

Abstract
PKR, a member of the eukaryotic initiation-factor 2α (eIF-2α) kinase family, mediates the host antiviral response and is implicated in tumor suppression and apoptosis. Here we show that PKR is regulated by the heat shock protein 90 (Hsp90) molecular chaperone complex. Mammalian PKR expressed in budding yeast depends on several components of the Hsp90 complex for accumulation and activity. In mammalian cells, inhibition of Hsp90 function with geldanamycin (GA) during de novo synthesis of PKR also interferes with its accumulation and activity. Hsp90 and its co-chaperone p23 bind to PKR through its N-terminal double-stranded (ds) RNA binding region as well as through its kinase domain. Both dsRNA and GA induce the rapid dissociation of Hsp90 and p23 from mature PKR, activate PKR both in vivo and in vitro and within minutes trigger the phosphorylation of the PKR substrate eIF-2α. A short-term exposure of cells to the Hsp90 inhibitors GA or radicicol not only derepresses PKR, but also activates the Raf–MAPK pathway. This suggests that the Hsp90 complex may more generally assist the regulatory domains of kinases and other Hsp90 substrates.
Keywords: geldanamycin/Hsp90/MAPK/phosphorylation/ PKR
Introduction
PKR is an interferon-induced, serine/threonine protein kinase (Williams, 1999). It belongs to the family of kinases of the α-subunit of the eukaryotic initiation-factor 2 (eIF-2α) (de Haro et al., 1996), and its activity is dependent on double-stranded RNA (dsRNA) (Robertson and Mathews, 1996). Binding of dsRNA to the N-terminal domain of PKR has been shown to trigger a conformational switch of PKR, resulting in PKR dimerization, autophosphorylation, and phosphorylation of eIF-2α. This structural switch releases the restraint of the N-terminal domain on the C-terminal kinase region, which is then free to bind ATP and to autophosphorylate (Nanduri et al., 2000). The phosphorylation of eIF-2α inhibits the exchange of GDP for GTP and, as a consequence, blocks the formation of the eIF-2–GTP–MettRNA heterotrimeric complex and thus translation (reviewed in Hershey, 1990).
PKR plays an important role during viral infection and is of central importance to the antiviral defense by interferon (Katze, 1995); it has also been implicated in cell growth control and differentiation (Williams, 1999). It is believed to be a tumor suppressor since the expression of dominant-negative mutants of PKR in mouse NIH 3T3 fibroblasts causes malignant transformation (Koromilas et al., 1992; Donzé et al., 1995). Heterologous expression of PKR in yeast also increases phosphorylation of eIF-2α and results in the inhibition of cell growth (Chong et al., 1992). Overexpression of PKR leads to apoptosis through activation of the FADD–caspase-8 death signaling pathway (Balachandran et al., 1998; Donzé et al., 1999). In addition to its role in translation, PKR participates in several transcriptional signaling pathways. For example, PKR mediates the activation of NF-κB by dsRNA (Maran et al., 1994) and contributes to the induction of early genes such as c-fos and c-jun by platelet-derived growth factor (PDGF) (Mundschau and Faller, 1995).
Hsp90 is a highly conserved protein of the heat shock protein family that is expressed at high levels, even under non-stress conditions, and is required for viability in eukaryotes (for review see Buchner, 1999). Hsp90 can act as a molecular chaperone in vitro to promote the refolding of denatured proteins, to hold denatured proteins in a folding-competent state for other chaperones, and to prevent protein unfolding and aggregation (see for example Jakob et al., 1995; Freeman et al., 1996). Hsp90 fulfills its function together with other proteins termed co-chaperones. One of these proteins is an acidic protein called p23, which binds to Hsp90 in an ATP-dependent manner. Its association with the chaperone is prevented by the Hsp90 inhibitor geldanamycin (GA) (Johnson and Toft, 1995). A remarkably large subset of known Hsp90 substrates are signaling molecules, notably kinases and ligand-regulated transcription factors (for example see Schulte et al., 1995; Pratt and Toft, 1997; Louvion et al., 1998).
We have previously reported that proper regulation of the eIF-2α kinase Gcn2 in budding yeast depends on the Hsp90 chaperone complex (Donzé and Picard, 1999). In certain Hsp90 mutant strains, Gcn2 is constitutively activated, which suggests that Hsp90 might act as an inhibitor of Gcn2. Because of the notorious difficulty of isolating Gcn2 from yeast in its inactive form, we decided to investigate this issue in a system more amenable to biochemical analysis. Here, we report that the Hsp90 complex is not only required during the folding and/or maturation of PKR, but subsequently functions as a repressor of PKR. Our results reveal a novel and unexpected activity of the Hsp90 inhibitor GA. By inducing the release of the Hsp90 complex, it activates this kinase.
Results
The toxicity and stability of human PKR are reduced in yeast strains with defective Hsp90 chaperone activity
To investigate the potential role of the molecular chaperone Hsp90 (Hsp82 and Hsc82 in budding yeast) in the maturation and/or regulation of the human kinase PKR, we took advantage of budding yeast as a genetic ‘test tube’. PKR overexpression in Saccharomyces cerevisiae causes a dramatic inhibition of protein synthesis and growth (Chong et al., 1992). We postulated that if PKR is dependent on Hsp90 and its co-chaperones, its folding and thus its activity (inhibitory effect on growth) should be affected in strains carrying mutations in HSP90 or in Hsp90 co-chaperone genes.
The cDNAs encoding wild-type and a kinase-defective mutant (K296R) of human PKR were expressed under the control of the galactose-inducible GAL1 promoter in different yeast strains. To lower the strength of the promoter, cells were grown in galactose-containing medium supplemented with 0.1% glucose. Under these conditions, a PKR-dependent difference in growth was observed between the wild type (wt) and the Hsp90 mutant strains, suggesting that PKR activity is dependent on Hsp90 (Figure 1A). We extended the analysis to strains lacking different co-chaperones of Hsp90. The toxicity of PKR is markedly reduced (Figure 1A) in yeast strains lacking the yeast homolog of human p23 (Δsba1) (Bohen, 1998; Fang et al., 1998), with a temperature-sensitive (ts) mutant of Cdc37 (cdc37-34) (Kimura et al., 1997) and expressing the Hsp90 mutants G313S or A587T (Nathan and Lindquist, 1995).

Fig. 1. PKR toxicity and accumulation are reduced in yeast strains with defective Hsp90 chaperone activity. (A) Slow growth of yeast strains expressing PKRwt is attenuated by alterations in the Hsp90 machinery. The different yeast strains containing pYES/PKRwt were grown for 8 h at 30°C under repressed (2% glucose) or partially induced (2% galactose plus 0.1% glucose) conditions. Growth was monitored by measuring OD600 and for each strain normalized to the growth on 2% glucose. (B) Defective PKR accumulation in cdc37-34 and Δsba1 strains. Strains containing the plasmids pYES/PKRwt or PKRK296R were cultured for 16 h on 2% galactose to induce expression of the proteins. PKR and Hsp82 were revealed by western blotting with anti-PKR and Hsp82 antibodies, respectively. Lanes 1, 2, 4, 5, 7 and 8, strains transformed with the plasmid pYES/PKRwt; lanes 3, 6 and 9, strains transformed with the plasmid pYES/PKRK296R.
