

Abstract
In certain cancers, constitutive Wnt signaling results from mutation in one or more pathway components. The result is the accumulation and nuclear localization of β-catenin, which interacts with the lymphoid enhancer factor-1 (LEF)/T-cell factor (TCF) family of HMG-box transcription factors, which activate important growth regulatory genes, including cyclin D1 and c-myc. As exemplified by APC and axin, the negative regulation of β-catenin is important for tumor suppression. Another potential mode of negative regulation is transcriptional repression of cyclin D1 and other Wnt target genes. In mammals, the transcriptional repressors in the Wnt pathway are not well defined. We have previously identified HBP1 as an HMG-box repressor and a cell cycle inhibitor. Here, we show that HBP1 is a repressor of the cyclin D1 gene and inhibits the Wnt signaling pathway. The inhibition of Wnt signaling and growth requires a common domain of HBP1. The apparent mechanism is an inhibition of TCF/LEF DNA binding through a physical interaction with HBP1. These data suggest that the suppression of Wnt signaling by HBP1 may be a mechanism to prevent inappropriate proliferation.
Keywords: β-catenin/HBP1/HMG-box transcription factor/Wnt
Introduction
The Wnt pathway exemplifies the interplay of oncogenes and tumor suppressor genes in tissue development and cancer (reviewed in Eastman and Grosschedl, 1999; Morin, 1999; Polakis, 2000). The binding of Wnt ligand to the Frizzled receptor initiates a cascade that results in β-catenin stabilization and accumulation. Wnt signaling inactivates GSK3β, which would otherwise trigger β-catenin degradation. The accumulated β-catenin protein translocates to the nucleus as a co-activator for the lymphoid enhancer factor-1 (LEF) and T-cell factor (TCF) transcription factor family (hereafter referred as LEF/TCF). The signature motif of the LEF/TCF family is the HMG-box-type DNA binding domain (Eastman and Grosschedl, 1999). An output of Wnt–β-catenin signaling is the activation of cyclin D1, c-myc and other genes to establish the oncogenic phenotype (He et al., 1998; Shtutman et al., 1999; Tetsu and McCormick, 1999).
Diverse evidence links Wnt signaling to several cancers. Wnt-1 was among the first oncogenes discovered. Trans genic expression of Wnt-1 remains an established model of breast tumorigenesis (Tsukamoto et al., 1988; reviewed in Li et al., 2000). Several of the genes at the endpoint of the Wnt pathway are documented oncogenes with frequent overexpression in cancer (e.g. cyclin D1 and c-myc). However, the strongest evidence is that a constitutive Wnt pathway occurs in several cancers through genetic mutation of one or more components (e.g. β-catenin, APC, axin; reviewed in Morin, 1999; Polakis, 2000). The consequence of the mutations identified is increased stability and accumulation of β-catenin. Mutations of β-catenin within the GSK3β phosphorylation sites, which normally trigger protein degradation, now result in stable and accumulated protein. Increased levels of β-catenin or direct mutations have been associated with numerous cancers, including breast, liver and colorectal (Lin et al., 2000; reviewed in Bienz and Clevers, 2000; Polakis, 2000). In addition, the loss-of-function mutations in APC are frequent in colorectal cancer and compromise β-catenin degradation (Korinek et al., 1997; Morin et al., 1997; He et al., 1998). Finally, mutations in axin, which also regulate β-catenin levels, have been associated with hepatocellular cancer (Satoh et al., 2000). These findings indicate that the negative regulators of Wnt and β-catenin signaling are effective tumor suppressor genes.
While β-catenin stability is an essential regulatory point, the consequence of β-catenin stability is increased transcriptional activation. Therefore, another possible mode of negative regulation could be the repression of β-catenin-mediated transcriptional regulation. In lower organisms, HMG-box transcriptional repressors have important roles in Wnt signaling. For example, in Xenopus, Sox17β and Sox3 repress β-catenin/TCF/LEF signaling (Zorn et al., 1999). In Caenorhabditis elegans, inactivation of the POP1 repressor is an important facet of Wnt signaling (Meneghini et al., 1999). In Drosophila, TCF has been shown to be a repressor of transcription in the absence of β-catenin signaling (Bienz, 1998; Nusse, 1999).
Few repressors of Wnt signaling have been identified in mammals. There is now evidence that various LEF and TCF family members are dual activators and repressors (reviewed in Barker et al., 2000). While clearly identified as a transcriptional activator, a recent study with LEF1, which demonstrates the association with histone deacetylases now confers repression functions (Billin et al., 2000). Another candidate is the HMG-box repressor HBP1. We previously isolated HBP1 as an HMG-box protein that interacts with retinoblastoma (RB) family members. HBP1 is a transcriptional repressor of some oncogenes, including n-myc (Tevosian et al., 1997). We demonstrated that HBP1 was a cell cycle inhibitor that regulated progression through G1 (Tevosian et al., 1997; Shih et al., 2001). Our studies have also revealed that HBP1 is a regulator of cell differentiation (Shih et al., 1998). Thus, our evidence was consistent with the notion that HBP1 is part of normal barriers that prevent proliferation in differentiated tissues. In contrast to other LEF/TCF family members, HBP1 has ubiquitous tissue distribution and, therefore, may serve as a general repressor.
Two observations led to an investigation of HBP1 as a regulator of Wnt signaling. In our previous studies, a subset of HBP1 DNA binding sites were similar to DNA sequences for LEF/TCF proteins (Love et al., 1995; Tevosian et al., 1997) and suggested that HBP1 might repress LEF/TCF target genes. While investigating gene targets of HBP1, we found that the cyclin D1 promoter was regulated by HBP1. Two additional studies then established cyclin D1 as a Wnt pathway target gene (Shtutman et al., 1999; Tetsu and McCormick, 1999). With a common link to cyclin D1, we investigated whether HBP1 could inhibit Wnt signaling.
In this paper, we describe evidence that HBP1 could be a suppressor of Wnt signaling. First, HBP1 expression inhibited promoter activation by four distinct components of the Wnt pathway: Wnt, β-catenin, GSK3β and LEF/TCF. Secondly, HBP1 expression repressed Wnt–β-catenin activation of the cyclin D1 promoter, as well as the expression of endogenous cyclin D1 and c-myc genes, two targets of the Wnt pathway. Thirdly, our data support a mechanism in which HBP1 prevents DNA binding by LEF/TCF family members, apparently through a physical interaction with LEF/TCF proteins. Our data are consistent with a model in which HBP1 is a suppressor of Wnt–β-catenin signaling by inhibiting the LEF/TCF transcription factors.
Results
HBP1 inhibits Wnt–β-catenin gene targets
Because HBP1 and LEF-1 are both HMG-box proteins with opposite effects on transcriptional output, we asked whether HBP1 could repress LEF/TCF activity and, therefore, the Wnt pathway. As our first objective, we assessed the impact of HBP1 on Wnt signaling. The binding of Wnt to the Frizzled receptor triggers the stabilization of endogenous β-catenin to activate LEF- and TCF-dependent gene expression, as shown schematically in Figure 1A. To score the activation of LEF/TCF-dependent transcription, we used the activity of the TOPFLASH reporter as an endpoint of Wnt signaling (e.g. Korinek et al., 1997; Morin et al., 1997). TOPFLASH is a synthetic reporter consisting of three LEF/TCF sites fused to a minimal FOS promoter (a generous gift of Drs Bert Vogelstein and Ken Kinzler). In agreement with published work, expression of Wnt1 [Hsu et al., 1998; cytomegalovirus (CMV) Wnt 1, gift of Dr Dan Sussman] resulted in activation of the TOPFLASH reporter, but not the control FOPFLASH reporter containing mutant LEF/TCF sites (Figure 1B). Furthermore, co-expression of HBP1 efficiently inhibited Wnt-mediated promoter activation (Figure 1B), confirming the predicted HBP1 function in Wnt signaling.




