

Abstract
Elongation factor 2 kinase (eEF2k) phosphorylates and inactivates eEF2. Insulin induces dephosphorylation of eEF2 and inactivation of eEF2 kinase, and these effects are blocked by rapamycin, which inhibits the mammalian target of rapamycin, mTOR. However, the signalling mechanisms underlying these effects are unknown. Regulation of eEF2 phosphorylation and eEF2k activity is lost in cells in which phosphoinositide-dependent kinase 1 (PDK1) has been genetically knocked out. This is not due to loss of mTOR function since phosphorylation of another target of mTOR, initiation factor 4E-binding protein 1, is not defective. PDK1 is required for activation of members of the AGC kinase family; we show that two such kinases, p70 S6 kinase (regulated via mTOR) and p90RSK1 (activated by Erk), phosphorylate eEF2k at a conserved serine and inhibit its activity. In response to insulin-like growth factor 1, which activates p70 S6 kinase but not Erk, regulation of eEF2 is blocked by rapamycin. In contrast, regulation of eEF2 by stimuli that activate Erk is insensitive to rapamycin, but blocked by inhibitors of MEK/Erk signalling, consistent with the involvement of p90RSK1.
Keywords: elongation/phosphorylation/protein kinase/rapamycin/translation
Introduction
The control of mRNA translation in mammalian cells involves regulated phosphorylation of a number of components of the translational machinery (Proud and Denton, 1997; Rhoads, 1999). Phosphorylation of several of these proteins is under the control of the mammalian target of rapamycin, mTOR. These include: (i) the 70 kDa protein kinase (p70 S6k), which acts on ribosomal protein S6 and may be involved in controlling the translation of the set of mRNAs in mammalian cells that encode ribosomal proteins (Fumagalli and Thomas, 2000; Meyuhas and Hornstein, 2000); (ii) the initiation factor 4E binding protein, 4E-BP1, which regulates the formation of the eIF4F complexes that are required for the initiation of cap-dependent mRNA translation (Raught et al., 2000); and (iii) elongation factor 2 (eEF2), which mediates the translocation step of elongation (Redpath et al., 1996). The phosphorylation states of each of these proteins are regulated by insulin in a rapamycin-sensitive manner, demonstrating that signalling to these proteins requires mTOR activity. However, the molecular mechanisms linking them to mTOR remain obscure. mTOR is structurally related to lipid kinases, but displays protein rather than lipid kinase activity (Brunn et al., 1997; Burnett et al., 1998; Thomas and Hall, 1997; Isotani et al., 1999). It appears to play an important role not only in insulin and mitogen signalling, but also in the regulation of cellular function in response to nutritional cues (Kimball and Jefferson, 2000).
p70 S6k exists as at least two isoforms, encoded by different genes, in mammalian cells (Gout et al., 1998; Shima et al., 1998). The β-isoform was discovered only recently and shows both similarities to, and certain differences from, p70 S6kα (Gout et al., 1998; Shima et al., 1998; Lee-Fruman et al., 1999; Martin et al., 2001). p70 S6kα has been the subject of detailed investigation (Fumagalli and Thomas, 2000). Activation of p70 S6kα involves its phosphorylation on multiple sites (reviewed in Fumagalli and Thomas, 2000). The regulation of p70 S6kα involves inputs from signalling events linked to phosphatidylinositol 3-kinase (PI 3-kinase) that are not yet fully understood. Thr229 (a site conserved in p70 S6kβ) is phosphorylated by phosphoinositide-dependent kinase-1 (PDK1) (Alessi et al., 1998; Gout et al., 1998; Pullen et al., 1998; Shima et al., 1998; Balendran et al., 1999). mTOR has been reported to phosphorylate Thr389 and some of the Ser-Pro or Thr-Pro sites in the C-terminus of p70 S6kα (Burnett et al., 1998; Isotani et al., 1999). PDK1 also plays an important role in the regulation of other members of the so-called AGC family by phosphorylating residues in their ‘T-loop’ that are required for their activation, such as members of the protein kinase C (PKC) family (Chou et al., 1998; Dutil et al., 1998; Toker and Newton, 2000) and p90RSK1 (Jensen et al., 1999; Richards et al., 1999). Kinases acting at other sites in p70 S6kα remain to be identified.
4E-BP1 also undergoes phosphorylation at multiple sites in vivo, although again it is not known which kinases act on them. It is clear that rapamycin blocks phosphorylation of the sites that are important in regulating its association with eIF4E (Raught et al., 2000). Binding of 4E-BP1 to eIF4E blocks its ability to form functional initiation complexes with eIF4G, a scaffolding protein that also interacts with eIF4A and eIF3, the latter mediating interactions with the 40S ribosomal subunit (Raught et al., 2000). The signalling events that lie upstream of 4E-BP1 remain unclear; some reports have suggested that they involve PKB (Gingras et al., 1998). It has also been suggested that PKB plays a role in regulating mTOR itself (Scott et al., 1998; Navé et al., 1999).
eEF2 is inactivated by phosphorylation at Thr56 (reviewed in Proud, 2000). It undergoes dephosphorylation in response to insulin or serum in a rapamycin-sensitive manner (Redpath et al., 1996; Diggle et al., 1998; Wang et al., 2000), and this correlates with activation of elongation, which is also sensitive to rapamycin. Dephosphorylation of eEF2 in response to insulin or serum involves the inactivation of the kinase that phosphorylates eEF2 (Redpath et al., 1996; Wang et al., 2000), a highly specific enzyme that does not belong to the main kinase superfamily (Ryazanov et al., 1999). The regulation of eEF2 kinase (eEF2k) by insulin or mitogens is poorly understood and, in particular, it is not known how mTOR-dependent signalling is coupled to the regulation of its activity.
Here we have made use of cells engineered to lack specific protein kinases to explore the signalling pathways by which these mTOR-sensitive translational regulators are controlled. Our data demonstrate that PDK1, and therefore PKB, are not required for basal phosphorylation of 4E-BP1 or, by implication, the basal activity of mTOR, which appears to be maintained by amino acids in the cells’ growth medium. Most importantly, we demonstrate that PDK1 is required for the regulation of the phosphorylation of eEF2, and show that eEF2k is regulated by phosphorylation by two PDK1-dependent protein kinases, p70 S6k and p90RSK. These studies identify novel signalling connections involved in regulating mRNA translation.
Results
PDK1 is required for the regulation of the phosphorylation of eEF2
eEF2 is a target for mTOR signalling and is involved in regulating translation elongation (Proud, 2000). To study its phosphorylation, we used an antibody that detects eEF2 only when it is phosphorylated at Thr56. Serum-starved cells displayed significant phosphorylation of eEF2 and this was higher in the PDK1–/– than in the PDK1+/+ cells (Figure 1A). Treatment of almost (80%) confluent PDK1+/+ cells with insulin-like growth factor 1 (IGF1) elicited a decrease in the level of phosphorylation of eEF2 (Figure 1A). In contrast, IGF1 had no effect on eEF2 phosphorylation in PDK1–/– cells. PDK1–/– cells subjected to mild serum starvation showed high levels of eEF2 phosphorylation, which were not increased further either by rapamycin (Figure 1A) or by amino acid withdrawal (not shown). In PDK1+/+ cells that were less confluent, the initial level of eEF2 phosphorylation was submaximal, as confirmed by the increase seen when such cells were treated with rapamycin (Figure 1B). Withdrawal of amino acids from such cells led to increased phosphorylation of eEF2, similar to that observed after addition of rapamycin (Figure 1B). In contrast, in PDK1–/– cells, neither amino acid withdrawal nor rapamycin caused any further increase in eEF2 phosphorylation (Figure 1B).
