

Abstract
Purpose:
In 2015, the American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology (AMP) published consensus standardized guidelines for sequence-level variant classification in Mendelian disorders. To increase accuracy and consistency, the Clinical Genome Resource Familial Hypercholesterolemia (FH) Variant Curation Expert Panel was tasked with optimizing the existing ACMG/AMP framework for disease-specific classification in FH. In this study, we provide consensus recommendations for the most common FH-associated gene, LDLR, where >2300 unique FH-associated variants have been identified.
Methods:
The multidisciplinary FH Variant Curation Expert Panel met in person and through frequent emails and conference calls to develop LDLR-specific modifications of ACMG/AMP guidelines. Through iteration, pilot testing, debate, and commentary, consensus among experts was reached.
Results:
The consensus LDLR variant modifications to existing ACMG/AMP guidelines include (1) alteration of population frequency thresholds, (2) delineation of loss-of-function variant types, (3) functional study criteria specifications, (4) cosegregation criteria specifications, and (5) specific use and thresholds for in silico prediction tools, among others.
Conclusion:
Establishment of these guidelines as the new standard in the clinical laboratory setting will result in a more evidence-based, harmonized method for LDLR variant classification worldwide, thereby improving the care of patients with FH.
Keywords: ACMG/AMP, ClinGen, Familial hypercholesterolemia, LDLR, Variant classification
Introduction
Familial hypercholesterolemia (FH) (OMIM: 143890) is a common (approximately 1:250 individuals affected)1 genetic dyslipidemia characterized by lifelong exposure to elevated low-density lipoprotein (LDL) cholesterol (LDL-C) levels. Early identification and appropriate treatment are imperative for prevention of premature atherosclerotic cardiovascular disease; however, <10% of individuals with FH worldwide have been diagnosed.2,3
FH is predominantly caused by heterozygous variants in 1 of 3 genes: LDLR (>90% of molecularly defined cases), APOB (approximately 5%-8% of cases), or PCSK9 (approximately 1% of cases).4 A single variant in APOE (p.Leu167del) can cause autosomal dominant hypercholesterolemia, and this variant may explain the cause of hypercholesterolemia in 1% to 2% of patients with FH phenotype in some countries.5 Identification of a pathogenic variant in an FH-associated gene can strongly affirm a diagnosis, motivates and simplifies family-based cascade screening, has the potential to direct therapeutic strategy and/or promote adherence, and may impact insurance coverage of certain medications. Genetic testing has increasingly become a central part of diagnosing FH in many countries. Genetic testing in FH is recommended by the United Kingdom National Institute for Health and Clinical Excellence,6 both the European Atherosclerosis Society and International Atherosclerosis Society,2 and an international expert panel convened by the FH Foundation and American College of Cardiology,7 among others. The US Centers for Disease Control and Prevention Office of Public Health Genomics also recommends the use of genetic information in the care of FH.8 Moreover, the American College of Medical Genetics and Genomics (ACMG) lists LDLR, APOB, and PCSK9 among the 59 medically actionable genes,9 which has, in part, led to frequent inclusion of these genes on commercially available clinical panels10 and direct-to-consumer tests.
With the increasingly widespread implementation of genetic testing for FH, it is becoming ever more essential to establish a consensus, standardized method for the clinical classification of identified variants. In a 2018 study of >6500 FH-associated variants submitted to the ClinVar database, there were at least 12 different variant classification criteria (including internal laboratory-specific ACMG/AMP criteria or other classification methods) being used among 30 submitters from 14 different countries.11 This heterogeneity leads to discordance in variant classification. For instance, 379 unique FH variants had conflicting classifications in ClinVar.
Application of the ACMG/Association for Molecular Pathology (ACMG/AMP) guidelines12 has been a major advancement toward achieving a more critical and consistent approach to variant classification for many disorders, including FH.11 However, because these guidelines are meant to be generalizable to all Mendelian disorders, they include inherent ambiguities that may lead to differences in their classification and application among users. Indeed, 114 FH-associated variants in ClinVar have conflicting classifications despite each laboratory having cited the same ACMG/AMP guidelines as their applied criteria. Genespecific modifications to these guidelines are essential to provide the clarity required for standardized variant classification.
In 2013, the Clinical Genome Resource (ClinGen) Consortium was established as a centralized collaborative resource that aims to define the clinical relevance of genes and variants.13 Among their major initiatives is the commission of disease/gene expert panels to provide consensus specifications of ACMG/AMP variant classification criteria. The ClinGen FH Variant Curation Expert Panel (VCEP) has been tasked with providing gene-specific recommendations for LDLR, APOB, and PCSK9. In this study, we describe consensus ACMG/AMP specifications for the LDLR gene, where >2300 unique variants have been identified in patients with a clinical association of FH.11
Materials and Methods
ClinGen FH VCEP
FH VCEP membership includes clinicians, laboratory diagnosticians, research scientists, genomic medicine specialists, and genetic counselors who share expertise knowledge in FH. To achieve international harmonization of variant classification practice, additional emphasis was placed on global representation, with members coming from 12 countries (United States, Canada, Brazil, United Kingdom, Portugal, Spain, France, Netherlands, Czech Republic, Japan, Australia, and Israel). The FH VCEP is part of the larger ClinGen Cardiovascular Domain Working Group.
Specification of ACMG/AMP criteria
A core group of 11 FH VCEP members reviewed all criteria in the original ACMG/AMP guidelines and began to propose initial LDLR-specific modifications on the basis of expert opinion and prior publications.14 Proposed modifications were discussed frequently through conference calls, emails, and several in-person meetings at international conferences until consensus was reached. Proposed guidelines in various iterations were consistently evaluated in analyses using well-known variants ranging from pathogenic to benign. ClinGen’s Sequence Variant Interpretation (SVI) committee provided feedback and suggestions, which were incorporated in multiple rounds of revisions. Finalized criteria were ultimately voted on and approved by all members of the FH VCEP. Note that given differences in mechanisms of disease, prevalence, and penetrance, it was decided that APOB- and PCSK9-specific guidelines will be completed separately.
Validation and pilot testing
After guideline approval from the SVI committee, a formalized pilot study of 54 LDLR variants was performed in the ClinGen Variant Curation Interface (VCI; https://curation.clinicalgenome.org/). The VCI is a publicly available, comprehensive resource that systematically facilitates individual- and group-level curation activities in accordance with the ACMG/AMP guidelines.
Pilot study curations in the VCI were performed independently by 2 trained VCEP biocurators, followed by a review from 2 VCEP leadership members. Publicly available data used for curation were supplemented with internal case-level data from VCEP member laboratories. When applicable, internal laboratory data used in the classifications were uploaded and saved into the VCI. After independent curation of the 54 LDLR pilot variants, biocurators extracted the data from the VCI and sent it to the reviewers. Any discordance in the application of criteria codes or in the final classification for each variant was recorded. Discordances were resolved in discussion among the biocurators and reviewers. Final classifications were approved by the reviewers and were submitted to ClinVar under the FH VCEP affiliation. The ontology used for FH caused by LDLR variation was hypercholesterolemia, familial (MONDO:0007750) with semidominant inheritance (HP:0032113), and the reference sequence used for LDLR was NM_000527.5.
Rules for combining pathogenic and benign criteria follow the original ACMG/AMP scoring algorithm (Richards et al12) (Supplemental Table 1).
