

Abstract
Drosophila MEI-9 is the catalytic subunit of a DNA structure-specific endonuclease required for nucleotide excision repair (NER). The enzymatic activity of this endonuclease during NER requires the presence of a second, noncatalytic subunit called ERCC1. In addition to its role in NER, MEI-9 is required for the generation of most meiotic crossovers. To better understand the role of MEI-9 in crossover formation, we report here the characterization of the Drosophila Ercc1 gene. We created an Ercc1 mutant through homologous gene targeting. We find that Ercc1 mutants are identical to mei-9 mutants in sensitivity to DNA-damaging agents, but have a less severe reduction in the number of meiotic crossovers. MEI-9 protein levels are reduced in Ercc1 mutants; however, overexpression of MEI-9 is not sufficient to restore meiotic crossing over in Ercc1 mutants. We conclude that MEI-9 can generate some meiotic crossovers in an ERCC1-independent manner.
DURING meiosis, chiasmata form physical linkages between homologous chromosomes that ensure their proper segregation. Chiasmata result from a combination of sister-chromatid cohesion and recombinational exchange events called crossovers. Although the molecular mechanism of meiotic crossover formation is not fully known, analysis of recombination events has led to a model with several important features: (1) meiotic recombination initiates with a double-strand break on one chromatid, (2) recombination proceeds through the formation of a heteroduplex-containing intermediate structure with two four-way DNA junctions called Holliday junctions, and (3) crossovers arise through the cleavage of Holliday junctions (Stahl 1996).
In Drosophila melanogaster, several genes required for meiotic crossover formation have been identified (McKim et al. 2002). One such gene is mei-9, mutations of which abolish 90% of all meiotic crossovers. Molecular identification of mei-9 revealed that it encodes the Drosophila homolog of Rad1 in Saccharomyces cerevisiae and XPF in mammals (Sekelsky et al. 1995). Rad1/XPF is a DNA-structure-specific endonuclease that is required for nucleotide excision repair (NER), the primary pathway for repair of DNA damage induced by ultraviolet (UV) light. NER involves excision of an oligonucleotide containing the damaged bases and fill-in synthesis using the intact strand as a template. Rad1/XPF creates the excision nick 5′ to the damaged bases by recognizing a transition in the DNA from 5′ double stranded to 3′ single stranded (Bardwell et al. 1994; Park et al. 1995). Sensitivity of mei-9 mutants to various DNA-damaging agents demonstrates a requirement for MEI-9 in NER as well as in other DNA repair pathways (Boyd et al. 1976).
Identification of MEI-9 as a protein whose homologs have a DNA-structure-specific endonuclease activity provides a clue to the role MEI-9 may play in meiotic crossover formation. We previously proposed that MEI-9 acts as a DNA endonuclease on Holliday junctions during meiotic recombination to resolve the intermediate DNA structure into crossover products (Sekelsky et al. 1995). This proposal predicts that the MEI-9 endonuclease is modified during meiotic recombination to change the specificity from that of the NER substrate to that of a Holliday junction.
Rad1/XPF is the catalytic subunit of the NER 5′ endonuclease. Rad1/XPF forms a complex with a second protein called Rad10 in S. cerevisiae and with ERCC1 in mammals. This second, noncatalytic subunit is required for enzymatic activity (Davies et al. 1995). Mutations in either gene have identical phenotypes in both S. cerevisiae (Prakash et al. 1985; Schiestl and Prakash 1990; Ivanov and Haber 1995) and mouse (Weeda et al. 1997; Tian et al. 2004). The Drosophila homolog of Rad10/ERCC1 was identified by sequence homology and has been named ERCC1 (Sekelsky et al. 2000). Additionally, we recently identified a novel protein, MUS312, that physically interacts with MEI-9 to generate crossovers, but that is not required for NER (Yildiz et al. 2002). This leads to the interesting possibility that the substrate specificity of MEI-9 is affected by the partner protein(s) present. Two possible alternatives are: MUS312 may add to the MEI-9-ERCC1 endonuclease complex or MUS312 may replace ERCC1 in the complex to change the substrate specificity. To distinguish between these models, characterization of Drosophila ERCC1 is necessary.
We report here the generation of a Drosophila Ercc1 mutant by homologous targeting. Genetic characterization of this mutant reveals a hypersensitivity to DNA-damaging agents that is identical to that of mei-9. In contrast, Ercc1 mutants have a less severe meiotic phenotype than mei-9 mutants. We find that levels of MEI-9 protein are decreased in Ercc1 mutants; however, overexpression of MEI-9 protein in an Ercc1 mutant is not sufficient to restore meiotic crossing over. We conclude that MEI-9 can generate some meiotic crossovers in the absence of ERCC1. This is the first report of an ERCC1/Rad10-independent function for MEI-9/XPF/Rad1.
MATERIALS AND METHODS
Drosophila stocks and genetics:
Genetic loci not described in the text are described in FlyBase (Drysdale et al. 2005). Flies were reared on standard medium at 25°.
Sensitivity to killing by UV light was assessed by collecting embryos on grape agar plates overnight, aging them to third instar larvae (∼4 days), and then spreading larvae into a monolayer on chilled petri plates. Larvae were irradiated in a Stratalinker (Stratagene, La Jolla, CA). Sensitivity to methyl methanesulfonate (MMS) was assessed by allowing adults to lay eggs in plastic vials for 2 days and, after 1 additional day, adding 250 μl of 0.025, 0.05, or 0.075% solution of MMS in water to the food. For Ercc1X homozygous and hemizygous crosses, Ercc1X/CyO females were crossed to Ercc1X/Df(2R)knSA3 males. For mei-9A2 crosses, mei-9A2/FM7 females were crossed to mei-9A2 males. Percentage survival is calculated as the treated ratio of mutant to control flies divided by the untreated ratio of mutant to control flies. Means and standard deviations were determined from at least three independent experiments.