Fig. 1. HBP1 inhibits Wnt–β-catenin-activated gene expression. (A) Schematic diagram of Wnt signaling (adapted from Bienz and Clevers, 2000; Polakis, 2000). The asterisks mark Wnt pathway components with known mutation in cancer. (B) HBP1 inhibits Wnt1-activated gene expression. HEK293T cells were transfected with 1 µg of TOPFLASH, 2 µg of RSV–β-Gal, 5 µg of Wnt1, and 6 or 12 µg of pEFBOS-HBP1. One microgram of FOPFLASH with and without 5 µg of Wnt1 was also transfected as a negative control. TOPFLASH contains three LEF/TCF sites followed by the minimal TATA and luciferase. FOPFLASH contains three mutated LEF/TCF sites followed by the minimal TATA and luciferase. Cells were harvested 48 h after transfection, and luciferase and β-Gal activity were measured. The results are normalized for transfection efficiency and are expressed as a relative ratio of luciferase to β-Gal activities (denoted as relative activity). (C) HBP1 inhibits LiCl-activated gene expression. HEK293T cells were transfected with 1 µg of TOPFLASH, 2 µg of RSV–β-Gal, and 3, 6 or 12 µg of pEFBOS-HBP1 (indicated by ramp). Cells were treated with 15 mM LiCl for 12 h. One microgram of FOPFLASH was also transfected as a negative control. Cells were then harvested at 48 h after transfection. The results are normalized for transfection efficiency and are expressed as a ratio of luciferase to β-Gal activities. (D) HBP1 inhibits β-catenin activation through LEF/TCF DNA binding sites. HEK293T cells were transfected with 2 µg of RSV–β-Gal, 1 µg of TOPFLASH or 1 µg of FOPFLASH as indicated, 2 µg of β-catenin and 7.5 µg of pEFBOS HBP1 as indicated. Cells were harvested 48 h after transfection, and luciferase and β-Gal activity were measured. The results are normalized for transfection efficiency and are expressed as a relative ratio of luciferase to β-Gal activities (denoted as relative activity).
While HBP1 was able to repress Wnt-mediated promoter activation efficiently, we next asked whether HBP1 could also repress further downstream in the pathway. We next showed that lithium chloride (LiCl) activation of TOPFLASH was also inhibited by HBP1 (Figure 1C). LiCl mimics Wnt signaling by inhibiting GSK3β activity (which provides increased β-catenin stability and transcriptional activation) (e.g. Chen et al., 2000). The control FOPFLASH reporter was not affected. Thus, the data in Figure 1C suggest that HBP1 inhibited the Wnt pathway by affecting events downstream of GSK3β. Finally, we examined the effect of HBP1 on β-catenin-induced gene activation. A β-catenin expression vector was transfected with TOPFLASH in the absence and presence of HBP1. As expected from published work, β-catenin activated the TOPFLASH reporter. As shown in Figure 1D, expression of HBP1 suppressed β-catenin activation through LEF/TCF sites, as exemplified by reduced TOPFLASH activity. No effect was observed on the control FOPFLASH reporter that contained mutated LEF/TCF sites. These data suggest that HBP1 inhibited the Wnt signaling pathway, as shown by the ability to repress transcriptional output of Wnt, GSK3β inhibition and β-catenin.
We then examined the impact of HBP1 on the activation of known Wnt target genes. The cyclin D1 promoter is upregulated by β-catenin through activation of LEF/TCF family members (Shtutman et al., 1999; Tetsu and McCormick, 1999). Cyclin D1 expression is upregulated in numerous tumor types in which the Wnt pathway has been implicated (Weinstat-Saslow et al., 1995; Hosokawa and Arnold, 1998). Therefore, we examined the effect of HBP1 on both β-catenin and LEF activation of the –963 cyclin D1 promoter construct. As shown in Figure 2A, LEF1 expression strongly activated the cyclin D1 promoter. Importantly, co-expression of HBP1 inhibited LEF activation, indicating that HBP1 could directly oppose a LEF signal. Next, we showed that co-transfected HBP1 could repress the cyclin D1 promoter that was activated by β-catenin expression (Figure 2B). Notably, repression was maximal at the lowest β-catenin concentrations and less at high β-catenin, suggesting that the ratio of activating to repressing HMG-box factors contributed to the transcriptional output. These results are independent of cell type, as all experiments have been repeated multiple times in several cell types (e.g. NIH 3T3, 293T, HCT116, Caco 2) with identical results. In addition, using epitope-tagged β-catenin, HBP1 did not affect expression of β-catenin and indicated that this was not the reason for the reduction in TOPFLASH activity (data not shown).



Fig. 2. HBP1 inhibits expression of cyclin D1. (A) Lef-1 and HBP1 have opposite effects on the cyclin D1 promoter. HEK293T cells were transfected with 1 µg of –963 cyclin D1 promoter, 2 µg of RSV–β-Gal, CMV–Lef-1 (1, 2 or 4 µg), pEFBOS-HBP1 (5, 7.5 or 10 µg). Cells were harvested 48 h after transfection, and luciferase and β-Gal activity were measured. The results are normalized for transfection efficiency and are expressed as a ratio of luciferase to β-Gal activities. (B) HBP1 inhibits β-catenin activation of the cyclin D1 promoter. HEK293T cells were transfected with 1 µg of –963 cyclin D1, 2 µg of RSV–β-Gal, indicated amounts of β-catenin, and 7.5 µg of pEFBOS-HBP1. Cells were harvested 48 h after transfection, and luciferase and β-Gal activity were measured. The results are normalized for transfection efficiency and are expressed as a ratio of luciferase to β-Gal activities. (C) HBP1 inhibits endogenous expression of two β-catenin/LEF/TCF targets, cyclin D1 and c-myc. Ten micrograms of pEFBOS-HBP1 were transiently transfected into HCT116 cells along with 2 µg of pMACS Kk. Transfected cells were selected and RNA isolated. As a control, RNA from cells transfected only with pMACS Kk was also isolated. RT–PCR was performed using 2 µg of RNA from each cell population. Results of the RT–PCR using primers to cyclin D1, c-myc and 18S RNA were run on an agarose gel. RT–PCR cycle number for each primer-pair was optimized in order to calibrate into the linear range (see Materials and methods).
We next investigated whether HBP1 expression could repress the endogenous cyclin D1 and other Wnt target genes. To compare and extend our analysis, c-myc was chosen as an example of another Wnt target gene (He et al., 1998). Both the cyclin D1 and c-myc oncogenes are APC/β-catenin target genes that are frequently overexpressed in numerous cancers. HCT116 colon cancer cells were used as an example of a tumor cell with a constitutive Wnt–β-catenin pathway (through a stabilizing mutation in β-catenin). Thus, no exogenous β-catenin is necessary to activate cyclin D1 and other Wnt pathway genes in HCT116 cells. The expression of endogenous cyclin D1 and c-myc upon HBP1 expression was scored by RT–PCR. The success of the RT–PCR analysis for cyclin D1 depended upon uniform expression of HBP1 and, therefore, purified transfected cells were used [as described in Materials and methods and Watanabe et al. (1998) and Tetsu and McCormick (1999)]. As shown in Figure 2C, HBP1 expression suppressed the endogenously activated cyclin D1 and c-myc genes efficiently. HBP1 had no effect on 18S or actin (not shown) gene expression. All measurements were within the linear range of PCR detection (as described in Materials and methods). These data demonstrate that the mechanism of HBP1 suppression of Wnt signaling is directed at β-catenin-activated target promoters. Significantly, HBP1 repressed transcription regardless of whether endogenous or exogenous β-catenin was used to activate target genes such as cyclin D1 and c-myc.
HBP1-mediated inhibition of Wnt signaling does not require DNA binding
We next defined the regions on HBP1 that were necessary for repression of the Wnt pathway. As shown in Figure 3A, HBP1 contains both transcriptional repression and HMG-box DNA binding domains. Because of the similarity of HBP1/LEF DNA binding sites, we predicted that DNA binding by HBP1 would be required for repression, but were surprised by the actual results. Figure 3A and B shows the activity of several HBP1 mutants in repression of β-catenin activation and of Wnt-mediated transcriptional activation, respectively. Both signals use the endogenous LEF/TCF family members. Additionally, activation by Wnt would use endogenous levels of β-catenin and the relevant LEF/TCF family member. These data have been repeated in two to three different cell types with similar results. Furthermore, all experiments described below exhibit equal protein expression of wild-type and mutant HBP1, and at levels that were in the linear range of the transcriptional assays.


