

Abstract
We previously showed that in mitochondrial tRNALys with an A8344G mutation responsible for myoclonus epilepsy associated with ragged-red fibers (MERRF), a subgroup of mitochondrial encephalomyopathic diseases, the normally modified wobble base (a 2-thiouridine derivative) remains unmodified. Since wobble base modifications are essential for translational efficiency and accuracy, we used mitochondrial components to estimate the translational activity in vitro of purified tRNALys carrying the mutation and found no mistranslation of non-cognate codons by the mutant tRNA, but almost complete loss of translational activity for cognate codons. This defective translation was not explained by a decline in aminoacylation or lowered affinity toward elongation factor Tu. However, when direct interaction of the codon with the mutant tRNALys defective anticodon was examined by ribosomal binding analysis, the wild-type but not the mutant tRNALys bound to an mRNA– ribosome complex. We therefore concluded that the anticodon base modification defect, which is forced by the pathogenic point mutation, disturbs codon– anticodon pairing in the mutant tRNALys, leading to a severe reduction in mitochondrial translation that eventually could result in the onset of MERRF.
Keywords: anticodon/mitochondrial disease/mitochondrial translation/mitochondrial tRNA/post-transcriptional modification
Introduction
Mutations in tRNA genes encoded on mitochondrial DNA (mtDNA) are associated with a wide spectrum of human pathologies caused by mitochondrial disorders (Schon et al., 1997). A point mutation in the tRNALys gene at nucleotide position 8344, which is responsible for myoclonus epilepsy associated with ragged-red fibers (MERRF) (Fukuhara et al., 1980)—a major subgroup of the mitochondrial encephalomyopathies—was the first reported disease-related tRNA gene mutation (Shoffner et al., 1990). On the other hand, point mutations at nucleotide positions 3243 and 3271 in the tRNALeu(UUR) (R = A and G) gene were found in patients with another major subgroup—mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes (MELAS) (Goto et al., 1990, 1991; Kobayashi et al., 1990).
By establishing cybrid cell clones in which mutant mtDNA derived from patients was transferred intercellularly into human mtDNA-lacking (ρ0) cells (King and Attardi, 1989; Hayashi et al., 1991), the above three mutations (i.e. those at nucleotide positions 8344, 3243 and 3271) were revealed to be directly involved in mitochondrial dysfunction. In particular, the A to G transition at nucleotide position 8344 in the tRNALys gene was demonstrated to cause severe impairment of mitochondrial protein synthesis and respiratory deficiency in cultured myoblasts from MERRF patients (Boulet et al., 1992) and in ρ0 transformants (Chomyn et al., 1991; Yoneda et al., 1994). Peptide analysis carried out to try and explain the molecular mechanism suggested the occurrence of premature translation termination resulting from a decrease in aminoacyl-tRNALys (Enriquez et al., 1995). On the other hand, in tissue biopsies from MERRF patients harboring the 8344 mutation, neither the relative abundance nor the aminoacylation of the mutated tRNALys was found to be affected (Börner et al., 2000), which raises the question of whether the amount of aminoacyl-tRNALys significantly affects the molecular pathogenesis of MERRF.
Recently, we found that a modification normally present at the anticodon wobble nucleotide is absent in mitochondrial tRNALys with the MERRF mutation [tRNALys(A8344G)] (Yasukawa et al., 2000a), while the other modified nucleotides remain normal in the mutant tRNALys (Figure 1). In addition, modification at the wobble position is also defective in tRNALeu(UUR) with either the MELAS 3243 [tRNALeu(UUR)(A3243G)] or 3271 [tRNALeu(UUR)(U3271C)] mutation (Yasukawa et al., 2000b). The wild-type tRNAs have novel uridine derivatives at the wobble position, whose chemical structures have been identified recently (T.Suzuki, T.Suzuki, T.Wada, K.Saigo and K.Watanabe, in preparation). Both tRNAs have a taurinomethyl group at position 5 of the uridine, while tRNALys additionally has a thio-group at position 2 [tRNALyssU*UU and tRNALeu(UUR)U*AA, respectively]. Uridine modification at the wobble position has been studied in terms of its apparent importance in codon–anticodon interaction (Yokoyama and Nishimura, 1995; Takai et al., 1996; Hagervall et al., 1998; Krüger et al., 1998). Ashraf et al. (1999) have also shown that modification of the wobble uridine, particularly thiolation at position 2, is essential for tRNALyssU*UU species having 2-thiouridine derivatives at the wobble position (5-methylaminomethyl-2-thiouridine (mnm5s2U) in Escherichia coli tRNALys and 5-methoxycarbonylmethyl-2-thiouridine in human cytoplasmic tRNALys3, respectively) to interact with codons on ribosomes.

Fig. 1. Cloverleaf structures of human mitochondrial tRNALys from wild-type cells (left) and from MERRF patient-derived cells with the A8344G mutation (right). sU* indicates the modified uridine, and U on a round black background, unmodified uridine. G on a square black background represents the point mutation. The other modified nucleosides were determined previously: 1-methyladenosine (m1A), 2-methylguanosine (m2G), pseudouridine (Ψ) and N6-threonino carbonyladenosine (t6A) (Helm et al., 1998; Yasukawa et al., 2000a).
These findings prompted us to speculate that the MERRF-mutant tRNALys intrinsically loses its translational activity due to an impairment of codon–anticodon interaction, eventually leading to mitochondrial dysfunction. To verify our speculation, we estimated the translational ability of tRNA in vitro using mitochondrial translation components and were able to obtain clear experimental evidence that the mutant tRNALys with the unmodified anticodon UUU (tRNALysUUU) is actually unable to translate its cognate codons AAR. Furthermore, taking advantage of ribosomal binding analysis, we show that the mutant tRNALys does not bind to AAA- programmed ribosomes. Our findings lead us to conclude that the wobble modification defect is primarily responsible for dispossessing the mutant tRNALys of its cognate codon binding affinity, forcing the mutant tRNALysUUU to become translationally inactive, which subsequently results in mitochondrial dysfunction. This is the first evidence that a post-transcriptional modification deficiency causes a human disease.
Results
Mitochondrial translation and oxygen consumption in MERRF-mutant cybrid cells
The cybrid cell lines used were constructed previously by the intercellular transfer of mtDNA from a MERRF patient, or from fetal human fibroblasts as a control, to ρ0 HeLa cells (King and Attardi, 1989; Hayashi et al., 1991). A mutant cybrid clone (ME1-4) exclusively harboring mtDNA with the A8344G MERRF mutation consumed oxygen at a significantly lower rate (1.7 ± 0.2 fmol/min/cell) than the control cybrid (Ft2-11) with the wild-type mtDNA (5.3 ± 0.8 fmol/min/cell) when the oxygen consumption was measured as described by King and Attardi (1989). This finding is consistent with previous observations (Chomyn et al., 1991; Yoneda et al., 1994). Figure 2A shows the mitochondrial translation products labeled with [35S]methionine for 50 min in the presence of a cytoplasmic translation inhibitor, emetine, in which the polypeptides were identified as reported previously (Hayashi et al., 1993). The labeling efficiency of all the polypeptides was markedly reduced, indicating that the overall rate of protein synthesis in ME1-4 cells was very slow as compared with that in Ft2-11 cells. A faint but reproducible additional band was detected (indicated by an arrow in Figure 2A), which probably corresponds to the premature translation termination product of cytochrome c oxidase subunit I (COI) as has been reported (Chomyn et al., 1991; Boulet et al., 1992; Yoneda et al., 1994; Enriquez et al., 1995). The intensity of the abnormal band may depend on the nuclear background of the cybrid cells. Pulse labeling for 30 min and 2 h gave similar results (not shown). Western blot analysis (Figure 2B) revealed severely decreased steady-state levels of not only mitochondrially encoded cytochrome c oxidase subunits I and II (COI and COII) but also of nuclearly encoded subunit IV (COIV) in the mutant cells. The steady-state levels of internal control mitochondrial proteins, dihydrolipoamide succinyltransferase (DLST) (Nakano et al., 1993) and a translocase of the outer mitochondrial membrane (TOM 40) (Pfanner and Geissler, 2001), were comparable in the mutant and wild-type cybrids.