For the X chromosome nondisjunction assay, female flies of the appropriate genotype were crossed to C(1;Y)1, v f B/O males. Progeny from normal disjunction are Bar females (X/C(1;Y)1, v f B) and non-Bar males (X/O). Half of the diplo-X progeny survive as non-Bar females (X/X) and half die (X/X/C(1;Y)1, v f B). Half of the nullo-X progeny survive as Bar males (C(1;Y)1, v f B/O) and half die (O/O). The percentage of X chromosome nondisjunction is calculated as twice the number of viable exceptional progeny divided by the total of normal progeny plus twice the number of exceptional progeny.
Direct measurements of crossing over on the third chromosome were performed in flies of the appropriate genotype and heterozygous for mutations in Ly st ry and e. Map distance is calculated as 100 times the number of recombination events in the interval divided by the total number of flies. For zygotic Ercc1X and mei-9A2 mutants, mutant females tested were derived from heterozygous mothers. For maternal/zygotic mutants, mutant females tested were derived from homozygous mutant mothers.
P-element constructs and germline transformation:
The transformation vector pP{Target} was built from pCaSpeR4 (Pirrotta 1988) and carries the w+mC marker from that vector. The flipase recombinase target (FRT) sequences are derived from oligonucleotides containing the 34-bp minimal sequence. For the derivative pP{TargetB}, the 3′ half of white was replaced with cDNA sequences, resulting in removal of introns 3–5 and of sequences downstream from the translation termination site. The vectors and complete sequences are available from the Drosophila Genomic Resources Center (http://dgrc.cgb.indiana.edu).
For the targeting construct pP{Ercc1X}, the Ercc1 region was amplified as two PCR products. The left product contained −2158–291 (using the ATG at the beginning of the Ercc1 coding region as +1). The primer within Ercc1 included 2 extra base pairs to generate an XhoI site, which was used to clone this fragment into a plasmid vector. Similarly, the right product extended from 288 to 2614, and the primer within Ercc1 included 2 extra base pairs to generate an XhoI site. When cloned into the construct carrying the left half, a genomic region of 4.8 kb was produced, with a 2-bp insertion in Ercc1 generating a frameshift mutation marked by an XhoI site. An I-SceI recognition sequence was inserted into the unique NsiI site in the 3′ untranslated region of Ercc1 by annealing two oligonucleotides that generated the desired sequence and overhangs compatible with NsiI-digested DNA. This fragment was cloned into pP{Target}.
To generate P{WUF9}, a 1886-bp fragment of the 5′-end of ubiquitin, containing the first noncoding exon, the first intron, and the first 11 bp of the second exon, was cloned into pBluescript KS+. Oligonucleotides encoding an initiator methionine and the FLAG epitope (DYKDDDDK) were added to generate pBUF (Bluescript-ubiquitin-FLAG). The complete coding sequence from a mei-9 cDNA (as reported in Sekelsky et al. 1995) was cloned into this vector, and the entire module was then transferred into pCaSpeR4 to generate pP{WUF9} (white-ubiquitin-FLAG-mei-9). The mei-9 cDNA used to create this construct encodes a 926-amino-acid protein. Version 3.2 of the Drosophila genome reports a mei-9 cDNA that encodes a 961-amino-acid protein, with 35 amino acids added at the N terminus; however, these 35 amino acids have no homology to other proteins related to MEI-9, and this P{WUF9} construct rescues mei-9 mutant phenotypes (data not shown), indicating that the extra amino acids are not essential to MEI-9 function.
Homologous gene targeting:
We conducted gene targeting according to the method of Rong et al. (2002), with the following modifications: females carrying the targeting construct and the I-SceI and FLP transgenes were crossed to males homozygous for a transgene expressing FLP constitutively in bottles. Rare progeny with colored eyes were then crossed to generate stocks and to map the insertion to a chromosome. Of three inserts on chromosome 2, one was determined by PCR to be within Ercc1 (Figure 3). The other two were determined genetically to be unlinked to Ercc1.
Figure 3.—

Molecular analysis of tandem duplication and Ercc1X reduction. Allele-specific PCR was performed using either an Ercc1-specific primer (+) or a Ercc1X-specific primer (X) and a reverse primer complementary to sequence outside of the targeting sequence. PCR reactions were run on a standard agarose gel for analysis. The expected 2.4-kb product is seen with the “+” primer in wild type; however, the expected 2.4-kb product is missing with the X primer in Ercc1X and instead a 1.2-kb product is seen. A 800-bp product is seen with the X primer in the duplication, confirming the presence of the 1.6-kb deletion.
Molecular analysis of Ercc1X:
To generate the Ercc1X allele, we recovered viable reductions of the targeted duplication over a deficiency for the region and then analyzed these events by PCR followed by XhoI digestion. Fly DNA was prepared by homogenization in buffer and incubation with proteinase K (Gloor et al. 1993). PCR was done using a forward primer at −67 (numbers are relative to the start of Ercc1 translation) (5′-GTGCCTTCGTCACCTGATA-3′) and a reverse primer at 700 (5′-GGCAGCATCAGTCTTGTTC-3′). Following digestion, products were analyzed on agarose gels. The presence of Ercc1 generates a 769-bp PCR product, while the presence of Ercc1X generates two cleaved products of 414 and 355 bp and the presence of both alleles generates all three products. One of the four viable reductions recovered had only the two smaller products and so has only the Ercc1X allele.
Allele-specific PCR was designed with primers to the region of Ercc1 in which the XhoI site was inserted. The forward primer at 274 to amplify Ercc1 is 5′-GATTATGTGGTCGGTCGAAC-3′ and to amplify Ercc1X is 5′-GATTATGTGGTCGGCTCGAG-3′. These primers were used in combination with a reverse primer at 2655 (5′-GCCACACTCCGGATCTTCTG-3′) on wild-type, targeted duplication, and Ercc1X flies. We confirmed that the Ercc1X primer does not amplify Ercc1, that the targeted duplication contains an ∼1.6-kb deletion downstream of the Ercc1X allele, and that Ercc1 is not present in Ercc1X; however, we also unexpectedly recovered an ∼1.2-kb product from Ercc1X flies. Sequencing of this product revealed a vector sequence expected to have been removed by the reduction.