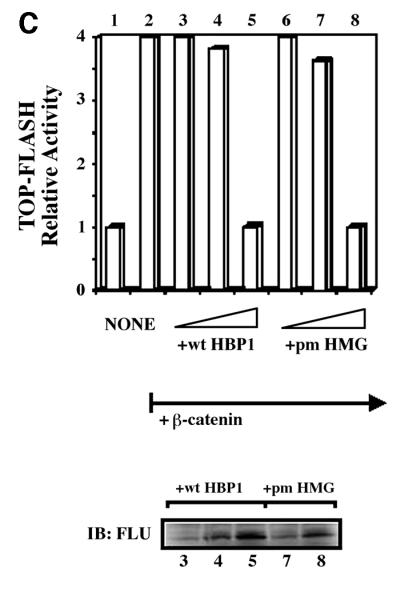



Fig. 3. HBP1 repression of Wnt–β-catenin-activated gene expression requires the repression domain but not DNA binding. (A) HBP1 domain requirement for repression of LEF/TCF-dependent gene expression. HEK293T cells were transfected with 1 µg of TOPFLASH, 2 µg of RSV–β-Gal, 2 µg of β-catenin, 10 µg of pEFBOS-HBP1 or the indicated HBP1 mutant. Cells were harvested 48 h after transfection and results were scored as a relative ratio of luciferase to β-Gal activities (denoted as relative activity). (B) HBP1 domain requirement for repression of Wnt1 gene activation. HEK293T cells were transfected with 1 µg of TOPFLASH, 2 µg of RSV–β-Gal, 5 µg of Wnt1, and 6 µg of pEFBOS-HBP1 or the indicated HBP1 mutant. Cells were harvested 48 h after transfection. The results are normalized for transfection efficiency and are expressed as a relative ratio of luciferase to β-Gal activities. (C) WT HBP1 and pmHMG/HBP1 both suppress β-catenin-induced gene expression. WT HBP1 and pmHMG were transfected in equal amounts to titrate the repression response to β-catenin activation of TOPFLASH. HEK293T cells were transfected with 1 µg of TOPFLASH, 2 µg of RSV–β-Gal, 2 µg of β-catenin, and 3, 6 or 12 µg of pEFBOS-HBP1 or pmHMG (indicated by the ramp). Cells were harvested 48 h after transfection. Luciferase and β-Gal activity were measured. The results were normalized for transfection efficiency and are expressed as a ratio of luciferase to β-Gal activities. Anti-FLU western blotting for HBP protein level is shown for transfected samples below. (D) HBP1 does not bind the TOPFLASH LEF/TCF DNA binding site. GST–HBP1(HMG), GST–HBP1(pmHMG) and GST–TCF4(HMG) proteins were used to score the ability of GST–HBP1(HMG) to bind the LEF/TCF site present in TOPFLASH and the cyclin D1 promoter. Two hundred nanograms of each protein were used in each gel-shift reaction with radioactively labeled TOPFLASH probe or the high affinity (HA) HBP1 site probe as a control for HBP1 DNA binding. A 100-fold molar excess of wild-type and mutant competitors was used in all cases. GST–TCF4(HMG) was used as a positive control for binding TOPFLASH. (E) Repression of the n-myc promoter by HBP1 requires both DNA binding and a repression domain. C33A cells were trans fected with 3 µg of PUC–n-myc–CAT, 2 µg of RSV–β-Gal and 10 µg of pEFBOS-HBP1 or the indicated HBP1 mutant. Cells were harvested 48 h after transfection and results were scored as CAT activity normalized with RSV–β-Gal activity. (F) Repression of the cyclin D1 promoter by HBP1 requires a repression domain, but not an intact DNA binding domain. The –963 cyclin D1–luciferase construct was analyzed as in Figure 2B, except that β-catenin was omitted. Both wild-type and the indicated HBP1 mutants were used.
Regardless of whether Wnt signaling or β-catenin was used, two consistent results emerged from Figure 3A–C. One is that the HBP1 DNA binding domain was not required for repression, despite the apparent similarity in the HMG domains of LEF and HBP1. As shown in Figure 3B, a mutant with the N-terminal 330 amino acids of HBP1 that lacks the HMG box (denoted N-330) still represses transcription. Similarly, the pmHMG mutant of HBP1, which is also defective in DNA binding because of three mutations in three common residues that bind DNA (Love et al., 1995; Tevosian et al., 1997), still represses transcription (Figure 3A and B). To extend this observation we further titrated the wild-type HBP1 and pmHMG to compare the repression of β-catenin-activated TOPFLASH. Both the wild type and pmHMG exhibited similar specific activity, further supporting the view that DNA binding was not required for the observed repression of Wnt signaling on β-catenin activation (Figure 3C). In Figure 3D, the lack of HBP1 DNA binding to the high affinity LEF/TCF sites of TOPFLASH and cyclin D1 was confirmed by in vitro gel-shift assays using purified, recombinant protein. Figure 3D further shows that glutathione S-transferase (GST)–TCF4–HMG expectedly binds well to the high affinity LEF/TCF sites, but not to the high affinity HBP1 sites. Similarly, GST–HBP1–HMG binds well to its own high affinity site, but undetectably to the high affinity LEF/TCF sites of TOPFLASH. The pmHMG mutant of HBP1 binds to neither high affinity LEF/TCF nor HBP1 DNA probes. While there is some homology, the HMG boxes of TCF4 and HBP1 have different high affinity DNA binding sites. Together, Figure 3A–D shows that DNA binding by HBP1 is not needed for suppression of Wnt signaling.
The second consistent result is that the HMG box alone (mutant 1242, amino acids 401–513) was fully defective in repression (Figure 3A and B). An HBP1 mutant with the N-terminal 191 amino acids (denoted N191) also did not inhibit β-catenin-activated TOPFLASH reporter activity (Figure 3B). In contrast, mutant 660 (containing amino acids 220–513) or N330 (containing the N-terminal 330 amino acids) had near wild-type repression activity (Figure 3B). Together, these data indicate that the Wnt–β-catenin suppression region lies within amino acids 191–401 and that DNA binding is not required. Two independent repression regions may also be present within the region from 191 to 401, as demonstrated by normal repression activity by mutants N-330 and del 660. This observation eliminates a model in which both LEF and HBP1 simply compete for occupancy of DNA elements. Thus, HBP1 repression of Wnt and β-catenin signaling is independent of its DNA binding activity. Similar results were obtained regardless of whether endogenous or exogenous β-catenin participated in gene activation.
To highlight the difference between the mechanisms of Wnt suppression by HBP1 and sequence-specific repression, HBP1 regulation of the n-myc promoter was compared. We had previously shown that the n-myc promoter was an example of sequence-specific repression by HBP1 (Tevosian et al., 1997). As shown in Figure 3E, pmHMG was defective in the repression of the n-myc promoter. Like the repression of Wnt signaling, mutant 660, but not mutant 1242, provided full repression of the n-myc promoter. Thus, for sequence-specific repression, the repression domain also lies between amino acids 220 and 401. The N330 and N191 mutants were not tested, but a deletion mutant with the N-terminal 393 amino acids was defective for repression of the n-myc promoter (not shown). The comparison with the n-myc promoter provides a contrast to the apparent DNA binding-independent repression of Wnt signaling by HBP1 (Figure 3E).
In a direct comparison with the –963 cyclin D1 promoter, the repression by HBP1 did not require DNA binding, but did require the defined repression domains (Figure 3F). Thus, the requirements for HBP1 on the cyclin D1 and n-myc promoters differ significantly in the requirement for DNA binding, but share a common repression domain. The data in Figure 3E and F highlight the two modes of repression for cyclin D1 and the n-myc promoters.
HBP1 binds and inhibits TCF4–β-catenin complexes
We next examined the mechanism behind the lack of requirement for DNA binding by HBP1 in suppression of Wnt–β-catenin activation of gene expression. Because promoter repression still occurred in the absence of HBP1 DNA binding, inhibition of TCF DNA binding by HBP1 provided a plausible explanation for the observed transcriptional repression. To address this hypothesis, we selected the TCF4 family member to test possible regulation by HBP1. TCF4 is the most abundant family member in colon cells in which the Wnt pathway has been well studied, but is also present in other cell types (Barker et al., 1999). The experimental design was to examine the consequence of HBP1 expression on TCF–β-catenin DNA binding activity, as measured by gel-shift assays with the high affinity LEF/TCF site. As shown in Figure 4A, complexes that corresponded to TCF alone and TCF– β-catenin were observed (Ishitani et al., 1999). These bands exhibited the appropriate specificity in competition with mutant and wild-type LEF/TCF sites (denoted as FOP and TOP, respectively, in Figure 4A). Consistent with the results in Figure 3A, wild-type HBP1, pmHMG, del 660 and N330 were fully effective in disrupting TCF4 DNA binding, but the del 1242 and N191 mutants were ineffective. The notable absence of a prominent new HBP1 binding species further supports the model that HBP1 DNA binding is not necessary for the transcriptional repression and inhibition of TCF DNA binding through these specific LEF/TCF DNA elements. Thus, the inhibition of both TCF4 DNA binding and of transcriptional activation by Wnt and β-catenin exhibited good correlation with the HBP1 repression domain between amino acids 220 and 401, and no correlation with an intact HBP1 DNA binding region.



