

Abstract
Here we show that exposure of aphidicolin-arrested Chinese hamster ovary (CHO) cells to the protein-kinase inhibitors 2-aminopurine or caffeine results in initiation of replication at successively later-replicating chromosomal domains, loss of the capacity to synthesize DNA at earlier-replicating sites, release of Mcm2 proteins from chromatin, and redistribution of PCNA and RPA from early- to late-replicating domains in the absence of detectable elongation of replication forks. These results provide evidence that, under conditions of replicational stress, checkpoint controls not only prevent further initiation but may also be required to actively maintain the integrity of stalled replication complexes.
In eukaryotes, chromosomes replicate from many origins that are programmed to initiate replication in a precise temporal sequence throughout S phase1–5. Whenever cells are prevented from completing synthesis of early replicons, either by damage-inducing agents or by inhibitors that stall replication forks, such as hydroxyurea or aphidicolin, late-firing origins are prevented from initiating replication6–9. In Saccharomyces cerevisiae, the block to late initiation in cells exposed to mutagens10 or to the elongation inhibitor hydroxyurea8,9 requires the cell-cycle checkpoint genes rad53 and mec1. This ‘intra-S-phase’ checkpoint prevents perpetuation of DNA damage by inhibiting replication in the presence of potentially harmful metabolites, and prevents cells from entering mitosis before completion of DNA replication under conditions of replicational stress11–13.
We are interested in what regulates the replication-timing programme in mammalian cells. In principle, an intra-S-phase checkpoint could influence this programme if replication forks that are normally assembled early in S phase trigger an inhibitory signal that delays initiation at later-firing origins. If this were the case, then checkpoint inhibition would be expected to significantly accelerate initiation at late-replicating origins even in the absence of replicational stress. In S. cerevisiae rad53 mutants, a modestly advanced initiation time has been reported for ARS609 (ref. 10), whereas another study did not detect any change in the late initiation programme for ARS305 (ref. 8). In any case, a response that requires the presence of preformed replication forks must function after S phase has begun, and thus cannot be responsible for establishing the order in which origins will fire but can only modulate a pre-existing programme. Indeed, in both S. cerevisiae14 and mammalian4 cells, the replication-timing programme is established very early in G1 phase.
Here we investigate the extent to which checkpoint control contributes to the replication-timing programme in mammalian cells. Mammalian cells arrested with inhibitors of DNA synthesis will enter mitosis when treated with protein-kinase inhibitors such as 2-aminopurine (2-AP)15,16 or caffeine17. Caffeine has been shown to inhibit the ATM (ataxia-telangiectasia-mutated) and ATR (ataxia-telangiectasia-related) genes18–20, which are the mammalian homologues of mec1 and rad53. Here we determine whether these kinase inhibitors might also interfere with the checkpoint mechanism that prevents initiation within late-replicating sequences and whether interference with this checkpoint can alter the normal replication-timing programme. To do so, we have developed ways of monitoring the dynamics of whole groups of coordinately replicated chromosomal domains, rather than individual replication origins4,21. Our results support the existence of such a checkpoint pathway in mammalian cells, and also indicate that the temporal program for replication may coordinate the assembly of replication factories at later-replicating sites with the disassembly of replication factories at earlier-replicating sites.
Results
Exposure of aphidicolin-arrested cells to 2-AP results in initiation at late origins.
DNA replication takes place at discrete sites that can be visualized by pulse-labelling cells with 5-bromo-2′-deoxyuridine (BrdU) and staining nuclei with fluorescent antibodies against BrdU22. The spatial pattern of replication sites reveals their temporal position during S phase. The progression of these spatial patterns during the course of S phase in CHO (CHOC 400) cells has previously been described in detail4. Briefly, during the first 6 h of S phase, replication takes place within many foci distributed throughout the euchromatic regions in the nuclear interior (type I/II patterns). Several sets of these early-replicating foci are activated over the course of the first half of S phase, each completing DNA synthesis in ~60 min. In mid-S-phase, replication takes place at the nuclear periphery and in nucleolar regions for 2–3 h (type III). In the final few hours of S phase (9–12 h), replication occurs first within several relatively large foci that are present throughout the nucleus (Type IV) and subsequently within a very few large internal or peripheral foci (Type V).
To determine whether the administration of checkpoint inhibitors to cells arrested in early S phase with aphidicolin would allow the initiation of replication at later-replicating sites, we synchronized CHO cells in mitosis and allowed them to accumulate at the G1/S border in the presence of aphidicolin for 12–14 h. Aphidicolin inhibits the processive elongation of nascent DNA strands but does not prevent the initiation of replication and the formation of short (100–500-base-pair (bp)) primers23. Thus, cells treated in this manner accumulate with replication forks arrested close to their sites of initiation. The primers and the proteins that carry out DNA synthesis remain at these sites, which can subsequently be revealed by removing the aphidicolin and allowing limited extension of nascent DNA strands in the presence of nucleotide analogues. When aliquots of these cells were washed free of aphidicolin, briefly pulse-labelled with BrdU, and stained with anti-BrdU antibodies, >80% of nuclei were labelled and 100% of labelled nuclei exhibited the type I/II pattern (Fig. 1a (0 h) and 1c). In the continued presence of aphidicolin, we treated half of the cultures with 2-AP and then cultured cells for a further 26 h. At various time points, we washed the aphidicolin from aliquots of both cell populations and pulse-labelled them with BrdU. In the absence of 2-AP, the percentage of cells labelled and the pattern of BrdU labelling remained constant throughout the 26-h time period (Fig. 1a, c). In contrast, in the presence of 2-AP, late replication patterns appeared after 9–12 h and after 15 h, nearly all labelled nuclei had initiated DNA synthesis within late-replicating type III, IV and V chromosomal domains (Fig. 1b). After 20 h in the presence of 2-AP, cell nuclei began to bleb (Fig. 1b) and chromatin became condensed. At this time, the percentage of nuclei that were labelled with BrdU dropped markedly (Fig. 1b, c) and many nuclei were fragmented or pulverized, possibly indicating an apoptotic response, triggered by the entry of cells into G2/M-phase in the presence of unreplicated genomes. To verify that aphidicolin effectively arrested DNA synthesis throughout the duration of these experiments, we analysed the DNA content of each cell population by flow cytometry (Fig. 1d). These results showed that, at the aphidicolin concentration typically used to arrest cells at the G1/S border (10 μg ml−1), many cells were able to synthesize a detectable amount of DNA over the course of the further 26 h of incubation. However, when the aphidicolin concentration was increased to 50 μg ml−1, cells were effectively prevented from synthesizing DNA to an appreciable extent. As the sequence of initiation events shown in Fig. 1a, b was indistinguishable at these two aphidicolin concentrations, we used the higher concentration for all further experiments. These results indicate that treatment of CHO cells with 2-AP allows late-replicating chromosomal domains to initiate replication in the absence of detectable DNA synthesis.