Fig. 2. (A) [35S]Methionine incorporation into the Ft2-11 wild-type cybrid (Wt) versus the ME1-4 MERRF-mutant cybrid (MERRF). CO, cytochrome c oxidase subunit; ND, NADH-ubiquinone oxidoreductase subunit; A, ATP synthase subunit. The arrow indicates the reported premature translation termination product of COI (Enriquez et al., 1995). (B) Western blots of cytochrome c oxidase subunits (COI, COII and COIV), dihydrolipoamide succinyltransferase (DLST) and translocase of the outer membrane 40 (TOM 40) in Ft2-11 and ME1-4 cybrids.
Steady-state amounts and extent of aminoacylation of mutant and wild-type tRNALys in cybrid cells
Northern blot analysis showed no reduction in the steady-state amount of the mutant tRNALys(A8344G) in the ME1-4 cybrid compared with that of the wild-type tRNALys in the Ft2-11 cybrid (Figure 3, lanes 1 and 4, and Figure 4 at time 0). The lysylation of the wild-type and mutant tRNAsLys was also examined by separating aminoacylated and uncharged tRNAs using acid urea polyacrylamide gel electrophoresis followed by northern hybridization (Varshney et al., 1991). Although the amount of uncharged tRNALys in the mutant cybrid was more than that in the control, the extent of lysylation in the mutant tRNALys appeared not to be markedly reduced (Figure 3), being 80% in the mutant cybrid and 93% in the control as determined by careful and repeated quantification.

Fig. 3. Quantitative analysis of aminoacyl-tRNALys in cells. The upper and lower bands correspond to aminoacyl and uncharged tRNALys, respectively. Lanes 2 and 3 depict RNA samples prepared under acidic conditions at 4°C from Ft2-11 and ME1-4 cybrids, respectively; lanes 1 and 4 show the Ft2-11 RNA and ME1-4 RNA, respectively, both treated with alkali.
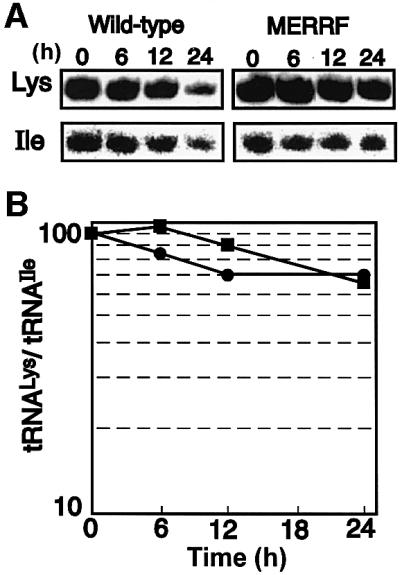
Fig. 4. Time-dependent degradation of tRNA in the respective cybrid clones in the presence of a potential inhibitor of mitochondrial transcription. (A) Examples of northern hybridization for tRNALys and tRNAIle in the wild-type cybrid (left panels) and in the MERRF cybrid (right panels). (B) Time courses of quantitative analysis for the wild-type tRNALys (filled circles) and the mutant tRNALys (filled squares), normalized with tRNAIle in the respective cybrid clones. The average values at the starting time were defined arbitrarily as 100. Each set of data represents the average of at least three independent northern blotting experiments (SD bars are within the circles and squares).
Stability of mutant tRNALys and its wild-type counterpart in cells
To verify the finding of comparable steady-state amounts of tRNALys in mutant and wild-type cybrid cells, we further examined the degradation rates of the tRNAsLys and, as a control, of tRNAIle in the presence of a potential mitochondrial transcription inhibitor, ethidium bromide (EtBr) (Hayashi et al., 1990). Both the wild-type tRNALys and tRNAIle in the Ft2-11 cells appeared to degrade slightly faster than their respective counterparts in the ME1-4 cells (Figure 4A), which might result from a difference in sensitivity to EtBr between the two cell lines. However, the amounts of remaining tRNALys normalized at each time point by that of the remaining tRNAIle in the respective cybrid cells were comparable (Figure 4), showing that the point mutation occurring in the TΨC loop of tRNALys affects neither the stability of the tRNA nor the steady-state level in vivo.
Aminoacylation efficiency and affinity for elongation factor Tu of mutant tRNALys in vitro
Aminoacylation and elongation factor Tu (EF-Tu)-dependent hydrolysis protection assays were performed in vitro to observe the aminoacylation efficiency and affinity of tRNAsLys toward EF-Tu, respectively. For the following in vitro experiments, both the wild-type and mutant tRNAsLys were purified homogeneously by a solid-phase probing technique (Wakita et al., 1994) from respective mass cultures of wild-type and mutant cells. The initial rates of tRNALys lysylation were determined using mitochondrial lysyl-tRNA synthetase (KRSmt) purified from bovine liver by a conventional purification method (see Materials and methods). As shown in Table I, the kinetic parameters for the lysylation of the wild-type and mutant tRNAsLys did not differ significantly.
Table I. Kinetic parameters in aminoacylation of wild-type and MERRF-mutant tRNALys.
| Substrate | Km (µM)a | Vmax (µM/s) | Vmax/Km (relative) |
|---|---|---|---|
| Wild-type tRNALys | 0.71 | 2.9 × 10–3 | 1 |
| MERRF tRNALys(A8344G) | 0.18 | 2.4 × 10–4 | 0.32 |
aThe apparent Km values are given since the KRSmt used was a partially purified fraction.
For the hydrolysis protection assay, recombinant bovine mitochondrial EF-Tu (EF-Tumt) was used and preparative aminoacylation of native tRNALys with or without the A8344G mutation was carried out with sufficient amounts of KRSmt and [3H]lysine. The stability of the ternary complex (Lys-tRNALys⋅EF-Tumt⋅GTP) against non-enzymatic deacylation under an alkaline pH condition was estimated by measuring the remaining radioactivity of [3H]Lys-tRNALys in the complex (Pingoud et al., 1977). Although the half-life of the wild-type tRNALys was about double that of the mutant tRNALys in the presence of each amount of EF-Tumt used (Figure 5C), the plots in Figure 5A and B show that both tRNAsLys were recognized efficiently by EF-Tumt.