Many, if not all, Hsp90-dependent proteins are destabilized and degraded by proteasomes when the activity of the Hsp90 complex is impaired (Nathan and Lindquist, 1995; Stancato et al., 1997). Therefore, we monitored the levels of PKR by immunoblot analysis. In a wild-type strain, wild-type PKR (PKRwt) is detected, albeit at low levels, upon galactose induction (Figure 1B, lane 2), whereas the kinase-defective mutant of PKR (PKRK296R) is expressed at higher levels (lane 3). This difference in expression is due to the down-regulatory effect of PKR on its own translation (Thomis and Samuel, 1992). In the Δsba1 and cdc37-34 strains, the levels of both wild-type and mutant PKR are lowered compared with the wt strain, as seen by immunoblot analysis (Figure 1B). Taken together, these data strongly suggest a role for Hsp90 and its cohort of co-chaperones in the folding, maturation and/or stabilization of human PKR in yeast.
GA decreases levels and activity of PKR in mammalian cells
PKR is an exogenous Hsp90 substrate in yeast. By using the specific Hsp90 inhibitor GA, we examined whether Hsp90 is also required for the maturation of PKR in different mammalian cell lines. GA strongly binds to the ATP binding pocket of Hsp90 (Stebbins et al., 1997), thereby blocking its ATPase activity and causing the misfolding and degradation of a variety of its substrates (Smith et al., 1995; Roe et al., 1999). As shown in Figure 2A, the addition of 1 µM GA to HeLa cells for 16 h leads to a decrease of endogenous PKR to almost undetectable amounts. Since the half-life of PKR is 6–7 h (Hovanessian et al., 1987), this long treatment mainly affects PKR made de novo.
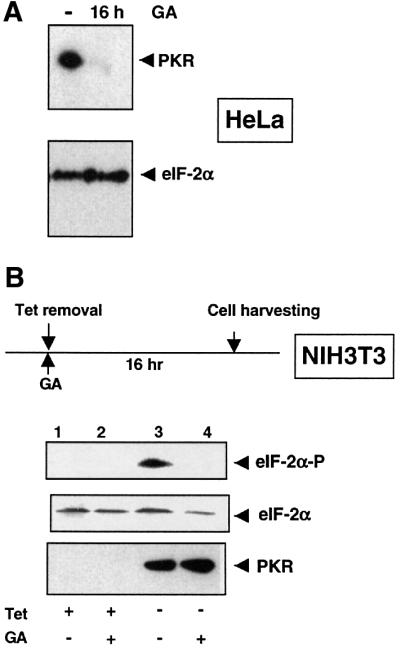
Fig. 2. GA decreases PKR levels and activity in mammalian cells. (A) Reduction of PKR protein levels upon GA treatment. HeLa cells were cultured for 16 h with or without 1 µM GA. Endogenous PKR and eIF-2α protein levels were revealed by immunoblot analysis with antibodies against PKR and eIF-2α, respectively. (B) Inhibition of PKR activity by GA in vivo. Overexpression of human PKR was induced in NIH 3T3-PKRwt in the presence or absence of 1 µM GA. Aliquots of cell lysates were examined by immunoblotting for the phosphorylation of eIF-2α with a phosphospecific anti-eIF-2α antibody, for total eIF-2α with an anti-eIF-2α antibody and for the presence of human PKR with an anti-PKR antibody. Tet, tetracycline.
To study the effect of GA on in vivo PKR activity, we monitored the phosphorylation of its natural substrate, eIF-2α, by immunoblot analysis using an antibody specific for the phosphorylated form of eIF-2α. We took advantage of NIH 3T3 cell lines, which overexpress either wild-type or mutant (K296R) human PKR from a tetracycline-repressed promoter (Donzé et al., 1999). Removal of tetracycline for 16 h induces expression of PKR and phosphorylation of eIF-2α (Figure 2B, compare lanes 1 and 3). Addition of GA to the cells while PKR is being synthesized reduces PKR activity, as seen by the absence of phosphorylated eIF-2α (Figure 2B, compare lanes 3 and 4). In contrast to the endogenous PKR in HeLa cells (Figure 2A), degradation of the human PKR in the presence of GA is not apparent in NIH 3T3 cells at steady state, presumably because of its massive overexpression, and yet its activity is defective (Figure 2B). Thus, human PKR requires an intact Hsp90 machinery both for its stabilization and to become an active kinase.
PKR forms a complex with Hsp90 and p23 in vivo
The experiments performed in yeast and mammalian cells indicate that PKR is a ‘client’ of the molecular chaperone Hsp90. Several Hsp90-dependent kinases, including Raf-1, v-Src and Gcn2, have been reported to be associated with Hsp90 (Pratt, 1997; Donzé and Picard, 1999). We performed co-immunoprecipation experiments using a monoclonal antibody against Hsp90. As presented in Figure 3A, PKR co-immunoprecipitates with Hsp90. A Flag-tagged version of PKR overexpressed in 293T cells is also found in a complex with Hsp90 (Figure 3B), as is the kinase-defective mutant (PKRK296R) expressed in our NIH 3T3 cell line (data not shown). This result is specific for PKR since we could not observe any interactions between Hsp90 and other unrelated kinases (data not shown).

Fig. 3. Hsp90 and its co-chaperone p23 form a complex with PKR in vivo. (A) Hsp90 co-precipitates with endogenous (upper panel) and overexpressed PKR (lower panel). Hsp90 immunoprecipitates from HeLa cell extracts were analyzed by western blotting with an anti-PKR antibody. Plasmid CKF/PKRwt was used to overexpress PKRwt in 293T cells. (B) PKR co-precipitates with p23. Endogenous p23 was immunoprecipitated from HeLa cell extracts with a monoclonal antibody against p23, and PKR, Hsp90 or p23 was detected by immunoblot analysis using anti-PKR, Hsp90 or p23 antibodies, respectively (upper panel). NIH 3T3-PKRK296R cells expressing the kinase-defective mutant PKRK296R were grown in the absence of tetracycline for 16 h and then cell lysates were subjected to immunoprecipitation experiments using an anti-p23 antibody (lower panel). C, unrelated monoclonal antibody (anti-β-galactosidase) as a negative control for immunoprecipitation; IP, immunoprecipitated proteins; input, total proteins.
Hsp90, as well as endogenous PKR, co-immunoprecipitates with endogenous p23 from HeLa cell extracts (Figure 3B). The kinase-defective mutant PKRK296R, exogenously expressed in NIH 3T3 cells, is also found associated with p23 (Figure 3B, lower panel), as is transiently expressed Flag-PKR (data not shown). This set of interaction experiments indicates clearly that PKRwt or mutant is specifically associated in vivo with the Hsp90–p23 complex.
PKR dissociates from Hsp90–p23 upon activation with dsRNA
One of the best characterized activators of PKR is dsRNA produced during viral infection (Katze, 1995). Upon direct binding of dsRNA to PKR, major structural changes occur in PKR, leading to its dimerization and activation (Nanduri et al., 2000). To follow the fate of the endogenous PKR–Hsp90 complex upon dsRNA treatment in vivo, we treated HeLa cells for 15 min with a synthetic dsRNA (pIC) (Gunnery and Mathews, 1998). Remarkably, this strongly reduces the co-precipitation of endogenous PKR with Hsp90 (Figure 4A). We performed the same experiment using the anti-p23 antibody, and could also observe a dissociation of PKR and p23 in HeLa cells (data not shown). This also holds true for NIH 3T3 cells expressing the kinase-defective mutant PKRK296R under a tetracycline-regulated promoter, indicating that kinase activity of PKR is not required for this effect (Figure 4B and C). dsRNA at 1 µg/ml for 15 min already affects the complex, with a peak at ∼100 µg/ml dsRNA, a concentration that corresponds to the optimal dsRNA levels used for PKR activation (Offermann et al., 1995). To follow the kinetics of complex disruption, we treated the cells with dsRNA for different time periods, as indicated in Figure 4C. Within 2 min, we observed a decrease in PKRK296R binding to p23, with a maximal effect at 15 min. The kinetics is identical with PKRwt (data not shown). Interestingly, the kinetics of PKR activation by dsRNA, indicated by the phosphorylation of eIF-2α (Figure 7A), parallels the disruption of the PKR–molecular chaperone complex. PKR can also be activated by arsenite or H2O2, although the mechanisms of activation are unknown (Ito et al., 1999). We found that a 15 min incubation with 1 mM H2O2 disrupts the complex (data not shown). These data indicate that Hsp90 forms a complex with PKR in vivo that is released upon PKR activation.
