

Abstract
Epstein–Barr virus (EBV) replicates in its latent phase once per cell cycle in proliferating B cells. The latent origin of DNA replication, oriP, supports replication and stable maintenance of the EBV genome. OriP comprises two essential elements: the dyad symmetry (DS) and the family of repeats (FR), both containing clusters of binding sites for the transactivator EBNA1. The DS element appears to be the functional replicator. It is not yet understood how oriP-dependent replication is integrated into the cell cycle and how EBNA1 acts at the molecular level. Using chromatin immunoprecipitation experiments, we show that the human origin recognition complex (hsORC) binds at or near the DS element. The association of hsORC with oriP depends on the DS element. Deletion of this element not only abolishes hsORC binding but also reduces replication initiation at oriP to background level. Co-immunoprecipitation experiments indicate that EBNA1 is associated with hsORC in vivo. These results indicate that oriP might use the same cellular initiation factors that regulate chromosomal replication, and that EBNA1 may be involved in recruiting hsORC to oriP.
Keywords: DNA replication/EBNA1/Epstein–Barr virus/ORC/oriP
Introduction
Replication of eukaryotic genomes is a tightly controlled process that occurs once per cell cycle. Studies in prokaryotic and eukaryotic systems have shown that this process initiates at defined cis-elements. Origins of DNA replication are recognized by proteins that together promote the formation of replication-competent origins. In the past decade, a general pattern has emerged that argues for a conserved mechanism in all eukaryotic systems, although specific differences are described. According to this model, origins are recognized by the six-subunit origin recognition complex (ORC). ORC binding is essential but not sufficient to produce replication competence. During each G1 phase, a pre-replicative complex (pre-RC) is assembled by associating additional proteins with origins. A functional pre-RC is a prerequisite for the activation of origins.
The yeast Saccharomyces cerevisiae ORC contacts two different regions of the origin: domain A is recognized in a sequence-specific manner, whereas the other functional element, domain B1, has no conserved motif (Bell and Stillman, 1992; Rao et al., 1994; Rao and Stillman, 1995; Rowley et al., 1995; Lee and Bell, 1997; Kelly and Brown, 2000). Given the remarkable conservation of initiation proteins in eukaryotic systems, one could speculate that target sequences for ORC might exist in other species as well. Despite much effort, such a picture is only beginning to emerge. Therefore, it is a key challenge to characterize ORC–DNA interactions in eukaryotic organisms other than S.cerevisiae, and to provide functional evidence for this complex generating a functional replicator. Similarly, it is hypothesized that replication initiates at or close to ORC-binding sites. In the case of S.cerevisiae, this expectation was proved by Bielinsky and Gerbi (1998, 1999) who mapped the initiation start site of DNA synthesis close to the ORC-binding site in ARS1. Progress has been made recently with the identification of initiation start sites at ars1 of Schizosaccharomyces pombe and at the lamin B2 origin in human cells (Gomez and Antequera, 1999; Abdurashidova et al., 2000; Bielinsky and Gerbi, 2001). In addition, it was shown by genomic footprinting at the lamin B2 origin that cell cycle-dependent changes in protein–DNA complexes occur that resemble the pre-RC/post-RC pattern described in S.cerevisiae (Diffley et al., 1994; Abdurashidova et al., 1998). These experiments extend the yeast paradigm to higher eukaryotes, but ORC binding to this element has remained elusive so far.
The identification of origins in higher eukaryotes has been difficult. The few origins identified thus far are not numerous enough to deduce conserved consensus motifs (Todorovic et al., 1999; Bogan et al., 2000). Epstein–Barr virus (EBV) has long been known to carry an origin of DNA replication, oriP, that has served as a model system to study replication initiation since its identification (Yates et al., 1984). EBV is a human herpesvirus that immortalizes B lymphocytes in vitro and in vivo, to establish a latent infection in continuously proliferating cell clones. In these cells, the EBV genome is maintained as an episome and replicates synchronously with the cellular DNA (Adams, 1987; Yates and Guan, 1991). OriP is a 1.8 kbp region that initially was identified for its ARS activity and functions as a bidirectional replication origin (Yates et al., 1984; Gahn and Schildkraut, 1989; Little and Schildkraut, 1995). Putative start sites were mapped at the nucleotide level (Niller et al., 1995). OriP consists of two elements, the family of repeats (FR) and the dyad symmetry (DS) element (Figure 1A; Reisman et al., 1985). Both elements contain binding sites for the viral transactivator EBV nuclear antigen 1 (EBNA1). FR has 20 copies of a 30 bp repeat, each including an EBNA1-binding site (Rawlins et al., 1985). The binding of EBNA1 to these sites contributes to plasmid maintenance (Reisman et al., 1985; Wysokenski and Yates, 1989; Aiyar et al., 1998). The DS element contains four EBNA1-binding sites and functions as a replicator that presumably requires the presence of EBNA1 (Harrison et al., 1994; Aiyar et al., 1998; Shirakata and Hirai, 1998; Yates et al., 2000). Replication competence of oriP plasmids is dependent on the integrity of the DS element. In contrast, the DS element is dispensable in the context of the complete EBV genome, where replication can also initiate outside of oriP (Gahn and Schildkraut, 1989; Little and Schildkraut, 1995; Norio et al., 2000). This is an interesting observation with respect to the ongoing debate as to whether replication in higher eukaryotes initiates either in broad initiation zones or at distinct sites (for a review see Bogan et al., 2000).


Fig. 1. (A) The bipartite structure of oriP. The dyad symmetry (DS) and the family of repeats (FR) are the two essential elements of oriP. Each black circle (oval in the enlargement) represents a single binding site for an EBNA1 dimer. At the DS element, a spacing of 21 bp exists between neighbouring pairs of EBNA1-binding sites (E). The binding sites within the FR were determined experimentally (Ambinder et al., 1990). It consists of two blocks of 10 EBNA1-binding sites showing dyad symmetry. Rep* is a 298 bp DNA fragment that can partially substitute for the DS element if multimerized on a plasmid (Kirchmaier and Sugden, 1998). (B) Human primary B cells were immortalized with the 81 kbp derived mini-EBV plasmid 1478.A. Functional elements are shown on the outer circle. The latent viral genes EBNA1, EBNA2 and LMP1 are depicted. oriP, the latent origin of DNA replication, is shown with its two essential components, FR and DS, and the non-essential element Rep*. The second origin of DNA replication of EBV, oriLyt, is active only during the lytic phase. The two components are indicated as circles. The plasmid backbone of this episome stems from the F-factor plasmid pMBO132 (arrows). The terminal repeats and the W repeats, repetitive sequences within the episome, are shown. The terminal repeats are essential elements for packaging of the viral genome. The inner circle of the map indicates the location and designation of the fragments that were analysed by PCR amplification after immunoprecipitation (see Table I).
Two-dimensional gel electrophoresis and initiation start site mapping provided evidence that oriP is a functional replicator. Additional data support the hypothesis that oriP can be regarded as a quasichromosomal origin of DNA replication constituting a suitable model system to study initiation of replication in human cells. The latent EBV genome replicates once per cell cycle during S phase like the cellular genome (Adams, 1987; Yates and Guan, 1991). It has been shown recently that each round of replication requires passage through mitosis (Shirakata et al., 1999). This finding suggests that replication licensing is essential for oriP-mediated replication. Dimethylsulfate (DMS) footprinting of cells that had been arrested in G1/S and mitosis revealed small cell cycle-dependent modifications, indicating a possible change in the DNA–protein complex at oriP (Niller et al., 1995). EBNA1 is the only viral protein that is involved in latent replication of EBV, all other replication proteins are provided by the cellular host. Therefore, and because of its prominence at oriP, it has been speculated for a long time that EBNA1 might be a strong candidate for an initiator protein. However, no function associated with an initiator, e.g. ATPase and helicase activity, could be identified thus far (Frappier and O’Donnell, 1991; Middleton and Sugden, 1992).
In order to address how oriP mediates replication initiation, we asked whether cellular initiation proteins interact with oriP and EBNA1. Here, we have tested the hypothesis that hsORC is involved in oriP function using the chromatin immunoprecipitation assay (ChIP). In this study, we provide evidence that hsORC binds to or close to the DS element of oriP. In addition, this association is dependent on the DS element. Together with our finding that hsORC and EBNA1 interact with each other in vivo, our data suggest that EBNA1 is directly involved in recruiting hsORC to oriP.
Results
Generation of a mini-EBV-immortalized B cell
To study the binding of cellular replication initiation proteins to oriP, we used a B-cell line, A39, immortalized with a mini-EBV (Figure 1B). Mini-EBV plasmids contain all viral genes required to support the proliferation of in vitro immortalized B cells similarly to wild-type EBV efficiency (Kempkes et al., 1995a,b). This system has the advantage that mini-EBV plasmids can be genetically engineered in Escherichia coli with conventional techniques (O’Connor et al., 1989). More importantly for this study, mini-EBV plasmids lack the majority of viral genes that mediate the lytic or productive phase of EBV. Therefore, no spontaneous induction of lytic genes can take place that might blur the picture of latent DNA replication of EBV (Pfuller and Hammerschmidt, 1996). The A39 cell line used in this study was generated with the mini-EBV plasmid 1478.A (Figure 1B; Kempkes et al., 1995b), as described in Materials and methods. Single-cell clones were confirmed by PCR to contain only mini-EBV plasmid (data not shown). Mini-EBVs are maintained as episomes with an average copy number of 5–10 per cell. In the mini-EBV plasmid 1478.A, oriP is flanked by authentic EBV sequences of at least 20 kbp on both sides of the origin. Therefore, the local chromosomal structure of oriP is most probably indistinguishable from its natural composition.