Figure 1. Treatment of aphidicolin-arrested cells with 2-AP results in the appearance of late-replication patterns.
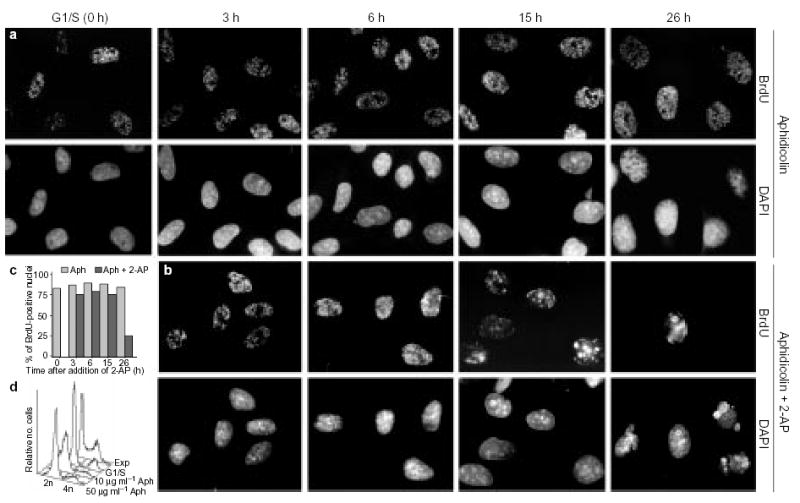
a, b, CHOC 400 metaphase cells were released in fresh medium and arrested at the G1/S border (0 h) in the presence of 50 μg ml−1 aphidicolin (Aph). At this point, 2-AP was added to half of the cell cultures (b), whereas the other half (a) was maintained in aphidicolin only. At the indicated time points, aliquots of cells were washed free of the drugs, pulse-labelled for 10 min with BrdU and stained for BrdU (see Methods). DNA was stained with 4′,6-diamidino-2-phenylindole (DAPI). c, Histogram showing the percentage of BrdU-positive nuclei in a (hatched bars) and b (black bars). d, Flow-cytometry analysis of the DNA content of asynchronously growing cells (Exp), cells arrested at the G1/S boundary, and cells incubated for a further 26 h in the presence of 2-AP and either 10 μg ml−1 or 50 μg ml−1 aphidicolin. As the results shown in a and b were indistinguishable at these two aphidicolin concentrations, the higher concentration was used for all further experiments to prevent any detectable DNA synthesis.
To verify that the late replication patterns observed were not a consequence of spatial rearrangement of early-replicating domains during drug treatment, we devised a protocol to distinguish between new initiation events in either early- and late-replicating domains (Fig. 2a). This protocol takes advantage of the ability to distinguish between DNA labelled either with 5-chloro-2′-deoxyuridine (CldU) or with 5-iodo-2′-deoxyuridine (IdU) using specific antibodies24. We tagged either the earliest- (onset of S phase) or the late-replicating (second half of S phase) DNA sequences by pulse-labelling CHO cells with CldU in one cell cycle4. We then arrested these separate populations of prelabelled cells at the subsequent G1/S border with aphidicolin and then incubated them in the presence or absence of 2-AP (in the continued presence of aphidicolin) for various time periods up to 30 h. After removal of aphidicolin from the medium, we pulse-labelled initiation sites activated during the aphidicolin block with IdU, and stained cells with anti-IdU (red) and anti-CldU (green) antibodies4,21,24. By comparing the pattern of IdU labelling to the distribution of sites of earliest or late replication that were tagged with CldU in the previous S-phase, we could determine whether the administration of 2-AP or caffeine during the aphidicolin arrest allowed the normally later-initiating replicons to fire. We have previously shown that a similar prelabelling protocol does not disturb the replication-timing programme or the selection of origin sites at the dihydrofolate reductase (DHFR) locus4.
Figure 2. Treatment of aphidicolin-arrested cells with 2-AP reveals the temporal program for replication.
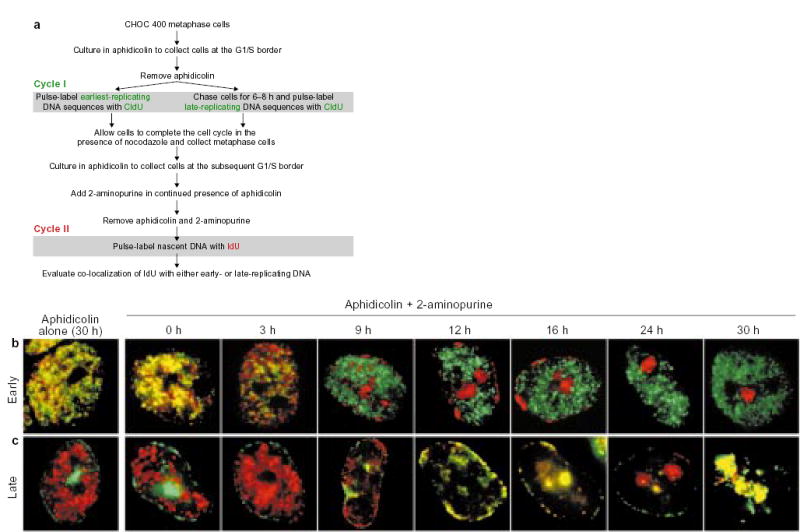
a, Summary of the protocol. b, c, Parallel cultures of CHOC 400 cells were prelabelled with CldU in cycle I and synchronized at the G1/S border of cycle II as outlined in a. Each culture was divided into two and 2-AP was added to one half (Aphidicolin + 2-aminopurine), whereas the other half was maintained in aphidicolin alone. At the indicated times, aliquots of cells were washed free of the drugs, labelled for 10 min with IdU, and then fixed and stained with anti-CldU (green) and anti-IdU (red) antibodies as described4. Images show typical nuclei observed at the indicated time points with cells that were prelabelled either early (b) or late (c) in the S phase of cycle I. As the cultures maintained in aphidicolin alone showed co-localization of IdU with the earliest-replicating sequences throughout the entire duration of the experiment (see Fig. 3c, black squares), only the 30-h time point is shown. Similar results were obtained in five independent experiments.