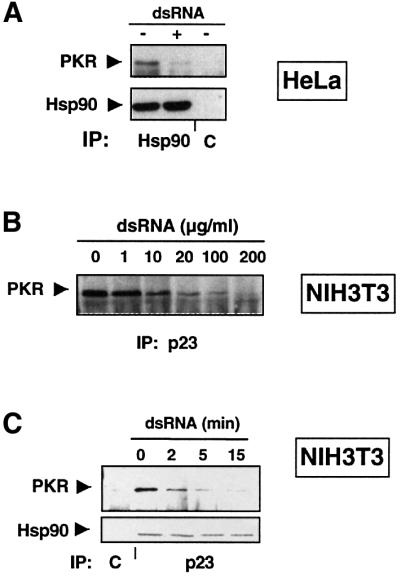
Fig. 4. dsRNA triggers the dissociation of PKR from Hsp90–p23. (A) Disruption of the Hsp90–PKR complex by dsRNA. HeLa cells were cultured with or without 100 µg/ml dsRNA for 15 min prior to lysis and immunoprecipitation with an anti-Hsp90 antibody. The immunoprecipitates were analyzed for the presence of Hsp90 and PKR by immunoblotting. (B) Dissociation of the PKR–p23 complex by dsRNA is dose dependent. After 16 h induction of PKRK296R, cells were treated for 15 min with increasing dsRNA concentrations as indicated. Cell extracts were immunoprecipitated with an anti-p23 antibody and PKR was detected by western blotting. (C) Time course analysis of the dissociation of the p23–PKR complex by dsRNA. PKRK296R was induced by removal of tetracycline. Cells were incubated with 100 µg/ml dsRNA for different periods of time as indicated, and processed as in (B).

Fig. 7. Hsp90 inhibitors activate PKR in vivo and in vitro. (A) GA and radicicol trigger eIF-2α phosphorylation. HeLa cells were incubated with 1 µM GA, 100 µg/ml dsRNA or 5 µM radicicol for different periods of time as indicated. The phosphorylation status of eIF-2α was examined by immunoblotting with a phosphospecific anti-eIF-2α antibody and total eIF-2α with a monoclonal antibody against eIF-2α. (B) GA induces PKR dimerization. 293T cells were transfected with the plasmids CKF/PKRK296R and CK/Nter encoding the Flag-tagged full-length kinase and the untagged N-terminal region, respectively. Prior to cell harvesting, dsRNA (100 µg/ml) or 1 µM GA was added for different periods of time as indicated. Cell lysates were immunoprecipitated with an anti-Flag antibody (Flag IP) and association with the PKR N-terminus was observed by immunoblot anaysis with an antibody against the N-terminus (N-18). (C) GA promotes PKR autophosphorylation. Extracts from 293T cells, 24 h after transfection with CKF/PKR, were immunoprecipitated with an anti-Flag antibody. Autophosphorylation of Flag-PKR was revealed using a phosphospecific anti-PKR antibody. Cells were treated with 1 µM GA or dsRNA (100 µg/ml) for 15 min prior to cell harvesting. The efficiency of immunoprecipitation was monitored with an anti-Flag antibody. (D) GA activates PKR in vitro. Flag-PKR expressed for 24 h in 293T cells was immunoprecipitated and subjected to an in vitro kinase assay in the absence or presence of GA (5 µM) or dsRNA (50 ng/ml). Coomassie, levels of Flag-PKR were visualized by Coomassie Blue staining; [32P] (kinase assay), labeled phosphate incorporation was detected by autoradiography.
GA triggers the dissociation of the PKR–chaperone complex
We investigated whether inhibition of Hsp90 by GA could affect the integrity of the PKR–Hsp90–p23 complex. Addition of 1 µM GA to HeLa cells 15 min prior to cell lysis is sufficient to dissociate Hsp90 from PKR (Figure 5A). Likewise, overexpressed PKRK296R dissociates from p23 upon short exposure to GA (Figure 5B).

Fig. 5. GA induces the dissociation of Hsp90–p23 from PKR. (A) Hsp90 dissociation from PKR in the presence of GA. HeLa cells were incubated with or without 1 µM GA for 15 min prior to lysis and immunoprecipitation with an anti-Hsp90 antibody. (B) Short- and long-term effects of GA on the p23–PKR complex. GA (1 µM) was added to the medium either during PKRK296R synthesis (for 16 h) or after completion of its synthesis (for 15 min). Immunoprecipitations were performed with an anti-p23 antibody, and PKR and Hsp90 were visualized by immunoblot analysis. C, control antibody.
In contrast, when PKR was synthesized in the presence of GA (16 h incubation) and immunoprecipitated using an anti-p23 antibody, we found PKR associated with p23 (Figure 5B). Under these conditions, immunoprecipitation of p23 did not reveal any bound Hsp90. This suggests that p23 may bind to misfolded PKR, as previously reported for the Hsp90 client reverse transcriptase from hepatitis B virus (Hu et al., 1997).
Hsp90 and p23 bind to the regulatory and kinase domains of PKR
In order to map the region of PKR required for Hsp90 binding, we constructed deletion mutants and tested them for their interaction with Hsp90 and p23. We generated an N-terminal variant (Flag-Nter; amino acids 1–243) containing the two dsRNA binding motifs (Schmedt et al., 1995), and deleted the N-terminal part of PKR to obtain the PKR kinase domain alone (Flag-Cter; amino acids 260–551). These truncated proteins, as well as the full-length, kinase-defective mutant PKRK296R, were expressed in 293T cells, immunoprecipitated with an anti-Flag antibody, and revealed, along with p23, by western blotting. All constructs are expressed at similar levels (Figure 6). Surprisingly, p23 and Hsp90 bind to the N-terminal domain of PKR as well as to full-length PKR (Figure 6 and data not shown). We have shown above that the Hsp90–p23 complex is released from PKR upon dsRNA treatment. Remarkably, this could be reproduced with the separated N-terminal domain of PKR (Figure 6A). Results discussed above suggested that the C-terminal moiety encompassing the kinase domain also depends on Hsp90 function. However, interaction of the C-terminal domain with the Hsp90–p23 complex must be of a different nature (see also Supplementary data available at The EMBO Journal Online). While it does not co-immunoprecipitate with p23 (Figure 6A) or Hsp90 (data not shown) with our standard buffer conditions, it does co-immunoprecipitate with Hsp90 in an ATP-sensitive fashion using a lysis buffer devoid of potassium (Figure 6B). Thus, Hsp90 differently associates with two structurally and functionally separate domains of PKR.

