization and forms no zipper motif.
The well-conserved residues of the PRD domain are found almost exclusively at either the hydrophobic core of the PRD subdomains or the dimer interface near the phosphorylatable positions. These residues are all inaccessible to eventual protein partners interacting with the activated state of the PRD. Interaction with the β′ subunit of the RNA polymerase was suggested for the E.coli BglG antiterminator (Nussbaum-Shochat et al., 1999). It was not proven that similar interactions take place for LicT. Since no conserved patches were observed at the surface of the activated mutant LicT dimer, no prediction can be made concerning possible contacts with the B.subtilis RNA polymerase or other protein partners.
Environment of the phosporylatable histidines and mutated aspartates
The PRD sequences are characterized by the presence of four well-conserved histidines, whose phosphorylation state determines the activation state of the protein. In our structure of the activated form of the LicT regulatory domain, the phosphorylatable histidines of PRD1 occupy the same position relative to secondary structure elements as their mutant aspartate equivalents in PRD2 (Figure 4B); His100 and Asp207 (replacing His207) are both located in the middle of the long α2 helix, whereas His159 and Asp269 (replacing His269) are closer to the C-terminus of helix α5. The folding of the individual PRD subdomains brings His100 close to His159 in PRD1 and Asp207 close to Asp269 in PRD2 (Figure 4B). Dimer formation then assembles the phosphorylation sites into an interacting cluster of four histidines at the PRD1–PRD1 interface and of four aspartates at the PRD2–PRD2 interface. The tertiary arrangement of the two clusters is markedly different, however, due to relative motions of the supporting helices: in PRD1, the distance between His100-Cα atoms of each monomer is 15.8 Å, whereas His159 is close to its dimer equivalent (9.9 Å between Cα atoms). In PRD2 by contrast, the sliding of the α2 helices brings the Asp207-Cα atoms to 10.7 Å whereas the Asp269-Cα atoms are at 16.7 Å.
In both PRD1 and PRD2, the side chains of the phosphorylatable residues are within hydrogen bonding distance of each other. His100-Nε2 is at 3 Å distance from His159-Nδ1. A number of well-defined water molecules contact His159, but these do not bridge the other monomer. His100-Nε2 interacts with Glu152-Oε1 in the same monomer and with Asn162-Oδ1 in the opposing monomer. Glu152 is also involved in a salt bridge with Arg107, bridging α2 and α5. Both residues are highly conserved in all PRD1 and PRD2 sequences (Figure 4A). The Oδ1 atom of Asp207 is at a distance of 2.45 Å from Asp269-Oδ1, suggesting a strong hydrogen bond and a different ionization state between these residues. Asp207-Oδ2 forms a salt bridge with Arg203 from the dimer partner. Asp269 interacts with the Tyr265 OH group of the other monomer via a tightly bound water molecule (not shown on the figure). Arg203 is completely conserved in all the PRD2 antiterminator sequences, but not in either the PRD1 or the transcriptional activators (Figure 4A). The double salt bridge between Arg203 and Asp207 is probably a crucial interaction for dimer formation.
The phosphorylation sites in both PRD1 and PRD2 are surrounded by a half ring of residues whose hydrophobic character is well conserved among the PRD sequences: Leu158, Phe155, Ile114 and Phe103 for PRD1; Ile268, Tyr265, Phe211 and Phe210 for PRD2 (Figure 5). This hydrophobic rim around the phosphorylation sites might provide the interaction surface for the PRD-phosphorylating proteins, HPr and BglP. Interestingly, the putative binding surface in PRD2 as well as in PRD1 has a concave shape that may complement the convex HPr active site surface, as seen in the complex between E.coli HPr and the N-terminal domain of EI (Garrett et al., 1999) or EIIAGlc (Wang et al., 2000).

Fig. 5. CPK presentation of the environment of the phosphorylatable histidines in PRD1 (A) and of the mutated aspartates in PRD2 (B). These are surrounded by conserved hydrophobic residues that probably constitute the interacting surface for (de)phosphorylating partners.