EBNA1 binds specifically to oriP
To investigate whether oriP is bound by cellular replication initiation proteins, we used the ChIP assay (see Figure 2A). To adapt the ChIP assay to our needs, we made use of the strong interaction between EBNA1 and oriP sequences, which seemed to be a good starting point to study protein–DNA interactions at oriP. It has been shown that EBNA1 binds both elements of oriP, FR and DS, in vivo, presumably throughout the cell cycle (Hsieh et al., 1993; Niller et al., 1995). The ChIP protocol was performed with logarithmically proliferating A39 cells as described in Materials and methods (Figure 2A). To avoid unspecific cross-links, cells were treated with a limited amount of formaldehyde (Göhring and Fackelmayer, 1997). To minimize the contamination with non-cross-linked material, nuclei were incubated with a high salt buffer. After lysis with N-laurylsarkosyl, the chromatin was purified on a caesium chloride gradient. The chromatin was dissolved at a concentration equivalent to 1 × 108 cells/ml and sonicated to shear the DNA for the qualitative analysis of protein binding. For the quantitative analysis and fine mapping of putative binding sites, an enzymatic digest with micrococcal nuclease (MNase) was added in late experiments. Such extracts were used for subsequent immunoprecipitations with chromatin preparations corresponding to 1–2 × 107 cells. The relative abundance of co-precipitated DNA was measured by conventional PCR in comparison with immunoprecipitates with control antibodies.

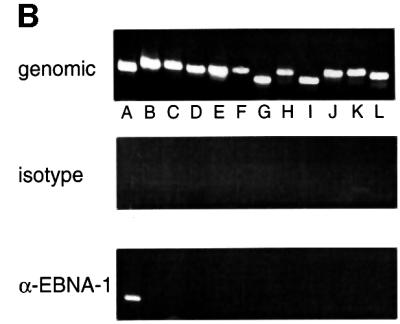

Fig. 2. In vivo association of EBNA1 and hsORC with oriP analysed by formaldehyde cross-linking. (A) Scheme of the ChIP protocol; see Materials and methods for details. (B) Asynchronously proliferating A39 cells were subjected to the ChIP protocol and, after cell lysis, nuclear extracts were divided into two parts. One half was immunoprecipitated with the 1H4 antibody directed against EBNA1 (bottom panel) and the other half was immunoprecipitated with an irrelevant antibody of the same isotype (middle panel). PCR was performed on sonicated chromatin, isolated after immunoprecipitation, or on 2 ng of sheared genomic DNA (top panel). The positions of the DNA fragments A–L are indicated within the centre of the mini-EBV genome. (C) ChIP assay with polyclonal antibodies directed against different subunits of the human ORC. Immunoblots performed with the indicated polyclonal rabbit antibodies and pre-immune sera are shown on the left. These antibodies were used for all ChIP experiments. They detect specific signals at the expected size for hsORC1 (105 kDa), hsORC2 (68 kDa) and hsORC3 (72 kDa). The pre-immune sera show no signal at the corresponding positions (left panel). The association of hsORC with the corresponding DNA fragments was analysed as described above. The top panel shows the amplification of all 12 fragments tested with sheared genomic DNA as template. A 300 pg aliquot of total genomic 1478.A DNA were used for each control reaction.
Immunoprecipitation of EBNA1 was performed with the monoclonal antibody 1H4 and a corresponding isotype control antibody (Grasser et al., 1994). To analyse the sequence-specific binding of EBNA1, we used 12 primer pairs to amplify DNA fragments that are evenly distributed over the entire mini-EBV episome (Figure 1B). The amplified fragments are 350–450 bp in length. Fragment A, used to detect oriP binding of EBNA1, encompasses the DS element. The amplification of FR itself was not feasible because of its repetitive sequences. All primer pairs used for the scanning analysis are ∼7 kbp apart from each other on average. Therefore, DNA was sonicated to an average length of 2–4 kbp (data not shown) to scan the entire mini-EBV plasmid. All 12 fragments from each locus were amplified from the sheared chromatin of A39 cells (Figure 2B, top panel). These reactions were always performed in parallel with immunoprecipitates obtained with the specific antibody and an isotype control.
As expected, the region encompassing oriP can be amplified with DNA immunoprecipitated with the 1H4 antibody directed against EBNA1. No other PCR fragment was amplified from these precipitates (Figure 2B, bottom panel). In addition, no DNA fragment was amplified from precipitates with a mock antibody of the same isotype as 1H4 (Figure 2B, middle panel). This result indicated that oriP-containing DNA fragments are associated exclusively with EBNA1 and precipitated only with an antibody of the respective specificity. This observation also documented that binding of EBNA1 is identical in the mini-EBV system and the EBV genome.
HsORC binds specifically to oriP
If cellular replication initiation proteins are involved in oriP-mediated replication, it is very likely that hsORC will be involved in this process. To address this question with the ChIP technology, we used antibodies directed against three different subunits of hsORC (hsORC1, hsORC2 and hsORC3). We generated polyclonal rabbit antibodies directed against the C-terminal domains of hsORC1 and hsORC3. The antibody directed against the hsORC2 subunit has already been described (Kreitz et al., 2001). These antibodies detect specific proteins that are not recognized by the pre-immune serum and migrate in the expected position of the respective hsORC subunit (Figure 2C, left panel). In addition, we raised monoclonal antibodies against the same immunogens. The antibodies 7A7 and 6D1 directed against hsORC1 and hsORC3, respectively, were characterized further and used for immunoblotting experiments later in this study (Figure 6A). The 7A7 antibody directed against hsORC1 does not cross-react with the partially homologous CDC6 protein (data not shown).
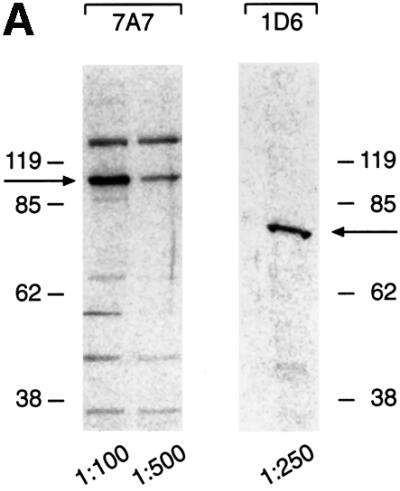
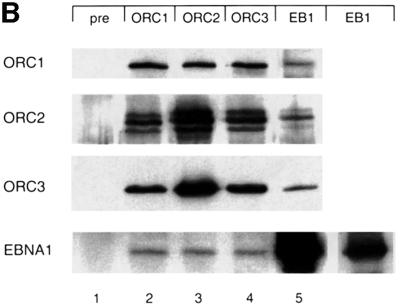
Fig. 6. Human ORC and EBNA1 associate. (A) Characterization of the monoclonal antibodies directed against hsORC1 and hsORC3. Bacterially expressed His-tagged C-terminal domains of hsORC1 and hsORC3 were used for the generation of antibodies in rats. Monoclonal 7A7 detects a protein of 105 kDa as the polyclonal hsORC1 antibody, whereas 6D1 detects a protein of the same migration pattern as the polyclonal hsORC3 antibody. The respective signals are indicated with an arrow. Extracts of 4 × 105 cells were loaded per lane, the dilutions used are indicated. (B) Native nuclear proteins that were released from the chromatin of logarithmically proliferating A39 cells were immunoprecipitated by using the antibodies indicated at the top: pre-immune serum (pre), hsORC1, hsORC2, hsORC3 and EBNA1 (EB1). HsORC immunoprecipitations were performed with the polyclonal antibodies used for the ChIP experiment, or with the monoclonal antibody 1H4 directed against EBNA1. The antibodies used for immunodetection are indicated on the left. The polyclonal antibody was used for the detection of hsORC2, and monoclonal antibodies 7A7 and 6D1 (see A) were used for the detection of hsORC1 and hsORC3, respectively. A shorter exposure of the EBNA1 immunoprecipitate detected with the 1H4 antibody is shown in the inset in the rightmost lower panel.
We used the ChIP procedure described above to test whether hsORC associates with the EBV genome, in particular with oriP. Chromatin extract was subjected to individual immunoprecipitations with the three polyclonal hsORC antibodies and pre-immune sera. PCR amplifications with all 12 primer pairs were performed in parallel with all immunoprecipitates and analysed by ethidium bromide-stained agarose gels. As shown in Figure 2C, fragment A, encompassing oriP or sequences close to it, was precipitated preferentially and amplified by PCR. No other DNA fragment was amplified specifically after immunoprecipitations with these antibodies. The immunoprecipitates obtained with all three antibodies directed against hsORC1, hsORC2 and hsORC3 gave rise to a PCR signal of comparable intensity. This observation indicates that all three hsORC subunits are present at oriP.