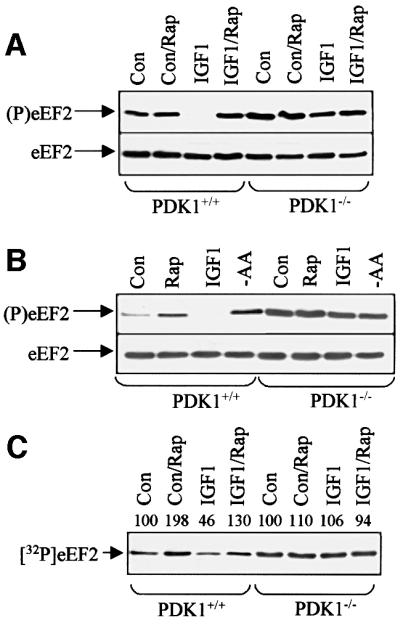
Fig. 1. Regulation of eEF2 and eEF2k in ES cells. (A) ES cells (PDK1+/+ or PDK1–/– as indicated, >80% confluent) were serum starved for 3 h and then treated with rapamycin (Rap) and/or IGF1 (40 min) as shown. Samples were subjected to SDS–PAGE and western blotting. Upper section: antibody against eEF2 phosphorylated at Thr56; lower section: blot with antibody detecting eEF2 irrespective of its phosphorylation state (loading control). (B) PDK+/+ or PDK1–/– ES cells (∼70% confluent) were serum starved for 3 h and then treated with rapamycin (Rap), IGF1 (20 min) or transferred to D-PBS/glucose for 1 h (-AA). Samples were subjected to SDS–PAGE/blotting with anti-(P)eEF2 antiserum or anti-eEF2. (C) Cell treatments as in (A). eEF2k activity was assayed in cell extracts using purified eEF2 as substrate. This figure is an autoradiograph. Signals were quantified by densitometry, activity for the control being set at 100. Con indicates control (untreated) cells.
To explore this further, we measured the activity of eEF2k in extracts of both cell lines before and after treatment with IGF1, using purified rabbit muscle eEF2 as substrate. IGF1 clearly decreased the activity of eEF2k in PDK1+/+ cells and this was inhibited by rapamycin (Figure 1C). In contrast, in PDK1–/– cells, IGF1 had no effect on eEF2k activity (Figure 1C). These data strongly imply that PDK1 is required for the regulation of eEF2k, either directly or through its action on another protein kinase, e.g. a member of the AGC family of kinases. eEF2k does not belong to this group of enzymes (Ryazanov et al., 1999) and we found no significant phosphorylation of recombinant eEF2k by PDK1 in vitro (data not shown). This is consistent with the lack, within the sequence of eEF2k, of a consensus site for PDK1. It seemed possible, therefore, that disruption of PDK1 might interfere with the function and/or regulation of mTOR; indeed, it has been suggested that PKB may play a role in the control of mTOR activity (Scott et al., 1998; Navé et al., 1999) or the regulation of other targets of mTOR signalling (Burgering and Coffer, 1995; Gingras et al., 1998; Kitamura et al., 1998).
4E-BP1 is highly phosphorylated in PDK1–/– cells
To assess whether mTOR was functional in PDK1–/– cells, we examined the phosphorylation of another target of mTOR signalling, 4E-BP1. Its phosphorylation state can be assessed by its mobility on SDS–PAGE, where more highly phosphorylated forms migrate more slowly. Extracts from PDK1+/+ or PDK1–/– embryonic stem (ES) cells (which had been starved of serum for 4 h) were subjected to SDS–PAGE and western blotting using anti-4E-BP1 antiserum. From this analysis, it is clear that 4E-BP1 is as highly phosphorylated in PDK1–/– cells as it is in PDK1+/+ cells (Figure 2A). As expected, pre-treatment of ES cells with rapamycin caused a shift in the migration of 4E-BP1 towards more mobile species in both cases. In other cell types, removal of amino acids induces dephosphorylation of 4E-BP1. To study this in ES cells, they were transferred to medium lacking amino acids for 1 h. This also resulted in a marked decrease in 4E-BP1 phosphorylation (Figure 2A). These data indicate that mTOR signalling is active in PDK1–/– cells, being maintained by the amino acids in the medium. These data thus demonstrate that neither PDK1 nor PKB is required for basal mTOR activity or for signalling downstream of mTOR to 4E-BP1.

Fig. 2. 4E-BP1 undergoes rapamycin-sensitive phosphorylation in PDK1–/– cells. (A) ES cells (PDK1+/+ or PDK1–/– as indicated) were treated with IGF1 (40 min) or transferred to amino acid-free medium (-AA; 1 h). Where shown (Rap), cells were pre-treated with rapamycin for 1 h. Extracts were subjected to immunoblotting using an antibody against 4E-BP1 that detects the protein irrespective of its phosphoryl ation state. Positions of the three electrophoretically distinct forms of 4E-BP1 (α–γ in order of increasing phosphorylation) are indicated. (B) Cell treatments as in (A) (but no amino acid withdrawal experiments). Cell extracts were subjected to affinity chromatography on m7GTP–Sepharose and the bound material was subjected to SDS–PAGE/western blotting using antisera for 4E-BP1, eIF4E and eIF4G (positions indicated). (C) ES cells were treated as in (B) and samples were analysed by SDS–PAGE/western blotting using phosphospecific antisera for the indicated sites in 4E-BP1. Con indicates control (untreated) cells.
Treatment of PDK1+/+ ES cells with IGF1 caused a small shift in the migration of 4E-BP1 towards the most phosphorylated γ-species (Figure 2A). This effect was blocked by either of two inhibitors of PI 3-kinase: LY294002 and wortmannin (data not shown). In contrast, IGF1 did not affect the mobility of 4E-BP1 in PDK–/– cells. These observations are consistent with the suggested role for PKB (Gingras et al., 1998) in the stimulation of 4E-BP1 phosphorylation by insulin. To assess whether IGF1 affected binding of 4E-BP1 to eIF4E, the latter was isolated from cell extracts on m7GTP–Sepharose, and the bound material was subjected to SDS–PAGE and western blotting to detect bound 4E-BP1 or eIF4G. Consistent with the high initial state of phosphorylation of 4E-BP1 in ES cells, relatively little was bound to eIF4E in serum-starved controls. Rapamycin treatment (Figure 2B) or withdrawal of amino acids (not shown) markedly increased the amount of 4E-BP1 bound to eIF4E in parallel with the decrease in its phosphorylation documented in Figure 2A (Figure 2B). In agreement with the low level of 4E-BP1 binding to eIF4E, the amount of eIF4G associated with eIF4E was high even in serum-starved controls (Figure 2B), for both control and knockout cells. Rapamycin treatment (Figure 2B) caused almost complete loss of eIF4G bound to eIF4E. The high basal level of eIF4F complexes resembles the situation in CHO cells (Campbell et al., 1999), where this also appears to be maintained by amino acids in the medium. Earlier reports suggested that the effects of amino acids on intracellular signalling pathways require PI 3-kinase (Wang et al., 1998; Peyrollier et al., 2000). However, the present data show that the effects of amino acids on mTOR-dependent eIF4F assembly are independent of PDK1. This concurs with earlier data (Campbell et al., 1999; Kimball and Jefferson, 2000; Peyrollier et al., 2000) indicating that amino acids do not activate PKB. Basal eIF4F formation is clearly also independent of PKB and other AGC kinases regulated by PDK1.
