

Abstract
The Fanconi anemia (FA) complementation group C gene product (FANCC) functions to protect hematopoietic cells from cytotoxicity induced by interferon-γ (IFN-γ), tumor necrosis factor-α (TNF-α) and double-stranded RNA (dsRNA). Because apoptotic responses of mutant FA-C cells involve activation of interferon-inducible, dsRNA-dependent protein kinase PKR, we sought to identify FANCC-binding cofactors that may modulate PKR activation. We identified the molecular chaperone Hsp70 as an interacting partner of FANCC in lymphoblasts and HeLa cells using ‘pull-down’ and co-immunoprecipitation experiments. In vitro binding assays showed that the association of FANCC and Hsp70 involves the ATPase domain of Hsp70 and the central 320 residues of FANCC, and that both Hsp40 and ATP/ADP are required. In whole cells, Hsp70–FANCC binding and protection from IFN-γ/TNF-α-induced cytotoxicity were blocked by alanine mutations located in a conserved motif within the Hsp70-interacting domain of FANCC. We therefore conclude that FANCC acts in concert with Hsp70 to prevent apoptosis in hematopoietic cells exposed to IFN-γ and TNF-α.
Keywords: FANCC/Fanconi anemia/Hsp70/IFN-γ and TNFα cytotoxicity/protective function
Introduction
Fanconi anemia (FA) is an autosomal recessive disease characterized by progressive bone marrow failure, variable congenital anomalies and a predisposition to malignant neoplasms (for reviews see D’Andrea and Grompe, 1997; Buchwald and Moustacchi, 1998). Cells from FA patients exhibit hypersensitivity to alkylating agents such as mitomycin C (MMC; Auerbach and Allen, 1991). Somatic cell fusion studies have shown that FA is genetically heterogeneous, with at least seven complementation groups (A–G) having been identified thus far (Joenje et al., 2000). The genes encoding the groups A (FANCA), C (FANCC), G (FANCG), D2 (FANCD2), E (FANCE) and F (FANCF) have been cloned (Strathdee et al., 1992; Lo Ten Foe et al., 1996; de Winter et al., 1998, 2000a,b; Timmers et al., 2001). Patient-derived inactivating mutations of FANCA, FANCC and FANCG proteins reduce assembly, stability and/or nuclear translocation of a multisubunit FA protein complex (Kupfer et al., 1997; Naf et al., 1998; Garcia-Higuera et al., 1999, 2000)
FANCC suppresses apoptotic responses of hematopoietic cells to a variety of environmental cues. For example, suppression of FANCC gene expression represses clonal growth of normal erythroid and granulocyte–macrophage progenitor cells, and disruption of the FANCC gene, in mice, renders hematopoietic progenitor cells hypersensitive to the apoptotic effects of interferon-γ (IFN-γ) and tumor necrosis factor-α (TNF-α) (Whitney et al., 1996; Rathbun et al., 1997, 2000; Haneline et al., 1998). Conversely, overexpression of FANCC suppresses apoptosis in human hematopoietic progenitor cell lines (Cumming et al., 1996), in CD34+ cells from FA-C patients (Walsh et al., 1994) and in hematopoietic progenitor cells from fancc knock-out mice (Wang et al., 1998). Consequently, it is likely that inactivating mutations of the FANCC gene account for the nearly universal development of bone marrow failure in FA patients.
Seeking to identify cellular proteins whose interaction with FANCC might provide protective function against cytotoxicity induced by IFN-γ and TNF-α, we used affinity chromatography with FANCC fused to glutathione S-transferase (GST) to obtain FANCC-interacting proteins. We found that the most prominent interacting protein was the molecular chaperone heat shock protein 70 (Hsp70). FANCC and Hsp70 protect cells against a variety of environmental factors, including oxidative stress, chemotherapeutic agents, radiation and TNF-α (Jaattela and Wissing, 1993; Simon et al., 1995; Mosser et al., 1997; Jaattela et al., 1998; Pang et al., 2000) and share the capacity to suppress both double-stranded RNA-dependent protein kinase (PKR) activity (Melville et al., 1999; Pang et al., 2001a) and caspase 3 activation (Rathbun et al., 2000; Li et al., 2000). Consequently, we sought to test formally the influence of FANCC on the anti-apoptotic activity of Hsp70. We confirmed the interaction of FANCC and Hsp70 by co-immunoprecipitation of the two proteins in cell extracts from lymphoblasts as well as from other cell types. Deletion mutants of both FANCC and Hsp70 revealed that the interaction of the two proteins requires specific structural domains and that these specific interactions are necessary for protection of hematopoietic cells from cytotoxicity induced by IFN-γ and TNF-α.
Results
Identification of the inducible chaperone Hsp70 as an FANCC-interacting protein
We hypothesized that the protective role exerted by FANCC against cytokine-induced apoptosis was mediated by its interaction with other cellular proteins. Using extracts of metabolically labeled lymphoblasts for affinity chromatography assays with GST–FANCC, we obtained several proteins ranging in size from 40 to 90 kDa bound specifically to GST–FANCC but not to GST alone (Figure 1A). We noted a prominent signal at 70 kDa that was absent from the GST control (lanes 1 and 2 versus lanes 3 and 4). Interaction of this 70 kDa protein with GST–FANCC apparently was IFN-γ independent (lane 3 versus lane 4). Also noted was another less intense band at ∼40 kDa, which we identified as co-chaperone Hsp40 in a subsequent analysis (see below).

Fig. 1. Identification of Hsp70 as an FANCC-interacting protein. (A) Proteins binding to GST–FANCC. GST alone or GST-fused wild-type FANCC (GST–FANCC) was incubated with 35S-labeled whole-cell lysates (WCLs) from JY lymphoblasts treated (+) or not (–) with IFN-γ (10 ng/ml) for 2 h. Bound proteins were analyzed by SDS–PAGE and autoradiography. (B) Amino acid sequences of three peptides derived from microsequencing of the 70 kDa protein. Parentheses indicate probable but not definite residues. (C) Association of FANCC with Hsp70. GST alone, GST–FANCC or GST-fused FA-C patient-derived mutant (GST–L554P) was incubated with WCLs from JY lymphoblasts treated (+) or not (–) with IFN-γ and TNF-α (10 ng/ml each), and the bound proteins were analyzed by immuno blotting with anti-Hsp70 antibody. The same WCL (100 µg) was electrophoresed directly as input control. NS, non-specific.
To obtain sufficient material for microsequencing analysis, we prepared whole-cell lysates (WCLs) from ∼109 cells, isolated binding proteins using GST–FANCC, separated them on a preparative SDS–PAGE, and transferred binding proteins to a PVDF protein transfer and sequencing membrane. The 70 kDa band was then excised and subjected to microsequence analysis. Three peptides derived from proteolytic digestion of the 70 kDa protein were found to be identical to predicted sequences of human Hsp70 (Hunt and Morimoto, 1985; Wu et al., 1985), with peptide 1 identical to residues 310–319, peptide 2 to residues 452–458 and peptide 3 to residues 517–523 (Figure 1B).
Hsp70 binds to GST–FANCC
To confirm that FANCC and Hsp70 interact specifically, we performed an in vitro binding assay with GST–FANCC fusion proteins and analyzed the bound products by immunoblotting with anti-Hsp70 antibody. In addition, since FA-C hematopoietic cells are hypersensitive to the inhibitory effects of both IFN-γ and TNF-α (Whitney et al., 1996; Rathbun et al., 1997, 2000; Haneline et al., 1998; Otsuki et al., 1999; Pang et al., 2000) and since Hsp70 has been shown to protect cells from TNF-α-induced cytotoxicity (Jaattela et al., 1993, 1998), we asked whether treatment by IFN-γ and TNF-α exposure might enhance interaction between FANCC and Hsp70. To this end, we used WCLs from the normal lymphoblasts stimulated with both IFN-γ and TNF-α in the binding assays. As shown in Figure 1C, Hsp70 did bind to GST–FANCC (top, lanes 5 and 6). Hsp70 also bound, albeit to a lesser degree, to GST–L554P that encodes a naturally occurring functionally inactivating point mutation (Gavish et al., 1993) (lanes 7 and 8) but not to GST alone (lanes 3 and 4). Furthermore, treatments with IFN-γ and TNF-α increased the association of Hsp70 with the normal FANCC (lane 6) but not with the mutant L554P (lane 8). Reprobing the blot with antibody to FANCC revealed similar amounts of GST-fused FANCC and L554P proteins in the binding reactions (Figure 1C, bottom).