Results and Discussion
Summary of specifications
FH VCEP specifications for LDLR variant classification in FH are summarized in Table 1. The type of LDLR-specific alterations we made to the original ACMG/AMP criteria codes can be categorized into the following: 14 disease-specific/strength-level changes, 13 disease-specific changes, 1 strength-level change, and 4 clarification changes (based on recent ClinGen recommendations). In addition, we found 6 criteria codes not applicable to LDLR. Key LDLR modifications include alteration of population data frequency thresholds, delineation of loss-of-function (LoF) variant types, functional study criteria specifications, cosegregation criteria specifications, and specific use and thresholds for in silico prediction tools.
Table 1.
Summary of ACMG/AMP guideline specifications for LDLR
| Gene | Disease | Transcript |
|---|---|---|
| LDLR | Hypercholesterolemia, familial (MONDO:0007750) | NM_000527.5 |
| Criteria | Criteria Description | LDLR Specification |
| Pathogenic Criteria | ||
| Very strong criteria | ||
| PVS1 | See PVS1 flow diagram (Figure 1). | Disease specific/strength |
| Strong criteria | ||
| PS1 | Missense variant at the same codon as a variant classified as pathogenic (by these guidelines) and predicts the same amino acid change. Caveat: there is no in silico predicted splicing impact for either variant. |
Clarification |
| PS2 | Variant is de novo in a patient with the disease and no family history. Follow SVI guidance for de novo occurrences: https://clinicalgenome.org/working-groups/sequence-variant-interpretation/ | Clarification |
| PS3 | Variant meets level 1 pathogenic functional study criteria. See Table 3. | Disease specific/strength |
| PS4 | Variant is found in ≥10 unrelated FH cases (FH diagnosis met by validated clinical criteria). Caveat: variant must also meet PM2. |
Disease specific/strength |
| PVS1_Strong | See PVS1 flow diagram (Figure 1). | Disease specific/strength |
| PM5_Strong | Missense variant at a codon with ≥2 missense variants classified as pathogenic (by these guidelines) and predicts a different amino acid change. | Strength |
| PP1_Strong | Variant segregates with phenotype in ≥6 informative meioses in ≥1 family. Must include ≥2 affected relatives (LDL-C > 75th centile) with the variant. | Disease specific/strength |
| Moderate criteria | ||
| PM1 | Missense variant located in exon 4, or a missense change in 1 of 60 highly conserved cysteine residues (listed in Supplemental Table 4). Caveat: variant must also meet PM2. |
Disease specific |
| PM2 | Variant has a PopMax MAF ≤ 0.0002 (0.02%) in gnomAD. Consider exceptions for known founder variants. | Disease specific |
| PM3 | This criterion can be used for a candidate LDLR variant observed in an individual with a homozygous FH phenotype when there is only 1 other pathogenic or likely pathogenic variant in LDLR (in trans), APOB, or PCSK9. Caveat: variant must also meet PM2. |
Disease specific |
| PM4 | In-frame deletion/insertions smaller than 1 whole-exon or in-frame whole-exon duplications not considered in any PVS1 criteria. Caveat: variant must also meet PM2. |
Disease specific |
| PM5 | Missense variant at the same codon as a variant classified as pathogenic (by these guidelines) and predicts a different amino acid change. | Clarification |
| PM6 | See PS2 above. | Clarification |
| PS3_Moderate | Variant meets level 2 pathogenic functional study criteria. See Table 3. | Disease specific/strength |
| PS4_Moderate | Variant is found in 6 to 9 unrelated FH cases (FH diagnosis made by validated clinical criteria). Caveat: variant must also meet PM2. |
Disease specific/strength |
| PP1_Moderate | Variant segregates with phenotype in 4 to 5 informative meioses in ≥1 family. Must include ≥2 affected relatives (LDL-C > 75th centile) with the variant. | Disease specific/strength |
| PVS1_Moderate | See PVS1 flow diagram (Figure 1). | Disease specific/strength |
| Supporting criteria | ||
| PP1 | Variant segregates with phenotype in 2 to 3 informative meioses in ≥1 family. Must include ≥1 affected relative (LDL-C > 75th centile) with the variant. | Disease specific/strength |
| PP3 | REVEL score ≥ 0.75 (missense variants) or predicted impact to splicing using MaxEntScan (see Figure 2 for suggested thresholds). | Disease specific |
| PP4 | Any LDLR variant identified in a patient with FH (diagnosis based on validated clinical criteria, eg, Dutch Lipid Clinic Network [≥6], Simon Broome [possible/definite], MEDPED), after alternative causes of high cholesterol are excluded. Caveat: variant must also meet PM2. |
Disease specific |
| PS3_Supporting | Variant meets level 3 pathogenic functional study criteria. See Table 3. | Disease specific/strength |
| PS4_Supporting | Variant is found in 2 to 5 unrelated FH cases (FH diagnosis made by validated clinical criteria). Caveat: Variant must also meet PM2. |
Disease specific/strength |
| Benign criteria | ||
| Standalone criteria | ||
| BA1 | Variant has a PopMax FAF ≥ 0.005 (0.5%) in gnomAD. | Disease specific |
| Strong criteria | ||
| BS1 | Variant has a PopMax FAF ≥ 0.002 (0.2%) in gnomAD. | Disease specific |
| BS2 | Variant is identified in ≥3 heterozygous or ≥1 homozygous well-phenotyped, untreated, normolipidemic adults (unrelated). | Disease specific |
| BS3 | Variant meets level 1 benign functional study criteria. See Table 3. |
Disease specific/strength |
| BS4 | Lack of segregation in ≥2 index case families (unrelated) when data are available for ≥2 informative meioses in each family. Caveat: must be ≥1 unaffected relative (LDL-C <50th centile) who is positive for the variant. |
Disease specific |
| Supporting criteria | ||
| BP2 | If a patient with FH with a heterozygous phenotype has a proven pathogenic variant in LDLR (in trans), APOB, or PCSK9, BP2 is applicable to any additional LDLR variants. | Disease specific |
| BP4 | REVEL score ≤ 0.5 (missense variants) and no predicted impact to splicing using MaxEntScan (see Figure 2 for suggested thresholds). | Disease specific |
| BP7 | Variant is synonymous. Caveat: variant must also meet BP4 (ie, no predicted impact on splicing). |
Disease specific |
| BS3_Supporting | Variant meets level 3 benign functional study criteria. See Table 3. |
Disease specific/strength |
PopMax refers to the gnomAD subpopulation with the highest allele frequency.
ACMG/AMP, American College of Medical Genetics and Genomics/Association for Molecular Pathology; FAF, filtering allele frequency; FH, familial hypercholesterolemia; gnomAD, Genome Aggregation Database; LDL-C, low-density lipoprotein cholesterol; LoF, loss-of-function; MAF, minor allele frequency; MEDPED, Make Early Diagnosis to Prevent Early Death; SVI, Sequence Variant Interpretation.
Population data (PM2, BA1, BS1)
The FH VCEP recommends using the Genome Aggregation Database (gnomAD) PopMax Filtering Allele Frequency (FAF) in evaluation of BA1 and BS1 codes,15 whereas evaluation of PM2 should be performed using the PopMax Minor Allele Frequency (MAF). Frequency thresholds specific for LDLR variants in FH are displayed in Table 2 and were calculated using the CardioDB metrics allele frequency web tool (https://www.cardiodb.org/allelefrequencyapp/) on the basis of prevalence, penetrance, and allelic/genetic heterogeneity. Allele frequency thresholds were equal to FAF ≥ 0.005 (0.5%) for BA1, FAF ≥ 0.002 (0.2%) and < 0.005 (0.5%) for BS1, and MAF ≤ 0.0002 (0.02%) for PM2. Note that if both exomes and genomes have an FAF/MAF value presented in gnomAD, consider the value corresponding to the higher number of alleles tested (ie, higher total allele number). When evaluating whole-exon deletions and duplications, which are a relatively common pathogenic variant type in LDLR,16 the gnomAD Structural Variant data set (gnomAD SV) should be queried, applying the same thresholds as defined earlier.