To determine the genomic sequence surrounding the remaining vector sequences, we performed PCR using a forward primer at 1990 (5′-CCCATTGATGGTCAAGTGCT-3′) and a reverse primer complementary to the I-CreI site in the vector sequence (5′- GACCAAACTGTCTCACGACGTTTTG-3′) and performed a second PCR using a forward primer complementary to the I-CreI site (5′-CGTCGTGAGACAGTTTGGT-3′) and the reverse primer at 2655 (see above). These PCR products were sequenced, showing that from 1990 through to the I-CreI site, the genomic sequence is identical to that of the targeted duplication, while from the I-CreI site through to 2655, 6279 bp of vector sequence, including most of the white gene, is deleted relative to the targeted duplication.
To ensure that Ercc1X, which we have shown to be the only Ercc1 allele present, is upstream of this remaining vector sequence, we did a PCR reaction using a forward primer at 217 (5′-CCCATCCTGAAATCCATACT-3′) and the reverse primer to the I-CreI site (see above). This reaction was sequenced and showed the presence of the inserted XhoI site. The resulting Ercc1X allele is pictured in Figure 2D.
Figure 2.—
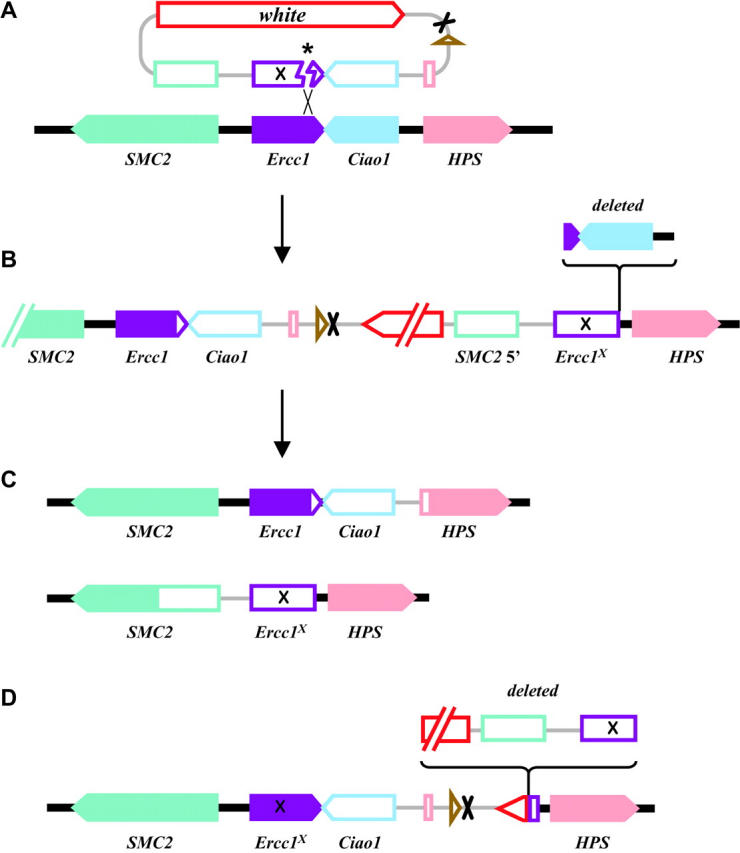
Targeting of Ercc1. (A) Genes from the Ercc1 genomic region are shown as solid arrows (indicating direction of transcription) on a solid line, and genes on the targeting DNA are shown as open arrows or boxes on a shaded line. The targeting DNA is shown after excision by FLP recombinase and cutting by I-SceI endonuclease, with an asterisk marking the site of the double-strand break. This targeting fragment is drawn to show alignment of sequences with homology between the targeting DNA and the genomic DNA. The XhoI site introduced into Ercc1 is indicated by an X. The mini-white marker gene and the FRT and I-CreI sites are as in Figure 1. (B) The predicted product of ends-in integration with sequences derived from the targeting DNA in open symbols and chromosomal sequences as solid symbols. The region deleted in the integration that we recovered is indicated. (C) Predicted products from reduction of the tandem duplication after cutting with I-CreI and repair by single-strand annealing. One product is completely wild type (top), and the other carries Ercc1X and the adjacent deletion of Ciao1 (bottom). (D) The structure of the mutation used in these studies. It is equivalent to the targeted integration depicted in B, except that most of mini-white and one copy of Ercc1 have been deleted, and the remaining copy of Ercc1 carries the XhoI mutation.
Western blotting:
Ovaries were dissected on ice from Drosophila females of the genotypes described in the text. The ovaries were then boiled and sonicated in SDS sample buffer prior to loading onto a polyacrylamide gel at the equivalent of one pair of ovaries per lane. After separation by electrophoresis, proteins were transferred to a polyvinylidene difluoride (PVDF) membrane. MEI-9 was detected with rabbit polyclonal anti-MEI-9 serum, using the ECL detection kit (Amersham, Arlington Heights, IL).
Yeast two-hybrid assay:
We used the two-hybrid system described in James et al. (1996) with a modification to the pGBD fusion plasmid that inserts a second multiple cloning site flanked by a Gal4 promoter and transcription and translation start sites and an Adh1 3′-UTR (pGBT61; M. Nichols, personal communication). This vector allows expression of an untagged protein that can act as a bridge in the yeast two-hybrid assay. A full-length Ercc1 cDNA sequence was cloned into the NcoI and PstI sites in pGBT61 to create pGBT61-Ercc1. A full-length mei-9 cDNA sequence (as reported in Sekelsky et al. 1995) was cloned into the StuI and KpnI sites in pGBT61-Ercc1 to create pGBT61-Ercc1 + mei-9. We obtained pGAD-mei-9 from the Drosophila Genomics Resource Center (Bloomington, IN). Full-length mus312 cDNA sequence was cloned into the NcoI and BamHI sites in pGAD (J. R. LaRocque, personal communication).