Phosphorylation of 4E-BP1 is complex, with up to six sites of phosphorylation that have differing effects upon the mobility of 4E-BP1 and its ability to bind eIF4E (Lawrence and Abraham, 1997; Mothe-Satney et al., 2000a,b; Raught et al., 2000). In order to study the phosphorylation of specific sites in 4E-BP1, we made use of antisera specific for these sites. Antisera are available for Thr36/45 (antibody cannot distinguish between these sites; Mothe-Satney et al., 2000b), Ser64 and Thr69, but not for Ser82 or Ser112. For the four sites studied, phosphorylation appeared similar in PDK–/– and PDK1+/+ cells (Figure 2C). Phosphorylation of Ser64 was markedly suppressed by rapamycin. This site is only phosphorylated in the γ-species of 4E-BP1, suggesting that its phosphorylation contributes to or causes the mobility shift from β to γ (see also Mothe-Satney et al., 2000a,b). Using the anti-(P)Thr69 or anti-(P)Thr36/45 antisera, two bands (β and γ) are observed in some cases and one in others (in rapamycin-treated cells), so this makes changes in overall intensity rather harder to discern here. However, it does appear that rapamycin also suppresses the phosphorylation of these sites (Figure 2C). Treatment of ES cells with IGF1 caused little, if any, change in the phosphorylation of any of these sites (Figure 2C). The above data indicate that mTOR signalling, e.g. in response to amino acids, is functional in cells lacking PDK1 and thus that neither PDK1 nor any of the AGC family kinases that it regulates is required for mTOR function or for its control by amino acids. Consistent with this, Hara and colleagues have found that mTOR activity (measured in vitro against p70 S6kα or 4E-BP1) is identical in extracts from PDK1+/+ or PDK1–/– cells (K.Hara, D.Alessi and K.Yonezawa, in preparation).
Analysis of the phosphorylation of ribosomal protein S6 and GSK3 confirmed that the PDK1–/– cells used here are devoid of PKB and S6 kinase activity, although p70 S6kα appeared to be partially phosphorylated in these cells (see Supplementary data available at The EMBO Journal Online).
eEF2k is phosphorylated at Ser366 by p70 S6kα and p90RSK1
Since the above data for 4E-BP1 indicate that there is no defect in mTOR signalling in PDK1–/– cells, it appeared likely that the absence of regulation of eEF2 and eEF2k in such cells was due to a role for a PDK1-activated member of the AGC kinase family in the control of eEF2k. To study the phosphorylation of eEF2k by different members of the subfamily of AGC kinases, we expressed human eEF2k in Escherichia coli as a glutathione S-transferase (GST) fusion protein. p90RSK1 and p70 S6kα readily phosphorylated GST–eEF2k, but PKA, PKB and MSK1 (all tested at 1 U/ml) only showed low or very low activity against GST–eEF2k (Figure 3A). Control experiments showed that, under the same conditions, the BAD [Bcl-2/Bcl-X(L)-antagonist, causing cell death] was phosphorylated with similar efficiency by PKA, p90RSK1, PKB and MSK1 (Lizcano et al., 2000), whereas the transcription factor CREB was phosphorylated to a similar extent by MSK1 and PKA, but at a vastly lower rate by p90RSK1 (Deak et al., 1998; Figure 3A). p70 S6kα and p90RSK1 (at 1 U/ml) each phosphorylated GST–eEF2k to 0.5–0.8 mol of phosphate/mol of protein after 60 min (Figure 3B and data not shown). Tryptic digestion of GST–eEF2k labelled in vitro by p70 S6kα and p90RSK1, followed by HPLC, revealed one major tryptic phosphopeptide, P1, eluting at 28% acetonitrile (Figure 3C and E). Phosphoamino acid analysis revealed that P1 contained only phosphoserine. During solid-phase sequencing, 32P radioactivity was released after the third cycle of Edman degradation (Figure 3D and F). The molecular mass of P1 determined by MALDI-TOF mass spectrometry (4608.9) was identical to that expected for the monophosphorylated tryptic phosphopeptide comprising residues 364–374. This was confirmed by gas-phase Edman sequencing of P1 (data not shown; sequence given in Figure 4D and F). These data demonstrate that p70 S6kα and p90RSK1 each phosphorylate human eEF2k at Ser366. This residue lies in a consensus sequence for these kinases, i.e. RXRXXS, and it, and the sequence immediately adjacent to it, are conserved in the three known sequences for mammalian eEF2k (Figure 4A). The main exception is that while the residue immediately C-terminal to Ser366 is Gly in rat and human eEF2k, it is replaced by Ser in mouse.

Fig. 3. Phosphorylation of eEF2k at Ser366 by AGC kinases. (A) GST–eEF2k, GST–CREB, GST–BAD and histone 2B (H2B) were incubated with the indicated AGC kinases in the presence of [γ-32P]ATP, and phosphorylation was analysed as described in Materials and methods. Similar results were obtained in two separate experiments. (B) GST–eEF2k was incubated with [γ-32P]ATP in the presence or absence of p70 S6kα, and after the times indicated reactions were terminated and the phosphorylation of eEF2k was determined. The stoichiometry of GST–eEF2k phosphorylation at each time point was measured. (C and E) GST–eEF2k that had been phosphorylated with p70 S6kα or p90RSK1 was digested with trypsin and chromatographed on a Vydac 218TP54 C18 column (Separations Group, Hesperia, CA) equilibrated in 0.1% (by vol) TFA in water. The column was developed with CH3CN (dashed line) at 0.8 ml/min and fractions (0.4 ml) were collected. Eighty per cent of the radioactivity applied to the column eluted with the major 32P-containing peptide (P1) at 28% CH3CN. (D and F) An aliquot of the 32P-labelled P1 peptide (from eEF2k phosphorylated by p70 S6kα or p90RSK1, respectively) was subjected to solid-phase Edman degradation. Release of 32P radioactivity was measured after each cycle. Sequences determined by gas-phase Edman degradation are indicated in (D) and (F).

Fig. 4. Phosphorylation of eEF2k at Ser366 inhibits its activity. (A) Sequence alignment around Ser366. The sequences from rat (Rn), mouse (Mm) and human (Hs) eEF2k around the equivalent of Ser366 (indicated by the arrow: human protein; 365 in rodents) are shown. (B) Wild-type (wt) or mutant (S366A) eEF2k proteins were expressed as GST fusions in E.coli and incubated with p70 S6kα in vitro in the presence of [γ-32P]ATP/MgCl2 for the times indicated. Samples were analysed by SDS–PAGE followed by autoradiography. The figure shows an autoradiograph. (C) Wild-type GST–eEF2k was pre-treated with (lower section) or without (upper section) p70 S6kα for 30 min in the presence of ATP/MgCl2. eEF2, CaCl2 (final concentration 100 µM), CaM and [γ-32P]ATP were then added, and the incubation was continued for the times indicated. Samples were analysed by SDS–PAGE/autoradiography to assess incorporation of radiolabel into eEF2. (D) Wild-type (wt) eEF2k or eEF2k in which Ser366 was mutated to Ala or Glu was expressed in E.coli and their activities were assessed using eEF2 as substrate in incubations containing [γ-32P]ATP/MgCl2, CaM and the indicated final concentrations of CaCl2 (upper section). eEF2k (wt or mutant as indicated) was pre-incubated with ATP/MgCl2 and with (lower section) or without (upper section) p70 S6kα in the absence of Ca ions or CaM prior to assay against eEF2 under the conditions described above. Samples were analysed by SDS–PAGE and autoradiography to assess incorporation of label into eEF2.