Table 2.
LDLR-specific population data frequency thresholds
| gnomAD Frequency | Prevalence | Penetrance, % | Allelic Het. | Genetic Het. | |
|---|---|---|---|---|---|
| BA1 | PopMax FAF ≥ 0.005 (0.5%)a |
1/250 | 50 | 1.0 | 1.0 |
| BS1 | PopMax FAF ≥ 0.002 (0.2%) and < 0.005 (0.5%) |
1/250 | 95 | 1.0 | 0.9 |
| PM2 | PopMax MAF ≤ 0.0002 (0.02%) |
1/250 | 95 | 0.1 | 0.9 |
PopMax refers to the gnomAD subpopulation with the highest allele frequency.
FAF, filtering allele frequency; gnomAD, Genome Aggregation Database; Het., heterogeneity; MAF, minor allele frequency.
BA1 metrics were equal to 0.4%; however, we conservatively increased the BA1 threshold to 0.5%.
It is important to keep in mind that both case and control gnomAD cohorts are expected to contain many individuals with FH, given that FH is relatively common in the general population (1 in 250 individuals, or an estimated approximately 34 million affected worldwide), and >90% of individuals are thought to be undiagnosed.2,3 Furthermore, there are multiple cardiac case cohorts included in gnomAD, such as those from the Framingham Heart Study, Jackson Heart Study, Multi-Ethnic Study of Atherosclerosis, and Myocardial Infarction Genetics Consortium studies.
LoF (PVS1) and in-frame indels (PM4)
LDLR satisfies ClinGen’s 3 requirements for applicability of PVS1:17 (1) it is a definitive gene for FH, (2) 3 or more LoF variants reach an ACMG/AMP classification of “Pathogenic” without PVS1 (Supplemental Table 2), and (3) >10% of variants associated with the phenotype are LoF (across more than 1 exon). In fact, frameshift variants alone represent approximately 20% of all unique FH-associated variants in ClinVar and are distributed throughout the gene.11
In accordance with the PVS1 flowchart outlined in Abou Tayoun et al,17 we have specified PVS1 (Figure 1) on the basis of well-established evidence in LDLR. Notably, any stop codon amino-terminal of amino acid 830 (NM_000527.5; located in exon 17) has been shown to remove a region known to be critical to protein function (ie, the NPXY sequence of the cytoplasmic tail, required for LDLR internalization).18 Note that alternative splicing of exons 1 to 18 from LDLR’s biologically relevant transcript is not known to occur. Therefore, PVS1 (at very strong) includes the following variants: (1) deletion of full gene, (2) deletion of single or multiple exons (exons 1-17) that lead to an out-of-frame consequence, (3) nonsense or frameshift variants causing a premature stop codon amino-terminal of amino acid 830 (NM_000527.5:p.Lys830), (4) variants in canonical ±1,2 GT/AG splice sites that predict a frameshift in exons 1 to 17, and (5) intragenic exon duplications proven to occur in tandem that predict a frameshift in exons 1 to 17. PVS1_Strong includes the following variants: (1) deletion of single or multiple exons (exons 1-17) that do not predict a frameshift, (2) variants in canonical ±1,2 GT/AG splice sites that predict in-frame deletions in exons 1 to 17, and (3) intragenic exon duplications presumed to occur in tandem that predict frameshifts in exons 1 to 17. PVS1_Moderate includes the following variants: (1) variants in the initiation codon, (2) whole-exon deletion of exon 18, and (3) nonsense/frameshift variants carboxy-terminal of amino acid 830 (NM_000527.5:p.Lys830). Furthermore, Supplemental Table 3 provides information on the phase of LDLR exons for determining in- or out-of-frame consequences for applicable variants.
Figure 1. LDLR-specific recommendations for application of PVS1.

NMD, nonsense-mediated decay.
In addition, in-frame deletions or insertions smaller than a whole-exon or in-frame whole-exon duplications not considered in the PVS1 criteria are applicable to PM4, if they also meet PM2.
Experimental studies (PS3, BS3)
Following the SVI recommendations for application of functional studies codes PS3/BS3,19 we have defined the mechanism of disease, evaluated the applicability of classes of assays used in the field, and evaluated individual instances of assays in determining appropriate strength levels.
In summary, LDLR is expressed at the cell surface, where it binds circulating plasma LDL particles. The LDLR–LDL (receptor–ligand) complex is internalized at clathrin-coated pits via receptor-mediated endocytosis. Once internalized (as part of the endosome), acidic conditions mediate release of the LDL ligand from its receptor, and the receptor is recycled back to the cell surface where it can repeat this process; a single LDLR protein can be recycled ≥100 times.20 Pathogenic variants may induce a LoF at any part of the LDLR cycle,21 disrupting LDLR activity and leading to FH caused by an inability to effectively clear LDL-C from the bloodstream. The most reliable functional assays are adapted from the Nobel Prize winning work of Drs Michael Brown and Joseph Goldstein22 and allow the characterization of the whole LDLR cycle, which can be evaluated sufficiently at 3 key steps: (1) LDLR expression/biosynthesis, (2) LDL particle binding, and (3) LDL internalization. Such assays compare LDLR activity in wild-type cells against cells harboring a specific variant and are currently performed by flow cytometry with fluorescently labeled LDL (commercially available or isolated from a wild-type individual) in (1) heterologous cells (with no endogenous LDLR) transfected with a mutant plasmid or (2) patient cells (fibroblasts, lymphocytes, or lymphoblasts). Older studies using radioactively labeled LDL (125I-LDL) (eg, Hobbs et al23) are also valid if they use these same cell types. A more in-depth analysis of the rationale and methodologies of LDLR functional assays is presented in Bourbon et al.24
We have determined 3 different strength levels of LDLR functional assays (Table 3) on the basis of the appropriateness of the methodology. Level 1 studies (set at PS3/BS3) are the most reliable; these include flow cytometry assays, which evaluate the whole LDLR cycle (ie, LDLR expression/biosynthesis, LDL binding, and LDLR–LDL internalization), performed in heterologous cells (with no endogenous LDLR) transfected with a mutant plasmid. Using heterologous cells with site-directed mutagenesis ensures that the assay is variant-specific. Supplemental Figure 1 demonstrates thresholds and controls used to validate flow cytometry assays in heterologous cells. Level 2 and level 3 studies (set at PS3_Moderate, PS3_Supporting/BS3_Supporting) represent additional techniques that allow for evaluation of only part of the LDLR cycle or that use less robust cells/materials. It is important to note that when using patient cells, DNA sequence analysis of LDLR should indicate that the assay is variant-specific (ie, no other candidate variants identified in LDLR, including whole-exon deletions/duplications). Although the historical Brown and Goldstein LDLR activity assays using patient cells were thoughtfully designed to be gene-specific (eg, ApoB-containing LDL particles used are always wild-type and LDLR is overexpressed in the cultured cells), patient-specific genetic factors may still modify outcomes. Finally, studies in compound heterozygous patient cells are not considered as valid functional assays because it is difficult to delineate the individual effect of each variant.
Table 3.