To verify whether the crystal structure of the PRD dimer is compatible with the phosphorylated form of the two regulatory wild-type histidines, we changed Asp207 and Asp269 into phosphohistidines by molecular modelling (phosphohistidine coordinates were retrieved from the NMR structure of the phosphorylated form of HPr, PDB code 1PFH). Although phosphohistidines are considerably bulkier than the aspartates, they could be accommodated easily at the interface by simple adjustment of the torsion angles of the phosphohistidines and of Arg203. The ease with which these substitutions could be introduced is explained by the presence of the large cavities at the dimer interface, providing space for accepting these modifications. From this modelling, it appears that Arg203 must play a crucial role in the stabilization of the phosphoryl groups at the interface, as was already suggested from its interaction with Asp207 in our crystal structure of the mutant.
Conformational changes upon LicT activation
One of the most intriguing aspects of the LicT-PRD-H207D/H269D crystal structure is that the phosphorylation sites are all deeply buried at the dimer interface (0% accessible surface as calculated with the CCP4 program SURFACE; CCP4, 1994). This activated form of LicT-PRD adopts a closed dimer in which His100 and His159 in PRD1 as well as Asp207 and Asp269 in PRD2 are totally inaccessible to the PTS protein partners that regulate LicT activity. This observation provides a structural explanation for the constitutive phenotype conferred by the double histidine to aspartate mutation in vivo (Figure 1): in LicT-H207D/H269D, the regulatory histidines buried at the PRD1–PRD1 interface cannot be phosphorylated, nor can they interact with BglP and, therefore, the inhibitory signals cannot be transmitted to the double mutant protein. In contrast, the single mutant antiterminators, LicT-H207D and LicT-H269D, remain sensitive to the induction mechanism, indicating that these proteins adopt a more open conformation in which the conserved histidines are still accessible to the (de)phosphorylating enzymes.
The total inaccessibility of the phosphorylation sites observed in the LicT-PRD-H207D/H269D crystal suggests that a conformational transition and/or quaternary structural rearrangement takes place upon LicT activation. This is in good agreement with the proposal that the activity of PRD-containing proteins is regulated by phosphorylation events modulating their oligomerization state (Amster-Choder and Wright, 1992). The finding that the activated mutant form of LicT-PRD we crystallized has a dimeric structure is also in good agreement with other studies indicating that the antiterminators of the BglG/SacY family interact with their RNA target as dimeric molecules (Manival et al., 1997; Declerck et al., 1999). However, an activation mechanism converting an inactive monomer into an active dimer is not supported by the observation that the wild-type regulatory domain of LicT already forms dimers (Declerck et al., 1999). From an exhaustive biochemical and biophysical study on LicT and its mutants (N.Declerck, unpublished), we concluded that the regulatory domain presents a different dimer arrangement in the native than in the mutant proteins. Even in the absence of the native, non-phosphorylated LicT-PRD structure, examination of our double mutant structure provides insights into the conformational changes induced by the activating mutations.
Although PRD1 and PRD2 have the same global structure (r.m.s. deviation of 1.6 Å for 66 Cα positions), we noted important differences in the relative orientations of the helices in each domain (Figure 6). Global superposition of PRD1 and PRD2 results in a good fit for the helices α3, α4 and α5, but important shifts are observed in the positions of helices α1 and α2. Interestingly, the conserved salt bridge between α2 and α5 (involving Arg107/Glu152 in PRD1, and Arg214/Glu262 in PRD2) is maintained in spite of the repositioning of these helices (Figure 4B). This is achieved through rotation of the glutamate side chain: Glu152 forms a hydrogen bond to His100 in PRD1 but Glu262 points away from the negative charge cluster of the mutated aspartates at the PRD2 interface. The relative movement of this glutamate is accompanied by a repositioning of the arginine partner, thus maintaining the salt bridge at both interfaces. The side chain flip of this totally conserved residue could be an important structural change induced by the negatively charged substituting side chains, provoking, in turn, the sliding of α1–α2. We suspect that the different positioning of the α1–α2 motif in PRD1 and PRD2 is caused by the introduction of the activating mutations and not by the overall sequence divergence of the two domains.