Mutant eEF2k proteins in which Ser366 was mutated to Ala (which cannot be phosphorylated) or Glu (an acidic residue that may mimic phosphoserine) were not phosphorylated by p70 S6kα (Figure 4B and data not shown), demonstrating that Ser366 is the only significant site of phosphorylation for p70 S6kα in eEF2k.
Phosphorylation of eEF2k by p70 S6kα inhibits its activity
To assess whether phosphorylation by p70 S6kα affected the activity of eEF2k, GST–eEF2k was pre-incubated with or without p70 S6kα, and its activity against eEF2 was assayed at a free calcium ion concentration similar to that used in our standard assays (100 µM). eEF2k activity was clearly decreased following pre-treatment with p70 S6kα [the activity of the phosphorylated enzyme was 39.8 ± 2.6% of the unphosphorylated control (n = 5)]. To examine the effect of phosphorylation on eEF2k activity at Ca2+ ion concentrations similar to those occurring in vivo, we repeated the experiments performing the assays at three different Ca2+ concentrations: 0.5, 1 and 10 µM (Figure 4D). Pre-treatment of eEF2k [but not eEF2k(S366A)] with p70 S6kα resulted in a very marked decrease in its activity when assayed at low calcium as compared with the control, i.e. eEF2k incubated only with ATP-Mg (Figure 4D, compare upper and lower sections). Mutation of Ser366 to Glu decreased eEF2k activity at low calcium in a similar way to phosphorylation of the wild-type enzyme by p70 S6kα (Figure 4D), and may also decrease its activity at high Ca2+ concentrations. Taken together, these data show that eEF2k is phosphorylated by p70 S6k at Ser366 and that this markedly inhibits its activity, especially at low micromolar calcium concentrations. This provides a mechanism by which insulin and other agents that activate p70 S6k may switch off eEF2k in an mTOR-dependent manner.
IGF1-induced eEF2 dephosphorylation is compromised in p70 S6kα–/– cells
To assess the role of p70 S6kα in regulating eEF2 phosphorylation in intact cells, we made use of ES cells in which both copies of the gene for p70 S6kα have been disrupted (p70 S6kα–/– cells; Kawasome et al., 1998), using the corresponding p70 S6kα+/+ cells as control. Western blot analysis revealed a clear signal for p70 S6kα in p70 S6kα+/+ cells, and in these cells IGF1 increase the phosphorylation of p70 S6kα, as shown by retardation of its mobility on SDS–PAGE (Figure 5A). As anticipated, there was no detectable p70 S6kα in the p70 S6kα–/– cells (Figure 5A). These cells also lack p70 S6kα activity (data not shown). Consistent with this, while IGF1 or serum caused a marked rapamycin-sensitive increase in the phosphorylation of ribosomal protein S6 in p70 S6kα+/+ cells, the signal was very substantially reduced in the p70 S6kα–/– cells (Figure 5A, lower section). However, serum or IGF1 still elicited a small but reproducible increase in S6 phosphorylation in these cells. Two isoforms of p70 S6k exist in mammals, encoded by different genes, so it seemed possible that the p70 S6kα–/– cells expressed some p70 S6kβ+/+, which was responsible for the residual phosphorylation of S6. Immunoblotting using an antibody specific for p70 S6kβ revealed similar levels of this kinase in p70 S6kα–/– and p70 S6kα+/+ cells (Figure 5B).

Fig. 5. Regulation of S6 phosphorylation, eEF2 and eEF2k in p70 S6kα+/+ and p70S6kα–/– ES cells. (A) ES cells (p70 S6kα+/+ or p70 S6kα–/– as indicated) were treated with rapamycin (Rap), serum (40 min) or IGF1 (times in minutes). Samples were analysed by SDS–PAGE, followed by western blotting with an antibody against p70 S6kα (upper section) or S6 [Ser235(P)]. The band running just below the position of p70 S6α in the samples from the p70 S6kα–/– cells represents a non-specific cross-reaction (band is also visible in other lanes). (B) Extracts of p70 S6kα+/+ or p70 S6kα–/– cells were subjected to SDS–PAGE and western blotting using antiserum for p70 S6kβ (position indicated). (C) ES cells were treated with rapamycin (Rap) and/or IGF1. Samples were prepared and analysed by SDS–PAGE/western blotting using antisera for phosphorylated eEF2 (upper part) or eEF2 irrespective of its state of phosphorylation (loading control, lower part). (D) ES cells were treated with rapamycin (Rap), serum or IGF1. Cell lysates were prepared and assayed for eEF2k activity using eEF2 as substrate. Signals were quantified by densitometry, activity for the control being set at 100 in each case. (E) Recombinant eEF2k or S366A mutant was incubated with p70 S6kα or p70 S6kβ and [γ-32P]ATP as indicated. Samples were analysed by SDS–PAGE and autoradiography. Con indicates control (untreated) cells.
In p70 S6kα+/+ cells, IGF1 induced the dephosphorylation of eEF2 (Figure 5C). In contrast, IGF1 had little, if any, effect on the level of phosphorylation of eEF2 in p70 S6k–/– cells (Figure 5C). Thus, loss of p70 S6kα greatly decreases the ability of IGF1 to cause dephosphorylation of eEF2. Similarly, the regulation of eEF2k was also almost abolished in p70 S6kα–/– cells: treatment of p70 S6k–/– cells with IGF1 or serum had little effect on eEF2k activity, whereas it markedly inhibited eEF2k in p70 S6kα+/+ cells (Figure 5D).
These data suggest that p70 S6kβ does not play a major role in regulating eEF2 phosphorylation, and we studied whether p70 S6kβ could phosphorylate eEF2k. p70 S6kβ (Gout et al., 1998) was expressed as an epitope (EE)-tagged fusion protein in HEK 293 cells. Its activity in EE immunoprecipitates was assayed against the peptide substrate used to assay p70 S6kα. Equal amounts of the two forms (by activity) were tested for their ability to phosphorylate recombinant eEF2k and the S366A mutant. Both forms phosphorylated the wild-type protein, although the β-isoform showed markedly less activity against eEF2k than the α-isoform (Figure 5E). Neither phosphorylated eEF2k S366A (Figure 5E), showing that both known isoforms of p70 S6k phosphorylate eEF2k at the same site. The relative inefficiency with which p70 S6kβ phosphorylates eEF2k compared with p70 S6kα probably explains why although low levels of S6 phosphorylation occur in p70 S6kα–/– cells, the regulation of eEF2 phosphorylation is almost abolished.