Fig. 5. Stability of the ternary complex [3H]Lys-tRNALys⋅EF-Tumt⋅GTP against non-enzymatic deacylation. Time courses of remaining radioactivity of (A) wild-type and (B) mutant [3H]Lys-tRNAsLys determined at each indicated time point in the presence of the following amounts of EF-Tumt: 0.5 pmol (open triangles), 5 pmol (open circles), 25 pmol (filled squares), 37.5 pmol (filled triangles) and 50 pmol (filled circles), and in the absence of EF-Tumt (open squares). Values at the starting time (0 min) were defined as 100. (C) Half-life values (T1/2) of the wild-type [3H]Lys-tRNALys (open circles) and mutant [3H]Lys-tRNALys (open squares) plotted against the ratio of EF-Tumt/[3H]Lys-tRNALys initially added to the reaction mixture.
Translational activities of tRNAs estimated in vitro using bovine mitochondrial translation components
We examined whether the MERRF mutant tRNALys (A8344G), as well as the MELAS mutants tRNALeu(UUR) (A3243G) and tRNALeu(UUR)(U3271C), all lacking the modification only at the wobble position, could function in the translation elongation process. The in vitro assay was performed basically according to the literature (Eberly et al., 1985; Takemoto et al., 1995) using bovine mitochondrial components. To compare the translational efficiencies, all the tRNAs used were purified from the respective cell lines and equal amounts of the 3H-labeled aminoacyl-tRNAs prepared were put into the reaction mixtures as described in Materials and methods. Since we wished to emphasize the codon reading efficiency of the wild-type and mutant tRNAs, instead of natural mRNAs we used synthetic RNAs containing 30 repeats of cognate (AAA and AAG) and non-cognate (AAC and AAU) codons for tRNALys, and of cognate (UUA and UUG) codons for tRNALeu(UUR), respectively. Figure 6A and B shows that the mutant tRNALysUUU(A8344G) could not translate AAA and AAG codons, whereas the translation reaction proceeded quite efficiently with the wild-type tRNALys. Neither the mutant nor the wild-type tRNALys showed translational activity for non-cognate codons under the conditions used (Figure 6C and D). In addition, the translational activities of the two MELAS-mutant tRNAsLeu(UUR) for the cognate codons also decreased to a greater or lesser extent (Figure 6E and F). This result clearly supports our hypothesis that the MERRF-mutant tRNALys loses its translational activity, at least in vitro.

Fig. 6. Summary of in vitro translational elongation activities. (A–D) Translational efficiencies of the wild-type tRNALyssU*UU (Wt) and mutant tRNALysUUU with the A8344G mutation (MERRF) toward codons AAA, AAG, AAC and AAU. (E and F) Translational efficiencies of the wild-type tRNALeu(UUR)U*AA (Wt) and two mutant tRNAsLeu(UUR)UAA with the A3243G (MELAS 3243) or U3271C (MELAS 3271) mutation toward codons UUA and UUG. The radioactivity of the [3H]aminoacyl-tRNA initially added to the reaction mixture was defined arbitrarily as 100. Each set of data represents the average of three independent experiments, with bars showing the SD.
Binding affinity of mutant tRNALysUUU for AAA-programmed ribosomal small subunits
Although the above experiment showed that the mutant tRNALysUUU almost completely lost its translational activity in vitro, it was essential to prove that this translational deficiency arose from the wobble modification defect itself but was not a direct result of the A to G replacement in the TΨC loop (Figure 1). For this purpose, we considered it appropriate to apply the binding assay of AAA-programmed E.coli ribosomal small subunits. Since a crystallographic analysis has shown that only the tRNA anticodon stem–loop (ASL) region, but not the TΨC loop, is located on the small subunits when tRNA binds to the ribosomes (Ogle et al., 2001; Yusupov et al., 2001), the possibility of the mutant nucleotide directly affecting codon–anticodon pairing in this binding assay can be excluded. As is clearly shown in Figure 7, the wild-type tRNALyssU*UU bound efficiently to AAA-programmed small subunits, but the mutant tRNALysUUU did not. This finding suggests that the defective translational activity of the MERRF-mutant tRNALys is caused by an inability to form codon (AAA)–anticodon (unmodified UUU) base pairs on the ribosomes.

Fig. 7. Binding of the wild-type tRNALyssU*UU (filled circles) and the MERRF-mutant tRNALysUUU (filled squares) to AAA-programmed ribosomal small subunits. Three independent experiments were performed and the average values are plotted on the graph, with the bars indicating the SD.
Discussion
As has been reported previously (Chomyn et al., 1991; Yoneda et al., 1994; Enriquez et al., 1995), the overall rate of mitochondrial protein synthesis was shown to be severely impaired in MERRF-mutant cybrid cells (Figure 2A). As expected, the steady-state amounts of mitochondrially encoded COI and COII proteins were very low (Figure 2B). The level of nuclear-encoded COIV was also found to decrease markedly in the mutant cybrid, which presumably can be accounted for by enhanced intramitochondrial degradation previously observed in ρ0 cells in which mitochondrial subunits were absent, and as a consequence cytochrome c oxidase could not be built up (Nijtmans et al., 1995). This might also be the case with NADH-ubiquinone oxidoreductase, because seven of the subunits are produced from mtDNA. It is possible to speculate that if a very small number of mitochondria exclusively had the wild-type tRNALys, while the vast majority had only the mutant counterpart, the faint radiolabeled protein bands in Figure 2A would be derived from normal mitochondrial translation provided by a small number of mitochondria.
Enriquez et al. (1995) found decreased availability of aminoacyl-tRNALys with the 8344 mutation, which they postulated most probably brought about the translational defect in their mutant osteosarcoma-derived cybrid clones. In contrast, we found that the stability and steady-state amount of the mutant tRNALys(A8344G) in the ME1-4 cybrid remained unchanged when compared with its wild-type counterpart in the Ft2-11 cybrid (Figure 4). Furthermore, the extent of aminoacylation of the mutant tRNALys was comparable to that of the wild-type in the respective cybrids (Figure 3). Consistent with our findings, Börner et al. (2000) reported that neither the relative abundance nor the aminoacylation of mutated tRNALys was reduced in tissue biopsies from MERRF patients. The reason for the discrepancy with the result reported by Enriquez et al. (1995) is unknown, but it may be related to the cell types used. Nevertheless, our findings and those of Börner et al. (2000) both indicate that even without a reduction of aminoacylation, respiration declines in MERRF patients’ cells.
While the above analysis is salient, it was still considered important to elucidate the reason for the slightly reduced aminoacylation level in the mutant cybrid (Figure 3). The apparent kinetic parameters determined by a standard in vitro aminoacylation assay using bovine KRSmt showed that the lysine accepting activities (Vmax/Km values) of the wild-type and mutant tRNAsLys were similar, suggesting that the reduced aminoacylation level has no pathogenic significance (Table I). Because bovine KRSmt had the activity to aminoacylate human tRNALys as well as its bovine counterpart and the kinetic parameters in these aminoacylations were comparable (data to be published elsewhere), the region around the mutation point may not include an identity element(s) of the human, or bovine, tRNALys toward the corresponding KRSmt. For these reasons, the parameters obtained using bovine KRSmt would appear to be reasonable.