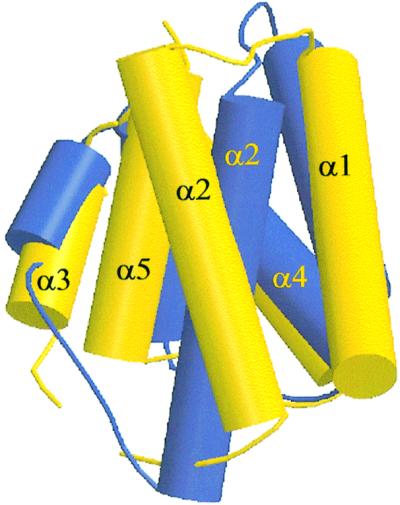
Fig. 6. Structure superposition of PRD1 (yellow) and PRD2 (blue). Helices are represented as solid cylinders. All Cα positions were used in the fit of the two modules. The helices α3, α4 and α5 of PRD1 and PRD2 superpose relatively well. The α1 helix in PRD2 is shifted over 5 Å along the helical axis in the N-terminal direction compared with α1 in PRD1, while α2 has pivoted by 30° around the centre of the helix compared with α2 in PRD2.
Since the linker between PRD1 and PRD2 (connecting α5 from PRD1 to α1 from PRD2) consists of only three residues, it can be expected that spatial movements of the PRD2 helices adjacent to this linker will be transmitted directly to PRD1. We attempted to model the inactivated state of PRD2, by replacing the α1–α2 motif of PRD2 with its conformation in PRD1 (data not shown). From this model, we conclude that a shift of α1 of 5 Å along its axis in PRD2 (as observed when superimposing PRD1 and PRD2) would have dramatic effects on the dimer association, especially at the PRD1–PRD1 interface. In contrast to the double mutant form, this modelled native LicT-PRD has an open dimeric conformation, providing access of the phosphorylatable histidines to the PTS regulatory proteins. Opening and closure of the LicT dimer may thus be the molecular process underlying the activation mechanism. This type of mechanism may apply to other PRD-containing regulators that are subjected to a carbon control by the PTS and whose activation requires multiple HPr-mediated phosphorylation. It remains to be determined by further experiments up to which level the double mutant mimics the phosphorylated form. The (inhibiting) phosphorylation of the BglG antiterminator by EII modulates a monomer–dimer equilibrium, indicating that different activation mechanisms may exist in other PRD transcriptional regulators (Amster-Choder and Wright, 1993).
Modelling of the intact protein
On sequence grounds, it was proposed that the region contained between residues 57 and 75 would form a linker between the CAT and the PRD domains. In our LicT-PRD crystal structure, the entire N-terminus has well-defined electron density and its conformation in the crystal could therefore be determined unambiguously. The first helix of the PRD1 domain starts at residue 66, and the linker region is therefore shorter than predicted. This linker region crosses over towards the opposing monomer, contributing to dimer contacts. The swapping of direction of the N-terminus of LicT-PRD is in agreement with what we observed for the C-terminus of the LicT-CAT domain (Declerck et al., 1999). In this crystal structure, the C-terminal part of the fourth strand in one CAT monomer crosses over to the other monomer.
Based on the crystal structures of LicT-CAT and LicT-PRD, we attempted to construct a model for the intact protein (Figure 7) that could guide us in our understanding of the communication between the two domains. CAT is a homodimer, each monomer consisting of an antiparallel four-stranded β-sheet covered on one side by a long loop between β3 and β4 and packed on the other side to the β-sheet of the other monomer. The RNA-interacting surface is made of residues from both monomers, located at the rim of the sheet and at the start of the long loop (Manival et al., 1997; van Tilbeurgh et al., 1997; Declerck et al., 1999). Since both CAT and PRD form symmetrical dimers, we used their 2-fold axes for aligning the two domains. As can be seen from Figure 7, interactions between CAT and PRD will probably be limited to PRD1. In this model, the RNA-binding site is situated opposite the PRD domain, quite far removed from PRD1. Nevertheless, because of the short length of the linker and the probable domain swapping between CAT and PRD1, any conformational changes taking place within PRD1 are likely to be communicated to the CAT dimer and thereby modify its affinity for RNA.

Fig. 7. Ribbon presentation of the model of the intact LicT protein using the coordinates of the crystal structures of the CAT domain (Declerck et al., 1999) and the PRD (this study). The individual monomers are in yellow and red. The N-terminus (N) of the PRD and the C-terminus (C) of the CAT domain are indicated. The model was created by first aligning the 2-fold axes dimer of the CAT and PRD domains and then approaching at best the C-terminus of CAT and the N-terminus of PRD. The RNA-binding surface as determined by NMR footprinting and site-directed mutagenesis is also indicated (Manival et al., 1997).