In contrast to the ChIP experiment with the EBNA1-specific monoclonal antibody, 32 cycles were required to obtain a decent signal with hsORC immunoprecipitates, as compared with 27 cycles with the EBNA1 precipitates. This observation might reflect different antibody affinities. Alternatively, it might be the result of ∼50 EBNA1 molecules binding to oriP, in contrast to a much lower number of hsORC molecules binding in close proximity. This observation indicated that the co-immunoprecipitations of oriP-containing DNA with anti-hsORC antibodies were much less efficient than immunoprecipitations with the antibody directed against EBNA1. Weak signals, occasionally observed in ChIP experiments with hsORC antibodies, i.e. fragment B with hsORC1, or fragment L with hsORC2 and hsORC3 immunoprecipitates, were also amplified in co-immunoprecipitates with pre-immune sera (i.e. lanes B and L in hsORC2 pre-immune). These signals varied not only between the different subunits, but also from experiment to experiment. At present, we cannot rule out that this reflects a weak and unspecific binding of hsORC to different regions of the mini-EBV episome.
HsORC binds at or near the DS element
The data presented above provide strong evidence for hsORC binding to a region at or close to oriP in vivo. In the next step, we aimed to define the putative hsORC-binding site(s) in more detail. Therefore, we changed the ChIP protocol in two respects. First, we reduced the size of the DNA fragments to an average size of 300–500 bp by incubating the purified and sonicated chromatin preparation with MNase (data not shown). We generated a new set of nine primer pairs, which amplify fragments of 250–350 bp in length. Secondly, to be in the position to quantify differences between the immunoprecipitated DNA fragments, we analysed the associated DNA fragments by real-time PCR technology. We chose real-time PCR, in contrast to conventional PCR techniques such as competitive PCR, because it allows precise and reproducible quantification over a wide dynamic range covering several orders of magnitude. Another advantage is that analysis is performed during the log phase of the reaction and is not based on end point analysis.
The analysis procedure for a given primer pair as a representative example of 10 primer pairs (psc10, Figure 4A) is outlined in Figure 3. We tested the overall sensitivity of each primer pair with a series of 10-fold dilutions using genomic DNA that had undergone the cross-linking procedure, but had not been subjected to immunoprecipitation (Figure 3A). A standard curve was generated to evaluate the amplification efficiency of the reaction (Econst; see Figure 3 legend for details). Standard curves were produced for all primer pairs. Econst was calculated from standard curves that cover at least four orders of magnitude (Figure 3B for psc10). To compare individual immunoprecipitation experiments and reactions with genomic DNA with each other, Econst has to be stable. To test how precisely a binding site can be mapped under these experimental conditions, and in order to control the reproducibility of our experiments, we performed three independent immunoprecipitations with the 1H4 antibody. Figure 3C shows as an example the result of one of the EBNA1 immunoprecipitations and the relevant isotype control, both analysed with the primer pair psc10. The difference between the crossing points is 6.4 cycles; the mean value of all three experiments was determined to be 4.9 ± 0.9 cycles (Figure 4C).



Fig. 4. HsORC binds at the DS element or nearby. (A) Enlarged view of oriP. The locations of the PCR fragments used to scan the binding sites of EBNA1 and hsORC are shown below the ruler (sc2–sc10). The ChIP experiment was performed with cross-linked A39 cells. Immunoprecipitations were executed with the chromatin fraction of 1–2 × 107 cells; 1/100 thereof was used for one PCR. The histogram shows the EBNA1 analysis. The difference between the crossing point of the EBNA1 immunoprecipitate and the isotype control is indicated on the y-axis. The graph shows the mean value and standard deviation of three independent experiments. (B) Histogram of DNA fragments accumulated in immunoprecipitates with polyclonal antibodies directed against the hsORC subunits 1, 2 and 3 (see Figure 2). Therefore, triplets are shown for each scanning PCR fragment to illustrate the data for hsORC1, 2 and 3. The mean values and standard deviations are again calculated from three independent experiments. (C) This table summarizes the data of the histograms and calculates the enrichment of the analysed fragments. Each individual Econst of the primer pairs was determined from standard curves of 10-fold dilutions of the immunoprecipitations, using the formula Econst = 10–1/slope (see Figure 3 for details). The cycle differences are shown as mean values of three independent experiments. The enrichment of each fragment was calculated as explained in Figure 3C using the mean values of Econst, and the difference of the crossing points between the specific immunoprecipitations and the pre-immune/isotype immunoprecipitations.

Fig. 3. Parameters for quantification of real-time PCR products. (A) The histogram shows the amplification profile of PCR products amplified from a series of 10-fold dilutions using cross-linked genomic DNA prior to any immunoprecipitation. Only one example with the primer pair psc10 is shown to demonstrate the principles of data collection and processing. To calculate the efficiency of the immunoprecipitation with the different antibodies, the concentration of the input genomic DNA was determined photometrically. To follow the amplification of the PCR product, a fluorescent dye was used in the real-time PCR. The fluorescence was indicative of the concentration of the amplified PCR fragments and was plotted against the cycle number (x-axis). Analysis of the PCR products was performed during the exponential phase (log phase) of the amplification and is not based on the end point of the reaction as in conventional PCR techniques. The log phase begins when sufficient product has accumulated to be detected above background and ends when the reaction enters the plateau. In theory, the PCR product is duplicated in a single PCR round and is described by the formula N = N0 × 2n (N, number of molecules; N0, number of starting molecules). In reality, the efficiency of the PCR is constant only during the log phase but usually is <2 and is described by the equation: N = N0 × (Econst)n. The log line of the exponential phase is calculated and set against the threshold line that subtracts the background (black lines in A). The threshold was determined automatically by the machine using the standard settings (second derivative method; for details, see LightCycler operator’s manual, version 3.5). The crossing of the log line and the threshold line defines the crossing point (Cp). (B) The standard curve of the given example is the linear regression line through the data points on a plot of Cp versus the logarithm of standard sample concentration. The purpose of the standard curve is 2-fold. It illustrates the range in which the collected data fit with the reconstruction of the initial template concentration. In addition, the standard curve also allows the calculation of the amplification efficiency of the reaction (Econst). Econst can be calculated from the slope of the standard curve using the formula Econst = 10–1/slope. In the given example, the slope is –3.74; the reaction efficiency is therefore 1.85.The amplification efficiency for each primer pair used in the scanning analysis was determined by dilution series and is listed in Figure 4C. (C) To determine the specific enrichment of a fragment after immunoprecipitation with a specific antibody, the difference between the crossing points of the specific immunoprecipitate and the isotype/pre-immune control was calculated. The graph shows the example of an EBNA1 immunoprecipitation; the Cp difference is 6.4 cycles. This number is the exponent to the basis Econst = 1.85. The enrichment of the EBNA1 immunoprecipitation is calculated by the equation: Nisotype – NEBNA1 = N0 × (Econst)n(isoytpe) – N0 × (Econst)n(EBNA1); in this example, the enrichment is: Nisotype/N0 – NEBNA1/N0 = 1.8528.8 – 1.8522.4 = 51-fold.
To analyse the binding of EBNA1 to oriP, real-time PCR was performed with scanning primer pairs covering the region of oriP (with the exception of FR) and including 2 kbp of its upstream and downstream flanks (Figure 4A, Table I). PCRs across FR are not possible, because of the highly repetitive character of this element. Therefore, we placed primer pairs immediately upstream and downstream of FR (sc3 and sc4, respectively). All real-time PCRs performed with the co-immunoprecipitated DNA were analysed as explained above. The data obtained with the EBNA1 immunoprecipitates clearly indicated that DNA fragments encompassing FR, DS and the region in between these two elements (sc4–sc7, domain A) were co-precipitated specifically and subsequently amplified by PCR. The histogram in Figure 4A elucidates two findings. First, it indicates a highly specific accumulation of DNA fragments containing oriP compared with the isotype control, confirming the strong association of EBNA1 with oriP (crossing point difference ΔCp: 11–12 cycles). Secondly, the distribution of the co-precipitated fragments did not follow an ideal Gaussian distribution. The enrichment of DNA fragments located more distally to oriP proved to be remarkably lower at the 5′-upstream region of FR compared with the 3′-downstream region of the DS element (compare sc3 and sc7/sc8). In addition, the primer pair psc3 showed a higher variability in comparison with others. This finding was surprising since FR was reported to be bound by the majority of EBNA1 molecules (Hsieh et al., 1993). A possible explanation for the lower amount of co-precipitated fragments upstream of the FR might stem from preferential cleavage by MNase within or adjacent to FR. On the contrary, the decrease of the PCR signals amplified downstream of the DS element was not as substantial as that of the upstream region of FR. This observation might reflect the enhancer function of EBNA1 on the BamC promoter, which is located downstream in close proximity to oriP (Figure 4A; Wysokenski et al., 1989).