Association of endogenous FANCC and Hsp70 in normal and FA cells
To demonstrate that the inducible interaction between FANCC and Hsp70 takes place in viable cells, we performed co-immunoprecipitation experiments. We first assessed a lymphoblast line derived from an FA-C patient, HSC536N that contains the L554P mutation. HSC536N cells are hypersensitive both to MMC (Marathi et al., 1996) and to combined treatments with both IFN-γ and TNF-α (see below). Cellular extracts from normal JY and HSC536N lymphoblasts containing vector alone (HSC536N/VEC) or the normal FANCC cDNA (HSC536N/FANCC) were immunoprecipitated with an antibody against FANCC protein (Hoatlin et al., 1998), and the immunoprecipitates were tested for the presence of Hsp70 by immunoblotting. As shown in Figure 2A, anti-FANCC antibody specifically brought down Hsp70 in all cell lines tested (lanes 3–8). Additionally, treatments of the normal JY and complemented HSC536N/FANCC cell with IFN-γ and TNF-α enhanced the association of the two proteins (Figure 2A, lanes 4 and 8). However, no enhanced association of FANCC with Hsp70 was observed in the mutant HSC536N/VEC cells treated with IFN-γ and TNF-α (lane 6). Hsp70 was not precipitated by pre-immune serum (lane 9).


Fig. 2. Co-immunoprecipitation of endogenous FANCC and Hsp70. (A) Normal JY and FA-C HSC536N lymphoblasts transduced with vector alone (HSC/VEC) or the wild-type FANCC cDNA (HSC/FANCC) were treated (+) or not (–) with IFN-γ and TNF-α (10 ng/ml each) for 2 h. WCLs (1 mg of total proteins) were immunoprecipitated (IP) with a polyclonal FANCC antibody (lanes 3–8) or pre-immune serum (lane 9). The immunocomplexes were then analyzed by western blotting with a monoclonal antibody specific for Hsp70. Lanes 1 and 2 are direct western blotting with 100 µg of WCLs from untreated (–) or treated (+) JY lymphoblasts. (B) WCLs prepared as described in (A) were immunoprecipitated with anti-Hsp70 and probed with anti-FANCC (top) and anti-Hsp70 (bottom). Input controls (lanes 1–6) were analyzed with 100 µg of WCL. (C) Stoichiometry of the FANCC–Hsp70 association. A 200 µg aliquot of 35S-labeled WCL was immunoprecipitated with antibodies for either FANCC or Hsp70. The amounts of Hsp70 co-immunoprecipitated by anti-FANCC (lanes 1–4) were compared with the amounts of total cellular Hsp70 immunoprecipitated directly with anti-Hsp70. The same comparison was applied to the FANCC protein (lanes 5–8).
To confirm the specific interaction between FANCC and Hsp70 in vivo, we also performed reverse co-immunoprecipitation experiments with anti-Hsp70 antibody. Immunoprecipitating Hsp70 with the antibody co-precipitated FANCC in cell extracts from all cell lines (Figure 2B, top, lanes 7–12). In cells treated with IFN-γ and TNF-α (lanes 8 and 12), the amount of co-precipitated FANCC was increased when compared with the mutant cells. This result is consistent with the results obtained with anti-FANCC co-immunoprecipitation (Figure 2A). Incubation of the same blot with anti-Hsp70 antibody demonstrated that both proteins had been precipitated specifically (Figure 2B, bottom). Expression of FANCC (top, lanes 1–6) or Hsp70 (bottom, lanes 1–6) was comparable in the three cell lines.
We also sought to demonstrate the relative proportions of FANCC and Hsp70 that were associated in FA-C and corrected lymphoblasts. Metabolically labeled lysates from HSC536N cells bearing vector alone or FANCC were immunoprecipitated with excessive amounts of antibodies specific for FANCC or Hsp70. Based in part on data shown in Figure 2C, we estimated that ∼10% of the total cellular Hsp70 that was immunoprecipitated directly with anti-Hsp70 was co-precipitated by the anti-FANCC antibody (lanes 1–4). Treatment of the corrected HSC536N/FANCC cells with IFN-γ and TNF-α slightly increased FANCC-associated Hsp70 (lane 4). Similarly, immunoprecipitation with anti-Hsp70 brought down ∼10% of the total cellular FANCC that was immunoprecipitated with anti-FANCC (lanes 5–8). These results indicate that significant fractions of FANCC and Hsp70 are associated with each other.
To determine whether FANCC–Hsp70 complexes formed in other cell types and in cells from FA patients with other complementation groups, we analyzed FANCC–Hsp70 interactions in lymphocytes from FA group A (FA-A) and G (FA-G) patients, fibroblasts from an FA group D (FA-D) patient, and HeLa cells. In all cell lines tested, formation of FANCC–Hsp70 complexes appeared to be normal and enhanced appropriately by treatment with IFN-γ and TNF-α (Figure 3).
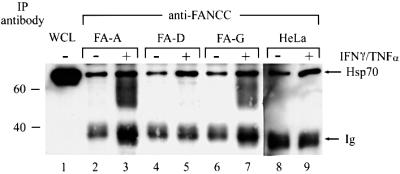
Fig. 3. Immunoprecipitation–western analysis of WCLs from FA-A and FA-D patient-derived lymphocytes, FA-G patient-derived fibroblasts and HeLa cells treated (+) or not (–) with IFN-γ and TNF-α (10 ng/ml each). Equal amounts of WCLs (1 mg of total proteins each) were immunoprecipitated with anti-FANCC, followed by western analysis with anti-Hsp70. A 100 µg aliquot of FA-A WCL was used as an input control (lane 1).
Location of the FANCC-interacting site to the ATPase domain of Hsp70
Hsp70 has two known functional domains, the N-terminal 44 kDa ATPase domain and the C-terminal 25 kDa substrate-binding domain (Chappell et al., 1987; Wang et al., 1993). To determine whether either of these domains interacts with FANCC, we generated in vitro transcription/translation constructs expressing full-length Hsp70, the ATPase domain (Hsp70A) or the substrate-binding domain (Hsp70S) (Figure 4A). [35S]methionine-labeled proteins were prepared by expression in a coupled transcription/translation system (Figure 4B, lanes 1–3). Purified GST–FANCC coated to glutathione–Sepharose beads was examined as an affinity reagent for the 35S-labeled Hsp70 proteins. When equal amounts of [35S]methionine-labeled Hsp70, Hsp70A and Hsp70S were passed individually through the glutathione– Sepharose columns containing GST–FANCC, no 35S-labeled Hsp70 proteins were retained (Figure 4B, lanes 4–6). We thus suspected that other cellular factor(s) might be required for the interaction between FANCC and Hsp70. Therefore, we incubated pairs of GST–FANCC and 35S-labeled Hsp70 proteins with 100 µg of WCLs prepared from IFN-γ/TNF-α-treated JY (normal) lymphoblasts. Under these conditions, full-length Hsp70 and Hsp70A were retained specifically in a complex with FANCC (lanes 6 and 7), whereas Hsp70S was not (lane 9). We also obtained association of FANCC with full-length Hsp70 and Hsp70A, albeit weaker, by incubation with untreated JY lysates (data not shown). It should be noted that binding of FANCC to the truncated Hsp70A was weaker than to full-length Hsp70 (compare lane 7 with lane 8). It is possible that deletion of the C-terminus of Hsp70 may have changed the conformation of the binding site leading to reduction in its binding capacity for FANCC. As a control, GST alone did not bind any of the Hsp70 proteins in the presence or absence of JY lysates (data not shown). That direct binding of FANCC to Hsp70 was not observed in the absence of JY lysates indicates that formation of FANCC–Hsp70 complexes is dependent upon certain additional factor(s) in the cell lysates.


Fig. 4. The ATPase domain of Hsp70 interacts with FANCC. (A) A diagram of full-length human Hsp70 (residues 1–640) and its deletion derivatives Hsp70A (residues 1–368 comprised of the ATPase domain) and Hsp70S (residues 386–640 comprised of the substrate-binding domain). (B) Interaction of FANCC with 35S-labeled Hsp70 proteins. GST–FANCC was absorbed to glutathione–Sepharose beads and incubated with 20 µl of [35S]methionine-labeled Hsp70 proteins. Eluted proteins were resolved by SDS–PAGE and analyzed by autoradiography. Lanes 1–3, 5 µl of each 35S-labeled full-length Hsp70, Hsp70A and Hsp70S as input controls; lanes 4–6, GST–FANCC incubated with the indicated 35S-labeled Hsp70 proteins; lanes 7–9, GST–FANCC incubated with the indicated 35S-labeled Hsp70 proteins plus 100 µg of WCL from IFN-γ/TNF-α-treated JY lymphoblasts.