Functional conclusions
In vivo and in vitro studies have suggested that at least two mechanisms are at work for controlling the activity of PRD-containing transcriptional regulators: (i) the influence of phosphorylations on the strength and probably the quaternary structure of the dimer; and (ii) tuning of the affinity for the nucleic acid target. Our structure of an activated mutant form of the LicT regulatory domain provides valuable insights into the molecular foundations of these mechanisms. In vivo, the His207-Asp and His269-Asp mutations activate the protein, as expected for the phosphorylation of LicT-PRD2. Assuming that the mutated aspartates behave structurally as phosphohistidines, we conclude that the LicT-PRD-H207D/H269D structure we observe must be closely related to the structure of the regulatory domain doubly phosphorylated on PRD2. The essential observations from this mutant PRD structure are: (i) the PRD in its active form is an extensive dimer; and (ii) the mutagenic aspartates (Asp207 and Asp269) and phosphate-accepting histidines (His100 and His159) are all deeply buried at the dimer interface and therefore inaccessible to phosphorylating enzymes. Despite the extensive buried surface, the nature of the dimer (occluding important cavities) is in accordance with the possibility of a reversible opening of the dimer interface to allow access to (de)phosphorylating partners. From the structure presented here and from biochemical solution studies (N.Declerck, unpublished results), we conclude that the introduction of activating mutations must induce considerable tertiary and quaternary rearrangements in the PRD, resulting in closure of the phosphorylation sites and formation of an active CAT dimer, competent for RNA binding. Similarly, phosphorylation of LicT-PRD2 upon carbon catabolite repression relief would convert an inactive dimer into an active dimer. LicT activation would arise primarily from a conformational switch of conserved residues interacting with the phosphoryl groups, thereby driving the movement of the motif containing helices α1 and α2, whose repositioning stabilizes the dimer interface. Structural dimer rearrangements constitute an elegant mechanism of communication between the protein modules. This type of mechanism may apply to other PRD-containing regulators that are subject to a carbon catabolite control by the PTS and which require HPr-mediated phosphorylation for activation. However, differences may exist in the structural and functional responses associated with the antagonistically acting phosphorylation events taking place in PRDs. Inhibition through EII-dependent phosphorylation, occurring in the absence of inducing sugar, may well proceed through a monomer–dimer switch mechanism, as proposed for the BglG antiterminator (Amster-Choder et al., 1993).
Materials and methods
Antitermination activity assays in B.subtilis
A 1.3 kb HindIII–PstI fragment comprising the wild-type licT gene was PCR amplified from pBGW3 (Schnetz et al., 1996) and cloned into a pBlueScriptII SK+ (Stratagene) derivative carrying the cat gene from pC194. The resulting plasmid (pIC515) was used as template for introducing the H207D mutation (pIC516), the H259D mutation (pIC517) or the H207D/H269D double mutation (pIC518). The nucleotide sequence of all the licT alleles was checked. The plasmids were integrated into the chromosome of B.subtilis strain BGC51 (Lindner et al., 1999), which carries a transcriptional bglP–lacZ fusion inserted at the amyE locus and deletions in sacXY, sacB, sacT and licTS. Transformants were grown with vigorous shaking at 37°C in liquid minimal medium containing glucitol as a carbon source. Expression of the bglP–lacZ fusion was induced by addition of salicin (0.5 g/l), together with glucose (5 g/l) when required. Cell extracts were prepared and β-gal was assayed as previously described (Piggot and Curtis, 1987). Details of the strains, plasmids and media used in this study will be published elsewhere (P.Tortosa, submitted).
Purification of His tag proteins from E.coli
The licT gene fragments encoding the double mutant regulatory domain of LicT, LicT-PRD-H207D/H269D (residues 57–277), were PCR amplified from pIC518 and cloned into QIAexpress pQE30 vector (Qiagen). The His tag protein was produced in E.coli M15-pREP4 and purified on NTA agarose (Qiagen) followed by gel filtration essentially as described previously for LicT-PRD (Declerck et al., 1999), except that the NaCl concentration was lowered from 300 mM for the wild-type to 50 mM for the mutant protein. The purified protein preparation was stored at 4°C after addition of 2 mM dithiothreitol (DTT) and 1 mM benzamidine. Se-Met-substituted protein was obtained by growing the E.coli strain in M9 minimal medium supplemented with appropriate antibiotics and 0.001% biotin. Se-Met (50 mg/ml) as well as amino acids inhibiting methionine synthesis (lysine, phenylalanine and threonine at 100 mg/ml; isoleucine, leucine and valine at 50 mg/ml) were added to the growing cells 30 min before induction with isopropyl-β-d-thiogalactopyranoside (IPTG). Production and purification of the Se-Met-subsituted protein were then performed using the same conditions as for the native protein. The substitution of methionine for Se-Met in the purified protein was verified by MALDI-TOF mass spectrometry performed on Voyager DE-RP.