Fig. 6. The N- and C-terminal domains of PKR have differential requirements for the Hsp90–p23 complex. (A) Mapping of the chaperone binding region of PKR. 293T cells were transfected with plasmids CKF, CKF/PKRK296R, CKF/Nter or CKF/Cter. Prior to cell harvesting (after 36 h), dsRNA (100 µg/ml) was added for 15 min where indicated. Cell extracts were immunoprecipitated with an anti-Flag (M2) antibody. Immunoprecipitates were examined for the presence of the different Flag-tagged variants and for p23 using anti-PKR and p23 antibodies, respectively. (B) Differential binding of the N- and C-terminal domains and full-length PKR to Hsp90. Extracts fom 293T cells transfected with plasmids CKF/PKRK296R, CKF/Nter or CKF/Cter were immunoprecipitated in a buffer without potassium (see Materials and methods) in the presence or absence of 1 mM ATP with an anti-Hsp90 antibody.
GA triggers eIF-2α phosphorylation, dimerization and autophosphorylation of PKR in vivo
Treating cells with dsRNA leads to the disruption of the PKR–Hsp90 complex concomitant with PKR activation. We wondered whether dissociation of the chaperone complex from the kinase is sufficient for its activation. To answer this question, we took advantage of the rapid dissociation that occurs upon addition of GA (Figure 5). We first monitored in vivo eIF-2α phosphorylation by incubating Hela cells with GA for short periods of time. A 5 min treatment with GA is sufficient to detect eIF-2α phosphorylation using an antibody specific for the phosphorylated form of eIF-2α (Figure 7A). The phosphorylation of the translation factor is even stronger than with dsRNA under the same conditions. To rule out an Hsp90-independent mechanism, we used a structurally and chemically different Hsp90 inhibitor, the macrocyclic drug radicicol, known to bind to Hsp90 with high specificity and to inhibit its ATPase activity (Roe et al., 1999; Schulte et al., 1999). A 5 min treatment with radicicol is sufficient to induce detectable eIF-2α phosphorylation as observed for GA (Figure 7A). Although consistent with the activation of PKR by GA and radicicol, any of the four mammalian eIF-2α kinases (HRI, Gcn2, PERK and PKR) (Kaufman, 1999) could contribute to the observed effect. We assessed the activation of PKR upon GA addition using two criteria: dimerization and autophosphorylation. The dimerization of PKR upon binding of dsRNA is a prerequisite for kinase activation (Tan et al., 1998), and requires the N-terminal domain of PKR (Cosentino et al., 1995). To monitor dimerization, we co-expressed the Flag-tagged full-length PKR with the untagged N-terminus of PKR (1–243) in 293T cells. After immunoprecipitation using the anti-Flag antibody, we followed association of the N-terminus with full-length Flag-PKR. As shown in Figure 7B, the N-terminal domain dimerizes with full-length Flag-PKR when cells are incubated with dsRNA for 5 min prior to immunoprecipitation. The association persists for at least 1 h. In the same experiment, we could also observe dimerization with endogenous PKR (data not shown). When cells are incubated with GA, dimerization of PKR is triggered with the same kinetics. The lower extent of activation could be explained by the absence of dsRNA, which is known to stabilize the dimer (Wu and Kaufman, 1997). Upon activation, PKR is autophosphorylated at several sites (Romano et al., 1998). Thr451 is a crucial phosphorylation site for PKR activity (Romano et al., 1998). Using the antibody specific for phosphorylated Thr451, we followed PKR activation upon treatment with dsRNA or GA. Twenty-four hours after transfection with CKF–PKR, 293T cells were incubated for 15 min with either dsRNA or GA and immunoblotting was then performed with the cell extracts. Surprisingly, immunoprecipitated Flag-PKR is not only autophosphorylated in the presence of dsRNA but also of GA (Figure 7C). Taken together, these results indicate that GA-induced inhibition and release of Hsp90 are sufficient to activate PKR. However, these experiments do not rule out the possibility that other eIF-2α kinases are also activated by this compound.
GA activates PKR in vitro
To confirm the unexpected activation of PKR by GA, we determined whether this compound can also activate PKR in vitro. We performed an in vitro kinase assay on immunopurified PKR. Flag-PKRwt, expressed for 24 h in transfected 293T cells, was immunoprecipitated with an anti-Flag antibody and subjected to a kinase assay in the presence of [γ-32P]ATP (Figure 7D). Without any treatment, the immunopurified kinase is weakly active. Addition of dsRNA during the kinase assay increases incorporation of the label. As observed in vivo, the Hsp90 inhibitor GA is a good activator of PKR in vitro. The result was confirmed with endogenous PKR from HeLa cells, immunoprecipitated with an anti-PKR polyclonal antiserum (data not shown).
Inhibitors of Hsp90 activate the Raf–MAPK pathway
Could the GA-induced release from Hsp90-mediated repression observed with PKR be extended to other kinases? We decided to investigate the mitogen-activated protein kinase (MAPK) signaling pathway that is defined by Raf, a MAPK kinase kinase known to be dependent on Hsp90 for stability and activity (Schulte et al., 1995). Upon activation by extracellular signals, Raf activates MAPKK, which in turn phosphorylates and activates the MAPKs ERK1 and ERK2 (Shields et al., 2000). Incubation for several hours with Hsp90 inhibitors such as GA (Stancato et al., 1997) or radicicol (Soga et al., 1998) leads to the destabilization and inactivation of Raf. As in the case of PKR, Hsp90 is known to be rapidly released from Raf upon GA treatment (Schulte et al., 1995). We incubated HeLa cells for different periods of time with either 5 µM GA or radicicol or 10 ng/ml epidermal growth factor (EGF), and monitored the activation of Raf indirectly using a phosphospecific anti-ERK antibody. Upon treatment with EGF, the MAPK pathway is activated within minutes (Figure 8). Surprisingly, GA (Figure 8) or radicicol (data not shown), in the absence of other stimuli, is sufficient to induce the phosphorylation of ERK. This effect is not due to PKR, since ERK is not activated by different PKR activators such as arsenite or dsRNA (data not shown and Harcourt and Offermann, 2001). These results indicate that the activation of PKR–eIF-2α by Hsp90 inhibitors is not peculiar to this signaling, but applies to other Hsp90-dependent pathways such as Raf–MAPK.

Fig. 8. GA activates the Raf–MAPK pathway. HeLa cells were incubated with 10 ng/ml EGF and 5 µM GA for different periods of time as indicated. The phosphorylation status of ERK was examined by immunoblotting with a phosphospecific anti-ERK antibody and total ERK with an anti-ERK antibody.
Discussion
Using genetic and biochemical approaches, we have demonstrated that the Hsp90–p23 complex has a dual function for PKR. It is first required for maturation and accumulation of the kinase, and it then represses the enzyme until its activation. Moreover, we demonstrate that the inhibitory role of Hsp90 applies to other Hsp90-dependent kinase pathways, such as the Raf–MAPK pathway.