Table I. Oligos used in this work for PCR analysis.
| Location | Oligo | Sequence | Oligo | Sequence |
|---|---|---|---|---|
| Domain A | A′′F6 | TTCGGGGGTGTTAGAGACAACC | F′′B5 | TTAGTCACAAGGGCAGTGGCTG |
| Domain B | B′′F10 | ACCTGATTCCCCCTGCTCATAC | B′′B5 | GCACTAACACAAGCCCAACAACACAC |
| Domain C | CF | GCCTACCGAACTTCAACCC | CB | CTGGTCTCCAAGGTCCACCGG |
| Domain D | DF1 | ATTCGGTGGCATCCCTGAAG | DB4 | CCCCCAAGAACACAAAGAGACAG |
| Domain E | EF30 | GGTGAGGGAACACGACCACG | EB35 | CAGATTACGGCGGTGATTCAAC |
| Domain F | FF | GAGACCCAAAGCAGCCAAGCC | FB | AGCCGAGCCTGGACACTCAGT |
| Domain G | GF24 | GGTCTCTCATCGCTCTCTTGGTG | GB25 | CAGTGGAAACTCCTATCCTCGTG |
| Domain H | HF1 | GCAAATCAGGAGGTCACGGATG | HB2 | ACCAGATGTTGGGAGGGCATTC |
| Domain I | IF26 | CGGTGAGATAGATTGGAGGCTG | IB12 | AGGCGTGGGTGTCAGACCTG |
| Domain J | J′F1 | CCGTGGGAAAAACTCCAGGTAG | J′B1 | AGGCATGCTCTCTCTGAAAATCG |
| Domain K | KFF1 | CGGAACCAGAAGAACCCAAAAG | KBB2 | ATGGACAACGACACAGTGATGAAC |
| Domain L | LF5 | ATTATGGTAAGGCTCTTCGGGC | LB6 | AAGGGCGTTGTCAAACAGGAC |
| Scanning 2 | sc2F | TCAACAGATAATCCACCCGCC | SC2B | CACAGGAGAGCCAGATGACGAC |
| Scanning 3 | SC3F | TCCATTATCCCGCAGTTCCAC | SC3B | GCCGAATACCCTTCTCCCAGAG |
| Scanning 4 | SC4F | TCGGCGTCCACTCTCTTTCC | SC4B | CAGTAAGGTGTATGTGAGGTGCTCG |
| Scanning 5 | SC5F | TGTCATAGCACAATGCCACCAC | SC5B | GGTCAGGATTCCACGAGGGTAG |
| Scanning 6 | SC6F | AAAGTCTGCTCCAGGATGAAAGC | SC6B | CCAGGGCGTTCTATTTTTACAGC |
| Scanning 7 | SC7F | TAGAACGCCCTGGAACTGCC | SC7B | TCTCTTAGGTCCCTCAACATTTGG |
| Scanning 8 | SC8F | CCTCTTGTGTTCTTGCCGCC | SC8B | TGCCCTCCCCACTTCTCTTC |
| Scanning 9 | SC9F | CAGATGCCCCCCACCAGTC | SC9B | CGAGCCCTGCGTCTTGAGC |
| Scanning 10 | SC10F | GCCTTATCTGGGAGGAGCGAC | SC10B | TGGGGGTCTTCGGTGTCC |
The data obtained with the EBNA1-specific antibody indicated that the ChIP protocol allowed the mapping of binding sites with a resolution of <1 kbp. To measure the association of hsORC with oriP in more detail, we applied the same procedure using the same hsORC-specific antibodies as before (Figure 4B). The relative accumulation of specifically co-precipitated DNA fragments was calculated as the difference in cycle numbers between the crossing points (ΔCp, Figure 3). The detailed analysis of immunoprecipitated DNA fragments demonstrated that DNA fragments containing the DS element and the Rep* element (Kirchmaier and Sugden, 1998) are both co-immunoprecipitated with hsORC antibodies (domain A, sc6 and sc7; Figure 4B). The spreading of co-precipitated DNA fragments showed a distribution resembling a Gaussian curve. To be in the position to compare the independent PCRs and different primer pairs with each other, the reaction efficiency Econst of each primer pair had to be determined (Figure 4C). To determine Econst, we analysed 10-fold dilutions using the specific immunoprecipitations. Standard curves were deduced as exemplified in Figure 3B and used to calculate Econst. The results are summarized in Figure 4C. As already indicated above, it was apparent that the enrichment of DS- and Rep*-containing fragments was many fold higher comparing hsORC-specific immunoprecipitations with pre-immune sera. Moreover, the concentration of these sequences was ∼18–45 times higher in relation to more distal DNA fragments (sc3 and sc10 compared with domain A, sc6 and sc7, in Figure 4B and C for example).
We also estimated the efficiency of the immunoprecipitations with the different antibodies. In EBNA1 immunoprecipitations, we recovered >90% of EBNA1 protein (data not shown) and about half of oriP DNA. For the different hsORC immunoprecipitations, we also precipitated >90% of hsORC protein present in the cell (data not shown). The recovery of oriP-containing DNA was estimated to be in the range of 1–3% based on the relative enrichment of EBNA1 versus hsORC immunoprecipitations, as shown in Figure 4C. This difference was in the same range as the five cycle difference found with conventional PCR (Figure 2B and C), assuming that the efficiency of conventional PCR is similar to that of real-time PCR. This finding presumably reflects the relative abundance of EBNA1 bound to oriP versus hsORC. Comparable results were obtained for oriP with ChIP experiments performed in the context of a full-size EBV genome (data not shown). Another important finding of this set of experiments was the almost identical amplification profile with all three hsORC subunits, supporting the idea that hsORC binds to oriP as a holocomplex, although this does not necessarily mean that the hsORC subunits bind in a 1:1 stoichiometry.
HsORC binding is dependent on the presence of the DS element
Two-dimensional gel electrophoresis indicated that replication initiates at or near the DS element of oriP (Gahn and Schildkraut, 1989; Little and Schildkraut, 1995). Recently, it has been shown that the deletion of the DS element in the context of the entire viral genome reduces the number of initiation events at oriP below the detection level of two-dimensional gel electrophoresis. This observation suggested that replication can initiate at sites other than oriP in the context of the full-size EBV genome (Norio et al., 2000). We took advantage of the P3-ΔDS-33 strain described by Norio et al. (2000) to see whether the DS element is required for the association of hsORC with oriP. This EBV genome carries a mutation within oriP that deletes the entire DS element, but leaves the remnants of this origin intact. We started this analysis by studying the binding of EBNA1 to the mutated oriP. Figure 5A shows that in P3-ΔDS-33 cells the peak of EBNA1-precipitated material was restricted to FR-containing DNA fragments, but was remarkably reduced at the region of the deleted DS element. This result was expected because EBNA1 cannot bind to this region in the absence of the DS element.


Fig. 5. ChIP experiment with P3-ΔDS-33 cells, which carry an EBV genome with a deletion of the DS element in the context of a full-size EBV genome. The experiment was performed and analysed as described in Figure 3 with (A) EBNA1 antibodies and (B) polyclonal antibodies directed against subunits 1, 2 and 3 of the human ORC, respectively.
By analysing the binding affinity of hsORC for oriP it became apparent that the binding of hsORC was dramatically reduced in P3-ΔDS-33 cells (Figure 5B), compared with hsORC binding to oriP in the wild-type context (Figure 4B and data not shown). Using quantitative PCR, it became clear that hsORC binding was reduced undetectably to background levels (see domain A, sc6 and sc7 in Figure 5B). This finding indicated that the DS element is essential for the association of hsORC with oriP. We are aware that replication also initiates at regions other than oriP, since the P3-ΔDS-33 genome initiates replication only outside of oriP (Norio et al., 2000). However, it is not understood how replication initiation is regulated at these positions.
EBNA1 and hsORC interact with each other
The data of Norio et al. (2000) revealed that the DS element of oriP is essential for DNA replication mediated by this origin. Our data obtained so far indicated that the DS element is critical for the association of hsORC with oriP. However, it is unclear whether hsORC binds to or close to the DS element or whether this is a function mediated by EBNA1 binding to DS. Previously published immunoprecipitation experiments indicated that hsORC subunits associate not only with each other but also with additional cellular proteins (Gavin et al., 1995; Quintana et al., 1997, 1998; Tugal et al., 1998; Dhar and Dutta, 2000; Thome et al., 2000; Kreitz et al., 2001). We wanted to analyse whether EBNA1 interacts with hsORC. To address this question, we performed immunoprecipitations with proteins that were released from chromatin under relatively stringent conditions (450 mM NaCl). The immunoprecipitates subsequently were analysed by immunoblotting experiments with antibodies directed against EBNA1 and different hsORC subunits (monoclonal antibodies 7A7 and 1D6 directed against hsORC1 and hsORC3, respectively; Figure 6A). We noted similar signal intensities in hsORC immunostainings, supporting the idea of an hsORC holocomplex (Figure 6B, lanes 2–4). We point out that the stoichiometry of metazoan ORC, in particular of human ORC, at origins has yet to be determined. In contrast to this finding, much less EBNA1 was detected in all immunoprecipitations with the different hsORC subunits. The signal obtained for EBNA1 itself in immunoprecipitations with an EBNA1-specific antibody was much stronger than the EBNA1 signal obtained in co-immunoprecipitations with hsORC antibodies (Figure 6B, bottom panel). This might indicate a rather weak contact between EBNA1 and hsORC. Alternatively, only a small proportion of EBNA1 molecules might actually interact with hsORC in vivo.