PS3/BS3 functional study criteria specifications for LDLR
| Criteria | LDLR Specification |
|---|---|
| Pathogenic | |
| PS3 (level 1) |
|
| PS3_Moderate (level 2) |
|
| PS3_Supporting (level 3) |
|
| Benign | |
| BS3 (level 1) |
|
| BS3_Supporting (level 3) |
|
Functional assays performed in compound heterozygous patient cells are not considered applicable in PS3/BS3 criteria because it is difficult to delineate the individual effect of each variant.
FH, familial hypercholesterolemia; LDL, low-density lipoprotein; MAVE, Multiplex Assays of Variant Effect; UTR, untranslated region; VCEP, Variant Curation Expert Panel.
Hotspot/well-established functional domains (PM1)
LDLR exon 4 is considered a mutational hotspot for missense variants in a well-established functional domain critical to protein function because it encodes LDLR type A repeats 3, 4, and 5, which compose the well-established ligand (LDL) binding domain25; exon 4 also has the highest number of FH-associated variants per nucleotide, with no variants proven benign by functional studies.14 In addition, LDLR contains 60 highly conserved cysteine residues (located throughout exons 2-8 and 14) critical to protein function; these 60 cysteine residues are involved in disulfide bond formation, essential for proper protein folding.26,27 Therefore, PM1 is applicable to any missense change in the amino acids of exon 4 (NM_000527.5:c.314-694 or p.105-232) that is also rare (ie, PM2 is met) or to any missense change in the 60 highly conserved cysteines, which we have listed in Supplemental Table 4.
Observed in healthy adults (BS2)
Pathogenic variants in LDLR are known to be highly penetrant, where affected status is typically identifiable as early as childhood28 through a simple and routine laboratory measure of plasma LDL-C level. Therefore, we have determined that BS2 is applicable for LDLR variants identified in ≥3 heterozygous or ≥1 true homozygous well-phenotyped, normolipidemic, untreated, unrelated adults. At a minimum, well-phenotyped refers to LDL-C measurements taken over multiple time points (≥2) with consistent results. Individuals considered in BS2 should not be taking any lipid-lowering therapy near the time of measurement and should have an LDL-C level below the ethnic- and country-specific 50th centile (adjusted for age and sex) (eg, Starr et al29). Because lipid-lowering therapies (eg, statins) are among the most widely prescribed medications in the general population and neither medication status nor LDL-C level are typically available in commonly used, publicly available resources such as gnomAD or Exome Aggregation Consortium, such resources must not be used for evaluation of BS2 in FH. Rather, we recommend evaluation of BS2 in well-phenotyped normolipidemic cohorts only, which are likely to be more available in internal laboratory settings. It is important to follow these caveats closely, given that BS2 is a strong-level criterion.
Specificity of phenotype (PP4) and case-control data (PS4)
There are a variety of validated clinical diagnostic criteria used for FH, which include the Dutch Lipid Clinic Network criteria,30 Simon Broome criteria,31 the United States Make Early Diagnosis to Prevent Early Death criteria,32 and other country-specific criteria. We have determined that PP4 is applicable to any rare (ie, PM2 is met) LDLR variant identified in a patient with a diagnosis of FH based on any validated clinical criteria; examples include a Dutch Lipid Clinic Network score ≥ 6, Simon Broome score of possible or definite FH, or a Make Early Diagnosis to Prevent Early Death diagnostic score of FH. Because all validated clinical criteria require extreme LDL-C levels to be present in the patient together with a family history positive for high LDL-C and/or premature coronary heart disease and because the LDLR gene is specific for FH (>90% of cases), we believe strongly in the appropriateness of PP4 for LDLR. However, in any case, PP4 is applicable only after alternative causes of high LDL-C are excluded. Alternative causes for high LDL-C are reviewed in Sturm et al7 and include polygenic dyslipidemia; elevated lipoprotein(a); nephrotic syndrome; obstructive liver disease; hypothyroidism; diabetes; FH caused by PCSK9, APOB, or APOE variants; or FH phenocopies caused by biallelic variants in LDLRAP1, LIPA, or ABCG5/8.
For the case-control criterion PS4, different strength levels may be applied depending on the number of unrelated FH cases with the rare variant. PS4 is applicable if the variant is found in ≥10 unrelated FH cases (FH diagnosis met using validated clinical criteria), PS4_Moderate is applicable if found in 6 to 9 unrelated FH cases, and PS4_Supporting is applicable if found in 2 to 5 unrelated FH cases. Note that in applying PS4-level criteria, the variant must also meet PM2.
Segregation data (PP1, BS4)
We have determined 3 strength levels for application of PP1 depending on the number of families/individuals studied. PP1_Strong is applicable when there is cosegregation of the variant with affected status in ≥6 informative meioses, PP1_Moderate is applicable when there are 4 to 5 informative meioses, and PP1 (at Supporting) is applicable when there are 2 to 3 informative meioses. Index cases should not be counted as positive cases for cosegregation results. When the same variant is identified in >1 family, data can be added to reach stronger evidence levels. Supplemental Figure 2 shows a typical example of cosegregation in a pedigree, with an explanation on informative meiosis for an FH-associated variant. Note that when an index case presents with a heterozygous FH phenotype and the hypercholesterolemia is associated with one branch of the family, individuals from the other branch should not be considered for cosegregation analysis.
BS4 is applicable when there is lack of cosegregation in ≥2 index case families (unrelated) and there are data on ≥2 informative meioses in each family. When applying BS4, there should be at least 1 instance where an unaffected family member carries the variant (ie, genotype-positive, phenotype-negative).
For cosegregation analysis, we consider an affected individual as one with an untreated total cholesterol (TC) or LDL-C level above the 75th centile adjusted for age and sex. Each country/region should preferably use their TC and LDL-C centile charts. Given the widespread use of lipid-lowering therapies in the general population, untreated TC or LDL-C measurements may not be obtainable for some individuals under consideration for PP1/BS4. For those with only known treated TC or LDL-C levels, several imputation factors may be applied for an estimation of untreated measurements, namely by specific medication and dose (preferred)33 or by the more general 0.8 and 0.7 correction factors corresponding to an estimated 20% TC and 30% LDL-C reduction on treatment, respectively.34 Unaffected family members should have untreated TC and LDL-C below the 50th centile adjusted for age and sex.
It is important to consider both affected and unaffected individuals when evaluating cosegregation. Alternative causes of high TC or LDL-C values, such as those described earlier, should be considered carefully given their ability to explain instances of hypercholesterolemia in genotypenegative family members. It is important to note that cholesterol concentrations are influenced by the coinheritance of common variants of small effect. Trinder et al35 have recently shown that individuals who have an LDL-C polygenic risk score in the lowest decile have LDL-C concentrations considerably lower than those in the highest decile (3.61 mmol/L vs 4.37 mmol/L, respectively). Finally, be aware that although rare, patients with FH with a pathogenic LDLR variant could also be positive for a rare monogenic cholesterol-lowering variant (possible in, eg, APOB or PCSK9 genes), as has been described in Emi et al36 and Motazacker et al.37 If identifiable, these individuals should not be considered for cosegregation analysis.