Fig. 4. HBP1 regulates LEF/TCF DNA binding activity. (A) HBP1 inhibits TCF4 and the TCF–β-catenin complex binding to DNA. HEK293T cells were transfected with 3 µg of Flag-TCF4, 7 µg of Flu–β-catenin, and 10 µg of HBP1 or the indicated HBP1 mutant. Cells were harvested 24 h after transfection. EMSA was performed using the 293T extract and the TOPFLASH probe. Both cold TOPFLASH and FOPFLASH were used as competitors at 100-fold molar excess. Where indicated by an asterisk, the Flag antibody was used to supershift the TCF4–β-catenin DNA binding complex. (B) Endogenous complex of HBP1 and TCF4. 293T and HCT116 cells were used to examine the endogenous interaction between HBP1 and TCF4. 293T and HCT116 cells were immunoprecipitated with anti-HBP1, anti-TCF4 or anti-IgG as a control. A SDS–PAGE gel was run, and to detect the presence of HBP1 the anti-HBP1 antibody was utilized in a western blot. As controls, 293T and HCT116 cellular extract untransfected (E), or 293T extract transfected (T) with HBP1 was used to visualize the mobility of HBP1. (C) The repression domain of HBP1 is necessary for TCF4 interaction. HEK293T cells were transfected with FLAG-TCF4 and pEFBOS-HBP1 or the indicated HBP1 mutant. Immunoprecipitations were performed using the FLAG antibody or normal mouse IgG (control). The immunoprecipitations were separated by SDS–PAGE and the associated HBP1 was detected by western blotting with the flu antibody (anti-HA.11; Babco). Total input of HBP1 and immunoprecipitated FLAG-TCF4 (lower panels) were also examined by western blotting as shown. The numbered lanes in the upper panel correlate with those in the lower panels for comparison.
Because both TCF4 and TCF4–β-catenin complexes were disrupted by HBP1 expression, we tested whether HBP1 might target TCF4 itself to inhibit DNA binding activity. In Figure 4B, an endogenous interaction between TCF4 and HBP1 was detected in both 293T and HCT116 cells. These data suggest that a complex of TCF4 and HBP1 may have general distribution in both colon and non-colon cells. The presence of an endogenous complex motivated a more detailed examination of the binding interaction of TCF4 and HBP1. Our specific focus was whether the interaction regions matched functionally important regions of both HBP1 and TCF4. As shown in Figure 4C, the HBP1 proteins (wild type, 660, pmHMG, N330) that support repression of β-catenin transcriptional activation and disruption of TCF4 DNA binding also physically interacted with TCF4, whereas the functionally inactive 1242 and N191 mutants failed to interact with TCF4. Together with the previous figures, these data suggested that there is good correlation between HBP1 regions necessary for repression of Wnt signaling, disruption of TCF4 DNA binding and for physical interaction with TCF4.
We next defined the HBP1 binding region on the TCF4 protein with the same co-immunoprecipitation assays. As shown in Figure 5A–C, through the analysis of targeted TCF4 mutants, HBP1 interacted with two regions of TCF4. One region included the TCF4 HMG box (within amino acids 327–400 of TCF4). The second region of HBP1 interaction (amino acid 53–171 of TCF4) represents an ill-defined region and awaits further investigation. The β-catenin binding region of TCF4 was specifically not involved in the interaction with HBP1. This is consistent with the results of Figure 4A in which HBP1 disrupted the DNA binding of both the TCF and TCF4–β-catenin complexes. As is summarized in Figure 5D, the repression domain of HBP1 and two regions in TCF4 all interact within a complex of TCF4 and HBP1. The physical interaction of HBP1 with the TCF4 HMG-box/N-terminal region supports a mechanism in which HBP1 disrupts TCF4 DNA binding activity, thus suppressing Wnt signaling.




Fig. 5. HBP1 interacts with two separate regions of TCF4. (A) HEK293T cells were transfected with pEFBOS-HBP1 and FLAG-TCF4 or the indicated FLAG-TCF4 mutant. Immunoprecipitations were performed using the FLAG antibody or normal mouse IgG (control). The immunoprecipitations were separated by SDS–PAGE and the associated HBP1 was detected by western blotting with the flu antibody (anti-HA.11; Babco). (B) Total input of HBP1 (lower panel) and immunoprecipitated FLAG-TCF4 or the indicated mutant (upper panel) was also examined by western blotting as shown. The numbered lanes correlate with the numbered lanes in (A) for comparison. (C) Schematic diagram of TCF4 mutants. (D) Summary of interactions on both TCF4 and HBP1. The rat and human HBP1 contain 513 and 509 amino acids, respectively. However, the indicated motifs lie at identical positions in both species.
HBP1 is an efficient growth suppressor
Because the Wnt pathway activates cell proliferation, we probed whether HBP1 could suppress growth in cells with a constitutively active Wnt pathway. The HCT116 and Caco-2A colon cancer cells have a mutation in APC and in both APC and β-catenin, respectively. Both have a constitutive Wnt pathway. In both HCT116 and Caco-2A cells, overexpression of cyclin D1 and/or hyper-activation of the TOPFLASH reporter have been reported (Morin et al., 1997; Tetsu and McCormick, 1999). Our objective was to determine whether growth suppression correlated with the regions necessary for transcriptional repression. We used colony formation assays to test whether HBP1 inhibited growth. The data with Caco-2A cells are shown in Figure 6, but identical results were obtained with HCT116 cells. As shown in Figure 6, HBP1 expression gave a reproducible reduction in colony formation. Similarly, del 660 and pmHMG also gave reproducible growth suppression. These mutants represented two of the several other HBP1 constructs that supported suppression of Wnt signaling. Importantly, the 1242 HBP1 mutant did not reduce colony formation. This mutant was defective in cyclin D1 repression, inhibition of TCF4 DNA binding and physical interaction with TCF4. The N191 and N330 mutants were not tested. The colony formation assays were repeated 7–10 times with good reproducibility, as illustrated by the small error bars. Like Wnt pathway suppression and LEF/TCF DNA binding inhibition, HBP1-mediated growth suppression in colon cancer cells required the repression domain, but not the DNA binding region.

Fig. 6. HBP1 suppresses colony formation in colon carcinoma cells. Caco-2a cells were transfected with 2 µg of pEFBOSHygro alone, or with 9 µg of pEFBOS-HBP1, or the indicated mutant. Transfected cells were selected in hygromycin media and after 10 days of selection stained with crystal violet. Positive colonies were counted, quantitated and normalized to the HYGRO-only transfection. The mean and SEM from five different experiments are plotted.
Discussion
In this work, we demonstrate that HBP1 can efficiently suppress gene activation by Wnt signaling. Our data are consistent with a model in which the apparent HBP1 mechanism is the inhibition of LEF/TCF transcriptional activity. Identical results were obtained regardless of whether gene activation was triggered by four separate Wnt pathway components: Wnt itself, β-catenin, inhibition of GSK3β or LEF/TCF. These results suggested a downstream blockade of the entire pathway by HBP1. Consistently, the expression of HBP1 inhibited endogenous Wnt-activated genes such as cyclin D1. The HBP1-mediated suppression of Wnt signaling was also consistent with growth suppression in colon cancer cells in which genes such as cyclin D1 are constitutively activated through mutations in the Wnt pathway. Thus, these data are consistent with HBP1 as both a transcriptional repressor of Wnt signaling and a suppressor of Wnt–β-catenin-regulated growth.