Figure 2b, c shows that, in the absence of 2-AP, cells were arrested at the G1/S border for at least 30 h without any evidence of progression through S phase or initiation within late-replicating domains. Under these conditions, IdU labelling immediately after release from the aphidicolin block resulted in the exclusive incorporation of IdU into the earliest-replicating domains, as shown by the nearly complete co-localization of the IdU label with the earliest CldU label (Fig. 2b) and by the complete lack of co-localization of the IdU label with the late-S-phase CldU label (Fig. 2c). However, if 2-AP was added during the aphidicolin block for various lengths of time, the IdU label was incorporated into progressively later-replicating domains, in their proper temporal order. In cells incubated with 2-AP for 2–9 h, incorporation of the IdU label took place within type I/II foci that were distinct from the earliest-replicating, CldU-labelled foci (Fig. 2b). Exposure of cells to 2-AP for >9 h resulted in incorporation of IdU label within type III, IV and V foci (Fig. 2b, c) and co-localization of the IdU label with late-replicating, CldU-labelled sequences (Fig. 2c).
Checkpoint inhibition results in temporally coordinated firing of replicons in the absence of DNA synthesis.
To quantitatively compare the time schedule of activation of progressively later replication foci under different conditions, we scored the percentage of S-phase nuclei (IdU-positive) that incorporated IdU into either early- (type I/II), middle- (type III) or late-replicating (type IV/V) patterns. In cells synchronized at the G1/S border with aphidicolin and then released (Fig. 3a), type III replication patterns were evident ~6 h after release, late patterns after 8–10 h and, after 24 h, cells had completed one cell cycle and were entering a subsequent S phase. When cells were released from the aphidicolin block and incubated in the presence of either 2-AP (Fig. 3b) or caffeine (Fig. 3d) alone, cells proceeded through S phase in a similar manner, although at about half the rate of cells incubated in the absence of 2-AP or caffeine. Most nuclei exhibited exclusively early replication patterns for the first 6–9 h, predominantly middle-replication patterns at 12–20 h, and, at 16–36 h, a significant number of nuclei exhibited late patterns. Strikingly, when cells were incubated in the continued presence of both aphidicolin and either 2-AP (Fig. 3c) or caffeine (Fig. 3e), the temporal order of initiation followed precisely the same schedule as was observed in cells released into S phase in the presence of 2-AP or caffeine alone. Two control experiments confirmed that the late replication observed was initiated without the completion of early replication. First, no incorporation of IdU was detected even after 30 min of labelling when aphidicolin was not removed from the medium (data not shown), whereas IdU pulses as short as 1 min were readily detected after removal of aphidicolin. Second, flow-cytometry analysis (Fig. 3f) showed that cells still had a DNA content characteristic of G1 phase, even after 26 h of treatment with 2-AP or caffeine in the presence of aphidicolin. These controls show that 2-AP and caffeine did not allow cells to escape the aphidicolin block and progress through a normal S phase. We carried out parallel experiments with the protein-kinase inhibitor roscovitine, which inhibits several cyclin-dependent kinases, including Cdk2, which has been implicated in the initiation of replication25. In contrast to the results with 2-AP and caffeine, when cells were incubated in the continued presence of aphidicolin and roscovitine, the ability to incorporate IdU label rapidly declined (Fig. 3c). Hence, administration of protein-kinase inhibitors known to inhibit checkpoint kinases, but not a CDK inhibitor, results in initiation of replication at late-firing origins in cells blocked at the onset of S phase with an inhibitor of DNA synthesis.
Figure 3. Temporally coordinated firing of replicons in the absence of DNA synthesis.
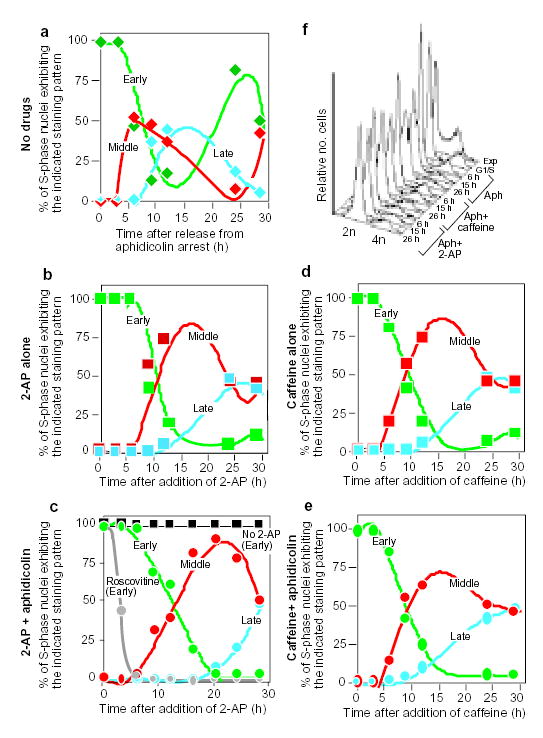
CHOC 400 cells were prelabelled with CldU in cycle I, synchronized at the G1/S border of cycle II as outlined in Fig. 2a, and then divided into aliquots that were treated in parallel as follows. a, Cells were washed free of aphidicolin and allowed to proceed into S-phase in the complete absence of drugs. b, d, Cells were washed free of aphidicolin and allowed to proceed into S phase in the presence of 2-AP (b) or caffeine (d) alone. c, e, Cells were maintained in the continuous presence of aphidicolin in combination with 2-AP (c) or caffeine (e). At the indicated times, cells were washed free of drugs, labelled for 10 min with IdU, and then fixed and stained with anti-CldU and anti-IdU antibodies as in Fig. 2. Percentages of S-phase (IdU-positive) nuclei showing co-localization with either early (type I/II, green), middle (type III, red) or late (type IV and V, blue) CldU-staining patterns were scored at each time point. As controls, aliquots of cells were maintained in aphidicolin alone (c, black squares), or in combination with roscovitine (c, grey circles). Cells maintained in aphidicolin alone showed the same labelling index and exclusively early replication patterns at all time points (see also Fig. 2b, c, aphidicolin alone). When roscovitine was used together with aphidicolin, only the earliest replication patterns were observed and the percentage of IdU-positive nuclei declined with time. f, Aliquots of the cells from a–e were collected at the indicated times and DNA content was measured by flow cytometry. Results were obtained from exponentially growing cells (Exp), cells at 0 h (G1/S), and cells from a (Aph), c (Aph + 2-AP) and e (Aph + caffeine) at the indicated time points. Similar results were obtained in three independent experiments.
Initiation at new sites is accompanied by disassembly of earlier replication factories.