Regulation of the phosphorylation of Ser366 in eEF2k in vivo
An important prediction of the finding that Ser366 is phosphorylated by p70 S6kα/β is that phosphorylation of this site should be sensitive to rapamycin. To study this, we generated antisera specific for eEF2k phosphorylated at Ser366. Since the sequences of human and mouse eEF2k differ slightly around Ser366 (Figure 4A), we generated two different antisera. We were unable to study phosphorylation of eEF2k in ES cells due to a combination of low levels of eEF2k and poor sensitivity of the antiserum for the mouse sequence. We therefore turned to human (HEK) 293 cells, which contain higher levels of eEF2k and where we could employ the alternative, better, antibody. IGF1 induced dephosphorylation of eEF2 in 293 cells and this was blocked by rapamycin (Figure 6A) and by the PI 3-kinase inhibitor wortmannin (not shown). To assess the specificity of the anti-phospho Ser366 antibody, we tested its ability to recognize unphosphorylated human eEF2k and eEF2k pre-treated with p70 S6kα, in the presence of the peptide used to raise the antibody or the corresponding non-phosphorylated peptide. As shown in Figure 6B, the antiserum did not recognize unphosphorylated eEF2k, and its ability to recognize the phosphorylated eEF2k was prevented by inclusion of the phosphopeptide. This confirms the specificity of the antibody. To study the regulation of phosphorylation of Ser366 in 293 cells, eEF2k was immunoprecipitated from the cell lysates and subjected to SDS–PAGE /immunoblotting using the anti-phospho Ser366 antibody (or, as a loading control, an antibody that detects eEF2k irrespective of its state of phosphorylation). Treatment of 293 cells with IGF1 caused a time-dependent increase in the phosphorylation of Ser366, as determined using the antibody designed for human eEF2k (Figure 6C). The increased phosphorylation caused by IGF1 was blocked either by rapamycin or LY294002, which inhibit activation of p70 S6k in 293 cells (Herbert et al., 2000). Furthermore, two structurally different inhibitors of MEK [PD184352 (Sebolt-Leopold et al., 1999) and U0126 (Favata et al., 1998)] failed to prevent the IGF1-induced increase in phosphorylation of Ser366 (Figure 6D). Immunoblots for activated phosphorylated Erk revealed that IGF1 does not activate Erk in HEK 293 cells (not shown), in agreement with earlier data (Herbert et al., 2000). These findings rule out a role for p90RSK1 in mediating the phosphorylation of this site in eEF2k, and also eEF2 phosphorylation, in response to IGF1. The data are entirely consistent with a role for p70 S6k in mediating these effects.

Fig. 6. Phosphorylation of eEF2 and eEF2k in response to IGF1. (A) HEK 293 cells were starved of serum for 16 h and then treated with rapamycin (Rap; 30 min) or DMSO, prior to addition of IGF1 for 20 min. Samples of cell lysate were subjected to SDS–PAGE/western blotting using anti-(P)eEF2 antiserum or (as loading control) anti-eEF2 as indicated. (B) Samples (6 ng) of untreated (–) recombinant eEF2k or eEF2k phosphorylated by p70 S6kα (+) were analysed by SDS–PAGE/western blotting using the anti-phospho Ser366 antibody. Where indicated, blots were performed in the presence of the competing phosphopeptide or the corresponding dephosphopeptide variant. The position of phosphorylated eEF2k is indicated (P-eEF2k). (C) Cell treatments as in (A), except that in some cases LY294002 (30 µM) was added 30 min before treatment with IGF1. Cell lysates (1 mg of protein) were subjected to immunoprecipitation using anti-(GST) eEF2k antibody, and samples analysed by SDS–PAGE/western blotting using anti-eEF2k P-Ser366 antibody or (loading control) anti-eEF2k antiserum. (D) Cell treatments were essentially the same as for (A) and (B), except that, where indicated, cells were pre-treated with PD184352 (1 h, 2 µM) or U0126 (1 h, 10 µM) prior to adding IGF1 for 20 min. Samples were analysed as in (B). Con indicates control (untreated) cells.
In contrast to insulin, the phorbol ester TPA potently activates Erk in HEK 293 cells. This was completely blocked by PD184352, but not affected by rapamycin (Figure 7A). TPA induced dephosphorylation of eEF2 in HEK 293 cells and this was blocked by PD184352 (Figure 7B). This indicates that MEK/Erk signalling is involved in the control of eEF2 phosphorylation in this situation; however, we have previously presented data showing that activation of p70 S6k by TPA in these cells is blocked by inhibitors of MEK (Herbert et al., 2000), which also block S6 phosphorylation in response to TPA (Figure 7C). It was, therefore, essential to use rapamycin to distinguish whether regulation of eEF2 was occurring through p90RSK1 or p70 S6k under these conditions. Rapamycin had little, if any, effect on TPA-induced dephosphorylation of eEF2 (Figure 7D). Since rapamycin does block activation of p70 S6k (Herbert et al., 2000) and S6 phosphorylation in response to TPA (Figure 7C), the data imply that eEF2 is not regulated by p70 S6k in response to TPA. Instead, they offer support to the operation of an alternative mechanism, by which eEF2k is phosphorylated and inactivated by p90RSK1. To explore this possibility further, we studied the effect of TPA on phosphorylation of Ser366 in eEF2k. TPA increased the phosphorylation of eEF2k at Ser366, and this was blocked by PD184352 but not rapamycin (Figure 7E and F). TPA activated p90RSK, activation being evident by 10 min and maximal at 30 min (38.3 ± 2.9 mU/mg protein). As expected, this was blocked by PD184352 (0.8 ± 0.08 mU/mg at 20 min), but not rapamycin (36.6 ± 1.4 mU/mg). Taken together, these data strongly suggest that TPA regulates eEF2k and eEF2 via MEK/Erk/p90RSK1, rather than through p70 S6k, which is, however, involved in the control of eEF2 by IGF1 (see above).

Fig. 7. Phosphorylation of eEF2 and eEF2k in response to TPA. (A) Serum-starved (16 h) HEK 293 cells were treated with TPA (times indicated). Where shown, cells were pre-incubated with rapamycin or PD184352 (2 µM) for 1 h prior to addition of TPA. Samples of cell lysate were subjected to SDS–PAGE/western blotting using either anti-(P)Erk or (loading control) anti-Erk as indicated. (B) 293 cells were treated with TPA for the indicated times; PD184352 (or DMSO, control) was added 1 h before TPA. Samples of cell extract (25 µg protein) were subjected to SDS–PAGE/western blotting using anti-(P)eEF2 antibodies. (C) Conditions as in (A), but immunoblotting employed anti-phospho-S6 antibody. (D) As in (B), except that rapamycin was used in place of PD184352. (E) Cell lysate (700 µg protein) from the same extracts as in (B) was immunoprecipitated with anti-eEF2k antiserum. Immunoprecipitates were subjected to SDS–PAGE/western blotting with the anti(P)Ser366 or phosphorylation-insensitive antisera for eEF2k. (F) Conditions as in (A). Samples were immunoprecipitated with anti-eEF2k antiserum and blots were probed with the anti-(P)Ser366 antibody. Loading controls showed equal amounts of eEF2k. In all cases, similar data were obtained in 3–4 separate experiments.
Discussion
This study addresses the upstream signalling events involved in the regulation of three components of the translational machinery, all of which are controlled in an mTOR-dependent manner. In particular, we delineate an mTOR-dependent mechanism by which the activity of eEF2k can be regulated in response to serum or other agents that cause the dephosphorylation and activation of eEF2. This involves the phosphorylation and inactivation of eEF2k by p70 S6k. We thus also identify a new substrate for both p70 S6kα and the recently discovered second isoform, p70 S6kβ. In addition, we show that eEF2 is subject to regulation in an mTOR-independent manner, probably via MEK/Erk/p90RSK1 (Figure 7), in response to phorbol esters, which activate the latter pathway.