In addition, our data are consistent with a mechanism in which HBP1 physically interferes with LEF/TCF DNA binding and transcriptional activity. Specifically, mutant HBP1 proteins that could not repress transcription also failed to inhibit TCF4 DNA binding. Furthermore, in an unexpected result, HBP1–DNA binding was not required for inhibition of either TCF4 DNA binding or of Wnt–β-catenin transcriptional activation. An endogenous complex of HBP1 and TCF4 was detected in both colon and non-colon cells, suggesting that HBP1 interaction with TCF4 might disrupt TCF4 function. Within HBP1, the necessary functional region for interacting with TCF4, inhibiting TCF4 DNA binding and TCF4 transcriptional activity was defined to amino acids 192–400, which contains the repression domain. Within TCF4, two HBP1 interaction regions (amino acids 53–171 and 327–400) were defined, of which the latter contains the HMG-box DNA binding region of TCF4. The relevance of the second region from amino acids 53 to 171 of TCF4 is unknown. Together, these data support a mechanism in which HBP1 blocks DNA binding in a physical interaction with the HMG box of TCF4. This study highlights the potential role of HBP1 in the repression of the Wnt–β-catenin pathway. The model for HBP1 function is summarized in Figure 7.

Fig. 7. The proposed role of HBP1 in the Wnt signaling pathway. As described in the background, the Wnt signaling pathway is conserved in evolution and has an intricate role in cancer. When Wnt proteins bind to Frizzled receptors, a signaling cascade is triggered where the β-catenin protein becomes stabilized and can enter the nucleus to activate transcription of target genes through binding target HMG-box proteins. Lef/TCF proteins directly bind the DNA, and when bound to β-catenin activate transcription of target genes such as cyclin D1 and c-myc (reviewed in Bienz and Clevers, 2000; Polakis, 2000). In normal tissues, the tumor suppressor protein, APC, along with a complex containing axin and GSK-3β, triggers the degradation of β-catenin when Wnt signaling is turned off. All three components are important for preventing accumulation of β-catenin; loss-of-function mutations in APC and axin are associated with colorectal and hepatocellular cancers. In addition, stabilizing mutations in β-catenin have been identified in many human cancers. With the literature of the Wnt pathway as a backdrop, the work in this paper supports a model in which HBP1 is a suppressor of Wnt signaling and is a negative regulator of proliferation in normal tissues. HBP1 can block Wnt signaling from four points in the pathway: Wnt itself, GSK3β inhibition, β-catenin and LEF/TCF. The endogenous cyclin D1 and c-myc target genes are also inhibited by HBP1 expression in a cell with a constitutive Wnt signaling pathway. Our data indicate that the probable mechanism is the inhibition of DNA binding by LEF and TCF through a physical interaction with HBP1. The same regions of HBP1 that are necessary for suppression of Wnt signaling are also required for growth inhibition in cells with a constitutive Wnt pathway. Thus, HBP1 may have a role in tumor suppression by inhibiting the Wnt signaling pathway.
Both our results and those of other studies reveal that negative regulation is a significant aspect of the Wnt–β-catenin pathway. While most studies have focused on negative regulation of β-catenin stability, transcriptional repression would be an effective mechanism to block gene expression that was activated by the stabilized β-catenin. Our work with HBP1 adds to possibilities for repressors of Wnt signaling in mammalian cells. We first identified HBP1 as an HMG-box transcriptional repressor and as an inhibitor of proliferation (Tevosian et al., 1997; Shih et al., 1998, 2001). While LEF and TCF family members have been largely associated with transcriptional activation, the newer results suggest dual activator and repressor functions for some members. For example, the interaction with the co-repressor HDAC confers repression functions to LEF1, a well-characterized activator (Billin et al., 2000). Secondly, the TCF1 family member may be a candidate repressor. Gene knockout studies of the TCF1 family member resulted in increased breast and intestinal tumor frequency in an APC +/– mouse background. While TCF1 may be a candidate tumor suppressor gene, the proper gene context for transcriptional repression has not been reported (Roose et al., 1999). Early reports indicated that TCF1 was an activator (Verbeek et al., 1995 and references therein). As described in the Introduction, most evidence for repression has emerged from lower organisms. Initially, the dual repressor–activator function was suggested in Drosophila with studies of CBP, which is paradoxically a co-repressor for TCF in this organism (Waltzer and Bienz, 1998). In C.elegans, Wnt signaling proceeds through the inactivation of POP1, an HMG-box repressor, by Lit1 (Meneghini et al., 1999). Lit1 is a MAP kinase that is necessary for the activation of the Wnt pathway in C.elegans by loss of POP1 repressor function. A mammalian Lit1 homolog, NLK, inhibited TCF– β-catenin binding to DNA and, therefore, its transcriptional activity. Expression of active NLK blocked TCF– β-catenin–DNA binding, but kinase-inactive NLK was ineffective (Ishitani et al., 1999). Finally, the closest functional analogy to HBP1 is the Xenopus Sox 17 repressor (Zorn et al., 1999). Like HBP1, Sox 17 inhibited β-catenin signaling that was independent of DNA binding. For Sox 17, the repression mechanism was β-catenin sequestration. For HBP1, we did observe weak, but specific binding to β-catenin (not shown). However, the functional inhibition correlated better with a physical interaction between HBP1 and TCF4. Nevertheless, the striking similarity is that the apparent inhibitory mechanism between Sox 17 and HBP1 was independent of DNA binding, although a perfectly functional HMG box was present in both proteins. It was suggested that yet-to-be defined targets must exist in which Sox 17 may function as a sequence-specific repressor (Zorn et al., 1999). The significance of the current work is that HBP1 is a negative regulator of Wnt signaling in mammalian cells from the aspects of transcriptional repression and growth suppression. Our work does not exclude a model in which HBP1 may somehow cooperate with other LEF/TCF proteins to enhance repression of Wnt target genes.
Because TCF4 has high expression in the intestine and is required for proper intestinal development (Korinek et al., 1998), TCF4 is a relevant family member for our studies that have used colon cancer cells. The observations in Figures 4 and 5 indicate that there are distinct interaction regions on both HBP1 and TCF4. These interactions utilize the HBP1 repression and TCF4 DNA binding regions. These data fully support a mechanism by which HBP1 interaction inhibits DNA binding to suppress Wnt signaling. An open question is the relationship of the TCF4–HBP1 and TCF4–β-catenin complexes in colorectal and other cancers. A possible model is that the HBP1–TCF4 complexes block Wnt signaling in normal cells. Upon constitutive mutation in APC or β-catenin, the concentration of the TCF4–β-catenin complex may exceed that of the TCF4–HBP1 complex. Under these circumstances, transcriptional activation of cyclin D1, c-myc and other growth control genes can then occur. Future work will investigate this hypothesis and test the general applicability to cancers in which constitutive Wnt signaling has a causative role (e.g. colon, liver and breast).
At this point, we have not fully defined whether RB family members play a role in the functions of HBP1 in the Wnt pathway. Notably, we first isolated HBP1 as a RB and p130 target protein, and defined the role of RB in the regulation of sequence-specific repression (Tevosian et al., 1997). HBP1 repression of Wnt–β-catenin gene activation appears to be independent of the RB family, as an HBP1 mutant that fails to bind RB and p130 still inhibits β-catenin activation (data not shown). Recently, we have identified other promoters in which HBP1 exhibits sequence-specific repression and a requirement for RB interaction (data not shown), further supporting two modes of repression with opposite requirements for DNA binding and for RB interaction. The simplest conclusion from existing data is that HBP1 has independent functions in the Wnt- and RB-mediated pathways.
Thus, an important aspect of future investigations is an elaboration of other HBP1 gene targets for repression that is independent of or dependent on DNA binding. The range of DNA elements through which HBP1 can inhibit or repress transcription is now unexpectedly complex with both sequence-independent and sequence-specific repression, as represented by the cyclin D1 and n-myc promoters. In the case of HBP1, n-myc is one of several sequence-specific targets (Tevosian et al., 1997; Zhuma et al., 1999; Lemercier et al., 2000; Lin et al., 2001). To add complexity, the histone H1(0) and myeloperoxidase promoters are apparently activated by HBP1 (Lemercier et al., 2000; Lin et al., 2001). This may be consistent with a previous report that showed a cryptic activation domain within HBP1 by Gal4 fusion experiments (Lavender et al., 1997). On the cyclin D1 and n-myc promoters, we have always observed transcriptional repression with the intact HBP1 protein and have never observed activation. We have not tested H1(0) or the myeloperoxidase promoters that have reported activation by HBP1. These reports raise the possibility that HBP1 may also have dual repressor and activator functions, which may be determined by promoter context and by cell type. This unexpected duality in HBP1 awaits future investigation.