In G1/S cells incubated with aphidicolin and either 2-AP or caffeine, DNA synthesis at progressively later-replicating sites was accompanied by a lack of DNA synthesis from previously initiated sites (Figs 1–3). This finding was unexpected, as earlier-replicating sites were expected to retain their preprimed templates as a result of the continued presence of aphidicolin. Thus, treatment with either caffeine or 2-AP not only allows late-replicating origins to fire but causes early replicons to lose the capacity to complete replication. To determine whether caffeine or 2-AP directly inhibits the progression of replication forks, we introduced the nuclei of CHO cells into an in vitro replication cocktail and determined the effect of these drugs on the rate of DNA synthesis. Neither caffeine, 2-AP nor roscovitine had any detectable effect on elongation of replication forks (Fig. 4a). Furthermore, treatment of cultured cells in vivo with 2-AP or caffeine for up to 6 h before in vitro replication had no effect on the rate of DNA synthesis (Fig. 4b, c). In contrast, nuclei from cells treated with roscovitine synthesized DNA poorly, indicating that they contained significantly fewer active replication forks, which is consistent with the results shown in Fig. 3c. These results show that 2-AP and caffeine have no direct effect on the progression of replication forks.
Figure 4. 2-AP and caffeine do not directly inhibit DNA synthesis.
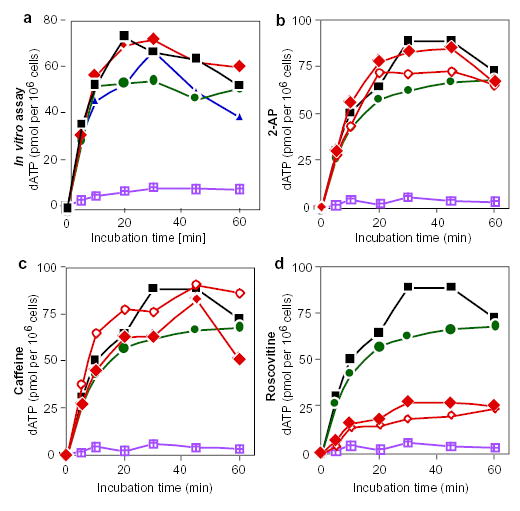
a, Replication run-on assay with drug treatment in vitro. Intact nuclei were prepared from an asynchronous culture of cells and incubated in a replication cocktail supplemented with [α-32P]-dATP in the presence of 2-AP (diamonds), caffeine (circles) or roscovitine (triangles). At the indicated time points, aliquots were removed and the amount of acid-precipitable [α-32P]-dATP was determined. Only extension of pre-existing replication forks occurs under these conditions. Black squares represent control samples without drugs; violet-hatched squares represent nuclei incubated in vitro in the presence of aphidicolin. Similar results were obtained in three independent experiments. b–d, Replication run-on assays after treatment with 2-AP (b), caffeine (c) or roscovitine (d) in vivo. Asynchronous cultures were incubated for 6 h in the presence of aphidicolin alone (circles), the indicate drug alone (filled diamonds), or the indicated drug in combination with aphidicolin (open diamonds). Intact nuclei were prepared from each cell culture and DNA synthesis was determined as in a. Black squares represent untreated control cultures; violet-hatched squares are reactions carried out in the presence of aphidicolin in vitro.
To investigate whether replication proteins are disassembled from arrested early replication forks when later replication origins fire, we monitored the assembly of RPA (Fig. 5a, b) and PCNA (data not shown) into replication foci in the presence of aphidicolin and of 2-AP or caffeine. Both RPA and PCNA have been shown to form distinct, Triton X-100-resistant foci minutes before initiation of detectable DNA synthesis and to co-localize exclusively with sites of DNA synthesis21,26. In the absence of 2-AP, release of the aphidicolin block always resulted in co-localization of RPA and PCNA exclusively with the earliest-replicating CldU-labelled sequences. Exposure of aphidicolin-arrested cells to 2-AP resulted in the assembly of RPA- and PCNA-containing replication factories at progressively later-replicating domains, following the schedule of DNA synthesis described in Fig. 3c. Consistent with the abandonment of earlier replication forks upon initiation within late-replicating domains, RPA and PCNA no longer co-localized with early-replicating domains in nuclei that exhibited mid- or late-replicating IdU labelling patterns. Similar results were obtained using caffeine-treated cells (data not shown).
Figure 5. Inhibition of the S-phase checkpoint is accompanied by redistribution of replication proteins.
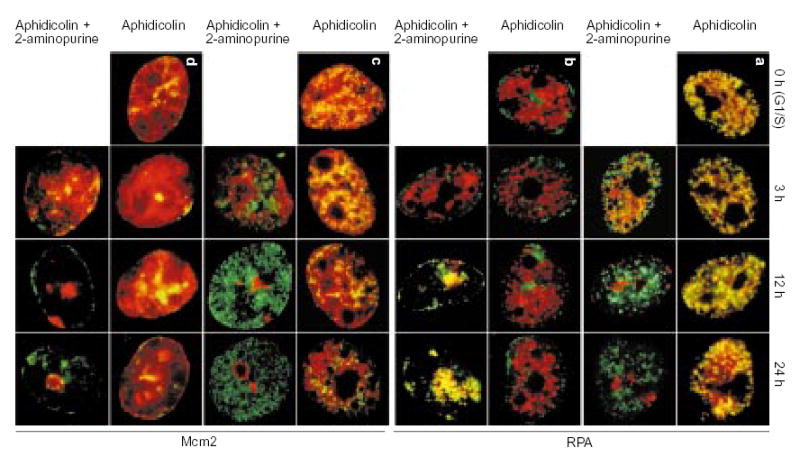
Aliquots of cell cultures, pre-abelled with CldU as in Fig. 2a, were synchronized at the G1/S border of cycle II and maintained in the continuous presence of aphidicolin with or without addition of 2-AP. At the indicated times, aliquots of cells were incubated for 10 min in free medium to match the pulse-labelling time in Fig. 2b, c, fixed and double-stained for CldU (green) and either RPA (a, b; red) or Mcm2 (c, d; red). The distribution of earliest-replicating, CldU-prelabelled chromosomal domains is shown in a and c; the distribution of of mid/late-replicating, CldU-prelabelled chromosomal domains is shown in b and d.
In mammalian cells, Mcm2 proteins, which are present in the prereplication complex (pre-RC), bind to both early- and late-replicating chromatin domains very early in G1 phase and are subsequently cleared from each set of domains shortly after initiation of replication21. This DNA cannot be re-replicated until new Mcm proteins are loaded back onto chromatin in the subsequent telophase, ensuring that DNA is replicated once per cell cycle. We have shown that Mcm2 proteins remain bound throughout an early-S-phase aphidicolin block and are not cleared from even the earliest-replicating chromatin until replication is allowed to proceed beyond this block21. To determine whether Mcm2 proteins were cleared from chromatin in the absence of replication when aphidicolin-arrested cells were treated with 2-AP, we stained the cells shown in Fig. 5a, b with Mcm2-specific antibodies (Fig. 5c, d). In the absence of 2-AP, Mcm2 remained bound to early-, middle-and late-replicating chromatin throughout the duration of aphidicolin arrest (although some clearing in the nuclear interior was observed at late time points). In contrast, after addition of 2-AP, Mcm2 was first cleared from the earliest-replicating chromatin and was subsequently cleared from middle- and late-replicating domains at the time of initiation. Similar results were obtained for caffeine (data not shown). Hence, when cells arrested with aphidicolin are treated with 2-AP or caffeine, they proceed through a series of molecular events that parallel the progression of normal S phase, including clearing of pre-RC proteins and redistribution of RPA and PCNA to sites at which replication is scheduled to take place.