In silico prediction (PP3, BP4)
For in silico classification of missense variants in LDLR, we suggest the use of REVEL, an ensemble method for pathogenicity prediction that combines predictions from 13 individual commonly used computational tools.38 Use of a single meta-predictor such as REVEL will eliminate discrepancies in which programs are used by curators and what specificities to account for when manually performing in silico analysis. REVEL was also selected because of its accessibility; REVEL scores are precomputed and automatically displayed in the ClinGen VCI (under the Variant Type tab) or are available for download (LDLR: https://rothsj06.u.hpc.mssm.edu/revel/revel_segments/revel_chrom_19_009082971-013246689.csv.zip). To determine LDLR-specific score thresholds for PP3/BP4, we evaluated REVEL scores for LDLR missense variants with (1) pathogenic/likely pathogenic or benign/likely benign classifications in ClinVar; (2) damaging or neutral results according to LDLR-specific PS3/BS3 functional study evidence; and (3) concordant in silico results for polymorphism phenotyping, Sorting Intolerant From Tolerant, Protein Variation Effect Analyzer, and MutationTaster (Supplemental Figure 3). Considering the PP3 threshold of ≥0.75 (as defined in Ioannidis et al38), the vast majority of variants with an association of pathogenic using any comparison (as described earlier) have REVEL scores above this threshold. Considering the BP4 threshold of ≤0.5, approximately half of variants with an association of benign from ClinVar classifications or other in silico predictors have REVEL scores below this threshold. Therefore, we recommend a REVEL score of ≥0.75 as supportive evidence of pathogenicity (PP3) and a REVEL score of ≤0.5 as supportive evidence of benign (BP4).
For in silico prediction of splicing effects, we recommend evaluation only if no functional data are available; furthermore, variants already considered in PVS1 (or modified strength) should not be further considered in PP3/BP4. We suggest the use of MaxEntScan,39 which is highly reputable and publicly available. We have defined distinct thresholds for MaxEntScan depending on the variant location, as described in Figure 2.
Figure 2. Familial Hypercholesterolemia Variant Curation Expert Panel suggestions for evaluating splicing effects using MaxEntScan dependent on Var location A, B, or C.
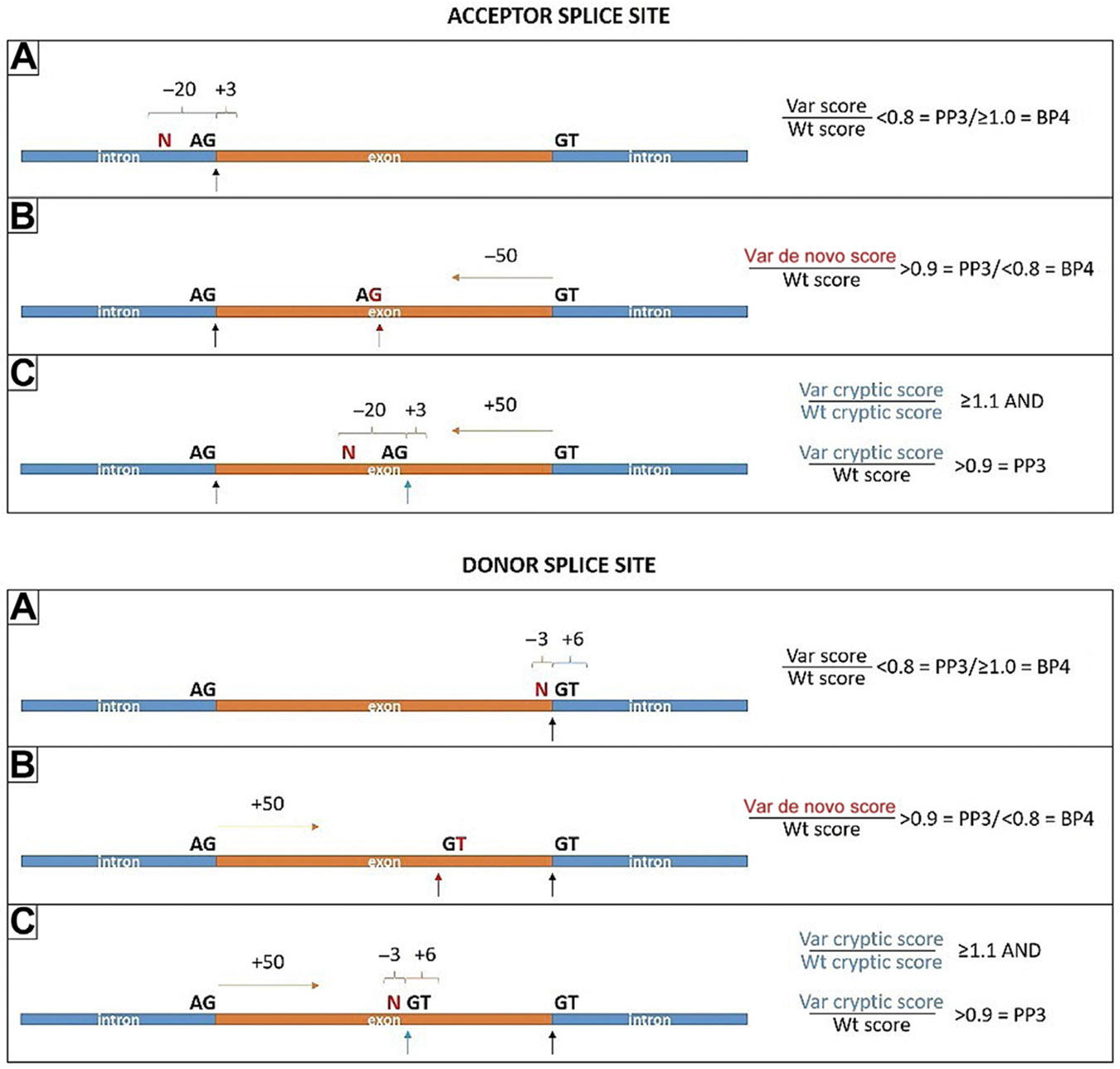
A. Var is located at −20 to +3 bases related to the authentic acceptor splice site or at −3 to +6 bases related to the authentic donor splice site. A result of authentic splice site strength Var/Wt score < 0.8 is supportive evidence of pathogenicity (PP3), whereas a score ≥ 1.0 is supportive evidence of benign (BP4). B. Var creates de novo acceptor splice site, which is at least 50 bases upstream of the authentic donor splice site, or creates de novo donor splice site, which is at least 50 bases downstream of the authentic acceptor splice site. A result of de novo splice site strength Var/authentic Wt score > 0.9 is applicable to PP3, whereas a score < 0.8 is applicable to BP4. C. Var is located at −20 to +3 bases relative to an intraexonic AG dinucleotide, which is at least 50 bases upstream of the authentic donor splice site, or at −3 to +6 bases relative to an intraexonic GT dinucleotide, which is at least 50 bases downstream of the authentic acceptor splice site. Results of both Var cryptic/Wt cryptic score > 1.1 and cryptic acceptor/authentic acceptor score or cryptic donor/authentic donor score > 0.9 is applicable to PP3. Note: BP4 is applicable to exonic Vars outside of the 50 base limits detailed earlier, given the unlikelihood of such Vars to impact splicing in LDLR. Var, variant; Wt, wild-type.
Finally, if both missense and splicing predictions are applicable, only 1 prediction of a damaging effect is sufficient in applying PP3; however, both need to predict a neutral effect in applying BP4.
Other variants in the same codon (PS1, PM5)
When there are other described variants in the same codon as a missense variant being classified, PS1 is applicable if at least 1 missense variant has a classification of pathogenic (classified using these LDLR-specific guidelines) and the variant predicts the same amino acid change. PM5 (at moderate) is applicable if there is 1 pathogenic missense variant that predicts a different amino acid in the same codon. Finally, PM5_Strong is applicable in the same context if there are ≥2 pathogenic missense variants that predict different amino acids in the same codon. Note that for these codes to be applied, the curated variant(s) should not already be considered in PM1 (hotspot/well-established functional domain) and have an in silico predicted splicing impact of benign. Combining PS1/PM5 with PM1 can be considered double-counting, ie, evaluating a variant under a similar premise twice, whereas investigating potential splicing impact provides greater confidence that pathogenicity is related to a predicted altered amino acid rather than creation of a de novo splice site or activation of cryptic splice site.