How do we envision the functions of HBP1 in terms of normal tissue homeostasis and tumor suppression? The essential issue is a blockade in proliferation to enforce the quiescence in normal tissues. HBP1 regulates in multiple contexts (e.g. Wnt- and RB-mediated networks), all of which may contribute to a lack of proliferation in most normal tissues. Based on known functions of HBP1 in suppressing cell cycle progression, we envision that HBP1 activity is necessary to enforce the blockade in proliferation through the repression or inhibition of expression of cyclin D1, c-myc and other relevant genes. In our model, the accumulation of β-catenin and activation of LEF/TCF-dependent transcription during Wnt signaling might overcome the constitutive repression by HBP1, thus activating genes necessary for proliferation. When Wnt signaling is complete and target gene expression must be inactivated, HBP1 could then re-establish the repression and inactivation of the Wnt target genes. The balance of transcriptional repression by HBP1 and transcriptional activation by LEF/TCF–β-catenin may ultimately determine the transcriptional output of critical genes such as cyclin D1, c-myc and other growth control genes.
Because activation of the Wnt–β-catenin pathway is widely associated with oncogenesis, negative inhibitory mechanisms are critical for tumor suppression and blockade of proliferation in normal tissues. The strongest argument is the prevalence of mutations in cancer that result in constitutive activation of the Wnt–β-catenin pathway. APC and axin negatively regulate β-catenin levels, and are both documented tumor suppressor genes that are mutated in colon and hepatocellular cancers, respectively (Korinek et al., 1997; Morin et al., 1997). β-catenin levels are increased in certain cancers of the breast and skin (Chan et al., 1999; Lin et al., 2000). Recently, the LEF1 gene has been associated with colorectal cancer (Hovanes et al., 2001).
In this paper, we highlight transcriptional inhibition/repression as an inhibitory mechanism for the Wnt pathway, and thus implicate HBP1 as a candidate growth and tumor suppressor gene. Through the human genome sequencing project, the availability of the full gene sequence and of the precise chromosomal location for HBP1 is informative. The HBP1 gene maps to chromosome 7q31.1, which is a region that is frequently mutated in cancer (http://www.ncbi.nlm.nih.gov/UniGene/clust. cgi?ORG=Hs&CID=10882; Zenklusen et al., 1994, 1995a,b; Bieche et al., 1997; Driouch et al., 1998; Liang et al., 1998; Koike et al., 1999). Gene deletion in cancer provides a clue for the existence of a tumor suppressor gene. In certain cancers, such as acute myelogenous leukemia, positional cloning efforts have identified two regions, of which one should contain the HBP1 gene (Liang et al., 1998). Interestingly, the cancer profile correlates with the spectrum of HBP1 expression in tissues. Public gene chip information (http://genome-www.stanford.edu/serum/serumsearch.html; UniGene cluster ID Hs.10882) has independently confirmed our observations that HBP1 RNA expression is decreased in proliferating cells, again suggesting that HBP1 could function in barriers to proliferation. Thus, a prediction for future studies is that HBP1 may be mutated in a variety of cancers. The studies in this paper demonstrate that the Wnt pathway is one functional context for assessing any tumor-derived HBP1 mutations and their possible roles in oncogenesis.
Materials and methods
Plasmids
Mammalian expression vectors used for transient transfection include hemagglutinin (HA)-tagged pEFBOS rat HBP1 and various mutants also in pEFBOS. These mutants were constructed by PCR and include pmHMG (Tevosian et al., 1997), 660, 1242, N330 and N191. 660 and 1242 contain N-terminal deletions of 220 and 401 amino acids, respectively. N330 and N191 contain C-terminal deletions of 330 and 191 amino acids, respectively. The pEFBOSHygro construct was obtained from Dr Fong Ying Tsai. pGEX2THBP1(HMG), pGEX2THBP1(pmHMG) and pGEX2TTCF4(HMG) were constructed by PCR within our laboratory. The FLAG-tagged TCF4E plasmid was constructed by PCR in our laboratory and cloned into the pEFBOS expression vector via XbaI sites. The mutant FLAG-TCF4 constructs were made using PFU (Stratagene) PCR. The PUC–n-myc–CAT reporter construct was constructed in our laboratory (Tevosian et al., 1997) and contains –981 to +178 of the n-myc promoter. The cyclin D1–luciferase reporter plasmid containing the –963 region of the cyclin D1 promoter was described previously (Albanese et al., 1995). TOPFLASH and FOPFLASH were gifts from Drs K.Kinzler and B.Voglestein (Morin et al., 1997). FluCMVβ-catenin and HACMVLEF-1 were gifts from Drs S.Byers and Rudolf Grosshedl, respectively. CMV Wnt-1 was a gift from Dr Dan Sussman.
Cell culture and transfection
C33A cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% calf serum, antibiotics and glutamine. Caco-2A cells were provided by Dr Doug Jefferson (GRASP Digestive Disease Center at New England Medical Center). Caco-2A, HCT116 and HEK293T cells were cultured in DMEM supplemented with 10% fetal calf serum (FCS), antibiotics and glutamine. FCS, DMEM, antibiotics and glutamine were purchased from Gibco-BRL. FCS was purchased from HYCLONE and was not heat inactivated before use. For LiCl treatment, the cells were treated with the indicated amounts of LiCl after 24 h of transfection, and harvested 24 h later.
HEK293T, C33A, HCT116 and Caco-2A cells were plated in 100-mm dishes. HEK293T and C33A cells were transfected by the calcium phosphate method (2× precipitation buffer was purchased from 5′-3′). HCT116 and Caco-2A cells were plated in 100-mm dishes and transfected via Lipofectamine-Plus (Gibco-BRL) according to the manufacturer’s protocol.
Reporter assays
Cells transfected with reporter constructs containing the luciferase or the CAT genes were harvested 48 h after transfection. For a 100-mm plate, 300 µl of Promega reporter lysis buffer were used, which was supplemented with 200 µM phenylmethylsulfonyl fluoride (PMSF), l µg/ml pepstatin and l µg/ml leupeptin. The cell lysates were rocked at 4°C for 30 min and then centrifuged at maximum speed for 20 min. The amount of luciferase enzyme present in the extracts was determined using the Luciferase Assay system (Promega) according to the manufacturer’s instructions. CAT ELISA assays were performed using the CAT ELISA kit from Boehringer Mannheim as described previously (Shih et al., 1998). All transcription data were normalized for transfection efficiency with Rous sarcoma virus β-galactosidase (β-Gal). β-gal activity was quantitated using an o-nitrophenyl-β-d-galactopyranoside assay.
Western blots and immunoprecipitations
Cells were lysed in Promega reporter lysis buffer according to the manufacturer’s instructions. The buffer was supplemented with 200 µM PMSF, 1 µg/ml pepstatin and 1 µg/ml leupeptin. For detection of transfected HBP1 and various mutants of HBP1, the HA11 antibody (Babco) was used. The β-catenin antibody was obtained from Upstate Biotechnology. The BioM2 flag antibody was purchased from Sigma for detection of FlagTCF4 and for the supershift of FlagTCF4. For detection and immunoprecipitation of endogenous TCF4, the 6H5-3 antibody from Exalpha was used. For detection and immunoprecipitation of endogenous HBP1, a polyclonal antibody raised against the HMG-box region of HBP1 was used. For immunoprecipitations, cell extracts were pre-cleared with protein A–Sepharose beads. Antibody and beads were added to pre-cleared cell lysates and incubated at 4°C for 2–4 h with gentle agitation. Following washes with reporter lysis buffer, the beads were boiled in SDS sample buffer for 15 min and analyzed on an SDS–PAGE gel. Western blotting was performed using enhanced chemiluminescence (ECL; NEN).