Hsp90 is required for maturation and activity of the kinase PKR
The first set of data presented in this study demonstrates that PKR requires the Hsp90 molecular chaperone complex to reach an activatable state. (i) PKR toxicity and accumulation are reduced in budding yeast with alterations in the Hsp90 machinery; (ii) incubation of mammalian cells with the Hsp90 inhibitor GA for several hours prevents accumulation and activation of PKR; (iii) PKR forms a complex with Hsp90 and its co-chaperone p23 in vivo; and (iv) this complex is rapidly disrupted upon incubation with GA. Similar observations have been reported for other Hsp90-dependent kinases such as Gcn2, Raf-1 and pp60v-src (Whitesell et al., 1994; Schulte et al., 1997; Donzé and Picard, 1999).
p50cdc37 is a co-chaperone that has been suggested to direct Hsp90 to kinases such as Raf-1, CDK4 (Stepanova et al., 1996; Grammatikakis et al., 1999) and Ste11 in yeast (Abbas-Terki et al., 2000). The observation that the toxicity of PKR in the cdc37 mutant strain is attenuated suggests a role for this co-chaperone in the folding and maturation of PKR. Likewise, the deletion of the p23 gene (Δsba1) in yeast leads to a failure of PKR to accumulate. This is consistent with the biochemical results obtained in mammalian cells. They indicate a role for the small acidic co-chaperone in PKR maturation. Upon a short (15 min) GA treatment, PKR, as well as Hsp90, dissociates from p23. In contrast, addition of GA throughout synthesis (16 h) inhibits PKR kinase function, disrupts the association with Hsp90 but not with p23, promotes its misfolding, and ultimately its degradation. The Hsp90-independent binding of p23 has already been observed for the hepatitis B virus reverse transcriptase (Hu et al., 1997). For PKR, we speculate that during the short treatment with GA, mature and completely folded PKR dissociates from both Hsp90 and p23 (see below), whereas the presence of GA during the de novo folding and maturation of PKR leads to the transient accumulation of misfolded PKR, which may be recognized by the chaperone p23 in the absence of Hsp90.
Hsp90 acts as a kinase repressor
For the first time, we present pharmacological evidence that Hsp90 is a repressor of a kinase. Addition of GA or dsRNA to cells leads to a rapid (within minutes) release of Hsp90 from PKR, concomitant with the activation (dimerization and autophosphorylation) of the kinase and phosphorylation of its substrate eIF-2α. To exclude the theoretical possibility that GA might act through a mechanism unrelated to Hsp90, we have demonstrated the induction of eIF-2α phosphorylation with radicicol, another specific Hsp90 inhibitor that is functionally and structurally distinct from GA (Roe et al., 1999). Most importantly, GA also activates immunopurified PKR, confirming that the observed activation in vivo occurs directly at the level of the kinase–chaperone complex. Moreover, dose–response experiments show that the optimal dsRNA concentrations used to disrupt the Hsp90–PKR complex (50–100 µg/ml) correspond to the amounts of dsRNA leading to maximal activation of PKR in vivo (Offermann et al., 1995). Finally, these results match perfectly with the data we reported recently on another member of the eIF-2α kinase family, Gcn2 of budding yeast: in certain Hsp90 mutant strains, Gcn2 is constitutively activated in the absence of its activating stimulus (amino acid starvation) (Donzé and Picard, 1999). We hypothesized that the activity of these Hsp90 mutants is only partially defective and still supports proper folding, but not repression.
The unexpected activation of PKR by GA was explored further in another Hsp90-dependent pathway, the Raf–MAPK pathway. For Raf, treatment of mammalian cells with GA for several hours has been reported to decrease kinase activity and induce rapid degradation by the proteasome (Schulte et al., 1997). Here we show that a short treatment with GA, known to dissociate Hsp90 from Raf (Schulte et al., 1995), activates the MAPK pathway leading to the phosphorylation of ERK. Together with the PKR results, these data demonstrate that the molecular chaperone Hsp90 not only plays a role in the maturation of these kinases, but subsequently acts as a repressor that dissociates from these kinases upon activation. A similar activating effect of GA on an Hsp90 substrate has previously only been reported for the heat shock transcription factor HSF1 (Zou et al., 1998).
Model
PKR binds Hsp90 and several co-chaperones during the maturation step (Figure 9). This interaction is most likely to be direct, but this remains to be established. Inhibition of the Hsp90 machinery during this step by addition of GA or by mutations of genes encoding the Hsp90 machinery interferes with the maturation of PKR. The protein is not folded properly and is degraded by the proteasome. After completion of folding (Figure 9, mature inactive), the kinase remains bound to Hsp90, maintained in a repressed state. Activation of PKR by dsRNA releases Hsp90 (Figure 9, activation step). dsRNA mediates a conformational change of PKR without a requirement for the kinase activity since the chaperone is released from the isolated N-terminal domain by dsRNA as well as from a kinase-defective mutant. Addition of GA to mature PKR mimics dsRNA by dissociating the kinase from Hsp90 and switches it to an active conformation.

Fig. 9. Model for Hsp90 regulation of PKR. GA blocks the maturation of PKR and activates the mature enzyme. See Discussion for details. dsRNA is represented as a double-stranded helix, and dsRNA binding motifs as small open boxes.
PKR contains two independent domains, the N-terminal domain containing two dsRNA binding motifs (dsRBDs) and the C-terminal kinase domain. A recent study suggests that dsRBD2 interacts with the catalytic domain, locking PKR into a closed and inactive conformation (Nanduri et al., 2000). Association of dsRNA with the dsRBDs releases PKR from its inhibitory state. This view of PKR activation by relieving repression of some inhibitory sequences present in cis in the N-terminal region is supported by several studies. Deletion of the entire N-terminal region is sufficient to activate PKR constitutively (Wu and Kaufman, 1997). In addition, ATP becomes associated with PKR only after exposure to dsRNA (Galabru and Hovanessian, 1987). Recently, Wek and collaborators have demonstrated that removal of small segments near the dsRBD2 is sufficient to activate PKR in the absence of dsRNA (Vattem et al., 2001). Finally, PKR undergoes a conformational change in the presence of dsRNA, acquiring a more elongated structure (Carpick et al., 1997). To our surprise, Hsp90 binding sites lie in the N-terminal regulatory region as well as in the kinase domain of PKR. We propose that Hsp90 enhances the inhibition by the PKR N-terminus, either by increasing the affinity between the N-terminal inhibitory region and the kinase domain or by covering the kinase moiety together with the N-terminal domain. However, Hsp90 is not directly involved in bridging the two domains, since this interaction occurs with recombinant domains purified separately (Nanduri et al., 2000). We postulate that this model also applies to the Gcn2 regulation scheme that has recently been proposed by Hinnebusch and collaborators (Dong et al., 2000). Hsp90 might help the bipartite tRNA binding domain of Gcn2 to inhibit the kinase moiety in vivo.
Hsp90 binds to many proteins involved in signaling, proliferation and cell cycle control, but the common structural motifs required for Hsp90 binding are still unknown (Buchner, 1999). Many Hsp90 substrates contain two functionally independent domains, e.g. a catalytic and a regulatory domain. We propose that Hsp90 assists regulatory domains in performing their tasks. It will typically be released from inhibitory domains upon activation. Alternatively, its continued assistance might be required to maintain a functionally competent conformation, as in the case of the hepatitis B virus reverse transcriptase (Hu and Anselmo, 2000).
In conclusion, we demonstrate that the kinase PKR is a client protein of Hsp90, and that the Hsp90 inhibitors and anti-cancer drugs GA and radicicol are kinase activators upon short-term exposure. Short-term activation and long-term inhibition of kinases by GA are thus only apparent discrepancies. However, they need to be studied further to reach a better understanding of the anti-tumorigenic effects of Hsp90 inhibitors. Since PKR activation can lead to apoptosis (Balachandran et al., 1998; Donzé et al., 1999), it will be interesting to determine whether the activation of PKR by GA contributes to the pro-apoptotic effects of GA in cancer treatment (Gan et al., 1998; Lopez-Maderuelo et al., 2001).