Allele data (cis/trans) (PM3, BP2)
LDLR variants show a semidominant pattern of inheritance on plasma cholesterol concentration such that the phenotypes in homozygous or compound heterozygous patients are significantly more severe than in heterozygotes. Because of this, both PM3 and BP2 criteria (observed in trans with a pathogenic variant) can be used when case-level data are available for individuals with >1 FH-associated variant. PM3 is applicable when a candidate LDLR variant is identified in a patient with a clear homozygous or compound heterozygous FH phenotype (defined here as untreated LDL-C ≥13 mmol/L or ≥500 mg/dL) who has an additional known pathogenic or likely pathogenic variant in LDLR (in trans), APOB, or PCSK9. The candidate variant must also meet PM2. PM3 must not be used if cis/trans status in LDLR has not been established. BP2 is applicable to any additional LDLR variants identified in a patient with a clear heterozygous FH phenotype (defined here as untreated, elevated LDL-C that is <8 mmol/L or <310 mg/dL in adults) who already has a known pathogenic variant in LDLR (in trans), APOB, or PCSK9.
For both PM3 and BP2, known pathogenic variants in LDLR must have been classified according to these guidelines, whereas known pathogenic variants in APOB or PCSK9 should have been formally assessed by general ACMG/AMP guidelines until these gene-specific guidelines have been established, at which time both variant classifications should be re-evaluated. Although, to the best of our knowledge, no formal studies evaluating the prevalence of double heterozygotes in these FH genes have been completed, they are uncommon in our experience.
De novo occurrence (PS2, PM6)
The FH VCEP recommends following the SVI recommendations for PM6 and PS2, which can be found at https://clinicalgenome.org/working-groups/sequence-variant-interpretation. These recommendations evaluate PM6/PS2 on the basis of a points system centered around 3 parameters: confirmed vs assumed status, phenotypic consistency, and number of de novo observations. Although data to address de novo occurrence in LDLR directly are lacking, we have no evidence to suggest that this a common feature in FH, given that to date, only 1 member of the FH VCEP has anecdotally observed a de novo occurrence in their clinical practice, and to the best of our knowledge, such cases have only been reported once in the literature.40 However, they are of course possible and should be considered.
Criteria not applicable (BP1, PP2, BP3, BP6, PP5, BP5)
BP1 (missense variant in a gene for which primarily truncating variants are known to cause disease) is not applicable because most of the FH-associated LDLR variants are missense variants. After SVI counsel regarding PP2 (missense variant in a gene that has a low rate of benign missense variation and where missense variants are a common mechanism of disease), PP2 is not applicable on the basis of a low z-score of 0.12 for LDLR in the gnomAD missense constraint table. BP3 (in-frame deletions/insertions in a repetitive region without a known function) is not applicable, given that there are no regions in LDLR without a known function. BP6 and PP5 (variant previously classified by a reputable source) have been advised by ClinGen not to be used and therefore are not applicable. Finally, the FH VCEP has decided to remove BP5 (alternative mechanism for disease), given that this premise is already evaluated in our specifications for BP2.
Pilot study
The pilot study of the final specifications was done on 54 LDLR variants in the ClinGen VCI. Variants, listed in Supplemental Table 5, were chosen to reflect LDLR variant variability and included 1 multiexon deletion, 40 missense, 7 intronic/splicing, 4 nonsense, and 2 synonymous variants. A total of 9 institutions, which included 6 clinical and 3 research laboratories, provided internal case-level data (via standardized template), supplementing classifications for 42 of 54 variants. For the remaining 12 variants, and whenever necessary, data from published literature were used to count number of cases and to evaluate cosegregation and functional evidence. Supplemental Figure 4 shows the impact of internally shared or literature-only case-level data in pilot variant classifications. Without internal case-level data sharing, 30 of 42 variants were classified as variant(s) of unknown significance (VUS); however, when considering shared case-level data, half of these variants were able to be upgraded/downgraded. In total, 14 variants were upgraded to likely pathogenic/pathogenic, whereas 1 was downgraded to benign.
Preliminary results had complete agreement (in both classification and individual criteria used) for 16 variants, agreement in classifications but not in each criterion used for 27 variants, and discrepancies in both classification and criteria used for the remaining 11 variants. Differences in classification were 8 counts of likely pathogenic vs VUS, 2 of likely pathogenic vs pathogenic, and 1 of benign vs VUS. A careful review of the discrepancies determined that most resulted from extracting different gnomAD MAF/FAF data from the VCI or from differences in applying PS4_Moderate vs PS4_Supporting owing to slight differences in case counts. Minor refinements were added to the guidelines to address these discrepancies; we clarified the use of MAF/FAF (exomes vs genomes feature) in gnomAD and designed a more efficient template for tracking case-level data. Consequently, finalized pilot results had complete agreement in both classification and criteria used for all 54 variants and represented 6 benign, 2 likely benign, 18 VUS, 15 likely pathogenic, and 13 pathogenic variants. The number of times each criterion was used is represented in Supplemental Figure 5. Reviewers approved the final classifications, which are now published in the ClinGen Evidence Repository and submitted to ClinVar under the FH VCEP affiliation.
Limitations
There are multiple criteria specifications that require diagnostic information and case-level data, which, if not readily available, may limit the classification of variants. For instance, PS4 and PP4 criteria require that cases considered are clinically diagnosed with FH on the basis of validated clinical criteria. PP1, PS2/PM6, PM3, BS2, BS4, and BP2 criteria require that further case-level data are available, including LDL-C measurements, genetic results, family history, and medication status. However, although this information may be difficult to ascertain in some settings, it is necessary to apply these criteria correctly. Whenever possible, we encourage curators to actively seek this information if it is not initially available. Because the FH VCEP works toward classifying all approximately 2300 LDLR variants currently in the ClinVar database using the guidelines presented in this study, we are hopeful we can overcome some of these limitations through internal data sharing efforts. We encourage any laboratory with internal data on LDLR variants in patients with FH to upload these data in ClinVar because these data can have a major impact on the proper classification of variants.
Conclusions and Future Directions
Here, the FH VCEP presents consensus recommendations for LDLR variant classification. Application of these guidelines will provide evidence-based, standardized classification of LDLR variants for use in clinical diagnostics and research. Future directions include sustained variant curation, with the aim of classifying all approximately 2300 unique LDLR variants at the 3-star status in ClinVar. A 3-star status indicates variants reviewed by an expert panel according to a set of ClinGen-approved VCEP-adapted ACMG/AMP criteria. It is noteworthy that in 2018, the ClinGen Variant Curation Expert Panel protocol was recognized by the US Food and Drug Administration, whereby variant classifications with 3-star status in ClinVar are now associated with a US Food and Drug Administration–recognized tag and can be used to support clinical validity of genetic tests. In the future, we expect that this may have implications for obtaining insurance coverage for certain medications, for enrollment in certain clinical trials or research studies, or in the feedback of incidental findings from exome or genome sequencing. Given these possible implications, we will prioritize the classification of LDLR variants with the greatest potential impact, such as those that are LoF variant types, those with many and/or conflicting submissions currently in ClinVar, or those known to be on clinically available arrays/panels. We are hopeful that the FH VCEP classification of all approximately 2300 LDLR variants is completed within the next 4 years; we also plan to review all classifications on a 2-year basis to ensure that recently emerging data are considered.