GST protein purification
MC1061 cells were transformed with pGEX2THBP1(HMG), pGEX2THBP1(pmHMG) and pGEX2TTCF4(HMG). Bacterial cultures were grown to an OD of 0.5 and isopropyl-β-d-thiogalactopyranoside (IPTG) was added at 1 mM for 5 h at 25°C. The cells were lysed in NETN (20 mM Tris pH 8, 1 M NaCl, 1 mM EDTA pH 8, 5% NP-40, 200 µM PMSF, 1 µg/ml pepstatin, 1 µg/ml leupeptin). Purification was carried out according to the Pharmacia Biotech protocol using glutathione beads.
Electrophoretic mobility assays
Electrophoretic mobility assays (EMSA) were performed with GST purified proteins (prepared as described above) or HEK293T cellular extract. HEK293Ts were lysed in NETN (20 mM Tris pH 8, 0.1 M NaCl, 1 mM EDTA pH 8, 0.5% NP-40) lysis buffer that was supplemented with 200 µM PMSF, 1 µg/ml pepstatin and 1 µg/ml leupeptin. The [α-32P]APT- and [α-32P]TTP-labeled gel-shift probes used were TOPFLASH, which contains three adjacent LEF/TCF sites (CCTTTGATC), or the high affinity HBP1 site, which contains three copies of the TTCA sequence. Mutant competitors were also used, including FOPFLASH (3mer-CCTTGGCC) and the mutant high affinity HBP1 site (3mer-GGCA). Twelve micrograms of cellular extract or 200 ng of the indicated GST proteins were used with 0.5 ng of probe. The binding reaction consisted of 5× gel-shift buffer (100 mM HEPES pH 7.6, 5 mM MgCl2, 0.5 mM EGTA, 0.1% azide, 200 mM KCl, 50% glycerol), 1 µg of poly(dI–dC) and 1 µg of salmon sperm DNA along with protein and gel-shift probe in a total volume of 15 µl. For supershifts, 2 µg of the Flag antibody were included in the reaction mixture. The reaction was incubated for 20 min at room temperature and then run on a 4% polyacrylamide gel at 350 V for 2 h. Protein–DNA complexes were visualized by autoradiography.
Cell selection and RT–PCR
Transfected cells were isolated using the MACSelect Kk transfected cell selection kit (Miltenyi Biotec). Briefly, the pMACS Kk plasmid was transfected along with HBP1, and transfected cells were isolated by the recognition of pMACS Kk-expressing protein on the cell surface. RNA was isolated using Trizol reagent (Gibco-BRL). Two micrograms of RNA were subjected to semi-quantitative RT–PCR using the Access RT–PCR System (Promega). The sequences of the primers for human cyclin D1 and c-myc are available upon request. 18S primers were obtained from Ambion and were utilized as a loading control. With these PCR conditions, the cyclin D1 and c-myc genes were in the linear range at cycle 50. For the 18S RNA control, the linear range was at cycle 17. These conditions were used for analysis. The products were examined by agarose gel electrophoresis and quantitated by computerized gel documentation (Bio-Rad).
Colony formation assays
Caco-2A cells were transiently co-transfected with pEF-BOSHBP1 or the indicated mutants and the hygromycin B resistance gene. Cells positive for transfection survived growth in media containing 250 µg/ml hygromycin B (Calbiochem). Colonies of hygromycin-resistant cells were visualized ∼10 days after selection using crystal violet staining.
Acknowledgments
Acknowledgements
We thank the members of the Yee and Paulson laboratories for many spirited discussions and suggestions. We thank Drs Bert Vogelstein, Kenneth Kinzler (β-catenin, TOPLASH) and Rudolf Grosschedl (LEF1) and Dan Sussman (CMV Wnt 1) for generously providing essential reagents in this study. The support of the GRASP Digestive Disease Center at New England Medical Center (P30 DK34928) and use of their core facilities is gratefully acknowledged. This project was also funded in part with federal funds from the USDA, Agricultural Research Service under contract 53-3K06-01 (K.E.P.). This work was supported by grants to K.E.P (NIH) and to A.S.Y. (NIH GM44634; Army BC990538). A.S.Y is an Established Investigator of the American Heart Association.
References
- Albanese C.J., Johnson,G., Watanabe,N., Eklund,D., Vu,D. and Pestell,R.G. (1995) Transforming p21 ras mutants and ets2 activate the cyclin D1 promoter through distinguishable regions. J. Biol. Chem., 270, 23589–23597. [DOI] [PubMed] [Google Scholar]
- Barker N., Huls,G., Korinek,V. and Clevers,H. (1999) Restricted high level expression of Tcf-4 protein in intestinal and mammary gland epithelium. Am. J. Pathol., 154, 29–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker N., Morin,P.J. and Clevers,H. (2000) The yin–yang of TCF/β-catenin signaling. Adv. Cancer Res., 77, 1–24. [DOI] [PubMed] [Google Scholar]
- Bieche I., Khodja,A., Driouch,K. and Lidereau,R. (1997) Genetic alteration mapping on chromosome 7 in primary breast cancer. Clin. Cancer Res., 3, 1009–1016. [PubMed] [Google Scholar]
- Bienz M. (1998) TCF: transcriptional activator or repressor. Curr. Opin. Cell Biol., 10, 366–372. [DOI] [PubMed] [Google Scholar]
- Bienz M. and Clevers,H. (2000) Linking colorectal cancer to Wnt signaling. Cell, 103, 311–320. [DOI] [PubMed] [Google Scholar]
- Billin A.N., Thirlwell,H. and Ayer,D.E. (2000) β-catenin–histone deacetylase interactions regulate the transition of LEF1 from a transcriptional repressor to an activator. Mol. Cell. Biol., 20, 6882–6890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan E.F., Gat,U., McNiff,J.M. and Fuchs,E. (1999) A common human skin tumour is caused by activating mutations in β-catenin. Nature Genet., 21, 410–413. [DOI] [PubMed] [Google Scholar]
- Chen R.H., Ding,W.V. and McCormick,F. (2000) Wnt signaling to β-catenin involves two interactive components. Glycogen synthase kinase-3 β inhibition and activation of protein kinase C. J. Biol. Chem., 275, 17894–17899. [DOI] [PubMed] [Google Scholar]
- Driouch K., Briffod,M., Bieche,I., Champeme,M.H. and Lidereau,R. (1998) Location of several putative genes possibly involved in human breast cancer progression. Cancer Res., 58, 2081–2086. [PubMed] [Google Scholar]
- Eastman Q. and Grosschedl,R. (1999) Regulation of LEF-1/TCF transcription factors by Wnt and other signals. Curr. Opin. Cell Biol., 11, 233–240. [DOI] [PubMed] [Google Scholar]
- He T.C., Sparks,A.B., Rago,C., Hermeking,H., Zawel,L., daCosta,L., Morin,P.J., Vogelstein,B. and Kinzler,K. (1998) Identification of c-myc as a target of the APC pathway. Science, 281, 1509–1512. [DOI] [PubMed] [Google Scholar]
- Hosokawa Y. and Arnold,A. (1998) Mechanisms of cyclin D1 (CCN1, PRAD1) overexpression in human cancer: analysis of allele-specific expression. Genes Chromosomes Cancer, 22, 67–71. [DOI] [PubMed] [Google Scholar]
- Hovanes K., Li,T.W., Munguia,J.E., Truong,T., Milovanovic,T., Lawrence Marsh,J., Holcombe,R.F. and Waterman,M.L. (2001) β-catenin-sensitive isoforms of lymphoid enhancer factor-1 are selectively expressed in colon cancer. Nature Genet., 28, 53–57. [DOI] [PubMed] [Google Scholar]
- Hsu S.C., Galceran,J. and Grosschedl,R. (1998) Modulation of transcriptional regulation by LEF-1 in response to Wnt-1 signaling and association with β-catenin. Mol. Cell. Biol., 18, 4807–4818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishitani T. et al. (1999) The TAK1-NLK-MAPK-related pathway antagonizes signalling between β-catenin and transcription factor TCF. Nature, 399, 798–802. [DOI] [PubMed] [Google Scholar]