Arrested replication forks lose the capacity to be elongated.
The results presented above indicate that a mechanism to maintain the integrity of stalled replication forks may be inhibited by the presence of 2-AP and caffeine, resulting in disengagement of replication proteins from the arrested early replication forks. To determine independently whether or not early replication forks remain arrested and lose the capacity to elongate nascent DNA upon initiation at later sites, we isolated nuclei from cells at various times after the administration of 2-AP and allowed them to synthesize DNA from preprimed templates in the presence of a replication cocktail containing [α-32P]dATP. We then hybridized the resulting radioactively labelled nascent DNA strands to a panel of probes derived first from various positions encompassing 120 kilobases (kb) of the CHO DHFR locus, and second from several repetitive sequences known to replicate at either early or late time points during the CHO S phase4,27 (Fig. 6). In accordance with earlier reports7,28,29, the amplified DHFR locus was found to replicate throughout S phase, with the maximum amount of replication intermediates hybridizing to DHFR probes at ~3 h into S phase (Fig. 6a, d). In the absence of 2-AP, DNA synthesis within aphidicolin-arrested nuclei at the G1/S border always took place within early-replicating chromatin and replication forks remained arrested close to the DHFR replication origins (Fig. 6b, f, h). However, several hours after administration of 2-AP, incorporation of label at the DHFR locus and at other early-replicating sequences diminished, an event that was simultaneous with the appearance of label in late-replicating DNA sequences (Fig. 6c, e). Notably, even as the amount of label began to diminish in the DHFR locus after addition of 2-AP (see, for example, Fig. 6c, e, 15 h), the remaining active replication forks were still arrested close to their sites of initiation (Fig. 6i). Hence, when aphidicolin-arrested cells were treated with 2-AP, replication forks were gradually abandoned by the enzymes required to elongate these forks.
Figure 6. Initiation at late-firing origins is accompanied by abandonment of primers at early origins.

a–c, Cells synchronized at the G1/S border were either released in drug-free medium (a), or maintained in the continuous presence of aphidicolin alone (b) or aphidicolin in combination with 2-AP (c). Nuclei were prepared from these cells at the indicated times and nascent DNA was briefly labelled with [α-32P]-dATP in a replication cocktail as described4,38. As a control, nuclei from exponentially growing cells (Exp) were similarly labelled. Radiolabelled DNA was isolated, sheared and hybridized to a panel of probes representing sequences in the CHOC 400 genome that replicate either early (DHFR), or late (LINES C1 and C3) in S phase. d, e, The relative amounts of hybridization to DHFR probe R (squares), as well as to the C1 (circles) and C3 (diamonds) probes at each time point in a (d) and c (e) were calculated and plotted (see Methods). f–i, Relative amounts of hybridization to all 17 DHFR probes38,39 for the 3-h and 15-h time points in b and c were plotted against the map position of each probe. The horizontal axis includes a diagram of the genomic region encompassed by these probes, including the positions of the DHFR and 2BE2121 loci37. Vertical shaded line shows the positions of probes B–R, which encompass the region of peak initiation activity.
ATM-deficient cells retain checkpoint control in response to stalled replication forks.
Caffeine is known to inhibit the checkpoint kinase ATM. Cell lines that express inactive forms of the ATM kinase have been shown to continue synthesizing DNA after γ-irradiation30. To determine whether ATM is responsible for the checkpoint that is activated in response to aphidicolin, we pulse-labelled ATM-deficient cells with CldU and then incubated them in the presence of aphidicolin for various lengths of time up to 60 h. We then released the cells from the aphidicolin block and pulse-labelled them with IdU. As shown in Fig. 7a, DNA synthesis in ATM-deficient cells arrested with aphidicolin was due to the continuation of replication forks established before the aphidicolin block, even after 60 h of incubation. As these cells are primary fibroblasts, it was important to show that they did not senesce during the incubation period. Hence, as a control, we incubated parallel CldU-labelled cultures for the same lengths of time in the absence of aphidicolin, and found them to proceed through more than 2 cell cycles within the 60-h incubation period (Fig. 7b). These results show that ATM is not required for the block to late initiation in the presence of replication forks that have been stalled by aphidicolin.
Figure 7. ATM is not involved in the replication checkpoint.

Two asynchronous cultures of AT8052 cells were pulse-labelled for 10 min with CldU, aphidicolin (Aph) was added to one of them (a), and both cultures were grown for a further 60 h. At various time points after CldU labelling (0, 6, 12, 24, 36, 48 and 60 h), aliquots of cells from each culture were washed free of drugs, pulse-labelled with IdU, and then fixed and stained with anti-CldU (green) and anti-IdU (red) antibodies as in Fig. 2. In the presence of aphidicolin, cells were arrested in S phase and the IdU label co-localized with the CldU label in nuclei at all stages of S phase (panels 1 and 3 show examples of early-S-phase nuclei; panel 2 shows a late-S-phase nucleus) throughout the entire chase period. As AT8052 is a primary cell line that could potentially enter into senescence, it was important to verify that cells would have continued to cycle during the chase period. In the absence of aphidicolin (b), the control cell culture grew normally. The size of the nuclei and the type of the IdU replication pattern were used as criteria to evaluate the progression of cells through the cell cycle. During cycle I, the CldU and IdU labelling patterns co-localized at 0 h (panel 4), but had become separated by 6 h (panels 5 and 6) and, at 12 h, large, IdU-negative G2 nuclei (panel 7) and mitotic figures (panel 8) were observed. The appearance of small, CldU-labelled, IdU-negative daughter nuclei (panel 9), often positioned adjacent to each other, indicated progression of cells into the G1 phase of cycle II by 24 h. Later, at 24–48 h, double-labelled S-phase nuclei (panels 10–12) and large, CldU-labelled G2 nuclei (panel 13) re-appeared as the cells moved through cycle II. The appearance of small, IdU-negative nuclei with gaps (arrows) in the CldU labelling pattern (panel 14) indicated that, by 48 h, cells had progressed into cycle III. Gaps correspond to the territories of unlabelled chromosomes40,41, which were generated after two cell divisions (the mechanism responsible for generation of unlabelled chromosome territories is schematically outlined in the two lower-right boxes). Nuclei with unlabelled chromosome territories were also found to have entered S phase (panels 15–17) and G2 phase (panel 18) of cycle III by the 60-h time point.