Our results demonstrate that PDK1 is not required for the basal activity through the mTOR pathway that is maintained by amino acids present in the medium. The data therefore imply that PDK1 is not important in amino acid signalling to 4E-BP1 or eIF4F, extending the conclusion of earlier studies that this does not involve changes in PKB activity (Campbell et al., 1999; Peyrollier et al., 2000). Amino acid regulation of 4E-BP1 phosphorylation is blocked by inhibitors of PI 3–kinase (Wang et al., 1998; Kimball and Jefferson, 2000); the present findings suggest that these effects are not mediated by PDK1/PKB signalling and imply the existence of an alternative PI 3-kinase-linked signalling pathway that responds to amino acids. The data also show that the activities of the (unknown) protein kinases that act on at least four of the five main phosphorylation sites in 4E-BP1 are not dependent on PDK1, although the ability of IGF1 to induce further phosphorylation of 4E-BP1 was compromised in PDK1–/– cells. Furthermore, the data appear to exclude a role for members of the PKC family that are also regulated by PDK1 in the regulation of 4E-BP1 phosphorylation: an earlier report indicated a role in this for PKCδ (Kumar et al., 2000). The only difference noted here between PDK1+/+ and PDK1–/– cells is a loss in the very small effect of IGF1 on the migration of 4E-BP1, but this does not appear to reflect a discernible difference in the phosphorylation of any of the four phosphorylation sites studied.
In contrast, all regulation of p70 S6kα or eEF2 phosphorylation by amino acids is abrogated in PDK1–/– cells, implying that a further input in addition to mTOR is required. In the case of p70 S6kα, this input is the phosphorylation of p70 S6kα by PDK1 itself, and for eEF2 this presumably reflects the role of p70 S6k in phosphorylating and inactivating eEF2k. PDK1–/– cells show no p70 S6k activity in the basal state and IGF1 has no effect on this, confirming that PDK1 is absolutely required for p70 S6k activity. This reflects the ability of PDK1 directly to phosphorylate p70 S6kα, at a site (Thr229) required for its activation (Alessi et al., 1998; Pullen et al., 1998).
A major finding of this study concerns the molecular mechanism underlying the mTOR-dependent regulation of eEF2 phosphorylation. The ability of IGF1 to affect eEF2 phosphorylation was lost in cells lacking PDK1 (and thus active PKB). This suggested that signalling from mTOR to eEF2 was defective in PDK1–/– cells. Since p70 S6k lies downstream of mTOR and is dependent upon PDK1 for activation, we studied its possible role in the regulation of eEF2k. p70 S6kα readily phosphorylates eEF2k and the site of phosphorylation was identified as Ser366. Phosphorylation of eEF2k by p70 S6kα resulted in inactivation of eEF2k, especially at low (physiologically relevant) calcium concentrations, but also at the higher Ca2+ concentrations used in our standard assays. [Owing to the difficulty of titrating Ca2+ ions accurately in the cell extracts used for our assays (which contain Ca2+ and various Ca2+ chelators, such as EGTA), our standard assays employ Ca2+ concentrations around 100 µM.] Treatment of eEF2k(S366A) with p70 S6kα had (as expected) no effect on its activity, while the S366E mutant showed low activity at low calcium, similar to the situation for eEF2k phosphorylated at this site. These data provide, for the first time, a link between mTOR and the regulation of eEF2k activity and eEF2 phosphorylation by stimuli such as insulin. Such stimuli activate p70 S6k, leading to phosphorylation and inactivation of eEF2k, resulting in the dephosphorylation of eEF2. This underlines the role of mTOR signalling in the co-ordinated activation of translation in response to this hormone. Through this pathway, insulin and other stimuli promote cap-dependent translation (through eIF4F), facilitate the translation of specific mRNAs (5′-TOP mRNAs, via p70 S6k) and accelerate elongation (via p70 S6k and inactivation of eEF2k).
Our data also reveal an alternative pathway through which agents that activate MEK/Erk can regulate eEF2k and eEF2. This involves the inactivation of eEF2k by phosphorylation by p90RSK1 at Ser366, the same site as is phosphorylated by p70 S6k. This allows agents that activate Erk signalling, e.g. mitogenic stimuli, to accelerate the elongation process. eEF2k thus lies at the convergence of two major signalling pathways that are activated by a wide range of stimuli that promote cell growth and protein synthesis (Figure 8). Recent data (Knebel et al., 2001) have identified a further phosphorylation site in eEF2k (Ser359) that is a target for a MAP-kinase related kinase termed SAPK4. Phosphorylation of this site also inactivates eEF2k. The only other phosphorylation site so far identified in eEF2k is Ser499, a target for cAMP-dependent protein kinase; this phosphorylation event renders eEF2k activity independent of Ca–calmodulin (Diggle et al., 2001). Interestingly, in contrast to the situation for IGF1, treatment of PDK1–/– cells with TPA still resulted in dephosphorylation of eEF2 (our unpublished data), suggesting the operation of an additional regulatory mechanism distinct from p70 S6k and p90RSK1. This might involve SAPK4 or other, as yet unidentified, regulatory events.

Fig. 8. Signalling connections involved in the regulation of eEF2k. This figure summarizes the signalling connections identified in this study in the context of other known or probable signalling events. Thick lines denote direct phosphorylation events, thin lines denote links that may or may not be direct and question marks indicate that the links are poorly understood or not certain. Broken lines indicate inhibition. The sites of action of the inhibitors used here are shown.
Materials and methods
Cells and cell culture
PDK1–/– ES cells were generated and maintained as described earlier (Williams et al., 2000). ES cells lacking p70 S6kα were grown as described earlier (Kawasome et al., 1998). Prior to hormone treatment, cells were starved of serum for 4 h. Where used, signalling inhibitors were added after 3 h and 1 h before addition of IGF1 (100 ng/ml), serum [10% (v/v)] or TPA (0.6 µM). Rapamycin (100 nM) was added 1 h before treatment of cells. In some cases, cells were deprived of amino acids by transferring them to Dulbecco’s phosphate-buffered saline (D-PBS) containing 5 mM d-glucose.
Phosphorylation of eEF2k by AGC kinases
PKA was purified from bovine heart by Dr C.MacKintosh in the MRC Protein Phosphorylation Unit, MSK1 and p90RSK1 were expressed as GST fusion proteins in E.coli, and purified from TPA-stimulated 293 cells. A mutant of p70 S6kα, which lacks the C-terminal 104 residues and in which Thr412 is mutated to Glu, and also has a His6 tag, was expressed in insect cells and activated in vitro by phosphorylation by PDK1 (Balendran et al., 1999). GST–eEF2k, GST–CREB, GST–BAD and histone H2B (all 1 µg) were incubated in a total volume of 40 µl at 30°C with 1 U/ml PKA, GST–MSK1, GST–p90RSK1, His-p70 S6kα in buffer B [50 mM Tris–HCl pH 7.5, 0.1 mM EGTA, 0.1 (v/v) β-mercaptoethanol containing 10 mM magnesium acetate, 100 µM [γ-32P]ATP (1000 c.p.m./pmol), 1 µM microcystin-LR]. After incubation for 15 min, incorporation of phosphate into GST–eEF2k, GST–CREB, GST–BAD and histone 2B was determined following the electrophoresis of samples on a NuPAGE Bis-Tris 4–12% gel and autoradiography. The peptide Crosstide (GRPRTSSFAEG; 30 µM) was used to assay the activity of PKB, GST–MSK1 or GST–p90RSK1, and Kemptide (LRRASLG; 30 µM) was used to assay the activity of PKA. One unit (U) of kinase activity is the amount of enzyme that phosphorylates 1 nmol of peptide in 1 min.