Identification of the Hsp70-interacting domain of FANCC
GST fusion proteins containing full-length FANCC, the N-terminal 200 amino acid (fanccN200), the central region 320 amino acid (fancc320) and the C-terminal 200 amino acid (fanccC200) polypeptides (Figure 5A) were expressed. The full-length and deletion mutants of FANCC were then examined, by incubation with WCLs from untreated or IFN-γ/TNF-α-treated JY lymphoblasts, for the appropriate interaction with Hsp70. As shown (Figure 5B), both full-length FANCC (lanes 3 and 4) and its central region (fancc320; lanes 7 and 8) retained Hsp70 from untreated and IFN-γ/TNF-α-treated JY lysates. Treatment with IFN-γ and TNF-α enhanced FANCC–Hsp70 interaction (lanes 4 and 8) but did not increase the expression of Hsp70 (lane 2). The two terminal constructs, fanccN200 and fanccC200, failed to bind to Hsp70 (lanes 5 and 6 and 9 and 10). These results suggest that the central 57% of FANCC is sufficient for interaction with Hsp70 and may be the nearly minimal domain required for the interaction, because two shorter peptides derived from the deletion construct fancc320 (residues 175–424 and residues 105–374) failed to bind Hsp70 (data not shown).


Fig. 5. The Hsp70-interacting domain is located in the central region of FANCC. (A) A schematic diagram of full-length and deletion mutants of FANCC. Full-length FANCC and truncated mutants fanccN200, fancc320 and fanccC200 are indicated as the regions of FANCC fused to GST. (B) Interaction of Hsp70 with the central region of FANCC. GST-fused full-length and truncated FANCC proteins were incubated with WCLs from JY lymphoblasts treated (+) or not (–) with IFN-γ and TNF-α (10 ng/ml each). The glutathione–Sepharose affinity precipitates were analyzed by SDS–PAGE followed by immuno blotting with anti-Hsp70 antibody (top) and reprobed with anti-FANCC to verify the input of the GST fusion proteins (bottom).
Formation of the FANCC–Hsp70 complex is Hsp40 dependent
FANCC–Hsp70 association requires additional cofactor(s) in the cell lysates (Figure 4B), and the minor band of ∼40 kDa pulled down with Hsp70 by GST–FANCC (Figure 1A) was a candidate for a factor that might facilitate or stabilize the FANCC–Hsp70 interaction. In addition, it has been shown that the activity of Hsp70 is regulated by chaperone cofactors including Hsp40 (Frydman et al., 1994), Hip (Hohfeld et al., 1995), Bag-1 (Luders et al., 2000) and Hsp90 (Bohen et al., 1995). We therefore investigated whether these components of the Hsp70 chaperone system existed in the FANCC–Hsp70 complex. Immobilized GST–FANCC was incubated with WCLs from untreated or IFN-γ/TNF-α-treated normal lymphoblasts and the retained proteins were subjected to immunoblot analysis (Figure 6A). Immunoblotting identified the minor 40 kDa protein as Hsp40. The constitutively expressed Hsc70 was also detected in the retained materials. However, other Hsp70 cofactors such as Hsp90, Hip or Bag-1 were not detectable by the corresponding antibodies.
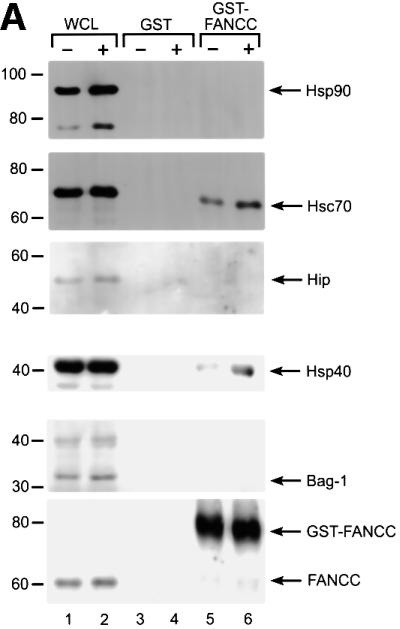
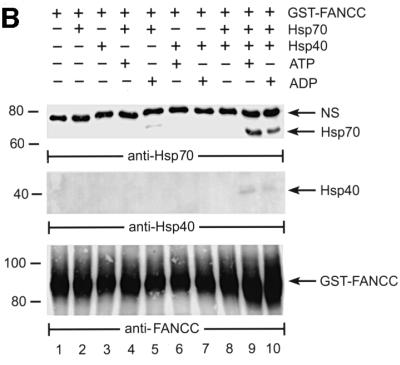
Fig. 6. Association of FANCC with Hsp70 is dependent on Hsp40. (A) Identification of Hsp40 as the factor required for FANCC–Hsp70 association. GST alone or GST–FANCC was incubated with WCLs from JY lymphoblasts treated (+) or not (–) with IFN-γ and TNF-α (10 ng/ml each), and the bound proteins were analyzed by immuno blotting with antibodies against the indicated proteins. (B) Hsp40-dependent association of FANCC with Hsp70. GST–FANCC (120 µM)–Sepharose beads were incubated with Hsp70 and/or Hsp40 (5 µM each) in the presence of ATP or ADP (1 mM each) for 15 min at 30°C. The Sepharose was washed and bound materials were analyzed by immunoblotting with antibodies for Hsp70 (top), Hsp40 (middle) or FANCC (bottom).
Having established Hsp40 as a component of the FANCC–Hsp70 complex, we performed an in vitro reconstitution experiment to determine whether association of FANCC with Hsp70 required this co-chaperone. As shown in Figure 6B, efficient binding of Hsp70 to immobilized GST–FANCC was obtained in the presence of Hsp40 and ATP (top, lane 9). We also observed less retention of Hsp70 on the GST–FANCC matrix when both proteins were incubated with ADP only (top, lane 5) or with both Hsp40 and ADP (top, lane 10). Consistent with these results, Hsp40 was detected in the FANCC–Hsp70 complex (middle, lanes 9 and 10). The bottom panel shows that input amounts of GST–FANCC were equivalent.
Repression of Hsp70 expression further sensitizes normal but not FA-C lymphoblasts to cytotoxicity induced by IFN-γ and TNF-α
Both normal JY and FA-C mutant HSC536N lymphoblasts express high levels of Hsp70 constitutively (Figure 7A, top, lanes 1 and 4). If a normal FANCC protein is absolutely required for the anti-apoptotic function of Hsp70, then suppression of Hsp70 expression should have a greater apoptotic effect on normal cells than on FA-C cells. To test this notion, we employed two different antisense Hsp70 cDNA constructs, asHsp-2 and asHsp-3, each of which has been shown to suppress Hsp70 expression in human ME-180 cells and thereby sensitize the cells to TNF-α-induced cell death (Jaattela et al., 1998). Hsp70 expression was measured by immunoblotting lysates of normal (JY) and FA-C mutant (HSC536N) lymphoblasts transduced with antisense Hsp70 retroviral vectors (Figure 7A, top). Both asHsp-2 and asHsp-3 effectively reduced the levels of Hsp70 protein in both JY and HSC536N cells (compare lanes 1 and 4 with lanes 2 and 3, and 5 and 6), and the reduction was independent of treatments of the cells with IFN-γ, TNF-α or both (lanes 7–9). To ensure equal loading, the immunoblot was reprobed with β-tubulin (bottom). Consequently, repression of Hsp70 expression by the antisense Hsp70 cDNAs also substantially reduced the formation of FANCC–Hsp70 complexes in JY and HSC536N cells (Figure 7B).
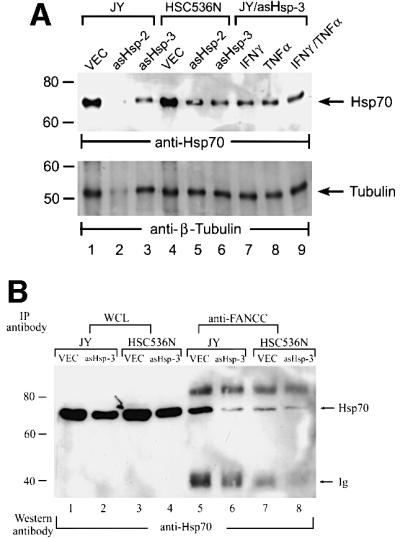

Fig. 7. Inhibition of Hsp70 expression sensitizes normal but not FA-C lymphoblasts to IFN-γ/TNF-α-mediated cytotoxicity. (A) Antisense Hsp70 constructs reduce Hsp70 expression. WCLs (50 µg of total proteins each) from JY and HSC536N lymphoblasts transduced with vector alone (VEC) or two antisense constructs (asHsp-2 or asHsp-3) was analyzed by immunoblotting with anti-Hsp70 (top) or anti-tubulin (bottom) antibody. The levels of Hsp70 in JY lymphoblasts carrying asHsp-3 treated with IFN-γ, TNF-α or both (10 ng/ml each) are indicated in lanes 7, 8 and 9, respectively. (B) Antisense Hsp70 constructs reduce FANCC–Hsp70 complexes. Equal amounts of WCLs (1 mg of total proteins each) from IFN-γ/TNF-α-treated JY and HSC536N lymphoblasts transduced with vector alone (VEC) or an antisense construct (asHsp-3) were immunoprecipitated with anti-FANCC followed by western analysis with anti-Hsp70. A 100 µg aliquot of the same WCL was run as input control (lanes 1–4). (C) Cell viability of JY (normal) and HSC536N (FA-C) lymphoblasts carrying vector alone (VEC) or antisense Hsp70 constructs (asHsp-2 or asHsp-3), in response to IFN-γ or/and TNF-α treatments. Data represent the means ± standard deviations of triplicate determinations.