These findings were confirmed by reciprocal immunoprecipitation experiments with the EBNA1-specific 1H4 antibody. All three hsORC subunits analysed by western blotting experiments were found in EBNA1 immunoprecipitates (lane 5). Again, we noted that only a subfraction of the hsORC subunits was found in the EBNA1 precipitates. Similar results were obtained in co-immunoprecipitation experiments performed with formaldehyde-cross-linked nuclear extracts (data not shown).
Discussion
The latent origin of DNA replication of EBV, oriP, accomplishes replication and maintenance with a single viral gene, EBNA1. oriP consists of two clusters of EBNA1-binding sites. The family of repeats and the dyad symmetry element are both required for stable replication of oriP plasmids. Biochemical studies suggest that DNA replication initiates at the DS element. It is, however, not understood how replication is initiated. Most studies indicate that oriP is regulated like a chromosomal origin that replicates once per cell cycle during S phase by using the cellular replication system. In this study, we provide evidence that supports this hypothesis by adding two important findings. First, we show that the human ORC binds to or close to the DS element. The association of hsORC is dependent on the integrity of the DS element. The second important result of this study is the finding that EBNA1 and hsORC interact with each other in vivo. This observation is the first hint of how oriP-mediated replication might be integrated into the cellular replication cycle.
The data provided here suggest that EBNA1 binding to the DS element might be required to direct hsORC to the EBV genome. It is an open issue, however, whether hsORC binds to the DS element itself or to an adjacent region. The resolution obtained with the ChIP analysis indicated that hsORC binds within the 650 bp fragment encompassing DS and Rep*. A more precise identification of a putative binding site does not seem possible by this method. The identification of a presumed transition point in the DS element would argue for hsORC binding within this element (Niller et al., 1995), if the replication initiation point lies immediately adjacent to the ORC-binding site analogously to the situation in S.cerevisiae. Here, replication initiation takes place next to the ORC-binding site of ARS1 (Bielinsky and Gerbi, 1998, 1999). Experiments at the human lamin B2 origin have identified a replication transition point in a region that shows a cell cycle-dependent protein–DNA interaction pattern (Abdurashidova et al., 2000; Bielinsky and Gerbi, 2001). These changes resemble the pre-RC/post-RC paradigm described for origins in S.cerevisiae (Diffley, 1994) Whether the DNase footprints at the lamin B2 origin are caused by the binding of hsORC and other components of the pre-RC will have to be determined. Footprinting experiments with EBV-positive cells have revealed that EBNA1 binds to the DS element throughout the cell cycle (Hsieh et al., 1993; Niller et al., 1995). In addition to this, three nonamer repeats have been identified, whose footprinting pattern might differ slightly between G1/S and mitotically arrested cells (Niller et al., 1995). The nonamers lie adjacent to the EBNA1-binding sites. The DMS footprints are difficult to interpret because they are superimposed by the prominent binding of EBNA1. It is still possible that ORC binds to oriP distal to the DS element, since no footprinting or start site mapping experiments have been performed outside of the 120 bp region of the DS element. It is possible, for example, that the DS element is required to direct hsORC to the adjacent Rep* element, ensuring specificity. Deleting this element results in loss of hsORC binding and thereby in hsORC-mediated replication (Norio et al., 2000). This loss might be compensated by multiple Rep* elements that are able partially to substitute DS function (Aiyar et al., 1998; Kirchmaier and Sugden, 1998). A potential role for Rep* in replication initiation can be tested by positioning multiple copies of Rep* distant to oriP in the context of the mini-EBV. Immunoprecipitation experiments will reveal whether ORC binds to the Rep* oligomer.
ORC binding seems to be dependent on the integrity of the DS element, which further supports the idea of the DS element as a functional replicator. We will address the question of whether the DS element is sufficient to mediate hsORC binding and possibly replication initiation. A second copy of the DS element positioned at a different locus within the mini-EBV genome will provide genetic access to address this question. The ChIP assay is suitable to test whether hsORC also binds to this second copy. Additional experiments might indicate whether the delocalized DS element itself can function as replicator. Analogously to the dislocation of the β-globin locus, this experiment will test directly the potential function of the DS element as a fully functional eukaryotic replicator (Aladjem et al., 1998).
Chromosomal replication is a tightly controlled process and is integrated into a number of other cellular events. ORC binding to origins is essential for replication initiation and a prerequisite for the formation of the pre-RC, as outlined in the Introduction. Therefore, it is not surprising that ORC interacts with a number of non-ORC proteins. Most of these interactions provide a link between replication and replication-related functions, i.e. cell cycle-dependent kinases such as cdc2–cyclin A of Xenopus laevis (Romanowski et al., 2000) and cdc2 of Schizosaccharomyces pombe (Leatherwood et al., 1996), to chromatin factors such as SIR1 of S.cerevisiae (Triolo and Sternglanz, 1996), HP1 of Drosophila melanogaster (Pak et al., 1997; Huang et al., 1998) or the human histone acetyltransferase HBO1 (Iizuka and Stillman, 1999). It is a widely accepted opinion that, as in yeast, all six ORC subunits are required to serve as initiator to recognize putative replicator elements. The formation of this complex in metazoan organisms is, to date, not fully understood. Metazoan ORC1, like hsORC6, seems to play a critical role in the formation of the functional holocomplex and is therefore of special interest (Vashee et al., 2001). ORC1 is either displaced from chromatin during mitosis or expressed in a cell cycle-dependent manner (Asano and Wharton, 1999; Natale et al., 2000; Tatsumi et al., 2000; Kreitz et al., 2001). Therefore, it is interesting that our immunoprecipitation data provide evidence that EBNA1 probably interacts with the hsORC holocomplex, including the largest subunit, hsORC1. Further experiments will have to prove the stoichiometry of the interacting complex and the subcomponent of hsORC that interacts with EBNA1.
EBNA1 is not only essential for latent replication of EBV, it is also involved in transcriptional activation, maintenance and segregation (Yates, 1996). To fulfil its diverse functions, it interacts with cellular partners such as EBP2 and P32/TAP (Wang et al., 1997; Shire et al., 1999; Huber et al., 2000; Kapoor et al., 2001). This study provides evidence that the DS element is required for ORC binding to oriP. Since EBNA1 interacts with hsORC in vivo, one could imagine that EBNA1 might direct hsORC to its target sequence. A simple explanation for this observation is that EBNA1 provides a link between the cellular replication machinery and the viral requirement to replicate the EBV genome. The low efficiency of oriP co-immunoprecipitations with hsORC subunits, in contrast to EBNA1 immunoprecipitations, might also be explained by an indirect interaction of hsORC with oriP. The interaction between EBNA1 and hsORC, the eukaryotic initiator, is a possible explanation for why EBNA1 seems to differ from other viral initiation proteins such as SV40 T-antigen in that no function of a typical replication initiator (i.e. ATPase activity, helicase activity) has been identified for EBNA1 so far.
Although it is widely believed that ORC is involved in determining metazoan origins, it is not known how specificity is achieved. This question is closely linked to the debate as to whether DNA replication initiates in zones or at distinct sites (for reviews see Bogan et al., 2000; Kelly et al., 2000). Even the binding of DmORC to the ACE3 element requires additional flanking regions to mediate DmORC binding (Austin et al., 1999). ACE3 is one element of the loci from which the chorion genes are amplified during the stage 10 endoreduplication process. The requirement for additional flanking regions might indicate the binding of auxiliary factors involved in targeting DmORC to specific sequences. This hypothesis raises the question of whether EBNA1 might take over the functions of some unidentified cellular protein(s) that directs ORC to metazoan origins. Still, it is highly speculative as to whether EBNA1 might substitute for an obligatory interaction of the EBV replicator with similar structures or proteins with analogous functions.
Materials and methods
Polyclonal antibodies
Antibodies (JDI 32 and 33) against hsORC1 were raised in rabbits using the ICRF animal facilities. The bacterially expressed recombinant His6-tagged fragment of hsORC1 encompassing amino acids 422–861 was cloned into pET15b (Novagen). The antibody against hsORC3 was raised in rabbits (Eurogentec) against the His6-tagged C-terminal part (amino acids 342–710) of this subunit created by cloning an AflII–BamHI fragment of hsORC into pET21b (Novagen).