Mapping the site on eEF2k labelled by p70 S6kα and p90RSK1
To map the site on eEF2k phosphorylated p70 S6Kα and p90RSK1, GST–eEF2k was incubated with these kinases as described above except that the reaction was performed for 60 min and a 10-fold higher specific activity of [γ-32P]ATP was used. Reactions were terminated by adding 1% (w/v) SDS and 10 mM dithiothreitol, and heating at 100°C for 1 min. After cooling, 4-vinylpyridine was added to 1% (v/v) and samples were left on a shaking platform for 30 min at room temperature to alkylate cysteine residues. The sample was subjected to electrophoresis on a 4–12% NuPAGE Bis-Tris gel, and the 32P-labelled band corresponding to GST–eEF2k was excised and cut into smaller pieces. Further analyses were performed as described in the Supplementary data.
Supplementary data
Supplementary data for this paper are available at The EMBO Journal Online.
Acknowledgments
Acknowledgements
We are grateful to Agnieszka Kieloch for maintaining the ES cells used in this study, and to Drs Ivan Gout, Axel Knebel, John C.Lawrence, Nicholas Redpath and Alexey Ryazanov for reagents. We thank members of the Division of Signal Transduction Therapy (Dundee) for purified kinases and Dr N.A.Morrice for performing peptide sequencing and mass spectrometry analyses. This work was supported by grants from the Wellcome Trust (046110) and BBSRC to C.G.P., and the MRC and Diabetes UK to D.R.A.
References
- Alessi D.R., Kozlowski,M.T., Weng,Q.-P., Morrice,N. and Avruch,J. (1998) 3-phosphoinositide-dependent protein kinase 1 (PDK1) phosphorylates and activates the p70 S6 kinase in vivo and in vitro. Curr. Biol., 8, 69–81. [DOI] [PubMed] [Google Scholar]
- Balendran A., Currie,R., Armstrong,C.G., Avruch,J. and Alessi,D.R. (1999) Evidence that 3-phosphoinositide-dependent protein kinase-1 mediates phosphorylation of p70 S6 kinase in vivo at Thr-412 as well as Thr-252. J. Biol. Chem., 274, 37400–37406. [DOI] [PubMed] [Google Scholar]
- Brunn G.J., Hudson,C.C., Sekulic,A., Williams,J.M., Hosoi,H., Houghton,P.J., Lawrence,J.C. and Abraham,R.T. (1997) Phosphorylation of the translational repressor PHAS-I by the mammalian target of rapamycin. Science, 277, 99–101. [DOI] [PubMed] [Google Scholar]
- Burgering B.M.T. and Coffer,P.J. (1995) Protein kinase B (c-Akt) in phosphatidylinositol-3-OH kinase signal transduction. Nature, 376, 599–602. [DOI] [PubMed] [Google Scholar]
- Burnett P.E., Barrow,R.K., Cohen,N.A., Snyder,S.H. and Sabatini,D.M. (1998) RAFT1 phosphorylation of the translational regulators p70 S6 kinase and 4E-BP1. Proc. Natl Acad. Sci. USA, 95, 1432–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell L.E., Wang,X. and Proud,C.G. (1999) Nutrients differentially modulate multiple translation factors and their control by insulin. Biochem. J., 344, 433–441. [PMC free article] [PubMed] [Google Scholar]
- Chou M.M., Hou,W., Johnson,J., Graham,L.K., Lee,M.H., Chen,C.-S., Newton,A.C., Schaffhausen,B.S. and Toker,A. (1998) Regulation of protein kinase Cζ by PI 3-kinase and PDK-1. Curr. Biol., 8, 1069–1077. [DOI] [PubMed] [Google Scholar]
- Deak M., Clifton,A.D., Lucocq,J.M. and Alessi,J.R. (1998) Mitogen- and stress-activated protein kinase-1 (MSK1) is directly activated by MAPK and SAPK2/p38 and may mediate activation of CREB. EMBO J., 17, 4426–4441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diggle T.A., Redpath,N.T., Heesom,K.J. and Denton,R.M. (1998) Regulation of protein synthesis elongation factor-2 kinase by cAMP in adipocytes. Biochem. J., 336, 525–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diggle T.A., Subkhankulova,T., Lilley,K.S., Shikotra,N., Willis,A.E. and Redpath,N.T. (2001) Phosphorylation of elongation factor-2k on serine 499 by cAMP-dependent protein kinase induces Ca2+/calmodulin-independent activity. Biochem. J., 353, 621–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutil E.M., Toker,A. and Newton,A.C. (1998) Regulation of conventional protein kinase C isozymes by phosphoinositide-dependent kinase 1 (PDK-1). Curr. Biol., 8, 1366–1375. [DOI] [PubMed] [Google Scholar]
- Favata M.F. et al. (1998) Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J. Biol. Chem., 273, 18623–18632. [DOI] [PubMed] [Google Scholar]
- Fumagalli S. and Thomas,G. (2000) S6 phosphorylation and signal transduction. In Sonenberg,N., Hershey,J.W.B. and Mathews,M.B. (eds), Translational Control of Gene Expression. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 695–717.
- Gingras A.-C., Kennedy,S.G., O’Leary,M.A., Sonenberg,N. and Hay,N. (1998) 4E-BP1, a repressor of mRNA translation, is phosphorylated and inactivated by the Akt(PKB) signalling pathway. Genes Dev., 12, 502–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gout I., Minami,T., Hara,K., Tsujishita,Y., Filonenko,V., Waterfield,M.D. and Yonezawa,K. (1998) Molecular cloning and characterisation of a novel p70 S6 kinase, p70 S6kβ containing a proline-rich region. J. Biol. Chem., 273, 30061–30064. [DOI] [PubMed] [Google Scholar]
- Herbert T.P., Kilhams,G.R., Batty,I.H. and Proud,C.G. (2000) Distinct signalling pathways mediate insulin and phorbol ester-stimulated eIF4F assembly and protein synthesis in HEK 293 cells. J. Biol. Chem., 275, 11249–11256. [DOI] [PubMed] [Google Scholar]
- Isotani S., Hara,K., Tokunaga,C., Inoue,H., Avruch,J. and Yonezawa,K. (1999) Immunopurified mammalian target of rapamycin phosphorylates and activates p70 S6 kinase α in vitro. J. Biol. Chem., 274, 34493–34498. [DOI] [PubMed] [Google Scholar]
- Jensen C.J., Buch,M.B., Krag,T.O., Hemmings,B.A., Gammeltoft,S. and Frodin,M. (1999) 90 kDa ribosomal protein S6 kinase is phosphorylated and activated by 3-phosphoinositide-dependent kinase-1. J. Biol. Chem., 274, 27168–27176. [DOI] [PubMed] [Google Scholar]
- Kawasome H., Papst,P., Webb,S., Keller,G.M., Johnson,G.L., Gelfand,E.W. and Terada,N. (1998) Targeted disruption of p70 S6 k defines its role in protein synthesis and rapamycin sensitivity. Proc. Natl Acad. Sci. USA, 95, 5033–5038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimball S.R. and Jefferson,L.S. (2000) Regulation of translation initiation in mammalian cells by amino acids. In Sonenberg,N., Hershey,J.W.B. and Mathews,M.B. (eds), Translational Control of Gene Expression. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 561–579.