Materials and methods
Plasmids
PKR plasmids for yeast. PKR was expressed in yeast from the galactose-inducible expression plasmid pYES2 (Invitrogen). The cDNAs for PKRwt and the kinase-defective mutant K296R were inserted into the BamHI–XhoI sites of pYES2 as PCR-generated BamHI–XhoI fragments from plasmids p10-3 PKRwt and PKRK296R (Donzé et al., 1999) (with the sequence GGATCCGAA preceding the AUG of PKR) to create constructs pYES/PKRwt and pYES/PKRK296R, respectively.
PKR plasmids for mammalian expression. PKR was expressed in mammalian cells from plasmids CK or CKF. The expression vector CK was created from pEGFP-C1 (Clontech) by excision of EGFP as a NheI–BglII fragment, treatment with Klenow and re-ligation. CKF is a version of CK containing a Flag epitope for N-terminal fusions. The Flag epitope (CAAAGCATGGACTACAAGGACGACGATGACAAGGGGATCC) was cloned into the SacI–EcoRI sites of CK. CKF/PKRwt and PKRK296R were created by ligation of blunt-ended BamHI–XhoI inserts from pYES/PKRwt and K296R, respectively, into the SmaI site of CKF. For expression of the Flag-tagged version of the N-terminal domain of PKR (CKF/Nter), a BglII fragment was removed from CKF/PKRwt, followed by blunt-ended re-ligation to create a truncated variant encoding amino acids 1–243 with two additional amino acids (Arg and Ser) at the C-terminus. To generate a vector for the untagged version of the Nter variant of PKR (called CK/Nter), a BamHI insert from CKF/Nter was ligated into the BamHI site of the vector CK. The expression vector for the Flag-tagged Cter kinase domain was generated by inserting a blunt-ended AccI–XhoI fragment of CKF/PKRwt into the SmaI site of CKF to create CKF/Cter.
Cell culture and transient transfection
293T, HeLa and NIH 3T3 cells were grown in Dulbecco’s modified Eagle’s medium (DMEM; Gibco) supplemented with 5% fetal calf serum (FCS; Gibco). For transient transfection, cells were passaged onto 6-cm plates 3 h prior to calcium phosphate transfection with 5 µg of plasmids. Cells were harvested and lysed 24–36 h later. The NIH 3T3 cells expressing PKR under a tetracycline-repressed promoter have been described (Donzé et al., 1999). To induce expression of PKR, cells were washed twice with Tris-buffered saline (TBS) to remove tetracycline (1 µg/ml) and fed with fresh DMEM containing 5% FCS. Where indicated, 1 µM GA (Drug synthesis and Chemistry Branch, NCI, NIH) or 5 µM radicicol (Sigma) dissolved in dimethylsulfoxide (DMSO) was added directly to the medium. DMSO was used as a negative control. PKR was activated with poly(I)⋅poly(C) (pIC; Sigma) added directly to the cell culture medium. For ERK activation, HeLa cells were cultured for 7 h in serum-free DMEM prior to treatment with 10 ng/ml EGF (Sigma) or 5 µM GA.
Yeast strains
pYES/PKRwt or pYES/PKRK296R was introduced into the wild-type (RMY326) and the non-isogenic mutant strains iA587T and iG313S, 8A7 and YNK234 containing mutated versions of HSP82 (Nathan and Lindquist, 1995), a ts mutation cdc37-34 (Fliss et al., 1997) and a deletion of SBA1 (Bohen, 1998), respectively. Plasmids were introduced into yeast by the LiAc/PEG method and selected for on appropriate minimal media.
Immunoprecipitation experiments
All immunoprecipitations were performed in the same lysis buffer A (20 mM Tris–HCl pH 7.5, 100 mM NaCl, 30 mM KCl, 20 mM sodium molybdate, 1 mM EDTA, 10% glycerol, 0.5% NP-40, protease inhibitor mix), except for the immunoprecipitations between Hsp90 and the Flag variants of PKR, where the buffer A′ (buffer A plus 130 mM NaCl and without KCl) was used. Five hundred micrograms to 1 mg of cell extracts were incubated with the appropriate antibodies: the monoclonal anti-Hsp90 (H90-10) and anti-human p23 (JJ3) were a kind gift from Dr David O.Toft (Mayo Clinic). Anti-Flag immunoprecipitations were carried out with the anti-M2 antibody (Sigma). PKR was detected using the polyclonal antibodies anti-PKR K17 or N-18, directed against the C- or the N-terminus, respectively (Santa-Cruz), or with a polyclonal anti-PKR antibody (a kind gift of Mike Mathews). The antibodies against the phosphorylated form of eIF-2α (Research Genetics) and against the phosphorylated Thr451 of PKR (Transduction Laboratories) were used as specified by the manufacturer. For control experiments, unrelated monoclonal antibodies were used: anti-β-galactosidase (Promega) or anti-estrogen receptor antibodies. Activation of ERK1/2 was monitored with the phosphospecific anti-Erk1/2 antibody (Promega) and total ERK2 proteins were revealed with an anti-ERK2 antibody (Santa-Cruz). Extracts were incubated at 4°C with the appropriate antibody for 2 h, followed by 1 h with protein G–Sepharose (Pharmacia). Immuno precipitates were washed three times for 1 min at 4°C with buffer A containing 0.5% Triton X-100, solubilized in SDS sample buffer, and loaded onto 8 or 10% SDS–polyacrylamide gels.
Kinase assays
PKR from HeLa cells was immunoprecipitated in buffer A as described above. The immune pellet bound to protein G–Sepharose beads was washed twice in activity buffer (20 mM Tris–HCl pH 7.5, 50 mM KCl, 2 mM MgCl2, 2 mM MnCl2, 5% glycerol). PKR bound to beads was tested for kinase activity in 40 µl of activity buffer containing 10 µM ATP and 10 µCi of [γ-32P]ATP. In the same experiment, 20 ng/ml dsRNA (pIC) or 10 µM GA was added to the kinase mix. Reactions were incubated at 30°C for 20 min.
Supplementary data
Supplementary data for this paper are available at The EMBO Journal Online.
Acknowledgments
Acknowledgements
We thank David Toft for Hsp90 and p23 antibodies, Michael Mathews for PKR antibody, Susan Lindquist, Avrom Caplan, Peter Piper and Sean Bohen for yeast strains, and Bruno Cenni for the construction of the mammalian expression plasmids CK and CKF. We are grateful to Peter Dudek for critical comments on the manuscript. This work was supported by the Swiss National Science Foundation and the Canton de Genève.