Since bovine EF-Tumt bound efficiently to human aminoacyl-tRNALys, as would be expected from the fact that bovine and human EF-Tusmt are 95% identical in their sequences (Woriax et al., 1995), we used bovine EF-Tumt instead of its human counterpart for an EF-Tu-dependent hydrolysis protection analysis. Figure 5C shows that the half-life of Lys-tRNALys was shortened in the case of the mutant tRNALys, which implies that the binding affinity of EF-Tumt toward the mutant tRNA decreased to some extent. This finding is intriguing in seeking an explanation for the slight decrease in the ratio of aminoacyl-tRNALys to total tRNALys in mutant cybrid cells (Figure 3). The reduced affinity of the aminoacylated mutant tRNALys toward EF-Tumt presumably could be a candidate for explaining the decreased aminoacylation in the osteosarcoma background cybrid reported by Enriquez et al. (1995). However, in this study, the difference in affinity toward EF-Tumt between the wild-type and mutant tRNAsLys would not affect the translational efficiency in vivo since the ratio of aminoacylated tRNALys to total tRNALys was not greatly reduced in the mutant compared with that in the control cells (Figure 3). Additionally, in the in vitro translation reaction mixture without mRNA, the rates of deacylation in the wild-type and mutant tRNALys were observed to be almost identical (not shown). Since, for technical reasons, we used bovine enzymes in the two in vitro experiments above, the possibility that the results were due to heterogeneous enzymes cannot be excluded completely. Nevertheless, taking the in vivo and in vitro analyses together, it is reasonable to consider that the amount of aminoacylated tRNA is not the main factor underlying mitochondrial dysfunction in MERRF-mutant cells.
We have already noted that the rate of mitochondrial protein synthesis was severely impaired in MERRF-mutant cybrid cells. At the same time, it has been proven that modification of a wobble uridine is indispensable for the tRNALyssU*UU species to bind poly(A) on ribosomal small subunits (Ashraf et al., 1999), and we recently found that the wobble modification is lacking in the MERRF-mutant tRNALys (Yasukawa et al., 2000a). Modifications of wobble bases are, in general, known to be very important for regulating the translational efficiency and accuracy of codon–anticodon base pairing. In addition, as shown in Figure 3, the absence of the wobble modification in the mutant tRNALys did not affect the aminoacylation, which is similar to the case in an in vivo study in which aminoacylation of E.coli tRNALys was shown to be unaffected by a specific lack of the wobble uridine modification (mnm5s2U) (Hagervall et al., 1998). There fore, we examined whether the mutant tRNALys is deprived of translational activity. For this purpose, we estimated the mutant tRNALysUUU activity in the translation elongation process in vitro using mitochondrial components and demonstrated that the mutant tRNALysUUU did indeed lose a significant amount of its essential function of translating lysine codons (Figure 6A and B). The magnitude of the decline in the translational activity of the mutant tRNA may have been overestimated to a certain extent, because unlike the actual circumstances in the mitochondrion, the mRNAs used contained repeats of the test codon. Furthermore, the in vitro conditions were heterogeneous. Nevertheless, we consider the in vitro assay appropriate to emphasize the defect of the mutant tRNA. Mistranslation of non-cognate codons was not observed in this system (Figure 6C and D). This finding is, unexpectedly, different from the cases of many mitochondrial and Mycoplasma tRNAs containing unmodified uridine at the wobble position (U34) that are specific for amino acids corresponding to four codon boxes (Samuelsson et al., 1983; Inagaki et al., 1995; Watanabe and Osawa, 1995).
To elucidate the molecular basis of the tRNALys dysfunction, we next examined in vitro the binding affinity of the tRNAs for mRNA-programmed ribosomal small subunits, which is a widely accepted method for examining codon–anticodon interactions (von Ahsen et al., 1997; Ashraf et al., 1999; Yarian et al., 2000). We excluded the possibility that the pathogenic point mutation (G at nucleotide position 8344) in the TΨC loop may affect the codon–anticodon pairing by using ribosomal small subunits. It is known that only the ASL region of tRNA interacts with mRNA on the small subunits, and the TΨC loop is not involved in the interaction (Ogle et al., 2001; Yusupov et al., 2001). Furthermore, ASL minihelices with appropriate modifications showed binding equivalent to intact tRNAs toward messenger-programmed small subunits (Ashraf et al., 1999; Yarian et al., 2000). We used E.coli small ribosomal subunits instead of the human mitochondrial counterparts since it has been shown that mitochondrial tRNA enables translation to proceed on E.coli ribosomes and, conversely, that E.coli tRNA does likewise on mitochondrial ribosomes (Schwartzbach and Spremulli, 1989; Kumazawa et al., 1991). Similar to the result shown in Figure 6, in an in vitro assay using E.coli ribosomes instead of their mitochondrial counterparts, the wild-type mitochondrial tRNALys allowed the translation reaction to proceed efficiently, whereas the MERRF-mutant tRNALys did not (data not shown). Therefore, this binding assay system permitted us to compare the physicochemical interactions of the wild-type and mutant tRNAs with AAA-programmed small subunits. In fact, the experiment clearly showed that while the wild-type tRNALys with the modified uridine bound efficiently to the cognate codon, the mutant tRNALysUUU did not under the same conditions (Figure 7). From this result, we infer that the defective translational activity of the mutant tRNALysUUU is derived from its inability to form codon– anticodon base pairs, at least in vitro, most probably due to the lack of the wobble modification.
On the other hand, there is a possibility that the base substitution in the TΨC loop could itself directly affect the translational activity of the tRNALys, because the mutation would have some influence on the interaction of the TΨC loop with intact ribosomes, thereby altering the translational activity of the mutant tRNA. Such an effect, if it exists, may not, however, be critical for the defective translation of the mutant tRNA observed in this study (Figure 6A and B), since the TΨC loop sequence is not conserved among mammalian mitochondrial tRNALys (Sprinzl et al., 1998). Moreover, as shown in Figure 7, the translational defect can be explained by the apparent inability for an interaction to occur between codon and the mutant tRNALys on the ribosomal small subunit.
Another possibility to be considered is that the observed translational deficiency may arise indirectly from a structural alteration, especially in the anticodon region, induced by the mutation. If a putative modification enzyme(s) of the wobble base is able to recognize the structure of the anticodon region for modifying the base, this possibility cannot be ruled out. However, structural alteration seems unlikely because the tRNA maturation, including processing and base modifications, was normal except for the specific lack of the wobble modification in the mutant tRNALys (Yasukawa et al., 2000a) and the aminoacylation and affinity toward EF-Tumt were not significantly affected (Table I; Figures 3 and 5). Moreover, the lifetime of the mutant tRNA was not changed (Figure 4), suggesting that the structural stability of the mutant tRNA was not influenced by the point mutation. Therefore, we conclude that the mutant tRNA could have the point mutation without incurring structural alteration and, even if such alteration does occur, the translational activity is likely to be affected only to a small extent.