Discussion
We have shown that administration of caffeine or 2-AP to S-phase-arrested mammalian cells results in initiation of replication at late-replicating origins. These two protein kinase inhibitors have been shown to override checkpoints in mammalian cells and, at least in the case of caffeine, to inhibit checkpoint kinases directly15–20. These results support the idea that an S-phase checkpoint in mammalian cells prevents the initiation of late-replicating sequences in the presence of stalled replication forks, as has been shown in S. cerevisiae8,9. Although we cannot be certain which gene products are targeted by 2-AP and caffeine to abrogate the checkpoint, recent evidence obtained from yeast indicates that the ‘intra-S-phase’ checkpoint may be mediated by the Rad53/Mec1 protein kinases9–11. Mec1 belongs to the same subfamily of proteins as the ATM gene product in humans, and both human ATM and yeast mec1 mutants exhibit a radioresistant DNA-synthesis phenotype31. However, when we arrested human ATM cells (AT8052) with aphidicolin, we found no evidence of initiation of replication in later replication domains (Fig. 7), showing that the block to S-phase progression when replication forks are stalled does not require ATM. In yeast, the checkpoints that respond to DNA damage and to stalling of replication forks are both transduced through Rad53 and Mec1 (refs 9, 10, 32). However, it is possible that, in mammalian cells, molecules that transduce checkpoint signals from damaged DNA may be different to those that transduce signals from stalled replication forks. Caffeine inhibits both ATM and the related ATR protein kinase18–20. Expression of a kinase-inactive ATR mutant in mammalian cells can render them deficient in regulated phosphorylation of p53 in response to ultraviolet and γ-irradiation33. In contrast, ATM-disrupted cells are unable to regulate p53 levels in response to γ-irradiation but not ultraviolet34. Thus, ATM and ATR may have overlapping, but not redundant, functions and may recognize different forms of DNA damage. Through a combination of recently developed in vitro assays of substrate recognition35 and the application of in vivo protocols, such as those described in this report, to the study of cell lines that harbour mutations in these and related proteins, the checkpoint pathways that respond to various types of DNA damage should soon be elucidated.
Intriguingly, we found that initiation of replication in late-replicating domains in the presence of 2-AP or caffeine was accompanied by a loss of the ability to synthesize DNA at earlier, previously initiated sites. In the absence of these inhibitors, replication forks remained poised for elongation. If checkpoint inhibition simply relieved the block to initiation of late-replicating origins, then it should have resulted in gradual accumulation of replication forks at early-, middle- and late-replicating chromosomal sites. Instead, checkpoint inhibition resulted in the removal of both pre-RC proteins and replication-fork proteins from earlier sites and the inability to extend nascent DNA strands that remained arrested close to their sites of initiation. In our experiments, checkpoint inhibition was mediated by fairly broad-based protein-kinase inhibitors. It is therefore possible that both 2-AP and caffeine act simultaneously on independent mechanisms that regulate initiation at late-replicating sites as well as the breakdown of replication forks at pre-existing sites. Alternatively, it is possible that 2-AP and caffeine act primarily to break down replication forks, resulting in loss of the inhibitory signal. We feel that these latter interpretations are unlikely for several reasons. First, caffeine is known to inhibit checkpoint kinases; second, neither 2-AP nor caffeine had a direct effect on the integrity of replication forks; third, cells completed S phase in the presence of 2-AP and caffeine (albeit at a reduced rate); and fourth, the programme for replication in the presence of either 2-AP or caffeine proceeded according to the same temporal plan with or without aphidicolin. We propose instead that checkpoint activation in the presence of stalled replication forks not only prevents initiation at new sites but also maintains the integrity of the stalled replication forks. If correct, such a mechanism would have important implications for maintaining genome stability under conditions of replicational stress. The disintegration of stalled replication forks, coupled with the loss of Mcm proteins from these sites, would completely prevent cells from replicating those portions of the genome. A means to maintain the primers and replication proteins at stalled replication forks would allow the cell to complete replication of the genome when favourable conditions are reinstated.
We found no evidence of advancement of the replication-timing programme after abrogation of checkpoint control with either caffeine or 2-AP in the absence of inhibitors of DNA synthesis. Hence, the ‘intra-S-phase’ checkpoint is unlikely to be a principal contributor to the delay between initiation events that is observed during the normal progression of S phase. These results are consistent with the previous finding9 that a late-firing yeast origin initiates replication at the same time in both wild-type and rad53 or mec1 mutants. However, this conflicts with the observation that the timing of initiation of another late-firing origin was modestly advanced in a rad53 mutant10. This discrepancy could be explained if experimental conditions imposed a replicational stress that delayed the firing of this origin in the wild-type strain, or if Rad53 has different effects on different origins. In this study, we examined whole groups of coordinately replicated chromosomal domains, giving a more general picture of the replication-timing programme. However, exceptional origins would have escaped our detection. Thus, our results indicate that the ‘intra-S-phase’ checkpoint may function to arrest a pre-established replication-timing programme under conditions of replicational stress, but may not normally have an important function in the timing of origin firing.
The data presented here also indicate potential new methods for mapping mammalian replication origins that initiate replication during late S phase. Mammalian genomes are complex, and many origin-mapping techniques rely on precise synchrony of cells at the time of origin firing36. The use of inhibitors of replication elongation to arrest cells at the onset of S phase has proven invaluable for studies of early-firing replication origins. However, as the ‘intra-S-phase’ checkpoint prevents initiation at later-firing origins in the presence of such inhibitors, these techniques have been limited to the study of early-firing replication origins. Hence, inhibition of this checkpoint may allow the staging of mammalian cells at the time of initiation of late origins, providing a potential means to use those origin-mapping techniques that rely on precise synchrony to the study of late-initiating origins. □
Methods
Cell culture, synchronization and immunofluorescent labelling.
CHOC 400 cells, a derivative of CHO cells in which the DHFR locus has been amplified ~500-fold by stepwise selection in methotrexate37, were maintained and synchronized in mitosis or at the G1/S border as described4,21. AT8052 cells were a gift from P. Hahn (SUNY Upstate Medical University) and were maintained in DMEM (Gibco) supplemented with nonessential amino acids and 20% FBS (Gibco) at 37 °C in a 5% CO2 atmosphere. For checkpoint-inhibition experiments, CldU-tagged cells grown on coverslips were arrested at the G1/S border of the second cell cycle in 10 μg ml−1 aphidicolin, as described4,21. At that time, the concentration of aphidicolin was increased to 50 μg ml−1, coverslips with cells were divided into two groups and 2-AP (10 mM; Sigma), caffeine (5 mM; Sigma) or roscovitine (100 μM; Calbiochem) were added to half of the cultures. At various times thereafter, coverslips with cells were transferred to warm drug-free medium supplemented with 10 μM IdU and incubated for 10 min. Cells were fixed and stained for CldU, IdU or replication proteins as described4,21,26. For the experiment shown in Fig. 1, cells were pulse-labelled with 30 μg ml−1 BrdU (Sigma) for 10 min. Immunofluorescence microscopy was carried out as described21.