Crystallization
The purified protein preparation was dialysed against 300 mM NaCl, 5 mM DTT and 10 mM Tris buffer pH 8.0, and concentrated to a final solution of 6 mg/ml. The protein was crystallized using the hanging drop vapour diffusion method. A 3 µl aliquot of protein stock was mixed with 4 µl of well solution containing 200 mM Mg(Ac)2, 100 mM cacodylate buffer pH 5.5, and 10% PEG8000. Crystals grew at room temperature over a few days as flat prisms typically of size 400 × 400 × 30 µm3. The space group is C2221 and the cell edges are: a = 78.25 Å, b = 129.71 Å, c = 50.71 Å. Isomorphous crystals from the Se-Met-substituted protein were obtained under the same conditions as the unsubstituted protein.
Data collection and resolution of the structure
Crystals were soaked for a few seconds in mother crystallization liquor containing extra 30% ethylene glycol and flash frozen under liquid nitrogen before data collection. The structure was solved by MAD phasing using three data sets collected to 2 Å resolution around the Se K edge at the ESRF beamline BM14 (Grenoble, France). Data were integrated using the DENZO image processing package (Otwinowski and Minor, 1997). The Patterson maps were deconvoluted using the program SOLVE (Terwilliger and Berendzen, 1999). The resulting 2 Å phases yielded excellent electron density maps. Model construction used the graphics program TURBO (Roussel and Cambillau, 1991). The model was refined against 1.55 Å resolution data collected on native crystals at the French CRG beamline BM30. These data were completed by a low resolution set collected on a home X-ray source. The refinement was assisted by molecular dynamics using CNS (Brünger et al., 1998). Data collection and refinement statistics are gathered in Table I. The structure has well-defined electron density, except for residues 222 and 223 in a connecting loop in PRD2. All Φ,Ψ angles map to the favoured allowed regions of the Ramachandran plot as defined in PROCHECK (Laskowski et al., 1993). Coordinates have been deposited with the Protein Data Bank (accession No. 1h99).
Acknowledgments
Acknowledgements
We are grateful to J.Janin and S.Aymerich for critical reading of the manuscript. We thank the staff of the BM14 and CRG BM30 beamlines at the ESRF for excellent technical assistance. This work was supported by an MPCV grant of the French Ministère de la Recherche.
References
- Amster-Choder O. and Wright,A. (1992) Modulation of the dimerization of a transcriptional antiterminator protein by phosphorylation. Science, 257, 1395–1398. [DOI] [PubMed] [Google Scholar]
- Amster-Choder O. and Wright,A. (1993) Transcriptional regulation of the bgl operon of Escherichia coli involves phosphotransferase system-mediated phosphorylation of a transcriptional antiterminator. J. Cell. Biochem., 51, 83–90. [DOI] [PubMed] [Google Scholar]
- Aymerich S. and Steinmetz,M. (1992) Specificity determinants and structural features in the RNA target of the bacterial antiterminator proteins of the BglG/SacY family. Proc. Natl Acad. Sci. USA, 89, 10410–10414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachem S. and Stülke,J. (1998) Regulation of the Bacillus subtilis GlcT antiterminator protein by components of the phosphotransferase system. J. Bacteriol., 180, 5319–5326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boss A., Nussbaum-Shochat,A. and Amster-Choder,O. (1999) Characterisation of the dimerisation domain in BglG, an RNA-binding transcriptional antiterminator from Escherichia coli. J. Bacteriol., 181, 1755–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brünger A.T. (1992) Free R value: a novel statistical quantity for assessing the accuracy of crystal structures. Nature, 355, 472–475. [DOI] [PubMed] [Google Scholar]
- Brünger A.T. et al. (1998) Crystallography and NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D, 54, 905–921. [DOI] [PubMed] [Google Scholar]
- Carson M. (1991) RIBBONS 2.0. J. Appl. Crystallogr., 24, 958–961. [Google Scholar]
- Collaborative Computational Project Number 4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D, 50, 760–763. [DOI] [PubMed] [Google Scholar]
- Crutz A.M. and Steinmetz,M. (1992) Transcription of the Bacillus subtilis sacX and sacY genes, encoding regulators of sucrose metabolism, is both inducible by sucrose and controlled by the DegS–DegU signalling system. J. Bacteriol., 174, 6087–6095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Declerck N., Vincent,F., Hoh,F., Aymerich,S. and van Tilbeurgh,H. (1999) RNA recognition by transcriptional antiterminators of the BglG/SacY family: functional and structural comparison of the CAT domain from SacY and LicT. J. Mol. Biol., 294, 389–402. [DOI] [PubMed] [Google Scholar]