The ability of an unmodified ASL minihelix of human cytoplasmic tRNALys3UUU to bind to AAA-programmed E.coli small ribosomal subunits was reported to be restored by the introduction of a single modified nucleotide, N6-threoninocarbonyladenosine (t6A), to position 37, 3′-adjacent to the anticodon third letter (Yarian et al., 2000). This does not agree with the case of the mutant mitochondrial tRNALysUUU, probably because the anticodon stem sequence is not at all conserved between the cytoplasmic tRNALys3 and the mitochondrial tRNALys (Figure 1).
The translational activity appeared to decrease more severely in the cases of AAG and UUG than AAA or UUA (Figure 6), indicating that G at the third position of the codon was recognized less efficiently than A by the unmodified U34 (Takai et al., 1996). The data in Figure 6 suggest that the modification at position 5 (taurino methyl group) of U34 in both tRNALyssU*UU and tRNALeu(UUR)U*AA is very important for codon–anticodon interaction and stabilization, particularly of the wobble pair, presumably because it is required for the anticodon to be arranged into an appropriate structure. The tendency observed in this study is in accordance with a report that lack of the modification at position 5 and thiolation at position 2 of the uridine base at the wobble position in E.coli tRNAGlu reduces the translation rate of codons ending with G (Krüger et al., 1998). Figure 6 also indicates that the MERRF mutation had the most drastic effect on translational ability, which may be related to the fact that in addition to the modification at position 5 of U34, thiolation at position 2 is necessary for tRNALys, but not for tRNALeu(UUR), to be functional. This postulation is supported by a report that thiolation at position 2 of U34 plays a crucial role in the ribosomal binding of tRNALys species (Ashraf et al., 1999) as well as by the finding that this modification is particularly important for the recognition of A at the third letter of the codon in the in vivo translation system of E.coli (Krüger et al., 1998). In contrast to the case of MERRF, the stability and aminoacylation of both mutant tRNAsLeu(UUR) were found to decrease (Yasukawa et al., 2000b), suggesting that the molecular pathogenesis of MELAS could be a combination of a lowered availability of aminoacyl-tRNALeu(UUR) and defective translation.
The defective translational activity of the MERRF-mutant tRNALysUUU can easily explain the exponential decrease in the rate of mitochondrial protein synthesis with increasing lysine content and premature termination of translation, which were elegantly described by Attardi and colleagues (Enriquez et al., 1995). Lysine codons in 12 mRNAs encoded on mtDNA seem to be distributed almost uniformly in each mRNA, and larger polypeptides tend to have more lysines (Anderson et al., 1981). Impairment of mitochondrial translation may result from stalling of ribosomes with an empty A-site at lysine codons due to a deficiency in the interaction of the mutant tRNALysUUU with lysine codons, leading to premature termination. Another possibility is that frameshifting may occur at lysine codons and abortive proteins might then be produced. Prematurely terminated proteins and polypeptides with abnormal sequences are probably not assembled in the complexes for the electron transport chain but instead are degraded rapidly by ATP-dependent mitochondrial proteases (Käser and Langer, 2000). Even if some peptides are produced in their full length (e.g. ND4L, which does not contain lysine), the nascent polypeptides are likely to be in danger of degradation—as happened with COIV (Figure 2B)—by proteases, since the loss of other peptides may disturb the normal assembly of intact subunits in the complex.
In conclusion, our study has unraveled the essential molecular mechanism causing mitochondrial dysfunction in MERRF pathology. A point mutation at nucleotide position 8344 in the mtDNA results in a modification defect at the anticodon first letter of the mutant tRNALys molecule, which subsequently disturbs codon–anticodon interaction, severely impairing mitochondrial translation. This is the first proof that a post-transcriptional modification deficiency causes a human disease.
Materials and methods
Cybrid cell lines
The mutant cybrid cell lines, which were constructed by the intercellular transfer of MERRF or MELAS patient mtDNA to ρ0 HeLa cells (EB8) (King and Attardi, 1989; Hayashi et al., 1991), were described previously (Yasukawa et al., 2000a,b). The ME1-4, ML2-2-2 and ML5-1-13 cell lines exclusively contain mtDNA with the A8344G, A3243G and T3271C point mutations, respectively. The Ft2-11 cell line, used as a wild-type control, harbors mitochondria containing wild-type mtDNA derived from fetal human fibroblasts (Hayashi et al., 1991). The cells were cultured in normal medium [Dulbecco’s modified Eagle’s medium/F-12 (1:1) (Gibco-BRL), 10% fetal calf serum (FCS)].
Oxygen consumption
The rate of oxygen consumption by 0.4–1 × 107 exponentially growing cells in 2 ml of the normal medium was measured at 37°C using a Clark-type electrode as described (King and Attardi, 1989).
Mitochondrial protein synthesis in vivo
[35S]methionine incorporation into mitochondrially encoded proteins was analyzed essentially as described (Chomyn et al., 1991; Hayashi et al., 1993). Semiconfluent cells were labeled with [35S]methionine (43.5 TBq/mmol) (American Radiolabeled Chemicals) containing 0.2% FCS in the presence of emetine (0.2 mg/ml) for 50 min. The mitochondrial fraction was separated by SDS–PAGE. The gel was dried and exposed to an imaging plate, and the labeled polypeptides were located with a bioimaging analyzer (BAS 1000, Fuji Photo Film).
Western blotting analysis
Western blotting was performed essentially as described (Towbin et al., 1979). Total mitochondrial protein was separated on an SDS– polyacrylamide gel and transferred to a polyvinylidene difluoride membrane (Immobilon P, Millipore). Subsequently, the proteins were visualized specifically using antibodies against COI, COII (Molecular Probes), COIV (Clontech Laboratories), DLST and TOM 40.
Evaluation of tRNA aminoacylation levels in cybrids
Total RNA from semiconfluent cultured cybrid cells was prepared under acidic conditions at 4°C in order to prevent deacylation. A portion of the RNA sample was incubated at 37°C for 3 h in a buffer containing 50 mM Tris–HCl pH 9.5 for deacylation of tRNA. The same amount of total RNA containing aminoacyl-tRNAs or forcibly deacylated tRNAs was electrophoresed at 4°C through an acid urea polyacrylamide gel to separate aminoacyl-tRNA and uncharged tRNA (Varshney et al., 1991) and the RNA was blotted onto a nylon membrane (Hybond N, Amersham Pharmacia Biotech). Northern hybridization for tRNALys was performed using a 32P-5′-end labeled probe specific for mitochondrial tRNALys: 5′-TCACTGTAAAGAGGTGTTGG-3′ (not including the mutation position). Aminoacyl- and non-acylated tRNAsLys were quantified by exposing the membrane to an imaging plate, followed by analysis with a Fuji BAS1000 bioimaging analyzer.
Analysis of tRNA stability in cybrids
The procedure was essentially identical to that described previously (Yasukawa et al., 2000b). Equal amounts of semiconfluent cells were put into four dishes and cultured in a medium containing EtBr (250 ng/ml) to block specifically mitochondrial transcription (Hayashi et al., 1990). At the indicated times, total RNA was isolated from one of the dishes by Isogen (Nippon Gene, Toyama, Japan). A sample (5 µg) of total RNA was electrophoresed on a denaturing polyacrylamide gel. Northern hybridization and quantification of RNAs were performed as described above. A 32P-5′-end labeled probe specific for mitochondrial tRNAIle (5′-TAG AAATAAGGGGGTTTAAGCTCCTATTAT-3′) was used to detect tRNAIle.