References
- Abbas-Terki T., Donzé,O. and Picard,D. (2000) The molecular chaperone Cdc37 is required for Ste11 function and pheromone-induced cell cycle arrest. FEBS Lett., 467, 111–116. [DOI] [PubMed] [Google Scholar]
- Balachandran S., Kim,C.N., Yeh,W.C., Mak,T.W., Bhalla,K. and Barber,G.N. (1998) Activation of the dsRNA-dependent protein kinase, PKR, induces apoptosis through FADD-mediated death signaling. EMBO J., 17, 6888–6902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohen S.P. (1998) Genetic and biochemical analysis of p23 and ansamycin antibiotics in the function of Hsp90-dependent signaling proteins. Mol. Cell. Biol., 18, 3330–3339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchner J. (1999) Hsp90 & Co.—a holding for folding. Trends Biochem. Sci., 24, 136–141. [DOI] [PubMed] [Google Scholar]
- Carpick B.W., Graziano,V., Schneider,D., Maitra,R.K., Lee,X. and Williams,B.R.G. (1997) Characterization of the solution complex between the interferon-induced, double-stranded RNA-activated protein kinase and HIV-I trans-activating region RNA. J. Biol. Chem., 272, 9510–9516. [DOI] [PubMed] [Google Scholar]
- Chong K.L., Feng,L., Schappert,K., Meurs,E., Donahue,T.F., Friesen,J.D., Hovanessian,A.G. and Williams,B.R. (1992) Human p68 kinase exhibits growth suppression in yeast and homology to the translational regulator GCN2. EMBO J., 11, 1553–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosentino G.P., Venkatesan,S., Serluca,F.C., Green,S.R., Mathews,M.B. and Sonenberg,N. (1995) Double-stranded-RNA-dependent protein kinase and TAR RNA-binding protein form homo- and heterodimers in vivo. Proc. Natl Acad. Sci. USA, 92, 9445–9449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Haro C., Mendez,R. and Santoyo,J. (1996) The eIF-2α kinases and the control of protein synthesis. FASEB J., 10, 1378–1387. [DOI] [PubMed] [Google Scholar]
- Dong J., Qiu,H., Garcia-Barrio,M., Anderson,J. and Hinnebusch,A.G. (2000) Uncharged tRNA activates GCN2 by displacing the protein kinase moiety from a bipartite tRNA-binding domain. Mol. Cell, 6, 269–279. [DOI] [PubMed] [Google Scholar]
- Donzé O. and Picard,D. (1999) Hsp90 binds and regulates Gcn2, the ligand-inducible kinase of the α-subunit of eukaryotic translation initiation factor 2. Mol. Cell. Biol., 19, 8422–8432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donzé O., Jagus,R., Koromilas,A.E., Hershey,J.W. and Sonenberg,N. (1995) Abrogation of translation initiation factor eIF-2 phosphorylation causes malignant transformation of NIH 3T3 cells. EMBO J., 14, 3828–3834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donzé O., Dostie,J. and Sonenberg,N. (1999) Regulatable expression of the interferon-induced double-stranded RNA dependent protein kinase PKR induces apoptosis and fas receptor expression. Virology, 256, 322–329. [DOI] [PubMed] [Google Scholar]
- Fang Y., Fliss,A.E., Rao,J. and Caplan,A.J. (1998) SBA1 encodes a yeast hsp90 co-chaperone that is homologous to vertebrate p23 proteins. Mol. Cell. Biol., 18, 3727–3734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fliss A.E., Fang,Y., Boschelli,F. and Caplan,A.J. (1997) Differential in vivo regulation of steroid hormone receptor activation by Cdc37p. Mol. Biol. Cell, 8, 2501–2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman B.C., Toft,D.O. and Morimoto,R.I. (1996) Molecular chaperone machines: chaperone activities of the cyclophilin Cyp-40 and the steroid aporeceptor-associated protein p23. Science, 274, 1718–1720. [DOI] [PubMed] [Google Scholar]
- Galabru J. and Hovanessian,A. (1987) Autophosphorylation of the protein kinase dependent on double-stranded RNA. J. Biol. Chem., 262, 15538–15544. [PubMed] [Google Scholar]
- Gan Y., Au,J.L., Lu,J. and Wientjes,M.G. (1998) Antiproliferative and cytotoxic effects of geldanamycin, cytochalasin E, suramin and thiacetazone in human prostate xenograft tumor histocultures. Pharm. Res., 15, 1760–1766. [DOI] [PubMed] [Google Scholar]
- Grammatikakis N., Lin,J.H., Grammatikakis,A., Tsichlis,P.N. and Cochran,B.H. (1999) p50(cdc37) acting in concert with Hsp90 is required for Raf-1 function. Mol. Cell. Biol., 19, 1661–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunnery S. and Mathews,M.B. (1998) RNA binding and modulation of PKR activity. Methods, 15, 189–198. [DOI] [PubMed] [Google Scholar]
- Harcourt J.L. and Offermann,M.K. (2001) Multiple signaling cascades are differentially involved in gene induction by double stranded RNA in interferon-α-primed cells. Eur. J. Biochem., 268, 1373–1381. [DOI] [PubMed] [Google Scholar]
- Hershey J.W. (1990) Overview: phosphorylation and translation control. Enzyme, 44, 17–27. [DOI] [PubMed] [Google Scholar]
- Hovanessian A.G., Galabru,J., Meurs,E., Buffet-Janvresse,C., Svab,J. and Robert,N. (1987) Rapid decrease in the levels of the double-stranded RNA-dependent protein kinase during virus infections. Virology, 159, 126–136. [DOI] [PubMed] [Google Scholar]
- Hu J. and Anselmo,D. (2000) In vitro reconstitution of a functional duck hepatitis B virus reverse transcriptase: posttranslational activation by hsp90. J. Virol., 74, 11447–11455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J., Toft,D.O. and Seeger,C. (1997) Hepadnavirus assembly and reverse transcription require a multi-component chaperone complex which is incorporated into nucleocapsids. EMBO J., 16, 59–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito T., Yang,M. and May,W.S. (1999) RAX, a cellular activator for double-stranded RNA-dependent protein kinase during stress signaling. J. Biol. Chem., 274, 15427–15432. [DOI] [PubMed] [Google Scholar]
- Jakob U., Lilie,H., Meyer,I. and Buchner,J. (1995) Transient interaction of Hsp90 with early unfolding intermediates of citrate synthase. Implications for heat shock in vivo. J. Biol. Chem., 270, 7288–7294. [DOI] [PubMed] [Google Scholar]
- Johnson J.L. and Toft,D.O. (1995) Binding of p23 and hsp90 during assembly with the progesterone receptor. Mol. Endocrinol., 9, 670–678. [DOI] [PubMed] [Google Scholar]
- Katze M.G. (1995) Regulation of the interferon-induced PKR: can viruses cope? Trends Microbiol., 3, 75–78. [DOI] [PubMed] [Google Scholar]
- Kaufman R.J. (1999) Stress signaling from the lumen of the endoplasmic reticulum: coordination of gene transcriptional and translational controls. Genes Dev., 13, 1211–1233. [DOI] [PubMed] [Google Scholar]
- Kimura Y., Rutherford,S.L., Miyata,Y., Yahara,I., Freeman,B.C., Yue,L., Morimoto,R.I. and Lindquist,S. (1997) Cdc37 is a molecular chaperone with specific functions in signal transduction. Genes Dev., 11, 1775–1785. [DOI] [PubMed] [Google Scholar]
- Koromilas A.E., Roy,S., Barber,G.N., Katze,M.G. and Sonenberg,N. (1992) Malignant transformation by a mutant of the IFN-inducible dsRNA-dependent protein kinase. Science, 257, 1685–1689. [DOI] [PubMed] [Google Scholar]