As shown in Figure 7C, FA-C lymphoblasts were hypersensitive to a combination of IFN-γ and TNF-α, with only 41% survival after a 48 h exposure (column ‘VEC’). The normal JY cells transduced with antisense Hsp70 cDNA constructs asHsp-2 and asHsp-3 exhibited substantially greater susceptibility to TNF-α or both IFN-γ and TNF-α than vector-transduced control cells (compare column VEC with column asHsp-2 or asHsp-3). Repression of Hsp70 expression did not sensitize the FA-C mutant HSC536N cells further to the cytotoxicity of IFN-γ and TNF-α. Specifically, when compared with control cells carrying vector alone, the viability of FA-C lymphoblasts expressing asHsp-2 or asHsp-3 remained virtually unchanged after a 48 h exposure to TNF-α or to both IFN-γ and TNF-α (compare column VEC with columns asHsp-2 and asHsp-3). That Hsp70 suppression has no added toxicity in IFN-γ/TNF-α-treated FA-C cells suggests that the capacity of FA-C cells to resist cyto toxicity is already fully compromised because a normal FANCC protein is required for Hsp70 to function as an anti-apoptotic factor under these particular circumstances.
Fully functional FANCC is required in the Hsp70 complex to protect hematopoietic cells from IFN-γ/TNF-α-induced apoptosis
The L554P mutation in FA-C HSC536N cells renders cells hypersensitive to IFN-γ and TNF-α (Figure 7C), yet this naturally occurring mutant protein retains its ability to interact with Hsp70 (Figures 1C and 2). This indicates that the formation of an FANCC–Hsp70 complex per se is not sufficient for cytoprotection and suggests that FANCC provides a functional capacity to Hsp70. To test this notion further, we sought to define the properties of FANCC in the presence or absence of FANCC–Hsp70 interaction. We previously identified three highly conserved motifs, located in the N- and C-termini and central region of FANCC (Figure 8A). We found that a highly conserved central motif was required for optimal activation of STAT1 (Pang et al., 2001b). Since this motif is located within the Hsp70-interacting domain of FANCC, we made alanine substitutions at the conserved residues S249 and E251 and determined whether the mutations affected FANCC–Hsp70 interaction and altered the cytotoxic response. As controls, we generated two additional alanine substitutions, F64A and Y531A, located in the conserved N-terminal motif I and C-terminal motif III, respectively. We then performed Hsp70–FANCC binding assays with GST fusion proteins encoding these alanine mutants using WCLs prepared from untreated and IFN-γ/TNF-α-treated JY cells. As shown in Figure 8B, the two terminally located alanine mutants, F64A and Y531A, associated normally with Hsp70 (top, lanes 5–8 and 13 and 14). Hsp70 showed a nearly normal interaction with GST-fused L554P, the expressed allele in HSC536N cells (lanes 15 and 16). However, the association of FANCC with Hsp70 was completely abolished with the two alanine mutants, S249A and E251A (lanes 9–12). Treatment with IFN-γ and TNF-α slightly enhanced FANCC–Hsp70 association with the normal FANCC and Y531A (lanes 6 and 14), but apparently not with F64A and L554P (lanes 8 and 16). All alanine-substituted mutants protected FA-C cells against MMC-induced toxicity (Pang et al., 2001b). Input amounts of GST fusion proteins were comparable in the binding reactions (Figure 8B, bottom).



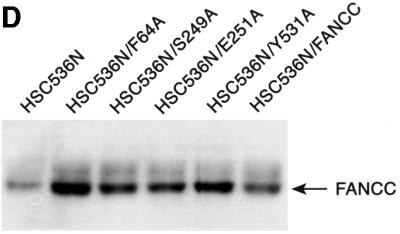


Fig. 8. Alanine-substituted mutations in the Hsp70-interactive domain of FANCC abolish FANCC–Hsp70 interaction and sensitize cells to IFN-γ and TNF-α. (A) Motifs conserved among FANCC homologs of human (hFANCC), mouse (mFANCC), bovine (bFANCC) and rat (rFANCC). Bold letters correspond to identical amino acids, and ‘X’ to non-conserved residues. The number of residues between the different motifs is indicated in parentheses. The asterisks indicate the positions at which alanine substitutions were made and the location of the FA-C patient-derived mutation L554P. (B) Effect of FANCC mutations on interaction between FANCC and Hsp70. WCL (1 mg of protein) of JY lymphoblasts treated with IFN-γ and TNF-α was incubated with GST fusion proteins bound to glutathione–Sepharose beads. The glutathione–Sepharose affinity precipitates were analyzed by SDS–PAGE followed by immunoblotting. Top: the western blot was probed with antibody to Hsp70; bottom, the same blot was reprobed with anti-FANCC to verify the input of the GST fusion proteins. Lanes 1 and 2 were WCLs (50 µg of total proteins each) to show the effects of IFN-γ and TNF-α on expression of Hsp70 and FANCC. (C) Association of alanine-substituted FANCC with Hsp70 in vivo. WCLs (1 mg of total proteins) from PD4 lymphoblasts transduced with vector alone (VEC), the wild-type FANCC (FANCC) or alanine-substituted FANCC cDNA were immuno precipitated (IP) with anti-Hsp70 antibody (lanes 2–7). The immunocomplexes were then analyzed by western blotting with anti-FANCC antibody. Lanes 1 is direct western blotting with 100 µg of WCL from PD4 lymphoblasts. (D) Expression of alanine-substituted FANCC mutant protein in HSC536N cells. WCLs (100 µg each) were subjected to SDS–PAGE and analyzed by immunoblotting with anti-FANCC antibody. (E) Flow cytometric analysis of fractional apoptotic responses in HSC536N lymphoblasts expressing FANCC mutations. Cells were incubated with IFN-γ (10 ng/ml) for 15 h before treatment with 10 ng/ml of TNF-α for 48 h and harvested for flow cytometric analysis. Treatments of HSC/VEC and HSC/FANCC with camptothecin were used as positive controls (50 mM for 9 h) and induced positivity of 65.1 and 22.5%, respectively. A total of 10 000 events for each sample were analyzed. (F) Cell viability of HSC536N lymphoblasts carrying vector alone (VEC), normal FANCC or alanine-substituted FANCC cDNA, in response to IFN-γ and TNF-α treatments. Data represent the mean ± SD of triplicate determinations.
To determine the capacity of the alanine-substituted FANCC proteins to bind Hsp70 in vivo, we expressed the mutant and normal FANCC proteins, by retroviral gene transfer, in an FA patient-derived lymphoblast line PD4, which does not express full-length FANCC (Pang et al., 2001b). WCLs from the resulting transduced cells were immunoprecipitated with anti-Hsp70 and immunocomplexes were analyzed using antibody for FANCC. Consistent with the in vitro binding results described above, the two terminal alanine mutants (F64A and Y531A) retained full capacity for binding to Hsp70 (Figure 8C, lanes 4 and 7). However, the Hsp70-binding ability of the two mutants in the middle motif (S249A and E251A) was either completely abolished or substantially impaired (lanes 5 and 6).