Purification of tRNAs from cybrid cells by the solid-phase probe method
About 1000–2000 A260 units (the actual amount varied depending on the cell line) of total RNA were obtained from a large-scale culture of the semiconfluent cell. A portion of the preparation for wild-type tRNAs was isolated from HeLa cells. The tRNALys and tRNALeu(UUR) with or without the mutations were purified homogeneously from the respective total RNA as described (Yasukawa et al., 2000a,b).
Partial purification of bovine mitochondrial Lysyl-tRNA synthetase and in vitro aminoacylation of tRNALys
Mitochondria were prepared from fresh bovine liver (Matthews et al., 1982; Schwartzbach et al., 1996), and fractionated on a DEAE–Sepharose Fast Flow column (Amersham Pharmacia Biotech) with a linear gradient of KCl between 20 and 300 mM in a buffer containing 20 mM Tris–HCl pH 7.6, 1 mM MgCl2, 0.1 mM EDTA, 6 mM β-mercaptoethanol and 10% glycerol. The fractions containing KRSmt activity were enriched at a protein concentration of 6 mg/ml with Centriprep (Millipore). All the procedures were performed at 4°C.
Aminoacylation reactions to determine the kinetic parameters were carried out at 37°C in a buffer consisting of 10 mM Tris–HCl pH 8.1, 15 mM MgCl2, 2 mM ATP, 12 mM KCl and 15.4 µM [14C]l-lysine (12 GBq/mmol) (Amersham Pharmacia Biotech). The initial rates of aminoacylation were determined by using five different concentrations of both tRNAsLys (0.1, 0.3, 0.5, 0.7 and 0.9 µM) at a fixed concentration of partially purified KRSmt, which gave reasonable kinetic plots for determining the apparent kinetic parameters.
Preparation of [3H]aminoacyl-tRNAs
Preparative aminoacylation of the wild-type or mutant tRNALys was performed with [3H]l-lysine (1.85 Mbq/mmol) (American Radiolabeled Chemicals) and a sufficient amount of KRSmt under the reaction conditions as described above. The charged tRNA was extracted by phenol and passed through an NAP-5 column (Amersham Pharmacia Biotech) to remove proteins, ATP and uncharged lysine. The purified [3H]Lys-tRNALys was stored in an acidic buffer at –80°C to prevent deacylation. The wild-type or mutant [3H]Leu-tRNALeu(UUR) was prepared by the same procedure except that [3H]l-leucine (1.85 Mbq/mmol) (American Radiolabeled Chemicals) and recombinant human mitochondrial leucyl-tRNA synthetase (LRSmt) were used. KIAA0028 (Nomura et al., 1994) containing the full-length human LRSmt gene was provided by Kazusa DNA Research Institute, Japan; the gene was cloned into pET 15b, expressed in E.coli and highly purified using a nickel affinity resin.
EF-Tu-dependent hydrolysis protection assay
The protection experiment was carried out basically as reported (Pingoud et al., 1977). A plasmid containing full-length bovine EF-Tumt cloned into pET-24c(+) was kindly provided by Dr L.L.Spremulli (University of North Carolina) (Woriax et al., 1995). The EF-Tumt was expressed in E.coli BL21(DE3) and highly purified using a nickel affinity resin. The reaction mixture (40 µl) contained 75 mM Tris–HCl pH 8.1, 75 mM NH4Cl, 15 mM MgCl2, 7.5 mM dithiothreitol (DTT), 0.1 mM GTP, 60 µg/ml bovine serum albumin (BSA), 2.25 mM phosphoenolpyrvate (PEP), 2.3 U/ml pyruvate kinase (PK) (Sigma), 0.5 pmol [3H]Lys-tRNALys and various amounts of EF-Tumt (0, 0.5, 5, 25, 37.5 and 50 pmol). The mixture was kept on ice for 6 min to allow formation of a ternary complex, and incubation was then started at 30°C.
In vitro translation using bovine mitochondrial translation components
Using fresh bovine liver, mitochondrial ribosomes were prepared according to the method of Matthews et al. (1982) with the modifications described by Eberly et al. (1985), and a partially purified mitochondrial EF-G (EF-Gmt) was obtained (Chung and Spremulli, 1990). Poly(AAN)30 and poly(UUR)30 (N = A, G, C or U) were synthesized in vitro using T7 RNA polymerase. The reaction mixture (20 µl) contained 50 mM Tris–HCl pH 8.6, 7 mM MgCl2, 5 mM KCl, 1 mM DTT, 2 mM spermine (SP), 2.5 mM PEP, 2.5 U/ml PK, 0.5 mM GTP, 15 pmol EF-Tumt, 0.25 µg/µl EF-Gmt, 2 pmol mitochondrial ribosome, 4 µg of poly(AAN)30 or poly(UUR)30 and 0.1 pmol [3H]Lys-tRNALys or [3H]Leu-tRNALeu(UUR), respectively. The amounts of aminoacyl-tRNAs were adjusted by measuring the radioactivity of the amino acid attached to tRNA. The mixture was incubated at 37°C for 20 min and the radioactivity of polymerized amino acids was measured as described (Gradner et al., 1962; Ravel and Shorey, 1971).
Binding of tRNALys to small ribosomal subunits
A portion of wild-type or mutant tRNALys was labeled at the 5′ end with 32P and mixed with the non-labeled respective counterpart. Escherichia coli small ribosomal subunits were prepared and activated according to Ericson et al. (1995). The non-enzymatic binding reaction was performed at 0°C for 1 h in 20 µl of a mixture consisting of 50 mM Tris–HCl pH 7.5, 30 mM MgCl2, 60 mM KCl, 1 mM DTT, 2 mM SP, 4 pmol 30S subunits, 0.1 µg/µl poly(A) (Sigma) and four different amounts (0.5, 1, 1.5 or 2 pmol) of either the wild-type or mutant tRNALys. The amount of bound tRNA was determined according to the method of Ashraf et al. (1999) with a slight modification.
Acknowledgments
Acknowledgements
We would like to express our thanks to Dr L.L.Spremulli (University of North Carolina) for kindly providing an EF-Tumt expression vector, to Dr J.-I.Hayashi (Tsukuba University) for giving us an original mutant cybrid cell line of the ME1-4 clone, and to Dr K.Mihara (Kyushu University) for kindly allowing us to use anti-TOM 40 antibody. We are also grateful to Dr P.F.Agris and Ms C.Yarian (North Carolina State University) for valuable discussions, to Dr T.Kanamori (Nippon Medical School) for providing anti-DLST antibody, and to Mr T.Hanada and Mr T.Suzuki (Tokyo University) for their technical assistance and helpful advice. This work was supported by a Grant-in-Aid for Scientific Research on Priority Areas from the Ministry of Education, Culture, Sports, Science and Technology (Japan).