RESULTS AND DISCUSSION
Vectors for gene targeting in Drosophila:
We used the gene targeting method developed by Rong and Golic (2000) to knock out Ercc1. In this method, the desired mutation is initially introduced into the genome as a P-element transgene. The genomic fragment to be used for targeting is flanked by FRTs and carries a recognition sequence for the I-SceI endonuclease. We built a pair of vectors to be used in generating such transgenes. pP{Target} is derived from pP{CaSpeR4} (Pirrotta 1988) and contains two FRTs flanking an I-CreI site, mini-white gene, and a multiple cloning site. As described below, we used pP{Target} to knock out Ercc1. We also built a derivative, pP{TargetB}, that is smaller (6377 bp) and has several additional cloning sites (Figure 1). We and others have successfully used this vector to target additional genes (Donaldson et al. 2004; M. D. Adams and J. Sekelsky, personal communication). These vectors have been made available at the Drosophila Genomic Resources Center (http://dgrc.cgb.indiana.edu).
Figure 1.—

Vectors for gene targeting in Drosophila. Schematic maps of pP{Target} and pP{TargetB} are shown. Both vectors carry a mini-white marker gene (shaded arrow), FRTs (stippled triangles), an I-CreI endonuclease recognition sequence (X), and P-element ends for transposition (solid arrows). The two vectors differ in size and in restriction sites available for cloning.
Targeted knockout of Ercc1:
We designed an Ercc1 targeting construct, pP{Ercc1X}, that carries a 4.7-kb genomic fragment. In addition to Ercc1, this fragment includes 1.8 kb of Smc2, all of CG12797, and the 5′-end of CG12855 (Figure 2A). The predicted protein product of CG12797 consists of seven WD40 repeats and is 60% identical to human Ciao1 over its entire length of 335 residues. Ciao1 was originally isolated on the basis of its interaction with the Wilms' tumor suppressor WT1 (Johnstone et al. 1998). The predicted protein product of CG12855 consists of 596 residues, with ∼200 residues in each of two regions that are 45–55% similar to human HPS. Mutations in HPS are associated with Hermansky-Pudlak syndrome, a recessive autosomal disorder of cytoplasmic organelles (Oh et al. 1996). We will refer to CG12797 as Ciao1 and CG12855 as HPS.
We inserted 2 bp into Ercc1 to generate a frameshift at codon 96 (of 259), upstream of the most highly conserved region. The insertion generates an XhoI site, which can be used as a diagnostic marker for the mutation. An I-SceI site was inserted 569 bp downstream of the XhoI site within the Ercc1 3′-untranslated region. We generated two insertions by germline transformation, one on the X chromosome and the other on chromosome 3. Additional autosomal insertions were generated by transposing the X-linked insertion.
We used the X-linked insertion of P{Ercc1X} and five autosomal insertions to generate putative targeted insertions, as described in Rong and Golic (2001). Of 16 insertions of the targeting DNA, three mapped to chromosome 2, and one of these was a homologous insertion into the endogenous Ercc1 locus. In the ends-in method of gene targeting, the result of integration of targeting DNA into the homologous target is a tandem duplication. Analysis of the targeted integration into Ercc1 that we recovered revealed that it was imprecise, containing a 1569-bp deletion on one copy of the duplication (Figure 2B and Figure 3). This deletion begins at the I-SceI cut site and removes the 3′-UTR of Ercc1 and all of Ciao1, ending 120 bp upstream of the beginning of the HPS protein-coding region.
When a double-strand break is introduced between the two copies of a tandem duplication, repair by single-strand annealing (SSA) results in reduction of the duplication to single copy with high efficiency (Ivanov et al. 1996). As described by Rong et al. (2002), we used the rare-cutting endonuclease I-CreI to generate reductions of our tandem duplication. We initially recovered 21 reductions. Eleven of these carried the wild-type Ercc1 and did not have the associated deletion; these were homozygous viable. The other 10 reductions carried the Ercc1X allele, but also had the adjacent deletion. All 10 of these were homozygous and hemizygous lethal. These two types of events are the predicted products of the SSA model for reduction (Figure 2C).
The lethality of the chromosome carrying Ercc1X and the adjacent deletion was rescued by crossing in a copy of the P{Ercc1X} targeting construct. The only functional gene on this construct is Ciao1, so we conclude that the lethality is due entirely to the deletion of Ciao1.
Although the Ercc1 mutation and the deletion were 100% linked in the original reductions that we recovered, we thought that we might be able to separate the two by generating a larger number of reductions. We devised a scheme in which putative reductions were recovered in trans to Df(2R)knSA3, which deletes the entire region, and we screened the hemizygous viable reductions for the presence of the Ercc1X mutation. We recovered one such event after analyzing only four reductions; however, the structure of this reduction was more complex than we had anticipated.
We performed a PCR with forward primers specific to either the wild-type Ercc1 locus or the Ercc1X mutation and a reverse primer within HPS just downstream of the sequence included in the original targeting vector. We expected a product of ∼2.4 kb in the wild-type genotype with the Ercc1-specific primer, a product of ∼800 bp in the targeted duplication with the Ercc1X-specific primer (as a result of the ∼1600-bp deletion), and a product of ∼2.4 kb in the Ercc1X genotype. We saw the first two expected products; however, instead of an ∼2.4-kb product, we saw an ∼1.2-kb product with the Ercc1X-specific primer in Ercc1X. Sequencing of this product led us to conclude that the Ercc1X-specific primer had annealed to the weakly complementary sequence surrounding an XhoI site downstream of the FRT in the original targeting vector and that the I-CreI site used to induce collapse of the duplication remains intact in the reduction. Further PCR and sequencing reactions (see materials and methods for details) show that no wild-type Ercc1 is present; only the XhoI-interrupted Ercc1 is present. There is also one wild-type copy of Ciao1; however, 1218 bp of extra sequence from the original targeting vector remains in the intergenic region between Ciao1 and HPS (Figure 2D). The recovered reduction, then, is mutant for Ercc1 and wild type for Ciao1.