We then sought to identify the functional consequence of the altered FANCC–Hsp70 interaction. We expressed mutant FANCC proteins in HSC536N lymphoblasts using retroviral-mediated gene transfer. Quantitative western blotting analysis of the alanine-substituted FANCC-expressing cells revealed that the total level of FANCC was more than twice that of non-transduced HSC536N cells and that at least 80% of FANCC derived from the retrovirally expressed proteins (Figure 8D). We then tested the transductants for sensitivity to IFN-γ and TNF-α using a flow cytometric method for caspase 3 activation. Figure 8E shows that the S249A and E251A mutants, defective in their capacity to associate with Hsp70 (Figure 8B and C), resulted in a >2-fold increase in apoptotic cells after treatment with IFN-γ and TNF-α, as compared with the corrected cells transduced with the normal FANCC (compare rows S249A and E251A with FANCC). Cells bearing either of these two mutant proteins were as hypersensitive to IFN-γ and TNF-α as the parent HSC536N cells (compare rows S248A and E251A with HSC536N). However, the levels of sensitivity to IFN-γ and TNF-α in cells expressing alanine substitutions in the N- and C-terminal motifs, F64A and Y531A, which were proficient Hsp70 interactors (Figures 8B and C), were comparable with those obtained with the normal FANCC-expressing cells (Figure 8E, compare rows F64A and Y531A with FANCC). We also determined the effect of these alanine substitutions on cell viability in response to IFN-γ and TNF-α. Indeed, cells expressing mutants S249A and E251A were more sensitive to IFN-γ/TNF-α cytotoxicity than those expressing the normal FANCC or either terminal alanine mutant (Figure 8F, compare columns S249A and E251A with columns FANCC, F64A and Y531A). Collectively, these data demonstrate that an induced interaction with a functional FANCC protein is necessary for Hsp70 to protect hematopoietic cells from apoptosis induced by IFN-γ and TNF-α.
Discussion
Through the use of affinity chromatography and protein microsequencing analysis, we have identified Hsp70 as an interacting partner of FANCC. We have investigated the protective role mediated by FANCC–Hsp70 interaction by assessing apoptosis in normal and FA hematopoietic cells. Taking into account that both FANCC and Hsp70 function to protect cells from apoptosis induced by mitogenic inhibitors such as TNF-α, and that both proteins are known to suppress activation of PKR and caspase 3 (Melville et al., 1999; Li et al., 2000; Rathbun et al., 2000; Pang et al., 2001a), our results suggest that the induced interaction between these two proteins is required for the anti-apoptotic activity of Hsp70.
Using site-directed mutagenesis, we have identified an evolutionarily conserved motif within the Hsp70-interacting domain of FANCC necessary for both the FANCC–Hsp70 interaction and the protective function of FANCC against IFN-γ and TNF-α-induced cytotoxicity (Figure 8). Secondly, reduced Hsp70 expression sensitizes normal lymphoblasts to IFN and TNF treatment but does not augment cytotoxicity further in FA-C HSC536N cells (Figure 7), consistent with the notion that a functional FANCC–Hsp70 is required for the protection.
FANCC forms complexes with at least three other FA proteins, namely FANCA, FANCF and FANCG (Kupfer et al., 1997; Garcia-Higuera et al., 1999; de Winter et al., 2000c). Such complexes are believed to function in response to genotoxic stress in part by permitting post-translational activation (monoubiquitination) of FANCD2 (a recently identified nuclear protein that complements FA-D2 cells), which can then translocate to nuclear damage foci (Garcia-Higuera et al., 2001). FA proteins, in particular FANCC, also participate in cytoplasmic signal transduction pathways. FANCC, for example, optimizes survival responses of hematopoietic cells to cytokines and growth factors (Pang et al., 2000) and protects cells from the cytotoxicity induced by IFN-γ and TNF-α (Whitney et al., 1996; Rathbun et al., 1997, 2000; Haneline et al., 1998; Otsuki et al., 1999). Therefore, FANCC forms functionally distinct protein complexes in response to diverse biological signals. The observations described here that FANCC interacts with the general anti-apoptotic protein Hsp70 and that the FANCC–Hsp70 interaction is enhanced by the treatment with IFN-γ and TNF-α in hematopoietic cells (Figures 1 and 2) further demonstrate that FANCC has the capacity to perform multiple functions in response to distinct biological and chemical signals by interacting with protein partners functioning in different biochemical pathways. An earlier report on the association of FANCC with the endoplasmic reticulum chaperone glycoprotein GRP94 suggested that FANCC served a non-nuclear co-chaperone function (Hoshino et al., 1998), but the survival signaling pathways that devolve from dysfunction of this particular interaction were not clear. However, the most compelling structural evidence for FANCC multifunctionality is found in results of our studies with alanine mutants. Specifically, two alanine mutants of FANCC complemented the MMC hypersensitivity of FA-C cells [reflective of an intact nuclear FA complex (Garcia-Higuera et al., 1999)] but did not correct the sensitivity of FA-C cells to IFN and TNF (Figure 8).
Using in vitro affinity protein chromatography, we have identified domains that appear to be responsible for mediating the interaction between FANCC and Hsp70. Formation of the FANCC–Hsp70 complex is mediated by the ATPase domain of Hsp70 (Figure 4). While this domain is necessary for the chaperone function of Hsp70, whether the FANCC–Hsp70 complex is an essential component of the chaperone machinery remains to be determined. Stable association of FANCC with Hsp70 required the presence of Hsp40 in our in vitro reconstitution assay (Figure 6B). This suggests that this co-chaperone may associate with Hsp70 and play a role in facilitating or stabilizing the FANCC–Hsp70 interaction. Hsp40 probably modulates the conformation of Hsp70 (Hsc70) in such a way that the ATP-binding domain becomes accessible for some Hsp70 cofactors such as Hip and Bag-1 (Hohfeld et al., 1995; Luders et al., 2000). Notwithstanding the role of Hsp40 in FANCC– Hsp70 association, the requirement that an active FANCC molecule bind to Hsp70 to optimize its anti-apoptotic activity suggests FANCC as a cofactor of Hsp70. Whether or not this function of FANCC resembles that of other co-chaperones, such as Hip and Hdj-1, which stimulate Hsp70 chaperone activity and promote the assembly of Hsp70 into multimeric protein complexes (Freeman et al., 1995; Hohfeld et al., 1995), remains to be tested.
The Hsp70-interacting domain was mapped to the central region comprising 320 amino acids (residues 105–424) of FANCC (Figure 5). There is no amino acid sequence homology between the two interacting domains of FANCC and Hsp70. However, the Hsp70-interacting domain of FANCC contains a highly conserved motif among FANCC homologs from human, mouse, rat and bovine origin (Ching Ying Wong et al., 1997). We suggest that this motif plays an important structural role in the FANCC–Hsp70 interaction. The in vitro binding assays (Figure 5B) indicate that FANCC–Hsp70 interaction does not require both terminal portions of the FANCC protein. However, our results do not exclude the possibility that these terminal parts of the protein play a role in maintaining a proper protein conformation necessary for the interaction or in recruiting other component(s) to the complex (see below).
An important implication of this study is that the FANCC–Hsp70 complex per se is necessary but not sufficient for protection of cells from the cytotoxicity mediated by IFN-γ and TNF-α. Alanine substitutions in the conserved central motif within the Hsp70-interacting domain of FANCC (S249A and E251A) not only completely abolished the FANCC–Hsp70 interaction but also eliminated the specific biological function of FANCC in protecting cells from apoptosis induced by IFN-γ and TNF-α (Figure 8). This suggests that this conserved motif is critical for the FANCC–Hsp70 interaction and that the interaction is absolutely necessary for the protective function. However, two findings suggest that other additional molecular elements are required for full functionality. First, an FA-C patient-derived mutation, L554P located near the C-terminus of the protein, retained its ability to interact with Hsp70 in the ground state but lost the capacity to increase the association in response to IFN-γ and TNF-α (Figures 1, 2 and 8B). In this regard, the FA-C HSC536N lymphoblasts carrying this L554P mutation exhibited hypersensitivity to IFN-γ/TNF-α-induced apoptosis (Figure 8E and F). This is compatible with the view that cofactors for induced association were incapable of effecting such a response in these cells and that cofactor function of FANCC derives from its capacity to interact with other enhancing factors. We speculate that the C-terminus of FANCC may play a role in the maintenance of a proper FANCC conformation, a conformation that may change in response to IFN-γ and TNF-α, perhaps by interacting with other proteins involved in cytoprotection. Secondly, the central domain of FANCC is also required for optimal STAT signaling. Therefore, other factors functioning in survival signal transduction pathways may be influenced by this domain of FANCC.