- Lopez-Maderuelo M.D., Fernandez-Renart,M., Moratilla,C. and Renart,J. (2001) Opposite effects of the Hsp90 inhibitor geldanamycin: induction of apoptosis in PC12 and differentiation in N2A cells. FEBS Lett., 490, 23–27. [DOI] [PubMed] [Google Scholar]
- Louvion J.F., Abbas-Terki,T. and Picard,D. (1998) Hsp90 is required for pheromone signaling in yeast. Mol. Biol. Cell, 9, 3071–3083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maran A., Maitra,R.K., Kumar,A., Dong,B., Xiao,W., Li,G., Williams,B.R., Torrence,P.F. and Silverman,R.H. (1994) Blockage of NF-κB signaling by selective ablation of an mRNA target by 2–5A antisense chimeras. Science, 265, 789–792. [DOI] [PubMed] [Google Scholar]
- Mundschau L.J. and Faller,D.V. (1995) Platelet-derived growth factor signal transduction through the interferon-inducible kinase PKR. Immediate early gene induction. J. Biol. Chem., 270, 3100–3106. [DOI] [PubMed] [Google Scholar]
- Nanduri S., Rahman,F., Williams,B.R. and Qin,J. (2000) A dynamically tuned double-stranded RNA binding mechanism for the activation of antiviral kinase PKR. EMBO J., 19, 5567–5574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathan D.F. and Lindquist,S. (1995) Mutational analysis of Hsp90 function: interactions with a steroid receptor and a protein kinase. Mol. Cell. Biol., 15, 3917–3925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Offermann M.K., Zimring,J., Mellits,K.H., Hagan,M.K., Shaw,R., Medford,R.M., Mathews,M.B., Goodbourn,S. and Jagus,R. (1995) Activation of the double-stranded-RNA-activated protein kinase and induction of vascular cell adhesion molecule-1 by poly (I)⋅poly (C) in endothelial cells. Eur. J. Biochem., 232, 28–36. [DOI] [PubMed] [Google Scholar]
- Pratt W.B. (1997) The role of the hsp90-based chaperone system in signal transduction by nuclear receptors and receptors signaling via MAP kinase. Annu. Rev. Pharmacol. Toxicol., 37, 297–326. [DOI] [PubMed] [Google Scholar]
- Pratt W.B. and Toft,D.O. (1997) Steroid receptor interactions with heat shock protein and immunophilin chaperones. Endocr. Rev., 18, 306–360. [DOI] [PubMed] [Google Scholar]
- Robertson H.D. and Mathews,M.B. (1996) The regulation of the protein kinase PKR by RNA. Biochimie, 78, 909–914. [DOI] [PubMed] [Google Scholar]
- Roe S.M., Prodromou,C., O’Brien,R., Ladbury,J.E., Piper,P.W. and Pearl,L.H. (1999) Structural basis for inhibition of the Hsp90 molecular chaperone by the antitumor antibiotics radicicol and geldanamycin. J. Med. Chem., 42, 260–266. [DOI] [PubMed] [Google Scholar]
- Romano P.R. et al. (1998) Autophosphorylation in the activation loop is required for full kinase activity in vivo of human and yeast eukaryotic initiation factor 2α kinases PKR and GCN2. Mol. Cell. Biol., 18, 2282–2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmedt C., Green,S.R., Manche,L., Taylor,D.R., Ma,Y. and Mathews,M.B. (1995) Functional characterization of the RNA-binding domain and motif of the double-stranded RNA-dependent protein kinase DAI (PKR). J. Mol. Biol., 249, 29–44. [DOI] [PubMed] [Google Scholar]
- Schulte T.W., Blagosklonny,M.V., Ingui,C. and Neckers,L. (1995) Disruption of the Raf-1–Hsp90 molecular complex results in destabilization of Raf-1 and loss of Raf-1–Ras association. J. Biol. Chem., 270, 24585–24588. [DOI] [PubMed] [Google Scholar]
- Schulte T.W., An,W.G. and Neckers,L.M. (1997) Geldanamycin-induced destabilization of Raf-1 involves the proteasome. Biochem. Biophys. Res. Commun., 239, 655–659. [DOI] [PubMed] [Google Scholar]
- Schulte T.W. et al. (1999) Interaction of radicicol with members of the heat shock protein 90 family of molecular chaperones. Mol. Endocrinol., 13, 1435–1448. [DOI] [PubMed] [Google Scholar]
- Shields J.M., Pruitt,K., McFall,A., Shaub,A. and Der,C.J. (2000) Understanding Ras: ‘it ain’t over ’til it’s over’. Trends Cell Biol., 10, 147–154. [DOI] [PubMed] [Google Scholar]
- Smith D.F., Whitesell,L., Nair,S.C., Chen,S., Prapapanich,V. and Rimerman,R.A. (1995) Progesterone receptor structure and function altered by geldanamycin, an hsp90-binding agent. Mol. Cell. Biol., 15, 6804–6812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soga S. et al. (1998) Radicicol leads to selective depletion of Raf kinase and disrupts K-Ras-activated aberrant signaling pathway. J. Biol. Chem., 273, 822–828. [DOI] [PubMed] [Google Scholar]
- Stancato L.F., Silverstein,A.M., Owens-Grillo,J.K., Chow,Y.H., Jove,R. and Pratt,W.B. (1997) The hsp90-binding antibiotic geldanamycin decreases Raf levels and epidermal growth factor signaling without disrupting formation of signaling complexes or reducing the specific enzymatic activity of Raf kinase. J. Biol. Chem., 272, 4013–4020. [DOI] [PubMed] [Google Scholar]
- Stebbins C.E., Russo,A.A., Schneider,C., Rosen,N., Hartl,F.U. and Pavletich,N.P. (1997) Crystal structure of an Hsp90–geldanamycin complex: targeting of a protein chaperone by an antitumor agent. Cell, 89, 239–250. [DOI] [PubMed] [Google Scholar]
- Stepanova L., Leng,X., Parker,S.B. and Harper,J.W. (1996) Mammalian p50Cdc37 is a protein kinase-targeting subunit of Hsp90 that binds and stabilizes Cdk4. Genes Dev., 10, 1491–1502. [DOI] [PubMed] [Google Scholar]
- Tan S.L., Gale,M.J.,Jr and Katze,M.G. (1998) Double-stranded RNA-independent dimerization of interferon-induced protein kinase PKR and inhibition of dimerization by the cellular P58IPK inhibitor. Mol. Cell. Biol., 18, 2431–2443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomis D.C. and Samuel,C.E. (1992) Mechanism of interferon action: autoregulation of RNA-dependent P1/eIF-2α protein kinase (PKR) expression in transfected mammalian cells. Proc. Natl Acad. Sci. USA, 89, 10837–10841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vattem K.M., Staschke,K.A., Zhu,S. and Wek,R.C. (2001) Inhibitory sequences in the N-terminus of the double-stranded-RNA-dependent protein kinase, PKR, are important for regulating phosphorylation of eukaryotic initiation factor 2α (eIF2α). Eur. J. Biochem., 268, 1143–1153. [DOI] [PubMed] [Google Scholar]
- Whitesell L., Mimnaugh,E.G., De Costa,B., Myers,C.E. and Neckers, L.M. (1994) Inhibition of heat shock protein HSP90–pp60v–src heteroprotein complex formation by benzoquinone ansamycins: essential role for stress proteins in oncogenic transformation. Proc. Natl Acad. Sci. USA, 91, 8324–8328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams B.R. (1999) PKR; a sentinel kinase for cellular stress. Oncogene, 18, 6112–6120. [DOI] [PubMed] [Google Scholar]
- Wu S. and Kaufman,R.J. (1997) A model for the double-stranded RNA (dsRNA)-dependent dimerization and activation of the dsRNA-activated protein kinase PKR. J. Biol. Chem., 272, 1291–1296. [DOI] [PubMed] [Google Scholar]
- Zou J., Guo,Y., Guettouche,T., Smith,D.F. and Voellmy,R. (1998) Repression of heat shock transcription factor HSF1 activation by HSP90 (HSP90 complex) that forms a stress-sensitive complex with HSF1. Cell, 94, 471–480. [DOI] [PubMed] [Google Scholar]