The insertion of 1218 bp of vector sequence is 120 bp upstream of the HPS start of translation. It is possible that this insertion disrupts HPS function in the Ercc1X reduction allele. We wanted to ensure that any meiotic phenotype seen using the reduction allele could be attributed to the targeted mutation of Ercc1, and not to the insertion of DNA upstream of HPS. Although we do not have an allele of HPS identical to that in the reduction allele in conjunction with wild-type Ercc1, we do have an allele of HPS that has an insertion of 7480 bp in the same location and is wild type for Ercc1, the original targeted duplication (Figure 2B). If the insertion of 1218 bp in the reduction allele affects HPS expression, it is likely that this larger insertion would mimic or exacerbate the effect. We assayed X nondisjunction in females homozygous for the original targeted duplication (see below for an explanation of the X nondisjunction assay). We did not see any X nondisjunction (n = 218), indicating that the insertion upstream of HPS likely does not contribute to the meiotic phenotype.
Ercc1 mutants are hypersensitive to UV and MMS:
mei-9 mutants are hypersensitive to several DNA-damaging agents, indicating a role for MEI-9 in DNA repair pathways, including NER (Boyd et al. 1976). To determine what role, if any, Drosophila ERCC1 plays in NER, we tested the sensitivity to killing of Ercc1X mutants in response to treatment with UV. UV induces primarily pyrimidine dimers and 6-4 photoproducts, both of which are repaired by NER.
Ercc1X mutants are equally as sensitive to UV treatment as mei-9 mutants, indicating that ERCC1 is indeed important for NER (Figure 4). Both Ercc1X homozygous and hemizygous (over Df(2R)knSA3, a deficiency for the region) mutants display an equal sensitivity to UV treatments over a variety of doses, indicating that Ercc1X is genetically null in this assay (Figure 4). We also see that Ercc1X mutants and mei-9 mutants are equally as sensitive to treatment with another type of DNA-damaging agent, MMS (data not shown).
Figure 4.—

Sensitivity to killing by UV light of Ercc1X and mei-9 mutants. Percentage survival, relative to wild-type controls, after exposure of larvae to 500–1000 erg/mm2 (1 erg/mm2 = 0.1 J/m2) of UV light is shown for Ercc1X homozygous and hemizygous mutants and mei-9A2 mutants. Bars indicate standard deviations.
Loss of ERCC1 function confers a meiotic phenotype that is less severe than that of loss of MEI-9 function:
MEI-9 is required to generate most meiotic crossovers. The chiasmata produced by crossovers are essential for proper segregation of homologs during meiosis I; therefore, in the absence of normal levels of crossovers there are high levels of meiosis I chromosome nondisjunction. In Drosophila, X chromosome nondisjunction can be measured by the use of appropriate genetic markers, giving an indirect assay for recombination defects. Wild-type levels of X nondisjunction are ∼0.3%, whereas null mei-9 mutants have ∼25–35% X nondisjunction (Yildiz et al. 2004). In Ercc1X mutants, there is only 16% X nondisjunction (Table 1). Although this is a 50-fold increase over wild type, indicating a severe meiotic defect, it is still only half the level of nondisjunction seen in mei-9 mutants.
TABLE 1.
X chromosome nondisjunction inErcc1 mutants
| Progeny
|
||||
|---|---|---|---|---|
| Genotype | Normal | Nullo-X | Diplo-X | X nondisjunction (%) |
| Wild type | 1473 | 2 | 0 | 0.3 |
| Zygotic mei-9A2a | 857 | 111 | 145 | 37 |
| Zygotic Ercc1X | 1848 | 91 | 82 | 16 |
| Zygotic Ercc1X/Df | 1223 | 23 | 37 | 9 |
| m/z Ercc1X | 1049 | 55 | 103 | 23 |
| P{WUF9}, m/z Ercc1X | 775 | 80 | 60 | 27 |
The mei-9 data are from Yildiz et al. (2004).
To assess more directly the recombination defect in Ercc1X mutants, we measured crossing over along the third chromosome. We examined three intervals from Ly, in the middle of the left arm, to e, in the middle of the right arm (Table 2). The total map distance was 35.0 MU in wild type, but only 8.5 MU in Ercc1X mutants, a reduction of 76%; however, map distance was 5.1 MU in mei-9A2 mutants, a reduction of 85% from the wild-type level. As seen in the X nondisjunction assay, Ercc1 mutants have a less severe defect in crossing over than do mei-9 mutants.
TABLE 2.
Meiotic crossing over inErcc1 andmei-9 mutants
| Exchange within the interval (MU) |
||||||
|---|---|---|---|---|---|---|
| Genotype | Ly-st | st-ry | ry-e | Total map distance (MU) |
% of wild type |
N |
| Wild type | 4.1 | 9.2 | 21.7 | 35.0 | 100 | 1887 |
| Zygotic Ercc1X | 0.9 | 1.6 | 6.0 | 8.5 | 24 | 3464 |
| m/z Ercc1X | 0.6 | 1.3 | 2.8 | 4.7 | 13 | 2007 |
| Zygotic Ercc1X/Df | 1.4 | 2.8 | 8.5 | 12.8 | 37 | 4226 |
| m/z Ercc1X/Df | 0.8 | 2.1 | 2.5 | 5.5 | 16 | 1063 |
| Zygotic mei-9A2 | 0.4 | 0.8 | 3.9 | 5.1 | 15 | 1862 |
| m/z mei-9A2 | 0.3 | 1.1 | 1.3 | 2.7 | 8 | 2006 |
One possible explanation for the less severe meiotic phenotype of the Ercc1X mutant is that the maternal ERCC1 protein deposited during oogenesis may perdure and partially compensate for the lack of ERCC1 protein produced in an Ercc1X mutant. To test this possibility, we measured X nondisjunction in Ercc1X mutants derived from Ercc1X mutant mothers. These maternal/zygotic (m/z) Ercc1X mutants have higher levels of X nondisjunction than zygotic Ercc1X mutants (Table 1), similar to the levels seen in mus312 mutants and in some mei-9 alleles (Yildiz et al. 2002, 2004). Direct measurements of crossing over also show that m/z Ercc1X mutants have a reduction in crossovers similar to zygotic mei-9 mutants; however, we also find that m/z mei-9 mutants have a stronger overall reduction in crossing over than either m/z Ercc1X or zygotic mei-9 mutants (Table 2). This indicates that there may be a maternal effect in mei-9 mutants as well as in Ercc1 mutants. We conclude that a maternal effect does not mask the true Ercc1X phenotype, but rather that the Ercc1X mutant truly causes a less severe reduction in crossovers than a mei-9 mutant does. These results also show that the residual crossovers in mei-9 mutants are not due to perdurance of maternal protein, but rather to a secondary, MEI-9-independent pathway.