Production of monoclonal antibodies (mAbs) against ORC1 and ORC3
A 50 µg aliquot of bacterially expressed and partially purified His6-tagged hsORC1 or hsORC3 fusion proteins was injected both intraperitoneally (i.p.) and subcutaneously (s.c.) into LOU/C rats. After a 2-month interval, a final boost was given i.p. and s.c. 3 days before fusion. Fusion of the myeloma cell line P3X63-Ag8.653 with the rat spleen cells was performed according to standard procedures. Hybridoma supernatants were tested in a solid-phase immunoassay using the hsORC1 or hsORC3 fusion proteins adsorbed to polystyrene microtitre plates. Following incubation with culture supernatants for 1 h, bound mAbs were detected using peroxidase-labelled goat anti-rat IgG + IgM antibodies (Dianova) and o-phenylenediamine as chromogen in the peroxidase reaction. An irrelevant His-tagged fusion protein was used as a negative control. The immunoglobulin subclass of the monoclonal antibodies was determined using biotinylated anti-rat IgG subclass-specific mAbs (ATCC). HsORC3 1D6 (rat IgG2b) and hsORC1 7A7 (rat IgG1) reacted specifically in immunoblot experiments and were therefore used in this study.
Generation of mini-EBV virions
Infectious mini-EBV virions were generated using the EBV packaging cell line, TR–2/293, derived from 293 cells stably transfected with a packaging-deficient EBV mutant (Delecluse et al., 1999). Mini-EBV plasmid 1478.A DNA (Kempkes et al., 1995b; Kilger et al., 1998), together with an equal amount of the expression plasmid pCMV-BZLF1, for the EBV lytic cycle inducer BZLF1, was transfected into semi-confluent packaging cells using Fugene reagent (Roche), as recommended by the manufacturer. Virion-containing supernatants were harvested on day 4 and again on day 5 and immediately used for infecting primary B cells or stored at –80°C.
Generation of a mini-EBV-immortalized B-cell line
The mini-EBV-immortalized B-cell line A39 was generated by infecting primary human B cells from adenoids with EBV virions containing 1478.A mini-EBV DNA. Primary B cells from adenoids were prepared as described (Zeidler et al., 1996). Adenoid tissue from routine adenoidectomies was washed with phosphate-buffered saline (PBS) and passed through a 100 µm cell strainer using the piston of a syringe and extensive washing with PBS. Sheep blood (Oxoid) was added to the resulting lymphocyte-rich cell suspension for T-cell rosetting. The cell suspension was purified by centrifugation on a Ficoll cushion (density 1.077; Biocoll). After washing, enriched B cells from the interphase were resuspended in RPMI/10% fetal calf serum and plated at 4 × 105 cells per well in 96-well plates onto a feeder cell layer consisting of irradiated (50 Gy) human WI-38 fibroblasts. Mini-EBV virion supernatant was added to one-third of the final volume in each well, and the cells were transferred to a 37°C, 5% CO2 incubator. Every 4–7 days, half of the supernatant was replaced by fresh medium. Outgrowth of immortalized cells was usually visible after 2–4 weeks. Cell lines from individual wells were expanded separately and checked by PCR for the presence of mini-EBV DNA sequences (chloramphenicol resistance gene) and the absence of wild-type EBV sequences not present on the mini-EBV plasmid (viral glycoprotein gp85; Kilger et al., 1999). Both amplifications were performed in the same vessel using a 95°C/45 s, 59°C/45 s, 72°C/45 s temperature cycle repeated 30 times.
Chromatin immunoprecipitation assay
The formaldehyde cross-linking was performed as described by Göhring and Fackelmayer (1997). A total of 5 × 108 cells were washed with PBS and cross-linked in serum-free medium for 10 min at 37°C with 1% formaldehyde. Cross-linked cells were harvested and washed twice with ice-cold PBS. After resuspension in 25 ml of RSB buffer (10 mM Tris–HCl, 10 mM NaCl and 3 mM MgCl2 pH 8.0), cells were homogenized with 10 strokes through a 23-gauge needle. Nuclei were washed twice with RSB. Unbound proteins were extracted in 25 ml of buffer E (10 mM Tris–HCl, 1 M NaCl, 0.1% NP40, 10 mM Na2S2O5 pH 8.0, protease inhibitors). After extraction, nuclei were washed again with buffer E, resuspended in 10.8 ml of buffer E and lysed by adding 1.2 ml of 20% Sarkosyl solution. The sample was divided into four parts and carefully layered onto a CsCl solution (1.57 mg/ml CsCl in 20 mM Tris–HCl, 0.5% Sarokosyl pH 8.0). The gradient was centrifuged for at least 24 h at 32 000 r.p.m. in an SW41 rotor. The chromatin was isolated and washed three times in 50 ml of ice-cold TE (10 mM Tris, 1 mM EDTA pH 8.0) supplemented with protease inhibitors for 30 min. After resuspension in 5 ml of TE, the DNA was sheared by sonication (Branson sonifier 250, 4 × 30 s, output control 5, duty cycle 50%). For the best results in immunoprecipitation experiments, freshly made DNA was used, but aliquots can also be frozen at –80°C.
For the optional MNase (Roche) digest, 2 mM CaCl2 and 8 U of MNase were added and the chromatin fraction was incubated for 10 min at 37°C. The reaction was stopped by adding 5 mM EGTA.
For immunoprecipitation, the extract was adjusted with 1/10 volume of 11× NET (1.65 M NaCl, 5.5 mM EDTA, 550 mM Tris–HCl pH 7.4, 5.5% NP-40). A 10 µg aliquot of the indicated antibody was added and incubated for 2 h. Protein A–Sepharose beads (Dianova) were added and incubated for another 1.5 h. For immunoprecipitation with the monoclonal 1H4 antibody, protein A–Sepharose beads were coupled to rabbit anti-rat antibodies (Dianova). The beads were washed six times with RIPA; at every second washing step, the content of the vial was transferred to a new tube with a 10 min incubation step. This transfer was applied in all subsequent wash steps. The washing procedure was as follows: four washes with LiCl buffer (10 mM Tris–HCl, 250 mM LiCl, 0.5% NP-40, 0.5% deoxycholate, 1 mM EDTA pH 8.0) and four washes with TE. The precipitate was incubated for 1 h or overnight at 4°C. They were washed again four times in RIPA, three times in LiCl buffer and three times in TE. Immunoprecipitated proteins and DNA were eluted twice with 200 µl of TE + 1% SDS for 10 min at room temperature. To isolate the precipitated DNA, proteins and antibodies were degraded by proteinase K treatment for 5 h at 55°C. This step also reverses the formaldehyde cross-link. The DNA was purified twice with phenol/chloroform/isoamylalcohol and precipitated with ethanol.
PCR analysis
A 1/50 or 1/100 aliquot of the purified DNA sample was used as template for PCR. For qualitative PCR analysis, Goldstar Taq polymerase (Eurogentec) was used with 5 mM MgCl2 for the PCR amplification. Primer pairs were designed to amplify the appropriate DNA fragments with the following conditions: 5 min initial denaturation, followed by cycles of 1 min at 94°C, 1 min at 56°C and 1 min at 72°C. Twenty-seven cycles were used for EBNA1 immunoprecipitates and 32 cycles for immunoprecipitates with antibodies directed against the hsORC subunits. Real-time PCR was perfomed with the LightCycler (Roche) according to the manufacturer’s instructions. The provided FastStart Reaction Mix was supplemented with MgCl2 to a final concentration of 2 mM. The amplification of PCR products was monitored on-line and usually stopped after 40 cycles. The following settings were used: 10 min at 95°C, cycles with 1 s at 95°C, 10 s at 62°C and 20 s at 72°C. The sequences of the primers used are shown in Table I.
Cell fractionation and immunoprecipitation
A total of 2 × 108 cells were washed with PBS and suspended in hypotonic buffer I [10 mM HEPES, 10 mM KCl, 1.5 mM MgCl2, 0.1 mM EGTA, 0.5 mM dithiothreitol (DTT), 1 mM ATP pH 7.9]. After 30 min on ice, the swollen cells were dounced with 10 strokes through a 23-gauge needle. Nuclei were isolated and washed with buffer I. To release chromatin-bound proteins, nuclei were incubated for 30 min in 2 ml of ice-cold buffer II (10 mM HEPES, 450 mM NaCl, 1.5 mM MgCl2, 0.1 mM EGTA, 0.5 mM DTT, 1 mM ATP pH 7.9). The supernatant was diluted with 2 ml of buffer I and divided into five aliquots.
For immunoprecipitation, the following amounts of antibodies were added: 15 µl of pre-immune, 15 µl of JDI 32 (α-ORC1), 20 µg of α-ORC2 (Kreitz et al., 2001), 15 µl of α-ORC3 and 100 µl of 1H4 (α-EBNA1; Grasser et al., 1994). For EBNA1 immunoprecipitates, the protein A beads were pre-incubated with rabbit α-rat antibodies (Dianova). Extracts were incubated for 2 h at 4°C. To isolate antibody-bound proteins, protein A–Sepharose was added for 2 h at 4°C. The beads were washed six times with buffer III (10 mM HEPES, 250 mM NaCl, 1.5 mM MgCl2, 0.1 mM EGTA pH 7.9), twice with NET buffer (50 mM Tris–HCl, 150 mM NaCl, 5 mM EDTA, 0.5% NP-40 pH 7.4) and twice with TE pH 7.4. An 80 µl aliquot of sample buffer was added and immunoprecipitates were incubated for 30 min at 60°C. For immunoblotting experiments, 20 µl were loaded per lane and separated on SDS–PAGE. Immunoblotting was performed according to standard procedures using the 1D6 and 7A7 rat mAbs to detect hsORC3 and hsORC1, respectively. The polyclonal hsORC2 and the monoclonal EBNA1 (1H4) antibodies have already been described (Grasser et al., 1994; Kreitz et al., 2001). Membranes were developed using the enhanced chemiluminescence system (ECL, Amersham).