Flow-cytometry analysis.
Cells were collected by trypsinization, washed with PBS, fixed with cold 70% ethanol and stored at 4 °C until needed. Immediately before analysis, cells were resuspended in PBS containing 1 mg ml−1 RNase A (Sigma) and incubated for 30–60 min at 37 °C. Cells were then collected by centrifugation, resuspended in propiduim iodide (20 μg ml−1; Sigma) and analysed on a FACScan (Becton Dickinson).
Replication run-on assays.
For the experiments described in Fig. 6a, aphidicolin-arrested cells were released from the block for the indicated times and cells were permeabilized with digitonin as described4. For those described in Fig. 6b, c, cells were maintained in the continuous presence of aphidicolin (Fig. 6b, 50 μg ml−1) or aphidicolin and 2-AP (Fig. 6c, 50 μg ml−1 and 10 mM, respectively), washed with PBS, collected without release from drug treatment and permeabilized as above. Replication intermediates in 107 nuclei from each time point were then radioactively labelled by in vitro run-on replication in the presence of [α-32P]-dATP as described4,38. Equal numbers of c.p.m. from each sample were hybridized to a panel of DNA plasmids (1 μg each) immobilized on nylon membranes (Hybond N+, Amersham). Relative c.p.m. values were obtained by phosphorimaging (Molecular Dynamics). For the plots in Fig. 3f–i, relative c.p.m. values for each probe were normalized to the corresponding values from parallel hybridizations with replication intermediates from exponentially growing CHOC 400 cells, labelled as described38. This was done to correct for differences in probe size, deoxyadenine content and hybridization efficiency. To express the relative capacity to extend replication forks in early- or late-replicating sequences at different time points (Fig. 3d, e), background c.p.m. values from DNA hybridized to the λ-phage probe were subtracted from c.p.m. values from DNA hybridized to the DHFR (R), C1 or C3 probes, respectively, and the resulting values for each probe were normalized to the lowest value among the different time points.
For run-on assays described in Fig. 4, 106 cells were used per assay and the in vitro replication cocktail38 was supplemented with 50 μCi [α-32P]-dATP (6,000 Ci mM−1; NEN, Boston, Massachusetts) and 50 μM dATP.
Acknowledgments
We thank J. Chen for technical assistance with flow cytometry, and P. Hahn, A. McNairn and B. Knox for critical reading of the manuscript. This work was supported by NIH grant GM57233-01 (to D.M.G.).
References
- 1.Gilbert DM. Temporal order of replication of Xenopus laevis 5S ribosomal RNA genes in somatic cells. Proc Natl Acad Sci USA. 1986;83:2924–2928. doi: 10.1073/pnas.83.9.2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hatton KS, et al. Replication program of active and inactive multigene families in mammalian cells. Mol Cell Biol. 1988;8:2149–2158. doi: 10.1128/mcb.8.5.2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ma H, et al. Spatial and temporal dynamics of DNA replication sites in mammalian cells. J Cell Biol. 1998;143:1415–1425. doi: 10.1083/jcb.143.6.1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dimitrova DS, Gilbert DM. The spatial position and replication timing of chromosomal domains are both established in early G1-phase. Mol Cell. 1999;4:983–993. doi: 10.1016/s1097-2765(00)80227-0. [DOI] [PubMed] [Google Scholar]
- 5.Diffley JF. Replication control: choreographing replication origins. Curr Biol. 1998;8:R771–R773. doi: 10.1016/s0960-9822(07)00483-6. [DOI] [PubMed] [Google Scholar]
- 6.Hamlin JL. Effect of damage to early, middle, and late-replicating DNA on progress through the S period in Chinese hamster ovary cells. Exp Cell Res. 1978;112:225–232. doi: 10.1016/0014-4827(78)90204-5. [DOI] [PubMed] [Google Scholar]
- 7.Larner JM, Lee H, Hamlin JL. Radiation effects on DNA synthesis in a defined chromosomal replicon. Mol Cell Biol. 1994;14:1901–1908. doi: 10.1128/mcb.14.3.1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Santocanale C, Sharma K, Diffley JF. Activation of dormant origins of DNA replication in budding yeast. Genes Dev. 1999;13:2360–2364. doi: 10.1101/gad.13.18.2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Santocanale C, Diffley JF. A Mec1- and Rad53- dependent checkpoint controls late-firing origins of DNA replication. Nature. 1998;395:615–618. doi: 10.1038/27001. [DOI] [PubMed] [Google Scholar]
- 10.Shirahige K, et al. Regulation of DNA-replication origins during cell-cycle progression. Nature. 1998;395:618–621. doi: 10.1038/27007. [DOI] [PubMed] [Google Scholar]
- 11.Desany BA, Alcasabas AA, Bachant JB, Elledge SJ. Recovery from DNA replicational stress is the essential function of the S-phase checkpoint pathway. Genes Dev. 1998;12:2956–2970. doi: 10.1101/gad.12.18.2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhao X, Muller EGD, Rothstein R. A suppressor of two essential checkpoint genes identified a novel protein that negatively affects dNTP pools. Mol Cell. 1998;2:329–340. doi: 10.1016/s1097-2765(00)80277-4. [DOI] [PubMed] [Google Scholar]
- 13.Weinberger M, et al. Induction by adozelesin and hydroxyurea of origin recognition complex-dependent DNA damage and DNA replication checkpoints in Saccharomyces cerevisiae. J Biol Chem. 1999;274:35975–35984. doi: 10.1074/jbc.274.50.35975. [DOI] [PubMed] [Google Scholar]
- 14.Raghuraman M, Brewer B, Fangman W. Cell cycle-dependent establishment of a late replication program. Science. 1997;276:806–809. doi: 10.1126/science.276.5313.806. [DOI] [PubMed] [Google Scholar]
- 15.Andreassen P, Margolis R. 2-aminopurine overrides multiple cell cycle checkpoints in BHK cells. Proc Natl Acad Sci USA. 1992;89:2272–2276. doi: 10.1073/pnas.89.6.2272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schlegel R, Belinsky GS, Harris MO. Premature mitosis induced in mammalian cells by the protein kinase inhibitors 2-aminopurine and 6-dimethylaminopurine. Cell Growth Differ. 1990;1:171–178. [PubMed] [Google Scholar]