- Garrett D.S., Seok,Y.J., Peterkofsky,A., Gronenborn,A.M. and Clore,G.M. (1999) Solution structure of the 40,000 Mr phosphoryl transfer complex between the N-terminal domain of enzyme I and HPr. Nature Struct. Biol., 6, 166–173. [DOI] [PubMed] [Google Scholar]
- Gouet P., Courcelle,E., Stuart,D.I. and Metoz,F. (1999) ESPript: analysis of multiple sequence alignments in PostScript. Bioinformatics, 15, 305–308. [DOI] [PubMed] [Google Scholar]
- Henkin T.M. (2000) Transcription termination control in bacteria. Curr. Opin. Microbiol., 3, 149–153. [DOI] [PubMed] [Google Scholar]
- Henstra S.A., Duurkens,R.H. and Robillard,G.T. (2000) Multiple phosphorylation events regulate the activity of the mannitol transcriptional regulator MtlR of the Bacillus stearothermophilus phosphoenolpyruvate-dependent mannitol phosphotransferase system. J. Biol. Chem., 275, 7037–7044. [DOI] [PubMed] [Google Scholar]
- Holm L. and Sander,C. (1993) Protein structure comparison by alignment of distance matrices. J. Mol. Biol., 233, 123–138. [DOI] [PubMed] [Google Scholar]
- Jones S. and Thornton,J. (1995) Protein–protein interactions: a review of protein dimer structures. Prog. Biophys. Mol. Biol., 63, 31–65. [DOI] [PubMed] [Google Scholar]
- Knezevic I., Bachem,S., Sickmann,A., Meyer,H.E., Stülke,J. and Hengstenberg,W. (2000) Regulation of the glucose-specific phosphotransferase system (PTS) of Staphylococcus carnosus by the antiterminator protein GlcT. Microbiology, 146, 2333–2342. [DOI] [PubMed] [Google Scholar]
- Kraulis, P.J. (1991) MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr., 24, 946–950. [Google Scholar]
- Krüger S., Gertz,S. and Hecker,M. (1996) Transcriptional analysis of bglPH expression in Bacillus subtilis: evidence for two distinct pathways mediating carbon catabolite repression. J. Bacteriol., 178, 2637–2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski R., MacArthur,M., Moss,D. and Thornton,J. (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr., 26, 283–291. [Google Scholar]
- Le Coq D., Lindner,C., Kruger,S., Steinmetz,M. and Stülke,J. (1995) New β-glucoside (bgl) genes in Bacillus subtilis: the bglP gene product has both transport and regulatory functions similar to those of BglF, its Escherichia coli homolog. J. Bacteriol., 177, 1527–1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindner C., Galinier,A., Hecker,M. and Deutscher,J. (1999) Regulation of the activity of the Bacillus subtilis antiterminator LicT by multiple PEP-dependent, enzyme I- and HPr-catalysed phosphorylation. Mol. Microbiol., 31, 995–1006. [DOI] [PubMed] [Google Scholar]
- Mahadevan S. and Wright,A. (1987) A bacterial gene involved in transcription antitermination: regulation at a rho-independent terminator in the bgl operon of E.coli. Cell, 50, 485–494. [DOI] [PubMed] [Google Scholar]
- Manival X., Yang,Y., Strub,M.P., Kochoyan,M., Steinmetz,M. and Aymerich,S. (1997) From genetic to structural characterization of a new class of RNA-binding domain within the SacY/BglG family of antiterminator proteins. EMBO J., 16, 5019–5029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Verstraete I., Charrier,V., Stülke,J., Galinier,A., Erni,B., Rapoport,G. and Deutscher,J. (1998) Antagonistic effects of dual PTS-catalysed phosphorylation on the Bacillus subtilis transcriptional activator LevR. Mol. Microbiol., 28, 293–303. [DOI] [PubMed] [Google Scholar]
- Nussbaum-Shochat A. and Amster-Choder,O. (1999) BglG, the transcriptional antiterminator of the bgl system, interacts with the β′ subunit of the Escherichia coli RNA polymerase. Proc. Natl Acad. Sci. USA, 96, 4336–4341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z. and Minor,W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol., 276, 307–326. [DOI] [PubMed] [Google Scholar]
- Piggot P.J. and Curtis,C.A. (1987) Analysis of the regulation of gene expression during Bacillus subtilis sporulation by manipulation of the copy number of spo–lacZ fusions. J. Bacteriol., 169, 1260–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postma P.W., Lengeler,J.W. and Jacobson,G.R. (1993) Phosphoenol pyruvate:carbohydrate phosphotransferase systems of bacteria. Microbiol. Rev., 57, 543–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roussel A. and Cambillau,C. (1991) TURBO FRODO, Silicon Graphics Geometry Partners Directory. Silicon Graphics.