Finally, please be aware that the LDLR-specific guidelines presented in this study are subject to change in response to emerging data and newly available resources, which will continually influence the evolving nature of variant classification methodology, both specific to LDLR and more broadly throughout the clinical genetics community. For this reason, please refer to the FH VCEP page in the ClinGen website (https://clinicalgenome.org/affiliation/50004) for the most currently accepted version.
Supplementary Material
Acknowledgments
Clinical Genome Resource (ClinGen) is primarily funded by the National Human Genome Research Institute through the following 3 grants: U41HG006834, U41HG009649, and U41HG009650. ClinGen also receives support for content curation from the Eunice Kennedy Shriver National Institute of Child Health and Human Development through the following 3 grants: U24HD093483, U24HD093486, and U24HD093487. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. J.R.C. acknowledges her PhD fellowship funded by the Science and Technology Foundation (SFRH/BD/108503/2015). L.T. and T.F. are supported by the Ministry of Health of the Czech Republic (grant NU20-02-00261). S.E.H. is an Emeritus British Heart Foundation Professor and is funded by PG08/008 and by the National Institute for Health Research University College London Hospitals Biomedical Research Centre. M.T. is supported by a Vanier Canada Graduate Scholarship. L.R.B. is a Michael Smith Foundation for Health Research scholar and a Canada Research Chair in Precision Cardiovascular Disease Prevention. R.A.H. is supported by the Jacob J. Wolfe Distinguished Medical Research Chair, the Edith Schulich Vinet Canada Research Chair in Human Genetics, the Martha G. Blackburn Chair in Cardiovascular Research, and operating grants from the Canadian Institutes of Health Research (Foundation Grant) and the Heart and Stroke Foundation of Ontario (G-18-0022147). J.W.K. is supported by the National Institutes of Health through grants P30DK116074 (to the Stanford Diabetes Research Center), R01 DK116750, R01 DK120565, and R01 DK106236 and by the American Diabetes Association (grant #1-19-JDF-108). Finally, the Familial Hypercholesterolemia Variant Curation Expert Panel would like to thank Drs Steven Harrison, Leslie Biesecker, Heidi Rehm, and the additional members from the ClinGen Sequence Variant Interpretation committee for their feedback and guidance in establishing these guidelines.
Footnotes
Ethics Declaration
Our study contains only the curation of already de-identified publicly available data, namely the curation of de-identified genetic variants already present in the ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/). As per ClinVar policy, variant submitters are expected to have obtained appropriate consent for data submission and sharing. Geisinger Health System provided institutional review board approval for Clinical Genome Resource submission of genetic variants to ClinVar. We have obtained ethics approval for our study by the Clinical Genome Resource Data Access, Protection, and Confidentiality Committee and adhere to their ethics policies.
Conflict of Interest
R.A.H. reports consulting fees from Acasti Pharma Inc, Aegerion Pharmaceuticals, Akcea/Ionis Pharmaceuticals, Amgen Inc, HLS Therapeutics Inc, Novartis AG, Pfizer Inc, Regeneron Pharmaceuticals Inc, and Sanofi. All other authors declare no conflicts of interest.
Additional Information
The online version of this article (https://doi.org/10.1016/j.gim.2021.09.012) contains supplementary material, which is available to authorized users.
Data Availability
All data generated or analyzed during this study are included in this manuscript (and its Supplemental Tables and Figures).
References
- 1.Akioyamen LE, Genest J, Shan SD, et al. Estimating the prevalence of heterozygous familial hypercholesterolaemia: a systematic review and meta-analysis. BMJ Open. 2017;7(9):e016461. 10.1136/bmjopen-2017-016461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nordestgaard BG, Chapman MJ, Humphries SE, et al. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: consensus statement of the European Atherosclerosis Society. Eur Heart J. 2013;34(45):3478–3490a. Published correction appears in Eur Heart J. 2020;41(47):4517. 10.1093/eurheartj/eht273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nordestgaard BG, Benn M. Genetic testing for familial hypercholesterolaemia is essential in individuals with high LDL cholesterol: who does it in the world? Eur Heart J. 2017;38(20):1580–1583. 10.1093/eurheartj/ehx136. [DOI] [PubMed] [Google Scholar]
- 4.Austin MA, Hutter CM, Zimmern RL, Humphries SE. Genetic causes of monogenic heterozygous familial hypercholesterolemia: a HuGE prevalence review. Am J Epidemiol. 2004;160(5):407–420. 10.1093/aje/kwh236. [DOI] [PubMed] [Google Scholar]
- 5.Cenarro A, Etxebarria A, De Castro-Orós I, et al. The p.Leu167del mutation in APOE gene causes autosomal dominant hypercholesterolemia by down-regulation of LDL receptor expression in hepatocytes. J Clin Endocrinol Metab. 2016;101(5):2113–2121. 10.1210/jc.2015-3874. [DOI] [PubMed] [Google Scholar]
- 6.NICE National Institute for Health and Care Excellence. Familial hypercholesterolaemia: identification and management. NICE National Institute for Health and Care Excellence. Published August 27, 2008. Updated October 04, 2019. https://www.nice.org.uk/guidance/CG71. Accessed November 2, 2020. [Google Scholar]
- 7.Sturm AC, Knowles JW, Gidding SS, et al. Clinical genetic testing for familial hypercholesterolemia: JACC scientific expert panel. J Am Coll Cardiol. 2018;72(6):662–680. 10.1016/j.jacc.2018.05.044. [DOI] [PubMed] [Google Scholar]
- 8.Khoury MJ, Bowen MS, Clyne M, et al. From public health genomics to precision public health: a 20-year journey. Genet Med. 2018;20(6):574–582. 10.1038/gim.2017.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kalia SS, Adelman K, Bale SJ, et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): a policy statement of the American College of Medical Genetics and Genomics. Genet Med. 2017;19(2):249–255. Published correction appears in Genet Med. 2017;19(4):484. 10.1038/gim.2016.190. [DOI] [PubMed] [Google Scholar]
- 10.Iacocca MA, Hegele RA. Recent advances in genetic testing for familial hypercholesterolemia. Expert Rev Mol Diagn. 2017;17(7):641–651. 10.1080/14737159.2017.1332997. [DOI] [PubMed] [Google Scholar]
- 11.Iacocca MA, Chora JR, Carrié A, et al. ClinVar database of global familial hypercholesterolemia-associated DNA variants. Hum Mutat. 2018;39(11):1631–1640. 10.1002/humu.23634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–424. 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rehm HL, Berg JS, Brooks LD, et al. ClinGen—the clinical genome resource. N Engl J Med. 2015;372(23):2235–2242. 10.1056/NEJMsr1406261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chora JR, Medeiros AM, Alves AC, Bourbon M. Analysis of publicly available LDLR, APOB, and PCSK9 variants associated with familial hypercholesterolemia: application of ACMG guidelines and implications for familial hypercholesterolemia diagnosis. Genet Med. 2018;20(6):591–598. 10.1038/gim.2017.151. [DOI] [PubMed] [Google Scholar]