References
- Anderson S. et al. (1981) Sequence and organization of the human mitochondrial genome. Nature, 290, 457–465. [DOI] [PubMed] [Google Scholar]
- Ashraf S.S., Sochacka,E., Cain,R., Guenther,R., Malkiewicz,A. and Agris,P.F. (1999) Single atom modification (O→S) of tRNA confers ribosome binding. RNA, 5, 188–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Börner G.V. et al. (2000) Decreased aminoacylation of mutant tRNAs in MELAS but not in MERRF patients. Hum. Mol. Genet., 9, 467–475. [DOI] [PubMed] [Google Scholar]
- Boulet L., Karpati,G. and Shoubridge,E.A. (1992) Distribution and threshold expression of the tRNALys mutation in skeletal muscle of patients with myoclonic epilepsy and ragged-red fibers (MERRF). Am. J. Hum. Genet., 51, 1187–1200. [PMC free article] [PubMed] [Google Scholar]
- Chomyn A., Meola,G., Bresolin,N., Lai,S.T., Scarlato,G. and Attardi,G. (1991) In vitro genetic transfer of protein synthesis and respiration defects to mitochondrial DNA-less cells with myopathy-patient mitochondria. Mol. Cell. Biol., 11, 2236–2244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung H.K. and Spremulli,L.L. (1990) Purification and characterization of elongation factor G from bovine liver mitochondria. J. Biol. Chem., 265, 21000–21004. [PubMed] [Google Scholar]
- Eberly S.L., Locklear,V. and Spremulli,L.L. (1985) Bovine mito chondrial ribosomes. Elongation factor specificity. J. Biol. Chem., 260, 8721–8725. [PubMed] [Google Scholar]
- Enriquez J.A., Chomyn,A. and Attardi,G. (1995) MtDNA mutation in MERRF syndrome causes defective aminoacylation of tRNALys and premature translation termination. Nature Genet., 10, 47–55. [DOI] [PubMed] [Google Scholar]
- Ericson G., Minchew,P. and Wollenzien,P. (1995) Structural changes in base-paired region 28 in 16S rRNA close to the decoding region of the 30S ribosomal subunit are correlated to changes in tRNA binding. J. Mol. Biol, 250, 407–419. [DOI] [PubMed] [Google Scholar]
- Fukuhara N., Tokiguchi,S., Shirakawa,K. and Tsubaki,T. (1980) Myoclonus epilepsy associated with ragged-red fibres (mitochondrial abnormalities): disease entity or a syndrome? Light- and electron-microscopic studies of two cases and review of literature. J. Neurol. Sci., 47, 117–133. [DOI] [PubMed] [Google Scholar]
- Goto Y., Nonaka,I. and Horai,S. (1990) A mutation in the tRNALeu(UUR) gene associated with the MELAS subgroup of mitochondrial encephalomyopathies. Nature, 348, 651–653. [DOI] [PubMed] [Google Scholar]
- Goto Y., Nonaka,I. and Horai,S. (1991) A new mtDNA mutation associated with mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes (MELAS). Biochim. Biophys. Acta, 1097, 238–240. [DOI] [PubMed] [Google Scholar]
- Gradner R.S., Wahba,A.J., Basilio,C., Miller,R.S., Lengyel,P. and Speyer,J.F. (1962) Synthetic polynucleotides and the amino acid code, VII. Proc. Natl Acad. Sci. USA, 48, 2087–2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagervall T.G., Pomerantz,S.C. and McCloskey,J.A. (1998) Reduced misreading of asparagine codons by Escherichia coli tRNALys with hypomodified derivatives of 5-methylaminomethyl-2-thiouridine in the wobble position. J. Mol. Biol., 284, 33–42. [DOI] [PubMed] [Google Scholar]
- Hayashi J., Tanaka,M., Sato,W., Ozawa,T., Yonekawa,H., Kagawa,Y. and Ohta,S. (1990) Effects of ethidium bromide treatment of mouse cells on expression and assembly of nuclear-coded subunits of complexes involved in the oxidative phosphorylation. Biochem. Biophys. Res. Commun., 167, 216–221. [DOI] [PubMed] [Google Scholar]
- Hayashi J., Ohta,S., Kikuchi,A., Takemitsu,M., Goto,Y. and Nonaka,I. (1991) Introduction of disease-related mitochondrial DNA deletions into HeLa cells lacking mitochondrial DNA results in mitochondrial dysfunction. Proc. Natl Acad. Sci. USA, 88, 10614–10618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi J., Ohta,S., Takai,D., Miyabayashi,S., Sakuta,R., Goto,Y. and Nonaka,I. (1993) Accumulation of mtDNA with a mutation at position 3271 in tRNALeu(UUR) gene introduced from a MELAS patient to HeLa cells lacking mtDNA results in progressive inhibition of mitochondrial respiratory function. Biochem. Biophys. Res. Commun., 197, 1049–1055. [DOI] [PubMed] [Google Scholar]
- Helm M., Brule,H., Degoul,F., Cepanec,C., Leroux,J.P., Giegé,R. and Florentz,C. (1998) The presence of modified nucleotides is required for cloverleaf folding of a human mitochondrial tRNA. Nucleic Acids Res., 26, 1636–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inagaki Y., Kojima,A., Bessho,Y., Hori,H., Ohama,T. and Osawa,S. (1995) Translation of synonymous codons in family boxes by Mycoplasma capricolum tRNAs with unmodified uridine or adenosine at the first anticodon position. J. Mol. Biol., 251, 486–492. [DOI] [PubMed] [Google Scholar]
- Käser M. and Langer,T. (2000) Protein degradation in mitochondria. Semin. Cell Dev. Biol., 11, 181–190. [DOI] [PubMed] [Google Scholar]
- King M.P. and Attardi,G. (1989) Human cells lacking mtDNA: repopulation with exogenous mitochondria by complementation. Science, 246, 500–503. [DOI] [PubMed] [Google Scholar]
- Kobayashi Y., Momoi,M.Y., Tominaga,K., Momoi,T., Nihei,K., Yanagisawa,M., Kagawa,Y. and Ohta,S. (1990) A point mutation in the mitochondrial tRNALeu(UUR) gene in MELAS (mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes). Biochem. Biophys. Res. Commun., 173, 816–822. [DOI] [PubMed] [Google Scholar]
- Krüger M.K., Pedersen,S., Hagervall,T.G. and Sørensen,M.A. (1998) The modification of the wobble base of tRNAGlu modulates the translation rate of glutamic acid codons in vivo. J. Mol. Biol., 284, 621–631. [DOI] [PubMed] [Google Scholar]
- Kumazawa Y., Schwartzbach,C.J., Liao,H.X., Mizumoto,K., Kaziro,Y., Miura,K., Watanabe,K. and Spremulli,L.L. (1991) Interactions of bovine mitochondrial phenylalanyl-tRNA with ribosomes and elongation factors from mitochondria and bacteria. Biochim. Biophys. Acta, 1090, 167–172. [DOI] [PubMed] [Google Scholar]
- Matthews D.E., Hessler,R.A., Denslow,N.D., Edwards,J.S. and O’Brien,T.W. (1982) Protein composition of the bovine mitochondrial ribosome. J. Biol. Chem., 257, 8788–8794. [PubMed] [Google Scholar]