- Rutberg B. (1997) Antitermination of transcription of catabolic operons. Mol. Microbiol., 23, 413–421. [DOI] [PubMed] [Google Scholar]
- Schirmer T., Bode,W., Huber,R., Sidler,W. and Zuber,H. (1985) X-ray crystallographic structure of the light-harvesting biliprotein C-phycocyanin from the thermophilic cyanobacterium Mastigocladus laminosus and its resemblance to globin structures. J. Mol. Biol., 184, 257–277. [DOI] [PubMed] [Google Scholar]
- Schnetz K., Stülke,J., Gertz,S., Kruger,S., Krieg,M., Hecker,M. and Rak,B. (1996) LicT, a Bacillus subtilis transcriptional antiterminator protein of the BglG family. J. Bacteriol., 178, 1971–1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stülke J. and Hillen,W. (2000) Regulation of carbon catabolism in bacillus species. Annu. Rev. Microbiol., 54, 849–880. [DOI] [PubMed] [Google Scholar]
- Stülke J., Arnaud,M., Rapoport,G. and Martin-Verstraete,I. (1998) PRD—a protein domain involved in PTS-dependent induction and carbon catabolite repression of catabolic operons in bacteria. Mol. Microbiol., 28, 865–874. [DOI] [PubMed] [Google Scholar]
- Terwilliger T.C. and Berendzen,J. (1999) Automated MAD and MIR structure solution. Acta Crystallogr. D, 55, 849–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson J.D., Higgins,D.G. and Gibson,T.J. (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res., 22, 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobisch S., Stülcke,J. and Hecker,M. (1999) Regulation of the lic operon of Bacillus subtilis and characterization of potential phosphorylation sites of the LicR regulator protein by site-directed mutagenesis. J. Bacteriol., 181, 4995–5003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tortosa P., Aymerich,S., Lindner,C., Saier,M.H.,Jr, Reizer,J. and Le Coq,D. (1997) Multiple phosphorylation of SacY, a Bacillus subtilis transcriptional antiterminator negatively controlled by the phosphotransferase system. J. Biol. Chem., 272, 17230–17237. [DOI] [PubMed] [Google Scholar]
- van Tilbeurgh H., Manival,X., Aymerich,S., Lhoste,J.M., Dumas,C. and Kochoyan,M. (1997) Crystal structure of a new RNA-binding domain from the antiterminator protein SacY of Bacillus subtilis. EMBO J., 16, 5030–5036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G., Louis,J.M., Sondej,M., Seok,Y.J., Peterkofsky,A. and Clore,G.M. (2000) Solution structure of the phosphoryl transfer complex between the signal transducing proteins HPr and IIA(glucose) of the Escherichia coli phosphoenolpyruvate:sugar phosphotransferase system. EMBO J., 19, 5635–5649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittekind M., Reizer,J., Deutscher,J., Saier,M.H. and Klevit,R.E. (1989) Common structural changes accompany the functional inactivation of HPr by seryl phosphorylation or by serine to aspartate substitution. Biochemistry, 28, 9908–9912. [DOI] [PubMed] [Google Scholar]