A second possible explanation for the less severe meiotic phenotype of Ercc1X mutants is that this allele is not null for meiotic function. Although this allele creates a frameshift approximately halfway through the coding region of Ercc1, there may be some readthrough that produces residual functional protein. If this were the case, then reducing the copy number of Ercc1X would result in a more severe meiotic phenotype. However, we find that this is not the case: X nondisjunction of Ercc1X hemizygotes is lower than that of Ercc1X homozygotes (Table 1, 9% vs. 16%). This evidence also argues against the possibility that the genetic background of the Ercc1X mutant is responsible for the less severe meiotic phenotype. The hemizygous case should dilute the background effects, resulting in a more severe meiotic phenotype than the homozygous, but the opposite is true.
The finding that a homozygous Ercc1X mutant has a more severe phenotype than a hemizygous Ercc1X mutant raises the concern that the Ercc1X allele encodes a protein that acts as a gain-of-function antimorph. To test whether Ercc1X encodes a dominant antimorph, we assayed X nondisjunction in the following genotypes: (1) one copy of Ercc1X and one copy of wild-type Ercc1, (2) one copy of a deficiency chromosome that removes Ercc1 and one copy of wild-type Ercc1, and (3) two copies of Ercc1X and one copy of wild-type Ercc1. If Ercc1X were acting as a dominant gain-of-function antimorph, we would expect to see wild-type levels of X nondisjunction in (2) and increased nondisjunction in (1) and (3); however, we saw wild-type levels in all three genotypes (data not shown). This shows that Ercc1X does not encode a dominant antimorphic form of ERCC1.
Although the Ercc1X allele does not act as a dominant antimorph, there remained the concern that the less severe meiotic defect of this allele is the result of a recessive gain-of-function antimorphic protein rather than the result of a loss of ERCC1 function. There are two possibilities: the Ercc1X protein antagonizes (1) the wild-type ERCC1 protein or (2) some other protein. To address these possibilities, we assayed X nondisjunction and meiotic recombination in zygotic and m/z hemizygous Ercc1X mutants. Because only zygotic mutants retain some ERCC1 protein, the first possibility predicts that the zygotic homozygous Ercc1X mutant would have a more severe meiotic defect than the zygotic hemizygous mutant (two doses of Ercc1X vs. one dose, equal doses of maternal ERCC1), while the two m/z mutants would have similar phenotypes (no ERCC1 to be antagonized). On the other hand, the second possibility predicts that the more ERCC1X protein present, the more severe we would expect the phenotype to be (i.e., m/z Ercc1X is more severe than zygotic Ercc1X, which is more severe than m/z hemizygous, which is more severe than zygotic hemizygous). Our results are consistent with the first possibility—that Ercc1X may encode an antimorphic protein that antagonizes the maternal ERCC1 protein (Table 2). An alternative interpretation of our results is that Ercc1X is a loss-of-function mutation and the difference in severity between the zygotic homozygous mutant and the zygotic hemizygous mutant reflects a background effect on the stability of maternally deposited ERCC1 protein.
Whether Ercc1X encodes a protein that antagonizes maternally deposited ERCC1 protein or whether the varying degrees of phenotype severity reflect a background effect, these results suggest that the m/z Ercc1X homozygous and hemizygous mutants represent the complete loss of ERCC1 function. The complete loss of ERCC1 function therefore confers a less severe meiotic defect than the complete loss of MEI-9 function (Table 2).
MEI-9 protein is unstable in the absence of ERCC1:
In mammalian XPF- or ERCC1-deficient cells, the respective partner protein is generally undetectable by Western blot, indicating that complex formation between these two proteins may be important for stability (Houtsmuller et al. 1999). We performed Western blots on whole-ovary extracts from Ercc1X mutants probed with an anti-MEI-9 polyclonal antibody. We find that MEI-9 protein is severely decreased, although still detectable, in m/z Ercc1X mutants (Figure 5). Knowing that complex formation with ERCC1 is important for the stability of MEI-9, we also investigated whether complex formation with MUS312 affects MEI-9 protein levels, because MUS312 must physically interact with MEI-9 to generate meiotic crossovers (Yildiz et al. 2002). The absence of MUS312 had no detectable effect on MEI-9 protein levels (data not shown).
Figure 5.—

Expression of MEI-9 in ovaries. Ovarian proteins were separated on a polyacrylamide gel, transferred to PVDF membrane, and detected with polyclonal anti-MEI-9 serum. Genotypes of ovaries are (1) wild type, (2) mei-9A2, (3) zygotic Ercc1X, (4) maternal/zygotic Ercc1X, and (5) P{WUF9} in maternal/zygotic Ercc1X. MEI-9 is indicated by a solid arrowhead (Mr = 125 kDa). The antiserum also detects unknown proteins of lower molecular weight; one of these is included as a loading control. Flag-tagged MEI-9 protein (open arrowhead) produced from the P{WUF9} transgene migrates slightly faster than untagged MEI-9 (Mr = 113 kDa). The mei-9 cDNA used to make this construct lacks 35 nonessential amino acids at the N terminus (see materials and methods for details).
It is possible that the meiotic defect in Ercc1X mutants is actually due to the decreased level of MEI-9, and not to the absence of ERCC1 per se. To test genetically whether low MEI-9 protein is responsible for the Ercc1X meiotic defect, we created a transgene, P{WUF9}, encoding FLAG-tagged MEI-9 under the control of the ubiquitin promoter. Insertions of P{WUF9} fully rescue mei-9 mutant phenotypes, including sensitivity to MMS and X chromosome nondisjunction (data not shown).