- 15.Whiffin N, Minikel E, Walsh R, et al. Using high-resolution variant frequencies to empower clinical genome interpretation. Genet Med. 2017;19(10):1151–1158. 10.1038/gim.2017.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Iacocca MA, Hegele RA. Role of DNA copy number variation in dyslipidemias. Curr Opin Lipidol. 2018;29(2):125–132. 10.1097/MOL.0000000000000483. [DOI] [PubMed] [Google Scholar]
- 17.Abou Tayoun AN, Pesaran T, DiStefano MT, et al. Recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criteria. Hum Mutat. 2018;39(11):1517–1524. 10.1002/humu.23626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen WJ, Goldstein JL, Brown MS. NPXY, a sequence often found in cytoplasmic tails, is required for coated pit-mediated internalization of the low density lipoprotein receptor. J Biol Chem. 1990;265(6):3116–3123. [PubMed] [Google Scholar]
- 19.Brnich SE, Abou Tayoun AN, Couch FJ, et al. Recommendations for application of the functional evidence PS3/BS3 criterion using the ACMG/AMP sequence variant interpretation framework. Genome Med. 2019;12(1):3. 10.1186/s13073-019-0690-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wijers M, Kuivenhoven JA, van de Sluis B. The life cycle of the low-density lipoprotein receptor: insights from cellular and in-vivo studies. Curr Opin Lipidol. 2015;26(2):82–87. 10.1097/MOL.0000000000000157. [DOI] [PubMed] [Google Scholar]
- 21.Soutar AK, Naoumova RP. Mechanisms of disease: genetic causes of familial hypercholesterolemia. Nat Clin Pract Cardiovasc Med. 2007;4(4):214–225. 10.1038/ncpcardio0836. [DOI] [PubMed] [Google Scholar]
- 22.Brown MS, Goldstein JL. A receptor-mediated pathway for cholesterol homeostasis (Nobel lecture). Angew Chem Int Ed Engl. 1986;25(7):583–602. 10.1002/anie.198605833. [DOI] [Google Scholar]
- 23.Hobbs HH, Brown MS, Goldstein JL. Molecular genetics of the LDL receptor gene in familial hypercholesterolemia. Hum Mutat. 1992;1(6):445–466. 10.1002/humu.1380010602. [DOI] [PubMed] [Google Scholar]
- 24.Bourbon M, Alves AC, Sijbrands EJ. Low-density lipoprotein receptor mutational analysis in diagnosis of familial hypercholesterolemia. Curr Opin Lipidol. 2017;28(2):120–129. 10.1097/MOL.0000000000000404. [DOI] [PubMed] [Google Scholar]
- 25.Russell DW, Brown MS, Goldstein JL. Different combinations of cysteine-rich repeats mediate binding of low density lipoprotein receptor to two different proteins. J Biol Chem. 1989;264(36):21682–21688. [PubMed] [Google Scholar]
- 26.Jeon H, Blacklow SC. Structure and physiologic function of the low-density lipoprotein receptor. Annu Rev Biochem. 2005;74:535–562. 10.1146/annurev.biochem.74.082803.133354. [DOI] [PubMed] [Google Scholar]
- 27.Russell DW, Lehrman MA, Südhof TC, et al. The LDL receptor in familial hypercholesterolemia: use of human mutations to dissect a membrane protein. Cold Spring Harb Symp Quant Biol. 1986;51(Pt 2):811–819. 10.1101/sqb.1986.051.01.094. [DOI] [PubMed] [Google Scholar]
- 28.van der Graaf A, Avis HJ, Kusters DM, et al. Molecular basis of autosomal dominant hypercholesterolemia: assessment in a large cohort of hypercholesterolemic children. Circulation. 2011;123(11):1167–1173. 10.1161/CIRCULATIONAHA.110.979450. [DOI] [PubMed] [Google Scholar]
- 29.Starr B, Hadfield SG, Hutten BA, et al. Development of sensitive and specific age- and gender-specific low-density lipoprotein cholesterol cutoffs for diagnosis of first-degree relatives with familial hypercholesterolaemia in cascade testing. Clin Chem Lab Med. 2008;46(6):791–803. 10.1515/CCLM.2008.135. [DOI] [PubMed] [Google Scholar]
- 30.Defesche JC, Lansberg PJ, Umans-Eckenhausen MA, Kastelein JJ. Advanced method for the identification of patients with inherited hypercholesterolemia. Semin Vasc Med. 2004;4(1):59–65. 10.1055/s-2004-822987. [DOI] [PubMed] [Google Scholar]
- 31.Scientific Steering Committee on behalf of the Simon Broome Register Group. Risk of fatal coronary heart disease in familial hypercholesterolaemia. Scientific Steering Committee on behalf of the Simon Broome Register Group. BMJ. 1991;303(6807):893–896. 10.1136/bmj.303.6807.893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Williams RR, Hunt SC, Schumacher MC, et al. Diagnosing heterozygous familial hypercholesterolemia using new practical criteria validated by molecular genetics. Am J Cardiol. 1993;72(2):171–176. 10.1016/0002-9149(93)90155-6. [DOI] [PubMed] [Google Scholar]
- 33.Ruel I, Aljenedil S, Sadri I, et al. Imputation of baseline LDL cholesterol concentration in patients with familial hypercholesterolemia on statins or ezetimibe. Clin Chem. 2018;64(2):355–362. 10.1373/clinchem.2017.279422. [DOI] [PubMed] [Google Scholar]
- 34.Baigent C, Keech A, Kearney PM, et al. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet. 2005;366(9493):1267–1278. Published correction appears in Lancet. 2005;366(9494):1358. Published correction appears in Lancet. 2008;371(9630):2084. 10.1016/S0140-6736(05)67394-1. [DOI] [PubMed] [Google Scholar]
- 35.Trinder M, Francis GA, Brunham LR. Association of monogenic vs polygenic hypercholesterolemia with risk of atherosclerotic cardiovascular disease. JAMA Cardiol. 2020;5(4):390–399. Published correction appears in JAMA Cardiol. 2020;5(4):488. 10.1001/jamacardio.2019.5954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Emi M, Hegele RM, Hopkins PN, et al. Effects of three genetic loci in a pedigree with multiple lipoprotein phenotypes. Arterioscler Thromb. 1991;11(5):1349–1355. 10.1161/01.atv.11.5.1349. [DOI] [PubMed] [Google Scholar]
- 37.Motazacker MM, Pirruccello J, Huijgen R, et al. Advances in genetics show the need for extending screening strategies for autosomal dominant hypercholesterolaemia. Eur Heart J. 2012;33(11):1360–1366. 10.1093/eurheartj/ehs010. [DOI] [PubMed] [Google Scholar]
- 38.Ioannidis NM, Rothstein JH, Pejaver V, et al. REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. Am J Hum Genet. 2016;99(4):877–885. 10.1016/j.ajhg.2016.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yeo G, Burge CB. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J Comput Biol. 2004;11(2-3):377–394. 10.1089/1066527041410418. [DOI] [PubMed] [Google Scholar]
- 40.Cassanelli S, Bertolini S, Rolleri M, et al. A ‘de novo’ point mutation of the low-density lipoprotein receptor gene in an Italian subject with primary hypercholesterolemia. Clin Genet. 1998;53(5):391–395. 10.1111/j.1399-0004.1998.tb02752.x. [DOI] [PubMed] [Google Scholar]
- 41.Thormaehlen A, Schuberth C, Won H-H, et al. Systematic cell-based phenotyping of missense alleles empowers rare variant association studies: a case for LDLR and myocardial infarction. PLoS Genet. 2015;11(2):e1004855. 10.1371/journal.pgen.1004855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Weile J, Roth FP. Multiplexed assays of variant effects contribute to a growing genotype–phenotype atlas. Hum Genet. 2018;137:665–678. 10.1007/s00439-018-1916-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed during this study are included in this manuscript (and its Supplemental Tables and Figures).