Although our results demonstrate that FANCC–Hsp70 interaction protects hematopoietic cells from IFN-γ/TNF-α-mediated cytotoxicity, the downstream consequences of the interaction are not yet clear. At least three alternative, not necessarily exclusive, mechanisms may be involved. First, recent studies suggest that the PKR inhibitor P58IPK recruits Hsp70 to a protein complex that inactivates PKR (Melville et al., 1999). Indeed, FANCC does function to suppress the activation of PKR induced by IFN-γ, TNF-α and double-stranded RNA (Pang et al., 2001a). Therefore, it is possible that by interacting with Hsp70, FANCC targets the chaperone to PKR, leading to its inhibition. Secondly, Hsp70 modulates caspase 3 activation (Li et al., 2000), at least in part by inhibiting apoptosome formation (Saleh et al., 2000), and it is known that caspase 3 activation is excessive in cytokine-stimulated FA-C cells (Rathbun et al., 2000). Consequently, FANCC may facilitate the apoptosome inhibitory activity of Hsp70. Thirdly, FANCC–Hsp70 interaction may promote the translocation of the FA complex into the nucleus. Several lines of evidence suggest that Hsp70 plays an important role in nuclear import of proteins (Shi and Thomas, 1992; Okuno et al., 1993; Yang and DeFranco, 1994). Moreover, Hsp70 has been proposed to facilitate the binding of nuclear localization signal (NLS)-containing karyophilic proteins by the NLS receptor, karyopherin α (Jeoung et al., 1991; Agostini et al., 2000). Among the four FA proteins that form a nuclear complex, only FANCA contains an NLS (Lo Ten Foe et al., 1996). While nuclear import of FANCF may occur independently, FANCC and FANCG putatively are co-transported into nuclei via stable interaction with NLS-containing FANCA (de Winter et al., 2000c). In fact, certain patient-derived mutations of FANCA can form a complex with FANCG but not FANCC, resulting in the retention of the mutant FANCA– FANCG complex in the cytoplasm (Garcia-Higuera et al., 2000). Therefore, the fact that nuclear translocation of the FA proteins requires a stable complex with FANCC suggests that FANCC–Hsp70 interaction could facilitate nuclear import of the FA complex by modulating the affinity of the importin–NLS interaction. Clearly, none of these potential mechanisms are mutually exclusive.
While the mechanisms by which FANCC interacts with Hsp70 to protect hematopoietic cells from cytotoxicity induced by IFN-γ and TNF-α are yet to be unraveled, it is clear that the anti-apoptotic function of Hsp70 in response to combined environmental cues of TNF and IFN requires physical contact with a central conserved motif of FANCC, and inactivating mutations of FANCC result in loss of the anti-apoptotic function of Hsp70. Moreover, the FANCC molecule that is bound must retain its survival signaling function. The characteristic hypersensitivity of FA-C cells to TNF and IFN can be accounted for, at least in part, by dysfunction of this anti-apoptotic pathway. Taking into account that conservative mutations of the Hsp70-interacting domain of FANCC resulted in loss of Hsp70-dependent anti-apoptotic signaling function as well as loss of the capacity of the mutant to bind to STAT1 in cytokine-stimulated cells without influencing the capacity of the mutant molecule to protect cells against MMC-induced damage (Pang et al., 2001b), our results define a molecular basis for multifunctionality of the FANCC protein.
Materials and methods
Cell culture, IFN-γ/TNF-α treatments and metabolic labeling
Normal JY and FA lymphoblasts were maintained in RPMI media 1640 (Life Technologies) supplemented with 15% heat-inactivated fetal calf serum (FCS). HeLa cells and FA fibroblasts were grown in Dulbecco’s modified Eagle’s medium (DMEM) with 10% FCS and α-minimal essential medium (αMEM) with 20% FCS, respectively. Cells were stimulated with recombinant human IFN-γ (10 ng/ml) and TNF-α (10 ng/ml) or both (R&D Systems) for the indicated time periods. For 35S labeling, 20 × 106 lymphoblasts were treated with IFN-γ (10 ng/ml) for 2 h. Cells were rinsed with methionine and cysteine-free RPMI medium, and labeling was performed in the same medium containing 50 µCi/ml of [35S]methionine–cysteine labeling mix (DuPont NEN) and IFN-γ or a combination of IFN-γ and TNF-α for 60 min at 37°C.
GST–FANCC fusion protein purification, in vitro binding assays, and Hsp70 isolation and microsequencing
The GST–FANCC and GST–L554P fusion constructs have been described elsewhere (Pang et al., 2000). Other GST fusion constructs encoding the alanine and deletion mutants were created by insertion of the indicated cDNAs into the BamHI and SmaI sites of the vector pGST (Nelson et al., 1996). The GST fusion proteins were purified by the protocol as described previously (Pang et al., 2000). [35S]methionine-labeled full-length Hsp70 and its truncated Hsp70A and Hsp70S were prepared by in vitro transcription and translation, using the TNT rabbit reticulocyte lysate system (Promega). For binding, GST fusion proteins bound to glutathione–Sepharose beads were incubated with [35S]methionine-labeled proteins and/or WCLs prepared in NP-40 lysis buffer (1% NP-40, 20 mM Tris–HCl pH 8.0, 137 mM NaCl and 10% glycerol) supplemented with 1% aprotinin, 1 µg/ml leupeptin, 1 mM phenylmethylsulfonyl fluoride (PMSF) and 2 mM sodium orthovanadate. After incubation at 30°C for 20 min, the beads were recovered, washed, and analyzed by SDS–PAGE and autoradiography or western blot analysis. To isolate the 70 kDa protein for sequence analysis, WCLs from ∼109 cells were passed through glutathione–Sepharose columns immobilized with GST–FANCC. After washing with 10 vols of buffer (20 mM Tris–HCl pH 8.0, 200 mM NaCl and 1 mM EDTA), the bound proteins were eluted in the same buffer containing 20 mM reduced glutathione. The eluted proteins were concentrated by Microcon-10 (Amicon), resolved by SDS–PAGE and transferred to a PVDF protein transfer and sequencing membrane (Schleicher & Schuell). The 70 kDa band was visualized by Ponceau staining, excised and subjected to protein microsequencing analysis (Shriners Hospital Core Facility, Portland, OR).
Construction of retroviral expression vectors and transduction of lymphoblasts
The normal FANCC (DDBJ/EMBL/GenBank accession No. X66893) and the FA-C patient-derived mutant L554P cDNAs (Gavish et al., 1993) cloned into the XhoI and BamHI sites of retroviral vector pLXSN have been described previously (Rathbun et al., 1997). Alanine (A) substitutions in the FANCC protein were introduced by recombinant PCR mutagenesis (Michael et al., 1990) using Pfu enzyme (Stratagene) with oligonucleotides encoding the following mutations. For F64A, the internal primers, encoding amino acid residues 61–69 of FANCC, were used to modify the protein at position 64 to alanine. The 314 bp PCR-generated fragments encoding the F to A substitution were used to replace the corresponding fragment in pLFACSN (Heinrich et al., 1998) to create the mutant F64A. To mutagenize the central and C-terminal motifs of FANCC, the 1452 bp HindIII to stop codon-containing XbaI fragment of FANCC was subcloned into the corresponding sites of pUC18 to create pUCFAC-D1. For S249A and E251A substitutions, the internal primers encoding amino acid residues 245–254 of FANCC, having a new codon in place of the codon for serine (S) or glutamic acid (E), were used to mutate residues 249 or 251 to alanine. The 290 bp PCR products digested with Bsu36I and ApaI were then ligated into Bsu36I- and ApaI-digested pUCFAC-D1. Finally, the HindIII–XbaI fragment from the resulting plasmids was used to replace the corresponding fragment in pLFACSN to create mutants S249A and E251A. The same technique was employed to create mutant Y531A. In this case, the internal primer encoded amino acid residues 522–534, and the PCR-generated 279 bp PstI–XbaI fragments were used to replace the corresponding fragments in pUCFAC-D1. All mutation-containing alleles were subjected to sequence analysis to confirm that the desired changes had occurred. The two antisense Hsp70 sequences, asHsp-2 and asHsp-3, were derived from the previously described antisense Hsp70 expression plasmids pcDNA-ashsp-2 and pcDNA-ashsp-3 (Jaattela et al., 1998), respectively. These FANCC cDNAs encoding alanine substitutions and antisense Hsp70 sequences were subcloned into the XhoI and BamHI sites of retroviral vector pLXSN. These pLXSN plasmids were transduced into lymphoblasts by a previously described protocol (Pang et al., 2000). Sets of isogenic lines were selected for G418 (300 µg/ml) resistance, then exposed to multiple doses of MMC to verify that the transduced cells were fully complemented using both cell survival and cytogenetic instability assays.