- Kitamura T. et al. (1998) Requirement for activation of the serine-threonine kinase Akt (protein kinase B) in insulin stimulation of protein synthesis but not of glucose transport. Mol. Cell. Biol., 18, 3708–3717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knebel A., Morrice,N. and Cohen,P. (2001) A novel method to identify protein kinase substrates: eEF2 kinase is phosphorylated and inhibited by SAPK4/p38δ. EMBO J., 20, 4360–4369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar V., Pandey,P., Sabatini,D., Kumar,M., Majumder,P.K., Bharti,A., Carmichael,G., Kufe,D. and Kharbanda,S. (2000) Functional interaction between RAFT1/FRAP/mTOR and protein kinase Cδ in the regulation of cap-dependent initiation of translation. EMBO J., 19, 1087–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence J.C. and Abraham,R.T. (1997) PHAS/4E-BPs as regulators of mRNA translation and cell proliferation. Trends Biochem. Sci., 22, 345–349. [DOI] [PubMed] [Google Scholar]
- Lee-Fruman K.K., Kuo,C.J., Lippincott,J., Terada,N. and Blenis,J. (1999) Characterisation of S6K2, a novel kinase homologous to S6K1. Oncogene, 18, 5108–5114. [DOI] [PubMed] [Google Scholar]
- Lizcano J.M., Morrice,N. and Cohen,P. (2000) Regulation of BAD by cAMP-dependent protein kinase is mediated via phosphorylation of a novel site, Ser155. Biochem. J., 349, 547–557. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Martin K.A., Schalm,S.S., Romanelli,A., Keon,K.L. and Blenis,J. (2001) Ribosomal S6 kinase 2 inhibition by a potent C-terminal repressor domain is relieved by mitogen-activated protein-extracellular signal-regulated kinase kinase-regulated phosphorylation. J. Biol. Chem., 276, 7892–7898. [DOI] [PubMed] [Google Scholar]
- Meyuhas O. and Hornstein,E. (2000) Translational control of TOP mRNAs. In Sonenberg,N., Hershey,J.W.B. and Mathews,M.B. (eds), Translational Control of Gene Expression. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 671–693.
- Mothe-Satney I., Brunn,G.J., McMahon,L.P., Capaldo,C.T., Abraham,R.T. and Lawrence,J.C. (2000a) Mammalian target of rapamycin-dependent phosphorylation of PHAS-1 in four (S/T)P sites detected by phospho-specific antibodies. J. Biol. Chem., 275, 33836–33843. [DOI] [PubMed] [Google Scholar]
- Mothe-Satney I., Yang,D., FaddenP., Haystead,T.A.J. and Lawrence,J.C. (2000b) Multiple mechanisms control phosphorylation of PHAS-I in five (S/T)P sites that govern translational repression. Mol. Cell. Biol., 20, 3558–3567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navé B.T., Ouwens,D.M., Withers,D.J., Alessi,D.R. and Shepherd,P.R. (1999) Mammalian target of rapamycin is a direct target for protein kinase B: identification of a convergence point for opposing effects of insulin and amino-acid deficiency on protein translation. Biochem. J., 344, 427–431. [PMC free article] [PubMed] [Google Scholar]
- Peyrollier K., Hajduch,E., Blair,A.S., Hyde,R. and Hundal,H.S. (2000) l-leucine availability regulates phosphatidylinositol 3-kinase, p70 S6 kinase and glycogen synthase kinase-3 activity in L6 muscle cells: evidence for the involvement of the mammalian target of rapamycin (mTOR) pathway in the l-leucine-induced up-regulation of System A amino acid transport. Biochem. J., 350, 361–368. [PMC free article] [PubMed] [Google Scholar]
- Proud C.G. (2000) Control of the elongation phase of protein synthesis. In Sonenberg,N., Hershey,J.W.B. and Mathews,M.B. (eds), Translational Control of Gene Expression. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 719–739.
- Proud C.G. and Denton,R.M. (1997) Molecular mechanisms for the activation of protein synthesis by insulin. Biochem. J., 328, 329–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pullen N., Dennis,P.B., Andjelkovic,M., Dufner,A., Kozma,S.C., Hemmings,B.A. and Thomas,G. (1998) Phosphorylation and activation of p70S6k by PDK1. Science, 279, 707–710. [DOI] [PubMed] [Google Scholar]
- Raught B., Gingras,A.-C. and Sonenberg,N. (2000) Regulation of ribosome recruitment in eukaryotes. In Sonenberg,N., Hershey,J.W.B. and Mathews,M.B. (eds), Translational Control of Gene Expression. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 245–293.
- Redpath N.T., Foulstone,E.J. and Proud,C.G. (1996) Regulation of translation elongation factor-2 by insulin via a rapamycin-sensitive signalling pathway. EMBO J., 15, 2291–2297. [PMC free article] [PubMed] [Google Scholar]
- Rhoads R.E. (1999) Signal transduction pathways that regulate eukaryotic protein synthesis. J. Biol. Chem., 274, 30337–30340. [DOI] [PubMed] [Google Scholar]
- Richards S.A., Fu,J., Romanelli,A., Shimamura,A. and Blenis,J. (1999) Ribosomal S6 kinase 1 (RSK1) activation requires signals dependent on and independent of the MAP kinase ERK. Curr. Biol., 9, 810–820. [DOI] [PubMed] [Google Scholar]
- Ryazanov A.G., Pavur,K.S. and Dorovkov,M.V. (1999) Alpha kinases: a new class of protein kinases with a novel catalytic domain. Curr. Biol., 9, R43–R45. [DOI] [PubMed] [Google Scholar]
- Scott P.H., Brunn,G.J., Kohn,A.D., Roth,R.A. and Lawrence,J.C. (1998) Evidence of insulin-stimulated phosphorylation and activation of mammalian target of rapamycin by a protein kinase B signaling pathway. Proc. Natl Acad. Sci. USA, 95, 7772–7777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebolt-Leopold J.S. et al. (1999) Blockade of the MAP kinase pathway suppresses growth of colon tumors in vivo. Nature Med., 5, 810–816. [DOI] [PubMed] [Google Scholar]
- Shima H., Pende,M., Chen,Y., Fumagalli,S., Thomas,G. and Kozma,S.C. (1998) Disruption of the p70S6k/p85S6k gene reveals a small mouse phenotype and a new functional S6 kinase. EMBO J., 17, 6649–6659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas G. and Hall,M.N. (1997) TOR signalling and the control of cell growth. Curr. Opin. Cell Biol., 9, 782–787. [DOI] [PubMed] [Google Scholar]
- Toker A. and Newton,A.C. (2000) Cellular signalling: pivoting around PDK1. Cell, 103, 185–188. [DOI] [PubMed] [Google Scholar]
- Wang L., Wang,X. and Proud,C.G. (2000) Activation of mRNA translation by insulin in rat cardiomyocytes involves multiple rapamycin-sensitive steps. Am. J. Physiol., 278, H1056–H1068. [DOI] [PubMed] [Google Scholar]
- Wang X., Campbell,L.E., Miller,C.M. and Proud,C.G. (1998) Amino acid availability regulates p70 S6 kinase and multiple translation factors. Biochem. J., 334, 261–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams M.R., Arthur,J.S.C., Balendran,A., van der Kaay,J., Poli,V., Cohen,P. and Alessi,D.R. (2000) The role of 3-phosphoinositide-dependent protein kinase 1 in activating AGC kinases defined in embryonic stem cells. Curr. Biol., 10, 439–448. [DOI] [PubMed] [Google Scholar]