- Koike M., Tasaka,T., Spira,S., Tsuruoka,N. and Koeffler,H.P. (1999) Allelotyping of acute myelogenous leukemia: loss of heterozygosity at 7q31.1 (D7S486) and q33–34 (D7S498, D7S505). Leuk. Res., 23, 307–310. [DOI] [PubMed] [Google Scholar]
- Korinek V., Barker,N., Morin,P.J., van Wichen,D., deWeger,R., Kinzler,K., Vogelstein,B. and Clevers,H. (1997) Constitutive transcriptional activation by a β-catenin–Tcf complex in APC–/– colon carcinoma. Science, 275, 1784–1787. [DOI] [PubMed] [Google Scholar]
- Korinek V., Barker,N., Moerer,P., van Donselaar,E., Huls,G., Peters,P.J. and Clevers,H. (1998) Depletion of epithelial stem-cell compartments in the small intestine of mice lacking Tcf-4. Nature Genet., 19, 379–383. [DOI] [PubMed] [Google Scholar]
- Lavender P., Vandel,L., Bannister,A.J. and Kouzarides,T. (1997) The HMG-box transcription factor HBP1 is targeted by the pocket proteins and E1A. Oncogene, 14, 2721–2728. [DOI] [PubMed] [Google Scholar]
- Lemercier C., Duncliffe,K., Boibessot,I., Zhang,H., Verdel,A., Angelov,D. and Khochbin,S. (2000) Involvement of retinoblastoma protein and HBP1 in histone H1(0) gene expression. Mol. Cell. Biol., 20, 6627–6637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Hively,W.P. and Varmus,H.E. (2000) Use of MMTV-Wnt-1 transgenic mice for studying the genetic basis of breast cancer. Oncogene, 19, 1002–1009. [DOI] [PubMed] [Google Scholar]
- Liang H., Fairman,J., Claxton,D.F., Nowell,P.C., Green,E.D. and Nagarajan,L. (1998) Molecular anatomy of chromosome 7q deletions in myeloid neoplasms: evidence for multiple critical loci. Proc. Natl Acad. Sci. USA, 95, 3781–3785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin K.M., Zhao,W.G., Bhatnagar,J., Zhao,W.D., Lu,J.P., Simko,S., Schueneman,A. and Austin,G.E. (2001) Cloning and expression of human HBP1, a high mobility group protein that enhances myeloperoxidase (MPO) promoter activity. Leukemia, 15, 601–612. [DOI] [PubMed] [Google Scholar]
- Lin S.Y., Xia,W., Wang,J.C., Kwong,K.Y., Spohn,B., Wen,Y., Pestell,R.G. and Hung,M.C. (2000) β-catenin, a novel prognostic marker for breast cancer: its roles in cyclin D1 expression and cancer progression. Proc. Natl Acad. Sci. USA, 97, 4262–4266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love J.J., Li,X., Case,D.A., Giese,K., Grosschedl,R. and Wright,P.E. (1995) Structural basis for DNA bending by the architectural transcription factor Lef-1. Nature, 376, 791–795. [DOI] [PubMed] [Google Scholar]
- Meneghini M.D., Ishitani,T., Carter,J.C., Hisamoto,N., Ninomiya-Tsuji,J., Thorpe,C.J., Hamill,D.R., Matsumoto,K. and Bowerman,B. (1999) MAP kinase and Wnt pathways converge to downregulate an HMG-domain repressor in Caenorhabditis elegans. Nature, 399, 793–797. [DOI] [PubMed] [Google Scholar]
- Morin P.J. (1999) β-catenin signaling and cancer. BioEssays, 21, 1021–1030. [DOI] [PubMed] [Google Scholar]
- Morin P., Sparks,A.B., Korinek,V., Barker,N., Clever,H., Vogelstein,B. and Kinzler,K. (1997) Activation of β-catenin–TCF signalling in colon cancer by mutations in B-catenin or APC. Science, 275, 1787–1790. [DOI] [PubMed] [Google Scholar]
- Nusse R. (1999) Wnt targets: repression and activation. Trends Genet., 15, 1–3. [DOI] [PubMed] [Google Scholar]
- Polakis P. (2000) Wnt signaling and cancer. Genes Dev., 14, 1837–1851. [PubMed] [Google Scholar]
- Roose J., Huls,G., van Beest,M., Moerer,P., van der Horn,K., Goldschmeding,R., Logtenberg,T. and Clevers,H. (1999) Synergy between tumor suppressor APC and the β-catenin–Tcf4 target Tcf1. Science, 285, 1923–1926. [DOI] [PubMed] [Google Scholar]
- Satoh S. et al. (2000) AXIN1 mutations in hepatocellular carcinomas and growth suppression in cancer cells by virus-mediated transfer of AXIN1. Nature Genet., 24, 245–250. [DOI] [PubMed] [Google Scholar]
- Shih H., Tevosian,S.G. and Yee,A.S. (1998) Regulation of differentiation by HBP1, a target of the retinoblastoma protein. Mol. Cell. Biol., 18, 4732–4743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih H., Berasi,S., Sampson,E.M., Leiter,A.B., Paulson,K.E. and Yee,A.S. (2001) HMG box transcriptional repressor HPB maintains a proliferation barrier in differentiated liver tissue. Mol. Cell. Biol., inpress. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shtutman M., Zhurinsky,J., Simcha,I., Albanese,C., D’Amico,M., Pestell,R. and Ben-Ze'ev,A. (1999) The cyclin D1 gene is a target of the B-catenin/LEF pathway. Proc. Natl Acad. Sci. USA, 96, 5522–5527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tetsu O. and McCormick,F. (1999) B-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature, 398, 422–426. [DOI] [PubMed] [Google Scholar]
- Tevosian S.G., Shih,H., Mendelson,K.G., Sheppard,K.A., Paulson,K.E. and Yee,A.S. (1997) HBP-1: a new transcriptional repressor that is targeted by the retinoblastoma family. Genes Dev., 11, 383–396. [DOI] [PubMed] [Google Scholar]
- Tsukamoto A.S., Grosschedl,R., Guzman,R.C., Parslow,T. and Varmus,H.E. (1988) Expression of the int-1 gene in transgenic mice is associated with mammary gland hyperplasia and adenocarcinomas in male and female mice. Cell, 55, 619–625. [DOI] [PubMed] [Google Scholar]
- Verbeek S. et al. (1995) An HMG-box-containing T-cell factor required for thymocyte differentiation. Nature, 374, 70–74. [DOI] [PubMed] [Google Scholar]
- Waltzer L. and Bienz,M. (1998) Drosophila CBP represses the transcription factor TCF to antagonize Wingless signalling. Nature, 395, 521–525. [DOI] [PubMed] [Google Scholar]
- Watanabe G., Albanese,C., Lee,R.J., Reutens,A., Vairo,G., Henglein,B. and Pestell,R.G. (1998) Inhibition of cyclin D1 kinase activity is associated with E2F-mediated inhibition of cyclin D1 promoter activity through E2F and Sp1. Mol. Cell. Biol., 18, 3212–3222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstat-Saslow D., Merino,M.J., Manrow,J., Lawrence,R.F., Bluth,K.D., Wittenbel,J.F., Simpson,J.F., Page,D.L. and Steeg,P.S. (1995) Overexpression of cyclin D mRNA distinguishes invasive and in situ breast carcinoma from non-malignant lesions. Nature Med., 1, 1257–1259. [DOI] [PubMed] [Google Scholar]
- Zenklusen J.C., Bieche,I., Lidereau,R. and Conti,C.J. (1994) (C-A)n microsatellite repeat D7S522 is the most commonly deleted region in human primary breast cancer. Proc. Natl Acad. Sci. USA, 91, 12155–12158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zenklusen J.C., Thompson,J.C., Klein-Szanto,A.J. and Conti,C.J. (1995a) Frequent loss of heterozygosity in human primary squamous cell and colon carcinomas at 7q31.1: evidence for a broad range tumor suppressor gene. Cancer Res., 55, 1347–1355. [PubMed] [Google Scholar]
- Zenklusen J.C., Weitzel,J.N., Ball,H.G. and Conti,C.J. (1995b) Allelic loss at 7q31.1 in human primary ovarian carcinomas suggests the existence of a tumor suppressor gene. Oncogene, 11, 359–363. [PubMed] [Google Scholar]
- Zhuma T. et al. (1999) Human HMG box transcription factor HBP1: a role in hCD2 LCR function. EMBO J., 18, 6396–6406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorn A.M., Barish,G.D., Williams,B.O., Lavender,P., Klymkowsky, M.W. and Varmus,H.E. (1999) Regulation of Wnt signaling by Sox proteins: XSox17 α/β and XSox3 physically interact with β-catenin. Mol. Cell, 4, 487–498. [DOI] [PubMed] [Google Scholar]