Immunoprecipitation and immunoblotting
WCLs (1 mg of total protein) were pre-cleared with 50 µl of 50% protein A–Sepharose suspension (Pharmacia Biotech Inc.) for 1 h at 4°C. After separation of the protein A–Sepharose, the lysate was incubated with anti-FANCC polyclonal antiserum (Hoatlin et al., 1998) or a monoclonal anti-Hsp70 antibody (StressGen) for 3–5 h at 4°C. Immunocomplexes were then bound to protein A–Sepharose beads, recovered by centrifugation and washed with NP-40 lysis buffer. For immunoblotting, samples were heated in Laemmli SDS sample buffer, separated by SDS–PAGE and transferred onto nitrocellulose membranes. Immunoblots were incubated with the indicated primary antibodies (other antibodies including anti-Hsp90, -Hip and -Bag-1 were purchased from StressGen) for 2 h at room temperature. After washing, the blots were incubated with appropriate secondary antibodies for 30 min at room temperature, and developed by using an enhanced chemiluminescence kit (Amersham).
In vitro reconstitution of FANCC–Hsp70 complexes
GST–FANCC (120 µM) was incubated at 30°C for 15 min with Hsp70 and Hsp40 (5 µM each, StressGen) in incubation buffer [50 mM KPO4 pH 7.2, 2 mM MgCl2, 150 mM NaCl, 1 mM dithiothreitol (DTT), 1 mM PMSF, 2 µg/ml aprotinin and 1 µg/ml leupeptin] containing 1 mM ATP or ADP (Sigma). Following three washes with incubation buffer, bound proteins were resolved on SDS–PAGE and analyzed by immunoblotting with the indicated antibodies.
Analysis of cell viability and apoptosis
Cell viability was measured by trypan blue exclusion analysis. To quantify apoptotic cells, we used a polyclonal antibody to the active form of caspase 3 in a flow cytometric assay to detect cells in the early stages of apoptosis. The flow cytometric assays were performed by the procedure described elsewhere (Pang et al., 2001a).
Acknowledgments
Acknowledgements
We thank Dr Manuel Buchwald for providing the lymphoblast cell line HSC536N from a type C Fanconi anemia patient, Dr A.D.Miller for the retroviral vector pLXSN, Dr David C.Hinkle for the pGST vector, Dr Marja Jaattela for the antisense Hsp70 expression plasmids pcDNA-ashsp-2 and pcDNA-ashsp-3, Dr Harm H.Kampinga for pcDNA-Hsp70 plasmid, and Tara Koretsky for valuable technical assistance. This work was supported by NIH Grant HL48546 and a Department of Veterans Affairs Merit Review Grant to G.C.B.
References
- Agostini I., Popov,S., Li,J., Dubrovsky,L., Hao,T. and Bukrinsky,M. (2000) Heat-shock protein 70 can replace viral protein R of HIV-1 during nuclear import of the viral preintegration complex. Exp. Cell Res., 259, 398–403. [DOI] [PubMed] [Google Scholar]
- Auerbach A.D. and Allen,R.G. (1991) Leukemia and preleukemia in Fanconi anemia patients. A review of the literature and report of the International Fanconi Anemia Registry. Cancer Genet. Cytogenet., 51, 1–12. [DOI] [PubMed] [Google Scholar]
- Bohen S.P., Kralli,A. and Yamamoto,K.R. (1995) Hold ‘em and fold ‘em: chaperones and signal transduction. Science, 268, 1362–1365. [DOI] [PubMed] [Google Scholar]
- Buchwald M. and Moustacchi,E. (1998) Is Fanconi anemia caused by a defect in the processing of DNA damage? Mutat. Res., 408, 75–90. [DOI] [PubMed] [Google Scholar]
- Chappell T.G., Konforti,B.B., Schmid,S.L. and Rothman,J.E. (1987) The ATPase core of a clathrin uncoating protein. J. Biol. Chem., 262, 746–751. [PubMed] [Google Scholar]
- Ching Ying Wong J., Alon,N. and Buchwald,M. (1997) Cloning of the bovine and rat Fanconi anemia group C cDNA. Mamm. Genome, 8, 522–525. [DOI] [PubMed] [Google Scholar]
- Cumming R.C., Liu,J.M., Youssoufian,H. and Buchwald,M. (1996) Suppression of apoptosis in hematopoietic factor-dependent progenitor cell lines by expression of the FAC gene. Blood, 88, 4558–4567. [PubMed] [Google Scholar]
- D’Andrea A.D. and Grompe,M. (1997) Molecular biology of Fanconi anemia: implications for diagnosis and therapy. Blood, 90, 1725–1736. [PubMed] [Google Scholar]
- de Winter J.P. et al. (1998) The Fanconi anaemia group G gene FANCG is identical with XRCC9. Nature Genet., 20, 281–283. [DOI] [PubMed] [Google Scholar]
- de Winter, J.P. et al. (2000a) Isolation of a cDNA representing the Fanconi anemia complementation group E gene. Am. J. Hum. Genet., 67, 1306–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Winter J.P. et al. (2000b) The Fanconi anaemia gene FANCF encodes a novel protein with homology to ROM. Nature Genet., 24, 15–16. [DOI] [PubMed] [Google Scholar]
- de Winter J.P. et al. (2000c) The Fanconi anemia protein FANCF forms a nuclear complex with FANCA, FANCC and FANCG. Hum. Mol. Genet., 9, 2665–2674. [DOI] [PubMed] [Google Scholar]
- Freeman B.C., Myers,M.P., Schumacher,R. and Morimoto,R.I. (1995) Identification of a regulatory motif in Hsp70 that affects ATPase activity, substrate binding and interaction with HDJ-1. EMBO J., 14, 2281–2292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frydman J., Nimmesgern,E., Ohtsuka,K. and Hartl,F.U. (1994) Folding of nascent polypeptide chains in a high molecular mass assembly with molecular chaperones. Nature, 370, 111–117. [DOI] [PubMed] [Google Scholar]
- Garcia-Higuera I., Kuang,Y., Naf,D., Wasik,J. and D’Andrea,A.D. (1999) Fanconi anemia proteins FANCA, FANCC and FANCG/XRCC9 interact in a functional nuclear complex. Mol. Cell. Biol., 19, 4866–4873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Higuera I., Kuang,Y., Denham,J. and D’Andrea,A.D. (2000) The Fanconi anemia proteins FANCA and FANCG stabilize each other and promote the nuclear accumulation of the Fanconi anemia complex. Blood, 96, 3224–3230. [PubMed] [Google Scholar]
- Garcia-Higuera I., Taniguchi,T., Ganesan,S., Meyn,M.S., Timmers,C., Hejna,J., Grompe,M. and D’Andrea,A.D. (2001) Interaction of the Fanconi anemia proteins and BRCA1 in a common pathway. Mol. Cell, 7, 249–262. [DOI] [PubMed] [Google Scholar]
- Gavish H., dos Santos,C.C. and Buchwald,M. (1993) A Leu554-to-Pro substitution completely abolishes the functional complementing activity of the Fanconi anemia (FACC) protein. Hum. Mol. Genet., 2, 123–126. [DOI] [PubMed] [Google Scholar]
- Haneline L.S., Broxmeyer,H.E., Cooper,S., Hangoc,G., Carreau,M., Buchwald,M. and Clapp,D.W. (1998) Multiple inhibitory cytokines induce deregulated progenitor growth and apoptosis in hematopoietic cells from Fac–/– mice. Blood, 91, 4092–4098. [PubMed] [Google Scholar]
- Heinrich M.C. et al. (1998) DNA cross-linker-induced G2/M arrest in group C Fanconi anemia lymphoblasts reflects normal checkpoint function. Blood, 91, 275–287. [PubMed] [Google Scholar]
- Hoatlin M.E., Christianson,T.A., Keeble,W.W., Hammond,A.T., Zhi,Y., Heinrich,M.C., Tower,P.A. and Bagby,G.C.,Jr (1998) The Fanconi anemia group C gene product is located in both the nucleus and cytoplasm of human cells. Blood, 91, 1418–1425. [PubMed] [Google Scholar]
- Hohfeld J., Minami,Y. and Hartl,F.U. (1995) Hip, a novel co-chaperone involved in the eukaryotic Hsc70/Hsp40 reaction cycle. Cell, 83, 589–598. [DOI] [PubMed] [Google Scholar]
- Hoshino T., Wang,M.P., Devetten,N., Iwata,S., Kajigaya,R.J., Wise,J.M., Liu,J.M. and Youssoufian,H. (1998) Molecular chaperone GRP94 binds to the Fanconi anemia group C protein and regulates its intracellular expression. Blood, 91, 4379–4386. [PubMed] [Google Scholar]