Acknowledgments
Acknowledgements
We are grateful to A.Moosmann for generating and providing the A39 cell line, and to M.Baack and R.Knippers for providing the rabbit α-ORC2 antibody prior to publication. This work was supported by grants HA1354/4-1 and HA1354/4-3 from the Deutsche Forschungs gemeinschaft to W.H and A.S., the AIDS-Stipendienprogramm to A.S., the Public Health Service (grant CA70723) to W.H., and by institutional grants.
Note added in proof
While the manuscript was under review, Wensing et al. published results showing that the region downstream of FR has an open chromatin structure.
Wensing,B., Stuhler,A., Jenkins,P., Hollyoake,M., Karstegl,C.E. and Farrell,P.J. (2001) Variant chromatin structure of the oriP region of Epstein–Barr virus and regulation of EBER1 expression by upstream sequences and oriP. J. Virol., 75, 6235–6241.
This might indicate a hypersensitivity for MNase and explain the variations for psc3 discussed in Figure 4.
References
- Abdurashidova G., Riva,S., Biamonti,G., Giacca,M. and Falaschi,A. (1998) Cell cycle modulation of protein–DNA interactions at a human replication origin. EMBO J., 17, 2961–2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdurashidova G., Deganuto,M., Klima,R., Riva,S., Biamonti,G., Giacca,M. and Falaschi,A. (2000) Start sites of bidirectional DNA synthesis at the human lamin B2 origin. Science, 287, 2023–2026. [DOI] [PubMed] [Google Scholar]
- Adams A. (1987) Replication of latent Epstein–Barr virus genomes in Raji cells. J. Virol., 61, 1743–1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aiyar A., Tyree,C. and Sugden,B. (1998) The plasmid replicon of EBV consists of multiple cis-acting elements that facilitate DNA synthesis by the cell and a viral maintenance element. EMBO J., 17, 6394–6403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aladjem M.I., Rodewald,L.W., Kolman,J.L. and Wahl,G.M. (1998) Genetic dissection of a mammalian replicator in the human β-globin locus. Science, 281, 1005–1009. [DOI] [PubMed] [Google Scholar]
- Ambinder R.F., Shah,W.A., Rawlins,D.R., Hayward,G.S. and Hayward,S.D. (1990) Definition of the sequence requirements for binding of the EBNA-1 protein to its palindromic target sites in Epstein–Barr virus DNA. J. Virol., 64, 2369–2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asano M. and Wharton,R.P. (1999) E2F mediates developmental and cell cycle regulation of ORC1 in Drosophila. EMBO J., 18, 2435–2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Austin R., Orr-Weaver,T. and Bell,S. (1999) Drosophila ORC specifically binds to ACE3, an origin of DNA replication control element. Genes Dev., 13, 2639–2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell S.P. and Stillman,B. (1992) Nucleotide dependent recognition of chromosomal origins of DNA replication by a multi-protein complex. Nature, 357, 128–134. [DOI] [PubMed] [Google Scholar]
- Bielinsky A.K. and Gerbi,S.A. (1998) Discrete start sites for DNA synthesis in the yeast ARS1 origin. Science, 279, 95–98. [DOI] [PubMed] [Google Scholar]
- Bielinsky A. and Gerbi,S. (1999) Chromosomal ARS1 has a single leading strand start site. Mol. Cell, 3, 477–486. [DOI] [PubMed] [Google Scholar]
- Bielinsky A.K. and Gerbi,S.A. (2001) Where it all starts: eukaryotic origins of DNA replication. J. Cell Sci., 114, 643–651. [DOI] [PubMed] [Google Scholar]
- Bogan J.A., Natale,D.A. and Depamphilis,M.L. (2000) Initiation of eukaryotic DNA replication: conservative or liberal? J. Cell. Physiol., 184, 139–150. [DOI] [PubMed] [Google Scholar]
- Delecluse H.J., Pich,D., Hilsendegen,T., Baum,C. and Hammerschmidt,W. (1999) A first-generation packaging cell line for Epstein–Barr virus-derived vectors. Proc. Natl Acad. Sci. USA, 96, 5188–5193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhar S.K. and Dutta,A. (2000) Identification and characterization of the human ORC6 homolog. J. Biol. Chem., 275, 34983–34988. [DOI] [PubMed] [Google Scholar]
- Diffley J.F.X. (1994) Eukaryotic DNA replication. Curr. Opin. Cell Biol., 6, 368–372. [DOI] [PubMed] [Google Scholar]
- Diffley J.F.X., Cocker,J.H., Dowell,S.J. and Rowley,A. (1994) Two steps in the assembly of complexes at yeast replication origins in vivo. Cell, 78, 303–316. [DOI] [PubMed] [Google Scholar]
- Frappier L. and O’Donnell,M. (1991) Overproduction, purification and characterization of EBNA1, the origin binding protein of Epstein–Barr virus. J. Biol. Chem., 266, 7819–7826. [PubMed] [Google Scholar]
- Gahn T.A. and Schildkraut,C.L. (1989) The Epstein–Barr virus origin of plasmid replication, oriP, contains both the initiation and termination sites of DNA replication. Cell, 58, 527–535. [DOI] [PubMed] [Google Scholar]
- Gavin K.A., Hidaka,M. and Stillman,B. (1995) Conserved initiator proteins in eukaryotes. Science, 270, 1667–1671. [DOI] [PubMed] [Google Scholar]
- Göhring F. and Fackelmayer,F.O. (1997) The scaffold/matrix attachment region binding protein hnRNP-U (SAF-A) is directly bound to chromosomal DNA in vivo: a chemical cross-linking study. Biochemistry, 36, 8276–8283. [DOI] [PubMed] [Google Scholar]
- Gomez M. and Antequera,F. (1999) Organization of DNA replication origins in the fission yeast genome. EMBO J., 18, 5683–5690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grasser F.A. et al. (1994) Monoclonal antibodies directed against the Epstein–Barr virus-encoded nuclear antigen 1 (EBNA1): immuno histologic detection of EBNA1 in the malignant cells of Hodgkin’s disease. Blood, 84, 3792–3798. [PubMed] [Google Scholar]
- Harrison S., Fisenne,K. and Hearing,J. (1994) Sequence requirements of the Epstein–Barr virus latent origin of DNA replication. J. Virol., 68, 1913–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh D.J., Camiolo,S.M. and Yates,J.L. (1993) Constitutive binding of EBNA1 protein to the Epstein–Barr virus replication origin, oriP, with distortion of DNA structure during latent infection. EMBO J., 12, 4933–4944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang D.W., Fanti,L., Pak,D.T., Botchan,M.R., Pimpinelli,S. and Kellum,R. (1998) Distinct cytoplasmic and nuclear fractions of Drosophila heterochromatin protein 1: their phosphorylation levels and associations with origin recognition complex proteins. J. Cell Biol., 142, 307–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber M.D., Dworet,J.H., Shire,K., Frappier,L. and McAlear,M.A. (2000) The budding yeast homolog of the human EBNA1-binding protein 2 (Ebp2p) is an essential nucleolar protein required for pre-rRNA processing. J. Biol. Chem., 275, 28764–28773. [DOI] [PubMed] [Google Scholar]
- Iizuka M. and Stillman,B. (1999) Histone acetyltransferase HBO1 interacts with the ORC1 subunit of the human initiator protein. J. Biol. Chem., 274, 23027–23034. [DOI] [PubMed] [Google Scholar]
- Kapoor P., Shire,K. and Frappier,L. (2001) Reconstitution of Epstein–Barr virus-based plasmid partitioning in budding yeast. EMBO J., 20, 222–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly T.J. and Brown,G.W. (2000) Regulation of chromosome replication. Annu. Rev. Biochem., 69, 829–880. [DOI] [PubMed] [Google Scholar]
- Kempkes B., Pich,D., Zeidler,R. and Hammerschmidt,W. (1995a) Immortalization of human primary B lymphocytes in vitro with DNA. Proc. Natl Acad. Sci. USA, 92, 5875–5879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kempkes B., Pich,D., Zeidler,R., Sugden,B. and Hammerschmidt,W. (1995b) Immortalization of human B lymphocytes by a plasmid containing 71 kilobase pairs of Epstein–Barr virus DNA. J. Virol., 69, 231–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilger E., Kieser,A., Baumann,M. and Hammerschmidt,W. (1998) Epstein–Barr virus-mediated B-cell proliferation is dependent upon latent membrane protein 1, which simulates an activated CD40 receptor. EMBO J., 17, 1700–1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilger E., Pecher,G., Schwenk,A. and Hammerschmidt,W. (1999) Expression of mucin (MUC-1) from a mini-Epstein–Barr virus in immortalized B-cells to generate tumor antigen specific cytotoxic T cells. J. Gene Med., 1, 84–92. [DOI] [PubMed] [Google Scholar]