- Nakano K. et al. (1993) Human dihydrolipoamide succinyltransferase: cDNA cloning and localization on chromosome 14q24.2–q24.3. Biochim. Biophys. Acta, 1216, 360–368. [DOI] [PubMed] [Google Scholar]
- Nijtmans L.G. et al. (1995) Altered kinetics of cytochrome c oxidase in a patient with severe mitochondrial encephalomyopathy. Biochim. Biophys. Acta, 1270, 193–201. [DOI] [PubMed] [Google Scholar]
- Nomura N. et al. (1994) Prediction of the coding sequences of unidentified human genes. I. The coding sequences of 40 new genes (KIAA0001-KIAA0040) deduced by analysis of randomly sampled cDNA clones from human immature myeloid cell line KG-1. DNA Res., 1, 27–35. [DOI] [PubMed] [Google Scholar]
- Ogle J.M., Brodersen,D.E., Clemons,W.M., Jr, Tarry,M.J., Carter,A.P. and Ramakrishnan,V. (2001) Recognition of cognate transfer RNA by the 30S ribosomal subunit. Science, 292, 897–902. [DOI] [PubMed] [Google Scholar]
- Pfanner N. and Geissler,A. (2001) Versatility of the mitochondrial protein import machinery. Nature Rev. Mol. Cell. Biol., 2, 339–349. [DOI] [PubMed] [Google Scholar]
- Pingoud A., Urbanke,C., Krauss,G., Peters,F. and Maass,G. (1977) Ternary complex formation between elongation factor Tu, GTP and aminoacyl-tRNA: an equilibrium study. Eur. J. Biochem., 78, 403–409. [DOI] [PubMed] [Google Scholar]
- Ravel J.M. and Shorey,R.L.A. (1971) GTP-dependent binding of aminoacyl-tRNA to Escherichia coli ribosomes. Methods Enzymol., 20, 306–316. [Google Scholar]
- Samuelsson T., Axberg,T., Boren,T. and Lagerkvist,U. (1983) Unconventional reading of the glycine codons. J. Biol. Chem., 258, 13178–13184. [PubMed] [Google Scholar]
- Schon E.A., Bonilla,E. and DiMauro,S. (1997) Mitochondrial DNA mutations and pathogenesis. J. Bioenerg. Biomembr., 29, 131–149. [DOI] [PubMed] [Google Scholar]
- Schwartzbach C.J. and Spremulli,L.L. (1989) Bovine mitochondrial protein synthesis elongation factors. Identification and initial characterization of an elongation factor Tu–elongation factor Ts complex. J. Biol. Chem., 264, 19125–19131. [PubMed] [Google Scholar]
- Schwartzbach C.J., Farwell,M., Liao,H.X. and Spremulli,L.L. (1996) Bovine mitochondrial initiation and elongation factors. Methods Enzymol., 264, 248–261. [DOI] [PubMed] [Google Scholar]
- Shoffner J.M., Lott,M.T., Lezza,A.M., Seibel,P., Ballinger,S.W. and Wallace,D.C. (1990) Myoclonic epilepsy and ragged-red fiber disease (MERRF) is associated with a mitochondrial DNA tRNALys mutation. Cell, 61, 931–937. [DOI] [PubMed] [Google Scholar]
- Sprinzl M., Horn,C., Brown,M., Ioudovitch,A. and Steinberg,S. (1998) Compilation of tRNA sequences and sequences of tRNA genes. Nucleic Acids Res., 26, 148–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takai K., Takaku,H. and Yokoyama,S. (1996) Codon-reading specificity of an unmodified form of Escherichia coli tRNASer1 in cell-free protein synthesis. Nucleic Acids Res., 24, 2894–2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takemoto C., Koike,T., Yokogawa,T., Benkowski,L., Spremulli,L.L., Ueda,T., Nishikawa,K. and Watanabe,K. (1995) The ability of bovine mitochondrial transfer RNAMet to decode AUG and AUA codons. Biochimie, 77, 104–108. [DOI] [PubMed] [Google Scholar]
- Towbin H., Staehelin,T. and Gordon,J. (1979) Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc. Natl Acad. Sci. USA, 76, 4350–4354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varshney U., Lee,C.P. and RajBhandary,U.L. (1991) Direct analysis of aminoacylation levels of tRNAs in vivo. Application to studying recognition of Escherichia coli initiator tRNA mutants by glutaminyl-tRNA synthetase. J. Biol. Chem., 266, 24712–24718. [PubMed] [Google Scholar]
- von Ahsen U., Green,R., Schroeder,R. and Noller,H.F. (1997) Identification of 2′-hydroxyl groups required for interaction of a tRNA anticodon stem–loop region with the ribosome. RNA, 3, 49–56. [PMC free article] [PubMed] [Google Scholar]
- Wakita K., Watanabe,Y., Yokogawa,T., Kumazawa,Y., Nakamura,S., Ueda,T., Watanabe,K. and Nishikawa,K. (1994) Higher-order structure of bovine mitochondrial tRNAPhe lacking the ‘conserved’ GG and TΨCG sequences as inferred by enzymatic and chemical probing. Nucleic Acids Res., 22, 347–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe K. and Osawa,S. (1995) tRNA sequences and variations in the genetic code. In Söll,D. and Rajbandary,U.L. (eds), tRNA: Structure, Biosynthesis and Function. ASM Press, Washington, DC, pp. 225–250.
- Woriax V.L., Burkhart,W. and Spremulli,L.L. (1995) Cloning, sequence analysis and expression of mammalian mitochondrial protein synthesis elongation factor Tu. Biochim. Biophys. Acta, 1264, 347–356. [DOI] [PubMed] [Google Scholar]
- Yarian C., Marszalek,M., Sochacka,E., Malkiewicz,A., Guenther,R., Miskiewicz,A. and Agris,P.F. (2000) Modified nucleoside dependent Watson–Crick and wobble codon binding by tRNALysUUU species. Biochemistry, 39, 13390–13395. [DOI] [PubMed] [Google Scholar]
- Yasukawa T., Suzuki,T., Ishii,N., Ueda,T., Ohta,S. and Watanabe,K. (2000a) Defect in modification at the anticodon wobble nucleotide of mitochondrial tRNALys with the MERRF encephalomyopathy pathogenic mutation. FEBS Lett., 467, 175–178. [DOI] [PubMed] [Google Scholar]
- Yasukawa T., Suzuki,T., Suzuki,T., Ueda,T., Ohta,S. and Watanabe,K. (2000b) Modification defect at anticodon wobble nucleotide of mitochondrial tRNAsLeu(UUR) with pathogenic mutations of mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes. J. Biol. Chem., 275, 4251–4257. [DOI] [PubMed] [Google Scholar]
- Yokoyama S. and Nishimura,S. (1995) Modified nucleotides and codon recognition. In Söll,D. and RajBhandary,U.L. (eds), tRNA: Structure, Biosynthesis and Function. ASM Press, Washington, DC, pp. 207–224.
- Yoneda M., Miyatake,T. and Attardi,G. (1994) Complementation of mutant and wild-type human mitochondrial DNAs coexisting since the mutation event and lack of complementation of DNAs introduced separately into a cell within distinct organelles. Mol. Cell. Biol., 14, 2699–2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yusupov M.M., Yusupova,G.Z., Baucom,A., Lieberman,K., Earnest,T.N., Cate,J.H. and Noller,H.F. (2001) Crystal structure of the ribosome at 5.5 Å resolution. Science, 292, 883–896. [DOI] [PubMed] [Google Scholar]