- Hunt C. and Morimoto,R.I. (1985) Conserved features of eukaryotic hsp70 genes revealed by comparison with the nucleotide sequence of human hsp70. Proc. Natl Acad. Sci. USA, 82, 6455–6459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaattela M. and Wissing,D. (1993) Heat-shock proteins protect cells from monocyte cytotoxicity: possible mechanism of self-protection. J. Exp. Med., 177, 231–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaattela M., Wissing,D., Kokholm,K., Kallunki,T. and Egeblad,M. (1998) Hsp70 exerts its anti-apoptotic function downstream of caspase-3-like proteases. EMBO J., 17, 6124–6134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeoung D.I., Chen,S., Windsor,J. and Pollack,R.E. (1991) Human major HSP70 protein complements the localization and functional defects of cytoplasmic mutant SV40 T antigen in Swiss 3T3 mouse fibroblast cells. Genes Dev., 5, 2235–2244. [DOI] [PubMed] [Google Scholar]
- Joenje H. et al. (2000) Complementation analysis in Fanconi anemia: assignment of the reference FA-H patient to group A. Am. J. Hum. Genet., 67, 759–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kupfer G.M., Naf,D., Suliman,A., Pulsipher,M. and D’Andrea,A.D (1997) The Fanconi anaemia proteins, FAA and FAC, interact to form a nuclear complex. Nature Genet., 17, 487–490. [DOI] [PubMed] [Google Scholar]
- Li C.-Y., Lee,J.-S., Ko,Y.-G., Kim,J.-I. and Seo,J.-S. (2000) Heat shock protein 70 inhibits apoptosis downstream of cytochrome c release and upstream of caspase-3 activation. J. Biol. Chem., 275, 25665–25671. [DOI] [PubMed] [Google Scholar]
- Lo Ten Foe J.R. et al. (1996) Expression cloning of a cDNA for the major Fanconi anaemia gene, FAA. Nature Genet., 14, 320–323. [DOI] [PubMed] [Google Scholar]
- Luders J., Demand,J., Papp,O. and Hohfeld,J. (2000) Distinct isoforms of the cofactor BAG-1 differentially affect Hsc70 chaperone function. J. Biol. Chem., 275, 14817–14823. [DOI] [PubMed] [Google Scholar]
- Marathi U.K., Howell,S.R., Ashmun,R.A. and Brent,T.P (1996) The Fanconi anemia complementation group C protein corrects DNA interstrand cross-link-specific apoptosis in HSC536N cells. Blood, 88, 2298–2305. [PubMed] [Google Scholar]
- Melville M.W., Tan,S.L., Wambach,M., Song,J., Morimoto,R.I. and Katze,M.G. (1999) The cellular inhibitor of the PKR protein kinase, P58(IPK), is an influenza virus-activated co-chaperone that modulates heat shock protein 70 activity. J. Biol. Chem., 274, 3797–3803. [DOI] [PubMed] [Google Scholar]
- Michael A.I., Gelfand,D.H., Sninsky,J.J. and White,T.J. (1990) PCR Protocols: A Guide to Method and Applications. Academic Press, San Diego, CA.
- Mosser D.D., Caron,A.W., Bourget,L., Denis-Larose,C. and Massie,B. (1997) Role of the human heat shock protein hsp70 in protection against stress-induced apoptosis. Mol. Cell. Biol., 17, 5317–5327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naf D., Kupfer,G.M., Suliman,A., Lambert,K. and D’Andrea,A.D. (1998) Functional activity of the Fanconi anemia protein FAA requires FAC binding and nuclear localization. Mol. Cell. Biol., 18, 5952–5960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson J.R., Lawrence,C.W. and Hinkle,D.C. (1996) Thymine–thymine dimer bypass by yeast DNA polymerase ζ. Science, 272, 1646–1649. [DOI] [PubMed] [Google Scholar]
- Okuno Y., Imamoto,N. and Yoneda,Y. (1993) 70-kDa heat-shock cognate protein colocalizes with karyophilic proteins into the nucleus during their transport in vitro. Exp. Cell Res., 206, 134–142. [DOI] [PubMed] [Google Scholar]
- Otsuki T., Nagakura,S., Wang,J., Bloom,M., Grompe,M. and Liu,J.M. (1999) Tumor necrosis factor-α and CD95 ligation suppress erythropoiesis in Fanconi anemia C gene knockout mice. J. Cell Physiol., 179, 79–86. [DOI] [PubMed] [Google Scholar]
- Pang Q., Fagerlie,S., Christianson,T.A., Keeble,W., Faulkner,G., Diaz,J., Rathbun,R.K. and Bagby,G.C. (2000) The Fanconi anemia protein FANCC binds to and facilitates the activation of STAT1 by IFN-γ and hematopoietic growth factors. Mol. Cell. Biol., 20, 4724–4735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang Q., Keeble,W., Diaz,J., Christianson,T.A., Fagerlie,S., Rathbun,R.K., Faulkner,G., O’Dwyer,M. and Bagby,G.C. (2001a) The role of double-stranded RNA dependent protein kinase (PKR) in mediating hypersensitivity of Fanconi anemia complementation group C cells to IFN-γ, TNF-α and double stranded RNA. Blood, 97, 1644–1652. [DOI] [PubMed] [Google Scholar]
- Pang Q., Christianson,T.A., Keeble,W. Diaz,J., Faulkner,G.R., Reifsteck,C., Olson,S. and Bagby,G.C. (2001b) The Fanconi anemia complementation group C (FANCC) gene product: structural evidence of multifunctionality. Blood, in press. [DOI] [PubMed] [Google Scholar]
- Rathbun R.K. et al. (1997) Inactivation of the Fanconi anemia group C gene augments interferon-γ-induced apoptotic responses in hemato poietic cells. Blood, 90, 974–985. [PubMed] [Google Scholar]
- Rathbun R.K., Christianson,T.A., Faulkner,G.R., Jones,G., Keeble,W., O’Dwyer,M. and Bagby,G.C. (2000) Interferon-γ-induced apoptotic responses of Fanconi anemia group C hematopoietic progenitor cells involve caspase 8-dependent activation of caspase 3 family members. Blood, 96, 4204–4211. [PubMed] [Google Scholar]
- Saleh A., Srinivasula,S.M., Balkir,L., Robbins,P.D. and Alnemri,E.S. (2000) Negative regulation of the Apaf-1 apoptosome by Hsp70. Nature Cell Biol., 2, 476–483. [DOI] [PubMed] [Google Scholar]
- Shi Y. and Thomas,J.O. (1992) The transport of proteins into the nucleus requires the 70-kilodalton heat shock protein or its cytosolic cognate. Mol. Cell. Biol., 12, 2186–2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon M.M., Reikerstorfer,A., Schwarz,A., Krone,C., Luger,T.A., Jaattela,M. and Schwarz,T. (1995) Heat shock protein 70 overexpression affects the response to ultraviolet light in murine fibroblasts. Evidence for increased cell viability and suppression of cytokine release. J. Clin. Invest., 95, 926–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strathdee C.A., Gavish,H., Shannon,W.R. and Buchwald,M. (1992) Cloning of cDNAs for Fanconi’s anaemia by functional com plementation. Nature, 356, 763–767. [DOI] [PubMed] [Google Scholar]
- Timmers C. et al. (2001) Positional cloning of a novel Fanconi anemia gene, FANCD2. Mol. Cell, 7, 241–248. [DOI] [PubMed] [Google Scholar]
- Walsh C.E., Nienhuis,A.W., Samulski,R.J., Brown,M.G., Miller,J.L., Young,N.S. and Liu,J.M. (1994) Phenotypic correction of Fanconi anemia in human hematopoietic cells with a recombinant adeno-associated virus vector. J. Clin. Invest., 94, 1440–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J. et al. (1998) Overexpression of the Fanconi anemia group C gene (FAC) protects hematopoietic progenitors from death induced by Fas-mediated apoptosis. Cancer Res., 58, 3538–3541. [PubMed] [Google Scholar]
- Wang T.F., Chang,J.H. and Wang,C. (1993) Identification of the peptide binding domain of hsc70. 18-Kilodalton fragment located immediately after ATPase domain is sufficient for high affinity binding. J. Biol. Chem., 268, 26049–26051. [PubMed] [Google Scholar]
- Whitney M.A. et al. (1996) Germ cell defects and hematopoietic hypersensitivity to gamma-interferon in mice with a targeted disruption of the Fanconi anemia C gene. Blood, 88, 49–58. [PubMed] [Google Scholar]
- Wu B., Hunt,C. and Morimoto,R. (1985) Structure and expression of the human gene encoding major heat shock protein HSP70. Mol. Cell. Biol., 5, 330–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J. and DeFranco,D.B. (1994) Differential roles of heat shock protein 70 in the in vitro nuclear import of glucocorticoid receptor and simian virus 40 large tumor antigen. Mol. Cell. Biol., 14, 5088–5098. [DOI] [PMC free article] [PubMed] [Google Scholar]