We crossed an insertion of P{WUF9} on the X chromosome into the Ercc1X background to determine whether overexpressing MEI-9 protein can rescue the Ercc1X meiotic defect. This transgene does provide an increased level of MEI-9 protein in ovaries (Figure 5); however, it has no effect on X chromosome nondisjunction levels of Ercc1X mutants (Table 1). These results suggest that the meiotic defect in Ercc1X mutants is likely due to a lack of ERCC1 protein rather than to low levels of MEI-9. One caveat to this experiment is that meiotic cells in pachytene (where recombination is occurring) make up a small percentage of the Drosophila ovary. It is possible that the increased MEI-9 protein in Ercc1X ovaries containing P{WUF9} is limited to the nonmeiotic cells of the ovary. This could explain the failure to rescue the Ercc1X meiotic defect; however, P{WUF9} is able to fully rescue the meiotic defects of mei-9 mutants (data not shown), indicating that this construct is able to express functional MEI-9 in pachytene.
It is not surprising that the decreased level of MEI-9 protein in Ercc1X mutants may not be the cause of the meiotic defect. MEI-9 protein is undetectable by Western blot in the ovaries of mei-9RT1 mutants, but these mutants have a very weak meiotic defect (2% X nondisjunction) (Yildiz et al. 2004), suggesting that very little MEI-9 protein may be necessary for generating meiotic crossovers. Once again, the caveat of this experiment is that the majority of ovary tissue is not undergoing meiotic recombination. MEI-9 protein may be decreased or absent in the majority of the cells of the mei-9RT1 ovary, but present at near wild-type levels in the pachytene cells, leading to the weak meiotic defect.
Taken together, the fact that little MEI-9 protein seems to be required for almost wild-type levels of meiotic recombination in mei-9RT1 mutants and that P{WUF9} expresses seemingly excess MEI-9 protein in the Ercc1X background, but is unable to rescue the meiotic phenotype, suggest that lack of ERCC1 protein is responsible for the lack of meiotic recombination in Ercc1X mutants.
MEI-9, MUS312, and ERCC1 physically interact by yeast two-hybrid:
Prior to this study, we had proposed two alternative scenarios: (1) MUS312 adds to the MEI-9-ERCC1 heterodimer or (2) MUS312 replaces ERCC1 to change the substrate specificity of MEI-9 from that of the NER substrate to a meiotic recombination intermediate, such as a Holliday junction. Although it has been shown that MEI-9 and ERCC1 (Yildiz et al. 2004) and MEI-9 and MUS312 (Yildiz et al. 2002) physically interact by yeast two-hybrid assay and that these interactions map to different regions of MEI-9 (Yildiz et al. 2004), evidence for all three proteins participating in a single complex, as predicted by the first scenario, was lacking.
We found that ERCC1 and MUS312 do not interact directly in a yeast two-hybrid assay; however, expression of untagged MEI-9 protein in the same yeast two-hybrid assay does allow ERCC1 and MUS312 to interact (Figure 6). This is the first evidence for all three proteins participating in a complex simultaneously.
Figure 6.—

Yeast two-hybrid assay with ERCC1, MUS312, and MEI-9. ERCC1 was expressed in yeast as a fusion protein with the Gal4 DNA-binding domain (GBD) in a vector that either did (2, 4, 6) or did not (1, 3, 5) also express MEI-9. Yeast were transformed with an empty Gal4 activation domain (GAD) vector (1, 2), a vector expressing GAD-MEI-9 fusion protein (3, 4), or GAD-MUS312 fusion protein (5, 6). Growth on the −HIS dropout media shown indicates an interaction between the GBD and GAD fusion proteins.
ERCC1 is required for a subset of meiotic crossovers:
We have shown that the Ercc1X somatic phenotype is identical to that of a mei-9 mutant, but that the meiotic phenotype is less severe than that of mei-9. Our two models for MEI-9 complex formation predicted that either ERCC1 would be required for meiotic recombination to the same extent as MEI-9 and MUS312 (MEI-9-ERCC1-MUS312 is the functional meiotic complex) or ERCC1 would not be required for meiotic recombination at all (MEI-9-MUS312 is the functional meiotic complex). Our results show that ERCC1 is required for generating some crossovers, but not as many as MEI-9. This suggests that MEI-9-ERCC1-MUS312 functions to generate most crossovers, but MEI-9-MUS312 is sufficient without ERCC1 to generate a small number of crossovers. It is unclear whether the crossovers that remain in an Ercc1 mutant represent a special class of crossovers that are normally generated by a MEI-9-MUS312 complex or whether these crossovers are normally generated by a MEI-9-ERCC1-MUS312 complex. We also cannot rule out the possibility that some meiotic crossovers may require ERCC1 but not MEI-9; however, we have determined that not all of the residual crossovers in mei-9 mutants require ERCC1 because in a mei-9 Ercc1 double mutant some crossovers remain (data not shown). Whether ERCC1 is required only for generating certain types of crossovers or whether MEI-9-MUS312 is able to function less efficiently without ERCC1 will be explored by investigating biochemical activities of these proteins.
Acknowledgments
We thank the members of the Sekelsky lab for helpful comments on the manuscript. S.J.R. was supported by a Thomas S. and Caroline H. Royster, Jr., Graduate Fellowship. This work was supported by a grant from the National Institute of General Medical Science to J.S. (GM61252).