- 17.Schlegel R, Pardee AB. Caffeine-induced uncoupling of mitosis from the completion of DNA replication in mammalian cells. Science. 1986;232:1264–1266. doi: 10.1126/science.2422760. [DOI] [PubMed] [Google Scholar]
- 18.Hall-Jackson CA, Cross DA, Morrice N, Smythe C. ATR is a caffeine-sensitive, DNA-activated protein kinase with a substrate specificity distinct from DNA-PK. Oncogene. 1999;18:6707–6713. doi: 10.1038/sj.onc.1203077. [DOI] [PubMed] [Google Scholar]
- 19.Blasina A, Price BD, Turenne GA, McGowan CH. Caffeine inhibits the checkpoint kinase ATM. Curr Biol. 1999;9:1135–1138. doi: 10.1016/s0960-9822(99)80486-2. [DOI] [PubMed] [Google Scholar]
- 20.Sarkaria JN, et al. Inhibition of ATM and ATR kinase activities by the radiosensitizing agent, caffeine. Cancer Res. 1999;59:4375–4382. [PubMed] [Google Scholar]
- 21.Dimitrova DS, Todorov IT, Melendy T, Gilbert DM. Mcm2, but not RPA, is a component of the mammalian early G1-phase pre-replication complex. J Cell Biol. 1999;146:709–722. doi: 10.1083/jcb.146.4.709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nakamura H, Morita T, Sato C. Structural organization of replicon domains during DNA synthetic phase in the mammalian nucleus. Exp Cell Res. 1986;165:291–297. doi: 10.1016/0014-4827(86)90583-5. [DOI] [PubMed] [Google Scholar]
- 23.Nethanel T, Kaufmann G. Two DNA polymerases may be required for synthesis of the lagging DNA strand of simian virus 40. J Virol. 1990;64:5912–5918. doi: 10.1128/jvi.64.12.5912-5918.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aten JA, Bakker PJM, Stap J, Boschman GA, Veenhof CHN. DNA double labelling with IdUrd and CldUrd for spatial and temporal analysis of cell proliferation and DNA replication. Histochem J. 1992;24:251–259. doi: 10.1007/BF01046839. [DOI] [PubMed] [Google Scholar]
- 25.Meijer L, et al. Biochemical and cellular effects of roscovitine, a potent and selective inhibitor of the cyclin-dependent kinases cdc2, cdk2 and cdk5. Eur J Biochem. 1997;243:527–536. doi: 10.1111/j.1432-1033.1997.t01-2-00527.x. [DOI] [PubMed] [Google Scholar]
- 26.Dimitrova DS, Gilbert DM. Stability and nuclear distribution of mammalian replication protein A heterotrimeric complex. Exp Cell Res. 2000;254:321–327. doi: 10.1006/excr.1999.4770. [DOI] [PubMed] [Google Scholar]
- 27.Holmquist GP, Caston LA. Replication time of interspersed repetitive DNA sequences in hamsters. Biochim Biophys Acta. 1986;868:164–177. doi: 10.1016/0167-4781(86)90019-9. [DOI] [PubMed] [Google Scholar]
- 28.Caddle MS, Heintz NH. The replication timing of the amplified dihydrofolate reductase genes in the Chinese hamster ovary cell line CHOC 400. Biochem Biophys Res Commun. 1990;170:134–139. doi: 10.1016/0006-291x(90)91250-v. [DOI] [PubMed] [Google Scholar]
- 29.Dijkwel PA, Hamlin JL. The chinese hamster dihidrofolate reductase origin consists of multiple potential nascent-strand start sites. Mol Cell Biol. 1995;15:3023–3031. doi: 10.1128/mcb.15.6.3023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Painter RB, Young BR. Radiosensitivity in ataxia-telangiectasia: a new explanation. Proc Natl Acad Sci USA. 1980;77:7315–7317. doi: 10.1073/pnas.77.12.7315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zakian V. ATM-related genes: what do they tell us about functions of the human gene? Cell. 1995;82:685–687. doi: 10.1016/0092-8674(95)90463-8. [DOI] [PubMed] [Google Scholar]
- 32.Santocanale C, Diffley JF. ORC- and Cdc6-dependent complexes at active and inactive chromosomal replication origins in Saccharomyces cerevisiae. EMBO J. . 1996;15:6671–6679. [PMC free article] [PubMed] [Google Scholar]
- 33.Tibbetts RS, et al. A role for ATR in the DNA damage-induced phosphorylation of p53. Genes Dev. 1999;13:152–157. doi: 10.1101/gad.13.2.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xu Y, Baltimore D. Dual roles of ATM in the cellular response to radiation and in cell growth control. Genes Dev. 1996;10:2401–2410. doi: 10.1101/gad.10.19.2401. [DOI] [PubMed] [Google Scholar]
- 35.Smith GC, et al. Purification and DNA binding properties of the ataxia-telangiectasia gene product ATM. Proc Natl Acad Sci USA. 1999;96:11134–11139. doi: 10.1073/pnas.96.20.11134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vassilev LT, DePamphilis ML. Guide to identification of origins of DNA replication in eukaryotic cell chromosomes. Crit Rev Biochem Mol Biol. 1992;27:445–472. doi: 10.3109/10409239209082569. [DOI] [PubMed] [Google Scholar]
- 37.Hamlin JL, Mosca PJ, Levenson VV. Defining origins of replication in mammalian cells. Biochim Biophys Acta. 1994;1198:85–111. doi: 10.1016/0304-419x(94)90008-6. [DOI] [PubMed] [Google Scholar]
- 38.Gilbert DM, Miyazawa H, DePamphilis ML. Site-specific initiation of DNA replication in Xenopus egg extract requires nuclear structure. Mol Cell Biol. 1995;15:2942–2954. doi: 10.1128/mcb.15.6.2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lawlis SJ, Keezer SM, Wu J-R, Gilbert DM. Chromosome architecture can dictate site-specific initiation of DNA replication in Xenopus egg extracts. J. Cell Biol. 1996;135:1–12. doi: 10.1083/jcb.135.5.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Visser AE, Aten JA. Chromosomes as well as chromosomal subdomains constitute distinct units in interphase nuclei. J Cell Sci. 1999;112:3353–3360. doi: 10.1242/jcs.112.19.3353. [DOI] [PubMed] [Google Scholar]
- 41.Jackson DA, Pombo A. Replicon clusters are stable units of chromosome structure: evidence that nuclear organization contributes to the efficient activation and propagation of S phase in human cells. J Cell Biol. 1998;140:1285–1295. doi: 10.1083/jcb.140.6.1285. [DOI] [PMC free article] [PubMed] [Google Scholar]