- Kirchmaier A.L. and Sugden,B. (1998) Rep*: a viral element that can partially replace the origin of plasmid DNA synthesis of Epstein–Barr virus. J. Virol., 72, 4657–4666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitz S., Ritzi,M., Baack,M. and Knippers,R. (2001) The human origin recognition complex protein 1 dissociates from chromatin during S phase in HeLa cells. J. Biol. Chem., 276, 6337–6342. [DOI] [PubMed] [Google Scholar]
- Leatherwood J., Lopez Girona,A. and Russell,P. (1996) Interaction of Cdc2 and Cdc18 with a fission yeast ORC2-like protein. Nature, 379, 360–363. [DOI] [PubMed] [Google Scholar]
- Lee D.G. and Bell,S.P. (1997) Architecture of the yeast origin recognition complex bound to origins of DNA replication. Mol. Cell. Biol., 17, 7159–7168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little R.D. and Schildkraut,C.L. (1995) Initiation of latent DNA replication in the Epstein–Barr virus genome can occur at sites other than the genetically defined origin. Mol. Cell. Biol., 15, 2893–2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middleton T. and Sugden,B. (1992) EBNA1 can link the enhancer element to the initiator element of the Epstein–Barr virus plasmid origin of DNA replication. J. Virol., 66, 489–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natale D.A., Li,C.J., Sun,W.H. and DePamphilis,M.L. (2000) Selective instability of Orc1 protein accounts for the absence of functional origin recognition complexes during the M–G1 transition in mammals. EMBO J., 19, 2728–2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niller H.H., Glaser,G., Knuchel,R. and Wolf,H. (1995) Nucleoprotein complexes and DNA 5′-ends at oriP of Epstein–Barr virus. J. Biol. Chem., 270, 12864–12868. [DOI] [PubMed] [Google Scholar]
- Norio P., Schildkraut,C.L. and Yates,J.L. (2000) Initiation of DNA replication within oriP is dispensable for stable replication of the latent Epstein–Barr virus chromosome after infection of established cell lines. J. Virol., 74, 8563–8574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connor M., Peifer,M. and Bender,W. (1989) Construction of large DNA segments in Escherichia coli. Science, 244, 1307–1312. [DOI] [PubMed] [Google Scholar]
- Pak D.T., Pflumm,M., Chesnokov,I., Huang,D.W., Kellum,R., Marr,J., Romanowski,P. and Botchan,M.R. (1997) Association of the origin recognition complex with heterochromatin and HP1 in higher eukaryotes. Cell, 91, 311–323. [DOI] [PubMed] [Google Scholar]
- Pfuller R. and Hammerschmidt,W. (1996) Plasmid-like replicative intermediates of the Epstein–Barr virus lytic origin of DNA replication. J. Virol., 70, 3423–3431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintana D.G., Hou,Z., Thome,K.C., Hendricks,M., Saha,P. and Dutta,A. (1997) Identification of HsORC4, a member of the human origin of the replication recognition complex. J. Biol. Chem., 272, 28247–28251. [DOI] [PubMed] [Google Scholar]
- Quintana D.G., Thome,K.C., Hou,Z., Ligon,A.H., Morton,C.C. and Dutta,A. (1998) Orc5l, a new member of the human-origin recognition complex, is deleted in uterine leiomyomas and malignant myeloid diseases. J. Biol. Chem., 273, 27137–27145. [DOI] [PubMed] [Google Scholar]
- Rao H. and Stillman,B. (1995) The origin recognition complex interacts with a bipartite DNA binding site within yeast replicators. Proc. Natl Acad. Sci. USA, 92, 2224–2228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao H., Marahrens,Y. and Stillman,B. (1994) Functional conservation of multiple elements in yeast chromosomal replicators. Mol. Cell. Biol., 14, 7643–7651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawlins D.R., Milman,G., Hayward,S.D. and Hayward,G.S. (1985) Sequence-specific DNA binding of the Epstein–Barr virus nuclear antigen (EBNA-1) to clustered sites in the plasmid maintenance region. Cell, 42, 859–868. [DOI] [PubMed] [Google Scholar]
- Reisman D., Yates,J. and Sugden,B. (1985) A putative origin of replication of plasmids derived from Epstein–Barr virus is composed of two cis-acting components. Mol. Cell. Biol., 5, 1822–1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanowski P., Marr,J., Madine,M.A., Rowles,A., Blow,J.J., Gautier,J. and Laskey,R.A. (2000) Interaction of Xenopus Cdc2⋅cyclin A1 with the origin recognition complex. J. Biol. Chem., 275, 4239–4243. [DOI] [PubMed] [Google Scholar]
- Rowley A., Cocker,J.H., Harwood,J. and Diffley,J.F.X. (1995) Initiation complex assembly at budding yeast replication origins begins with the recognition of a bipartite sequence by limiting amounts of the initiator, ORC. EMBO J., 14, 2631–2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirakata M. and Hirai,K. (1998) Identification of minimal oriP of Epstein–Barr virus required for DNA replication. J. Biochem., 123, 175–181. [DOI] [PubMed] [Google Scholar]
- Shirakata M., Imadome,K.I. and Hirai,K. (1999) Requirement of replication licensing for the dyad symmetry element-dependent replication of the Epstein–Barr virus oriP minichromosome. Virology, 263, 42–54. [DOI] [PubMed] [Google Scholar]
- Shire K., Ceccarelli,D.F., Avolio-Hunter,T.M. and Frappier,L. (1999) EBP2, a human protein that interacts with sequences of the Epstein–Barr virus nuclear antigen 1 important for plasmid maintenance. J. Virol., 73, 2587–2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatsumi Y., Tsurimoto,T., Shirahige,K., Yoshikawa,H. and Obuse,C. (2000) Association of human origin recognition complex 1 with chromatin DNA and nuclease-resistant nuclear structures. J. Biol. Chem., 275, 5904–5910. [DOI] [PubMed] [Google Scholar]
- Thome K.C., Dhar,S.K., Quintana,D.G., Delmolino,L., Shahsafaei,A. and Dutta,A. (2000) Subsets of human origin recognition complex (ORC) subunits are expressed in non-proliferating cells and associate with non-ORC proteins. J. Biol. Chem., 275, 35233–35241. [DOI] [PubMed] [Google Scholar]
- Todorovic V., Falaschi,A. and Giacca,M. (1999) Replication origins of mammalian chromosomes: the happy few. Front. Biosci., 4, D859–D868. [DOI] [PubMed] [Google Scholar]
- Triolo T. and Sternglanz,R. (1996) Role of interactions between the origin recognition complex and SIR1 in transcriptional silencing. Nature, 381, 251–253. [DOI] [PubMed] [Google Scholar]
- Tugal T., Zou-Yang,H., Gavin,K., Pappin,D., Canas,B., Kobayashi,R., Hunt,T. and Stillman,B. (1998) The ORC4 and ORC5 subunits of the Xenopus and human origin recognition complex are related to ORC1 and Cdc6. J. Biol. Chem., 273, 32421–32429. [DOI] [PubMed] [Google Scholar]
- Vashee S., Simancek,P., Challberg,M.D. and Kelly,T.J. (2001) Assembly of the human origin recognition complex. J. Biol. Chem., 276, 26666–26673. [DOI] [PubMed] [Google Scholar]
- Wang Y., Finan,J.E., Middeldorp,J.M. and Hayward,S.D. (1997) P32/TAP, a cellular protein that interacts with EBNA-1 of Epstein–Barr virus. Virology, 236, 18–29. [DOI] [PubMed] [Google Scholar]
- Wysokenski D.A. and Yates,J.L. (1989) Multiple EBNA1-binding sites are required to form an EBNA1-dependent enhancer and to activate a minimal replicative origin within oriP of Epstein–Barr virus. J. Virol., 63, 2657–2666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates J., Warren,N., Reisman,D. and Sugden,B. (1984) A cis-acting element from the Epstein–Barr viral genome that permits stable replication of recombinant plasmids in latently infected cells. Proc. Natl Acad. Sci. USA, 81, 3806–3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates J.L. (1996) Epstein–Barr virus DNA replication. In DePamhilis,M.L. (ed.), DNA Replication in Eukaryotic Cells. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 751–773.
- Yates J.L. and Guan,N. (1991) Epstein–Barr virus-derived plasmids replicate only once per cell cycle and are not amplified after entry into cells. J. Virol., 65, 483–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates J.L., Camiolo,S.M. and Bashaw,J.M. (2000) The minimal replicator of Epstein–Barr virus oriP. J. Virol., 74, 4512–4522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeidler R., Meissner,P., Eissner,G., Lazis,S. and Hammerschmidt,W. (1996) Rapid proliferation of B cells from adenoids in response to Epstein–Barr virus infection. Cancer Res., 56, 5610–5614. [PubMed] [Google Scholar]