References
- Bardwell, A. J., L. Bardwell, A. E. Tomkinson and E. C. Friedberg, 1994. Specific cleavage of model recombination and repair intermediates by the yeast Rad1-Rad10 DNA endonuclease. Science 265: 2082–2085. [DOI] [PubMed] [Google Scholar]
- Boyd, J. B., M. D. Golino, T. D. Nguyen and M. M. Green, 1976. Isolation and characterization of X-linked mutants of Drosophila melanogaster which are sensitive to mutagens. Genetics 84: 485–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies, A. A., E. C. Friedberg, A. E. Tomkinson, R. D. Wood and S. C. West, 1995. Role of the Rad1 and Rad10 proteins in nucleotide excision repair and recombination. J. Biol. Chem. 270: 24638–24641. [DOI] [PubMed] [Google Scholar]
- Donaldson, T. D., M. A. Noureddine, P. J. Reynolds, W. Bradford and R. J. Duronio, 2004. Targeted disruption of Drosophila Roc1b reveals functional differences in the Roc subunit of Cullin-dependent E3 ubiquitin ligases. Mol. Biol. Cell 15: 4892–4903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drysdale, R. A., M. A. Crosby and FlyBase Consortium, 2005. FlyBase: genes and gene models. Nucleic Acids Res. 33: D390–D395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gloor, G. B., C. R. Preston, D. M. Johnson-Schlitz, N. A. Nassif, R. W. Phillis et al., 1993. Type I repressors of P element mobility. Genetics 135: 81–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houtsmuller, A. B., S. Rademakers, A. L. Nigg, D. Hoogstraten, J. H. Hoeijmakers et al., 1999. Action of DNA repair endonuclease ERCC1/XPF in living cells. Science 284: 958–961. [DOI] [PubMed] [Google Scholar]
- Ivanov, E. L., and J. E. Haber, 1995. RAD1 and RAD10, but not other excision repair genes, are required for double-strand break-induced recombination in Saccharomyces cerevisiae. Mol. Cell. Biol. 15: 2245–2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov, E. L., N. Sugawara, J. Fishman-Lobell and J. E. Haber, 1996. Genetic requirements for the single-strand annealing pathway for double-strand break repair in Saccharomyces cerevisiae. Genetics 142: 693–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James, P., J. Halladay and E. A. Craig, 1996. Genomic libraries and a host strain designed for highly efficient two-hybrid selection in yeast. Genetics 144: 1425–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnstone, R. W., J. Wang, N. Tommerup, H. Vissing, T. Roberts et al., 1998. Ciao 1 is a novel WD40 protein that interacts with the tumor suppressor protein WT1. J. Biol. Chem. 273: 10880–10887. [DOI] [PubMed] [Google Scholar]
- McKim, K. S., J. K. Jang and E. A. Manheim, 2002. Meiotic recombination and chromosome segregation in Drosophila females. Annu. Rev. Genet. 36: 205–232. [DOI] [PubMed] [Google Scholar]
- Oh, J., T. Bailin, K. Fukai, G. H. Feng, L. Ho et al., 1996. Positional cloning of a gene for Hermansky-Pudlak syndrome, a disorder of cytoplasmic organelles. Nat. Genet. 14: 300–306. [DOI] [PubMed] [Google Scholar]
- Park, C.-H., T. Bessho, T. Matsunaga and A. Sancar, 1995. Purification and characterization of the XPF-ERCC1 complex of human DNA repair excision nuclease. J. Biol. Chem. 230: 22657–22660. [DOI] [PubMed] [Google Scholar]
- Pirrotta, V., 1988 Vectors for P-mediated transformation in Drosophila, pp. 437–456 in Vectors: A Survey of Molecular Cloning Vectors and Their Uses, edited by R. L. Rodriquez and D. T. Denhardt. Butterworths, Boston. [DOI] [PubMed]
- Prakash, L., D. Dumais, R. Polakowska, G. Perozzi and S. Prakash, 1985. Molecular cloning of the RAD10 gene of Saccharomyces cerevisiae. Gene 34: 55–61. [DOI] [PubMed] [Google Scholar]
- Rong, Y. S., and K. G. Golic, 2000. Gene targeting by homologous recombination in Drosophila. Science 288: 2013–2018. [DOI] [PubMed] [Google Scholar]
- Rong, Y. S., and K. G. Golic, 2001. A targeted gene knockout in Drosophila. Genetics 157: 1307–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rong, Y. S., S. W. Titen, H. B. Xie, M. M. Golic, M. Bastiani et al., 2002. Targeted mutagenesis by homologous recombination in D. melanogaster. Genes Dev. 16: 1568–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiestl, R. H., and S. Prakash, 1990. RAD10, an excision repair gene of Saccharomyces cerevisiae, is involved in the RAD1 pathway of mitotic recombination. Mol. Cell. Biol. 10: 2485–2491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekelsky, J., K. S. McKim, G. M. Chin and R. S. Hawley, 1995. The Drosophila meiotic recombination gene mei-9 encodes a homologue of the yeast excision repair protein Rad1. Genetics 141: 619–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekelsky, J., K. J. Hollis, A. I. Eimerl, K. C. Burtis and R. S. Hawley, 2000. Nucleotide excision repair endonuclease genes in Drosophila melanogaster. Mutat. Res. 459: 219–228. [DOI] [PubMed] [Google Scholar]
- Stahl, F., 1996. Meiotic recombination in yeast: coronation of the double-strand break repair model. Cell 87: 965–968. [DOI] [PubMed] [Google Scholar]
- Tian, M., R. Shinkura, N. Shinkura and F. W. Alt, 2004. Growth retardation, early death, and DNA repair defects in mice deficient for the nucleotide excision repair enzyme XPF. Mol. Cell. Biol. 24: 1200–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weeda, G., I. Donker, J. de Wit, H. Morreau, R. Janssens et al., 1997. Disruption of mouse ERCC1 results in a novel repair syndrome with growth failure, nuclear abnormalities and senescence. Curr. Biol. 7: 427–439. [DOI] [PubMed] [Google Scholar]
- Yildiz, Ö., S. Majumder, B. C. Kramer and J. Sekelsky, 2002. Drosophila MUS312 interacts with the nucleotide excision repair endonuclease MEI-9 to generate meiotic crossovers. Mol. Cell 10: 1503–1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yildiz, Ö., H. Kearney, B. C. Kramer and J. Sekelsky, 2004. Mutational analysis of the Drosophila repair and recombination gene mei-9. Genetics 167: 263–273. [DOI] [PMC free article] [PubMed] [Google Scholar]